

Abstract
PURPOSE: Foxhead box M1 (FOXM1) expression has been shown to be linked with human papillomavirus (HPV) 16/18–infected cervical cancer. However, the mechanism underlying the induction of FOXM1 in HPV 16/18–infected cancers remains elusive. EXPERIMENTAL DESIGN: The mechanistic actions of FOXM1 induced by the E6/NKX2-1 axis in tumor aggressiveness were elucidated in cellular and animal models. The prognostic value of FOXM1 for overall survival (OS) and relapse-free survival (RFS) in HPV-positive oral and lung cancers was assessed using Kaplan-Meier and Cox regression models. RESULTS: Herein, FOXM1 expression is upregulated by E6-mediated NKX2-1 in HPV-positive cervical, oral, and lung cancer cells. Induction of FOXM1 by E6 through the MZF1/NKX2-1 axis is responsible for HPV-mediated soft agar growth, invasiveness, and stemness through activating Wnt/β-catenin signaling pathway. In a nude mice model, metastatic lung tumor nodules in HPV 18 E6-positive GNM or HPV 16 E6-positive TL-1–injected nude mice were markedly decreased in both cell types with E6 knockdown, FOXM1 knockdown, or treatment with FOXM1 inhibitor (thiostrepton). Among the four subgroup patients, the worst FOXM1 prognostic value for OS and RFS was observed in HPV 16/18–positive patients with tumors with high-expressing FOXM1. CONCLUSIONS: Induction of FOXM1 by E6 oncoprotein through the MZF1/NKX2-1 axis may be responsible for HPV 16/18–mediated tumor progression and poor outcomes in HPV-positive patients.
Introduction
The high cancer risk of human papillomavirus (HPV) infection has been recognized as being associated with genital-related cervical, vulvar, and anal carcinomas [1], [2] and non–genital-associated head and neck squamous cell carcinomas (HNSCCs) [3], [4]. However, the involvement of HPV 16/18 infections in oral cavity squamous cell carcinoma (OCSCC) and non–small cell lung cancer (NSCLC) remains controversial [3], [4], [5], [6], [7], [8]. The E6 and E7 proteins in high-risk HPV act as primary transforming viral proteins to inactivate the p53 and Rb pathways that result in cell proliferation and resistance to apoptosis [9]. This leads to the accumulation of DNA damage and mutations that give rise to cell transformation and carcinoma development [9]. For example, the positive correlation of HPV 16/18 infection with the risk of epidermal growth factor receptor mutation occurrence has been shown in Taiwanese NSCLC patients [10]. Further insights into the mechanistic action of the E6 and E7 oncoproteins on tumor progression should be investigated.
The overexpression of Foxhead box M1 (FOXM1) is correlated with tumor progression and poor prognosis in various human carcinomas, including HNSCC [11], [12] and NSCLC [13], [14]. FOXM1 regulates cell cycle–related gene expression, such as cyclin B1, cyclin D1, and cdc25 expression, so as to promote cervical tumor progression [15]. However, the mechanism by which FOXM1 participates in HPV-mediated tumor progression is not fully understood. FOXM1 is upregulated by E2F1, which is released by Rb phosphorylation through p53 inactivation [16], [17]. FOXM1 interacts with HPV 16 E7 to promote the transformation of primary rat embryo fibroblasts [18]. Here, we provide evidence that E7 does not affect FOXM1 expression in HPV-positive cervical, oral, and lung cancer cells. Intriguingly, FOXM1 expression is upregulated by E6 through E6-mediated NKX2-1 expression. Consequently, FOXM1 induced by the E6/NKX2-1 axis is responsible for HPV-mediated tumor progression and metastasis through increased β-catenin nuclear translocation.
Materials and Methods
Study Subjects
OCSCC tumor specimens were collected from 110 patients with primary oral cancers in the Department of Otolaryngology, Chung Shan Medical University Hospital (Taichung, Taiwan) and the Department of Surgical Pathology, Changhua Christian Hospital (Changhua, Taiwan). Lung tumors were enrolled from 117 NSCLC patients who were treated with surgical resection at the Division of Thoracic Surgery, Taichung Veterans General Hospital (Taichung, Taiwan) between 1993 and 2004. This study is approved by the Institutional Review Board, Taipei Medical University Hospital (TMUH No. 201301051). The tumor type and stage of each collected specimen were histologically determined according to the World Health Organization classification system. Cancer relapse data were obtained by chart review and further confirmed by two clinical physicians.
Immunohistochemistry
Formalin-fixed and paraffin-embedded specimens were sectioned at a thickness of 3 μm. The detailed procedures were described previously [10]. For antigen detection, sections were heated in a microwave oven twice for 5 minutes in citrate buffer (pH 6.0) and then incubated with a monoclonal anti–NKX2-1 (SC-53136; at a dilution of 1:100) and FOXM1 antibody (GTX-100276; GeneTex, Irvine, CA; at a dilution of 1:100) for 60 minutes at 25°C.
Cell Lines and Culture Conditions
SiHa, C33A, H1299, and A549 cells were obtained from the American Type Culture Collection (Manassas, VA). The OECM-1 cells were kindly provided by Dr T. C. Lee (Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan). The GNM cells were kindly provided by Dr M. Y. Chou (Department of Dentistry, Chung Shan Medical University, Taichung, Taiwan). The TL-1 cells were established from patients’ plural effusions as described previously [7]. The SiHa, GNM, H1299, and A549 cells were maintained in Dulbecco’s modified Eagle’s medium. The TL-1, OECM, and C33A cells were maintained in RPMI 1640 containing 10% FBS supplemented with penicillin (100 U/ml) and streptomycin (100 mg/ml). Cells were grown in a 37°C humidified incubator with 5% CO2.
Western Blot Analysis
Cells were washed twice on ice with phosphate-buffered saline (PBS) before adding protein lysis buffer [1 × protease inhibitor cocktail (Roche, Basel, Switzerland), 1.5 mM EDTA, 1 mM DTT, 10% glycerol, 25 mM Hepes, pH 7.6]. The protein concentration was determined by the Bradford assay (BioRad, Hercules, CA) using BSA as a standard. Total protein (20 μg) was resolved by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) for subsequent Western blot analysis using antibodies against the following proteins [diluted in Tween–Tris-buffered saline: 0.02% Tween-20 in 100 mM Tris-Cl (pH 7.5), 1:1000 as indicated]: monoclonal anti-p53 (DO7; Dako, Carpinteria, CA), anti–HPV 16 E6 (sc-1586; Santa Cruz Biotechnology, Dallas, TX), anti–HPV 18 E6 (sc-1584; Santa Cruz Biotechnology), anti–HPV 16 E7 (sc-6981; Santa Cruz Biotechnology), anti–HPV 18 E7 (GTX40864; Gene Tex), anti-SP1 (sc-14027; Santa Cruz Biotechnology), anti–α-tubulin (sc-5286; Santa Cruz Biotechnology), anti-Rb (sc-102; Santa Cruz Biotechnology), anti–phosphorylated Rb (pRb; sc-12901; Santa Cruz Biotechnology), anti–β-catenin (sc-7199; Santa Cruz Biotechnology), anti–c-Myc (sc-40; Santa Cruz Biotechnology), anti-vimentin (sc-6260; Santa Cruz Biotechnology), anti-Snail (sc28199; Santa Cruz Biotechnology), anti-Nanog (sc-3769; Santa Cruz Biotechnology), anti-E2F1 (GTX11837; GeneTex), anti-Oct4 (100622; GeneTex), anti-FOXM1 (GTX100276; GeneTex), anti–NKX2-1(699P; Thermo Scientific, Pittsburgh, PA), anti–E-cadherin (80098; BD Biosciences, San Jose, CA), and MMP2 (Lot 804P703C; NeoMarkers, Woburn, MA). The gel was transferred to a Hybond-C Extra membrane (GE Healthcare Life Science, Piscataway, NJ) and immunoblotted with primary antibody, as indicated in the figure legends. Anti-mouse or rabbit IgG conjugated to HRP was used as the secondary antibody for detection using an ECL Western Blot Detection System.
Plasmid Construction
The NKX2-1 cDNAs were cloned into pcDNA3.1 Zeo(+) (Invitrogen, Carlsbad, CA). The FOXM1-Luc and NKX2-1-Luc plasmids were constructed by inserting KpnI/XhoI fragments into a KpnI/XhoI-treated pGL3 vector (Promega Corp, Madison, WI). The small hairpin NKX2-1 (shNKX2-1; TRCN0000275519), shMZF1 (TRCN0000017137), shLOX (TRCN0000045991), shFOXM1 (TRCN0000273982), shNEDD4 (TRCN0000004967), shβ-catenin (TRCN0000314991), shCaveolin1 (TRCN0000007999), and pLKO.1 vector plasmids were purchased from the National RNAi Core Facility, Academia Sinica. HPV 16 E6 and HPV 18 E6 cDNAs and HPV 16 E6 and HPV 18 E6 shRNAs were as previously described [19], [20]. HPV 16 E7 and HPV 18 E7 shRNA template were constructed using two complementary oligos that, when partially annealed, create a loop region with a sequence complementarity to HPV 16 E7 and HPV 18 E7 mRNA. The oligos contained 19 to 20 nucleotides of the HPV 16 E7 and HPV 18 E7 sequence, as follows: HPV 16 E7 shRNA forward, 5′-gatcaggaggatgaaatagatggtttcaagagaa TTTCAAGAGAA-3′; HPV 16 E7 shRNA reverse, 5′-agctaaaaaaggaggatgaaatagatggttctcttgaaa-3′; HPV 18 E7 shRNA forward, 5′-gatcgtgttgtaagtgtgaagcc ttcaagagag-3′; HPV 18 E7 shRNA reverse, 5′-agctaaaaa -gtgttgtaagtgtgaagcctctcttgaag-3′.The shRNA template was cloned into the pcDNA-HU6 vector as previously described [21]. The various concentrations of expression plasmids, as indicated, were transiently transfected into the lung cancer cells (1 × 106) using the Turbofect reagent (Fermentas, Pittsburgh, PA). After 48 hours, the cells were harvested, and the whole-cell extracts were assayed in subsequent experiments.
Soft Agar Colony Formation Assay
For the soft agar colony formation assay, the bottom gel consisted of 0.4% low-melting agarose (Cambrex, East Rutherford, NJ) in RPMI 1640 containing 10% FBS. When the bottom layer of agarose was solidified, 5 × 103 cells were resuspended in 2 × RPMI 1640 containing 20% FBS, mixed with 0.6% agarose, and then plated above this layer. Colonies larger than 100 μm in diameter were quantified by microscope after 12 days of culture.
Invasion Assay
A Boyden chamber with a pore size of 8 μm was used for the in vitro cell invasion assay. Cells (1 × 104) in 0.5% serum containing culture medium (HyClone, Ogden, UT) were plated in the upper chamber and 10% FBS was added to culture medium in the lower chamber as a chemoattractant. The upper side of the filter was covered with 0.2% Matrigel (Collaborative Research, Boston, MA) diluted in RPMI 1640. After 12 hours, cells on the upper side of the filter were removed and cells that adhered to the underside of the membrane were fixed in methanol and stained with 10% Giemsa dye. The number of invasive cells was counted. Ten contiguous fields of each sample were examined to obtain a representative number of cells that invaded across the membrane.
Sphere Assay
Single cells were suspended in Matrigel/serum-free RPMI 1640 (1:1) at a concentration of 104 cells per well in a total volume of 100 μl, in triplicate. Cells were further allowed to grow for 15 days, and the number of spheres was counted by a microscope.
Xenograft Tumors in Nude Mice
Female immunodeficient nude mice (BALB/c nu/nu mice) that were 5 weeks old and weighed 18 to 22 g were injected with PBS and the stable clones of TL-1/NC, TL-1/shE6, TL-1/shE6+FOXM1, TL-1/shFOXM1, GNM/NC, GNM/shE6, GNM/shE6+FOXM1, and GNM/shFOXM1 through the tail vein (106 cells in 0.1 ml of PBS). After 42 days, the mice were sacrificed, and their lungs were removed and fixed in 10% formalin. The number of lung tumor metastasis was counted under a dissecting microscope.
Statistical Analysis
The Student’s t test and Chi-square test were applied for continuous or discrete data analysis. This analysis was performed using SPSS software (version 13.0; SPSS Inc, Chicago, IL). For survival data, statistical differences were analyzed using the log-rank test. Survival curves were plotted using the Kaplan-Meier method, and the variables related to survival were analyzed using Cox’s proportional hazards regression model with SPSS software.
Results
FOXM1 Expression Is Regulated by E6, Not by E7, in HPV-Positive Cancer Cells
Western blot analysis showed that HPV 16 E6 and E7 were expressed in HPV 16–positive SiHa cervical and TL-1 lung cancer cells, as well as in HPV 18–positive GNM oral cancer cells (Figure 1A, upper panel). As expected, HPV 16/18 E6 and E7 were not detected in HPV 16/18–negative C33A cervical, H1299, A549 lung cancer, and OECM-1 oral cancer cells. MMP2, a downstream gene of FOXM1 expressed highly in HPV 16/18–positive SiHa, GNM, and TL-1 cells, has a low level of expression in HPV 16/18–negative C33A, A549, and OECM-1 cells (Figure 1A, upper panel). FOXM1 protein expression was consistent with its mRNA expression in these cells (Figure 1A, lower panel). These results suggest that E6 or E7 may contribute to increased FOXM1 expression at the transcription level. To explore whether E6 or E7 could regulate FOXM1 expression, E6 or E7 shRNAs were transfected into HPV-positive SiHa, GNM, and TL-1 cells. As expected, p53, Rb, and pRb were restored by E6 or E7 knockdown in these three cells when compared to parent cells transfecting with non-specific shRNA control (NC; Figure 1B, upper panel). Interestingly, FOXM1 expression was diminished by E6 knockdown to almost the degree seen in NC cells, but FOXM1 mRNA and protein expression were only slightly decreased by E7 knockdown in these three cell types (Figure 1B, upper and lower panels). E2F1 expression was unchanged by E6 or E7 knockdown. The interaction of E2F1 with Rb, when evaluated by immunoprecipitation, was expectedly increased by E6 or E7 knockdown and was especially increased by E6 plus E7 knockdown (Figure 1B, middle panel). These results suggest that FOXM1 expression may be upregulated by E6 at the transcriptional level, not by E7, in HPV-positive cancer cells.
Figure 1.
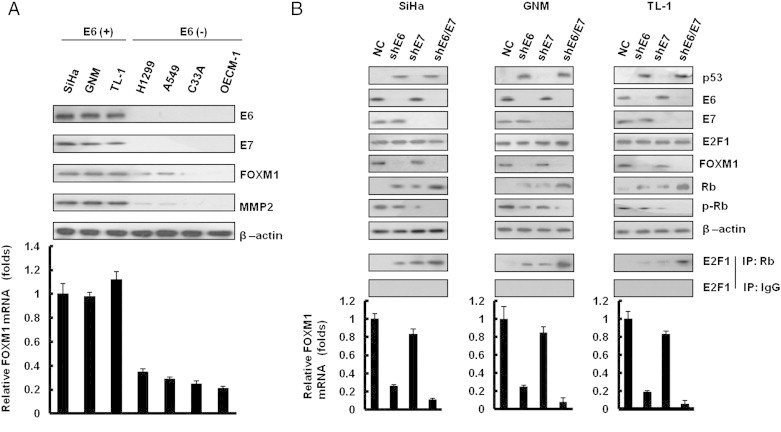
FOXM1 is upregulated by E6, not by E7, in HPV-infected cancer cells. (A) E6, E7, FOXM1, and MMP2 protein expressions were evaluated by Western blot analysis in a panel of cancer cells with or without HPV infection. (B) E6 and E7 expressions were depleted by shRNA in SiHa, GNM, and TL-1 cells. p53, E6, E7, E2F1, Rb, pRb, and FOXM1 were detected by immunoblot analysis with antibodies. These lysates were immunoprecipitated with anti–Rb-conjugated beads. The immunoprecipitates were analyzed through SDS-PAGE, followed by immunoblot analysis with anti-E2F1 antibody. Quantitative real-time PCR amplification of FOXM1 was performed using total cellular RNA extracts. β-Actin was used as a loading control for the whole-cell lysates. Values are given as means ± SEMs for triplicate samples.
FOXM1 Transcription Is Predominantly Upregulated by E6-Mediated NKX2-1
Software analysis (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3) predicted that the three putative binding sites for NKX2-1 (− 1342/− 923) were located upstream of the E2F1 binding sites on the FOXM1 promoter region (− 1436/+ 1; Figure 2A, upper panel). The possibility that NKX2-1 could play a more important role than E2F1 in FOXM1 transcription was assessed by constructing three FOXM1 promoters (P1: − 1436/+ 1, P2: − 766/+ 1, and P3: −1436/− 766; Figure 2A, lower panel). The luciferase reporter activity assays showed that compared to P1, ~ 80% of the P3 promoter activity was seen in E6-positive cells (TL-1, SiHa, and GNM), but this activity was not observed in E6-negative cells (A549, C33A, and OECM-1; Figure 2B). P3 promoter activity was significantly decreased by E6 knockdown in three E6-positive cell types (Figures 2, C and D, and 3E, lower panel), but it was markedly increased by E6 overexpression in three E6-negative cell types. Western blot analysis showed that NKX2-1 expression was reduced markedly in E6-knockdown SiHa, GNM, and TL-1 cells but elevated in E6-overexpressing C33A, OECM-1, and A549 cells (Figures 2, C and D, and 3E, upper panel). Chromatin immunoprecipitation (ChIP) analysis indicated that NKX2-1 was bound to its putative binding sites on the FOXM1 promoter in NKX2-1–overexpressing SiHa, GNM, and TL-1 cells transfected with shE6 and in NKX2-1–knockdown C33A, OECM-1, and A549 cells transfected with E6 (Figures 2, C and D, and 3E, middle panel). The binding activity of NKX2-1 on the putative binding sites of the FOXM1 promoter was increased or decreased in the respective cell types in a dose-dependent manner (Figures 2, C and D, and 3E, middle panel). These results clearly indicate that E6-mediated NKX2-1 plays a more important role than E2F1 does in the control of FOXM1 transcription in HPV-positive cancer cells.
Figure 2.
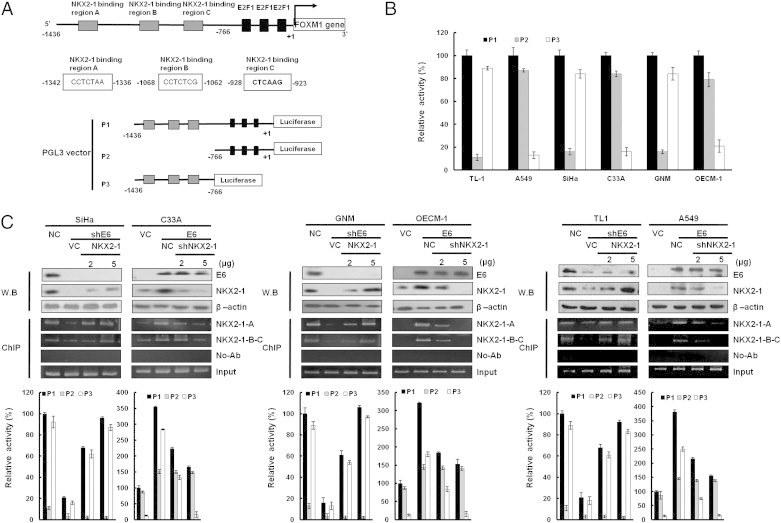
Up-regulation of FOXM1 transcription by E6 is through increased NKX2-1 binding to the FOXM1 promoter. (A) Schematic diagram of FOXM1 promoter–driven luciferase reporters: FOXM1 (− 1436/+ 1)-Luc, FOXM1 (− 766/+ 1)-Luc, and FOXM1 (− 1436/-766)-Luc. NKX2-1-A, NKX2-1-B, and NKX2-1-C possess putative NKX2-1 binding sites. (B) The three promoter constructs were co-transfected into SiHa, C33A, GNM, OECM-1, TL-1, and A549 cells for luciferase reporter assay after 48 hours. Luciferase activity was measured at 48-hour post-transfection. In all experiments, the relative luciferase activity was shown as the fold activation relative to that of the control cells. (C) SiHa, GNM, and TL-1 cells were transfected with various doses of E6-knockdown plasmid as indicated. C33A, OECM-1, and A549 cells were transfected with various doses of E6-overexpressing plasmid. The total amount of transfected DNA was kept constant by adding a control vector. The luciferase activity was measured at 48-hour post-transfection. In all experiments, the relative luciferase activity was shown as the fold activation relative to that of the control cells. SiHa, GNM, and TL-1 cells were transfected with various doses of E6-knockdown plasmid as indicated. C33A, OECM-1, and A549 cells were transfected with various doses of E6-overexpressing plasmid. In addition, SiHa, GNM, and TL-1 cells were co-transfected with NKX2-1-overexpressing (5 μg) and E6-knockdown (5 μg) plasmids. C33A, OECM-1, and A549 cells were co-transfected with shNKX2-1-knockdown (5 μg) and E6-overexpressing (5 μg) plasmids. These lysates were immunoprecipitated with anti–NKX2-1-conjugated beads. The immunoprecipitates were analyzed through SDS-PAGE, followed by immunoblot analysis with anti–NKX2-1 antibody. The input control was 30% of the cell extract without any treatment. The binding activity of NKX2-1 onto the FOXM1 promoter was evaluated through chromatin immunoprecipitation. The chromatin was isolated and immunoprecipitated with an antibody specific for NKX2-1.
Figure 3.
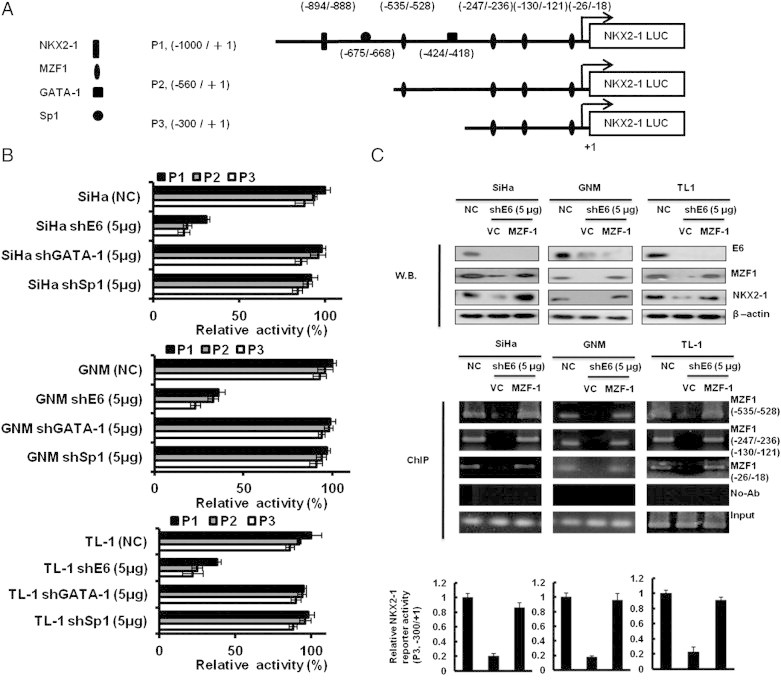
MZF1 is responsible for E6-mediated NKX2-1 transcription. (A) Schematic representation of the 5′ flanking region of NKX2-1 promoter with putative NKX2-1, GATA-1, SP1, and MZF1 binding sites. + 1, the transcription start site. The NKX2-1 (P1, − 1000 to + 1), NKX2-1 (P2, − 560 to + 1), and NKX2-1 (P3, − 300 to + 1) luciferase reporter constructs. (B) The NKX2-1 (P1, − 1000 to + 1), NKX2-1 (P2, − 560 to + 1), and NKX2-1 (P3, − 300 to + 1) luciferase reporter plasmids and shE6 (5 μg) plasmid were co-transfected into SiHa, GNM, and TL-1 cells. (C) SiHa, GNM, and TL-1 cells were transfected with shE6 (5 μg) and MZF-1 (5 μg) cDNA plasmids as indicated. The expression levels of E6, MZF1, and NKX2-1 were evaluated by Western blot analysis, and β-actin was served as protein loading control. The binding activity of MZF-1 onto the NKX2-1 promoter was evaluated by ChIP in SiHa, GNM, and TL-1 cells that were transfected with shE6 (5 μg) and MZF-1 (5 μg) cDNA plasmids as indicated. Chromatin was isolated and then immunoprecipitated with MZF-1–specific antibody. The level of NKX2-1 (P3, − 300 to + 1) luciferase reporter plasmids was evaluated.
MZF1 Is Responsible for E6-Mediated NKX2-1 Expression
We next explored the mechanistic connection between E6 and NKX2-1. We observed one NKX2-1, one SP1, one GATA-1, and four MZF1 putative binding sites in the NKX2-1 promoter region (− 1000/+ 1) by a software analysis (Figure 3A). Three NKX2-1 promoters (P1, P2, and P3) were constructed by deletion mutation to test which transcription factor could be more responsible for NKX2-1 transcription (Figure 3A). E6-positive SiHa, GNM, and TL-1 cells were collected to transfect with shE6 and then co-transfected with three NKX2-1 promoters for luciferase reporter assay. As shown in Figure 3B, the reporter activity of the NKX2-1 P3 promoter was similar with the NKX2-1 P1 and P2 promoter activities; however, the three promoter activities were almost reduced by E6 knockdown in these three E6-positive cells. As shown in Figure 3B, the reporter activity of these three NKX2-1 promoters did not change by GATA-1 or SP1 silencing. We thus suggest that GATA-1 and SP1 may play a minor role in NKX2-1 transcription. Therefore, GATA-1 or SP1 cannot increase NKX2-1 reporter activity. These results suggest that the three MZF1 putative binding sites located at the P3 promoter (− 300/+ 1) may be responsible for NKX2-1 transcription. Western blot showed that NKX2-1 and MZF1 were concomitantly reduced by E6 knockdown in SiHa, GNM, and TL-1 cells; however, NKX2-1 expression was restored by MZF1 overexpression in E6-knockdown SiHa, GNM, and TL-1 cells (Figure 3C, upper panel). ChIP analysis further indicated that MZF1 was indeed bound on the three MZF1 putative binding sites of the P3 promoter (− 300/+ 1) but not observed in E6-knockdown cells (Figure 3C, upper panel). The reporter activity of the P3 promoter was consistent with the observations of Western blot analysis and ChIP analysis (Figure 3C, lower panel). These results suggest that MZF1 may be responsible for E6-mediated NKX2-1 expression in HPV-positive cells.
Up-Regulation of FOXM1 by E6 through the MZF1/NKX2-1 Axis Is Responsible for HPV-Mediated Soft Agar Growth and Invasion
We explored the possibility that the up-regulation of FOXM1 by E6 through the MZF1/NKX2-1 could be responsible for HPV-mediated soft agar growth and invasion capability. As expected, FOXM1 expression was decreased by E6 knockdown in SiHa, GNM, and TL-1 cells and increased by E6 overexpression in OECM-1, C33A, and A549 cells (Figure 4, A–C, upper panel). In addition, MMP2 and E-cadherin expressions were concomitantly increased and decreased by E6-induced FOXM1 expression; however, both molecules were rescued by FOXM1 knockdown in E6-overexpressing C33A, OECM-1, and A549 cells. The representative soft growth on soft agar plate and invasive cell on Matrigel membrane are shown in Figure 4, A to C (middle panel). The soft agar growth and invasion capability were dose-dependently decreased and increased by E6 knockdown and E6 overexpression in these cells; however, both capabilities were nearly restored by FOXM1 knockdown in E6-overexpressing C33A, OECM1, and A549 cells (Figure 4, A–C, lower panel). These results clearly indicate that up-regulation of FOXM1 by E6 through the MZF1/NKX2-1 axis is responsible for HPV-mediated soft agar growth and invasiveness.
Figure 4.
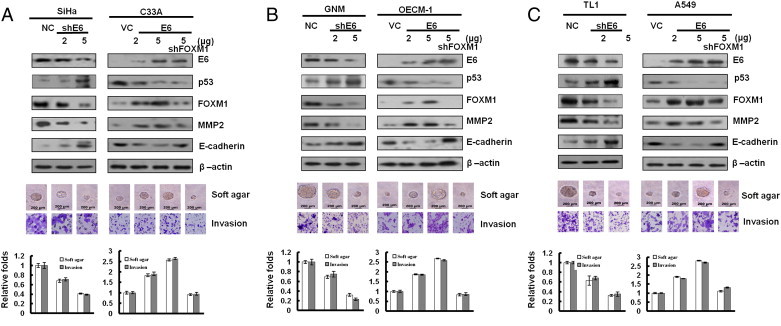
Induction of FOXM1 by E6 is responsible for colony formation and invasion capability. (A) E6 influenced FOXM1 expression in lung cancer cells. Upper: E6 of SiHa was transiently knocked down by using various doses (2 and 5 μg) of E6 shRNA for this experiment. The levels of the HPV 16 E6, p53, FOXM1, MMP2, E-cadherin, and β-actin proteins were evaluated through Western blot analysis. β-Actin was used as a protein loading control. E6 expression changes resulting from using various doses (2 and 5 μg) of E6-overexpressing plasmids were confirmed through Western blot analysis. In addition, C33A cells were co-transfected with shFOXM1-knockdown (5 μg) and E6-overexpressing (5 μg) plasmids. The cell lysates were analyzed through Western blot analysis. The representative soft agar colony sizes and invasion cell numbers are shown for cells. (B) GNM and OECM-1 cell lines. (C) TL-1 and A549 cell lines.
β-Catenin Activation Is Responsible for Invasiveness and Stemness Caused by the E6-Induced FOXM1 through the MZF1/NKX2-1 Axis
FOXM1 promotes β-catenin nuclear localization and controls Wnt target gene expression, stemness, and glioma tumorigenesis [22], [23], [24]. Therefore, activation of the Wnt/β-catenin signaling pathway is expected to be responsible for cell invasiveness and stemness mediated by E6-induced FOXM1 expression. Western blot analysis showed that nuclear β-catenin expression levels were elevated by ectopic FOXM1 expression in E6-knockdown GNM cells; however, nuclear β-catenin expression levels were reduced by FOXM1 silencing in E6-overexpressing OECM-1 cells (Figure 5A, middle panel). Wnt/β-catenin downstream gene—cyclin D1 and c-Myc—expressions were consistent with nuclear β-catenin expression in both cell types (Figure 5A, upper panel). The transcription factor 4 reporter activity (TOPFlash) was significantly elevated by FOXM1 overexpression in E6-knockdown GNM cells and reduced by FOXM1 knockdown in E6-overexpressing OECM-1 cells. However, the transcription factor 4 reporter activity (FOPFlash) was unchanged in both cell types with the same treatment (Figure 5A, lower panel). We examined whether an increase in nuclear β-catenin expression by E6-mediated FOXM1 could be responsible for cell invasiveness and stemness. As expected, cyclin D1 and c-Myc expressions were decreased by β-catenin silencing in E6-knockdown GNM cells with ectopic FOXM1 expression (Figure 5B, upper panel). A Boyden chamber assay showed that the invasiveness was markedly decreased by E6 knockdown; intriguingly, the invasiveness was nearly restored by ectopic FOXM1 expression in E6-knockdown GNM cells, as compared with VC cells (Figure 4B, upper right panel). Moreover, the increase in invasiveness caused by ectopic FOXM1 expression was rescued by β-catenin silencing in E6-knockdown GNM cells with ectopic FOXM1 expression. Interestingly, stemness-related c-Myc, Nanog, and Oct4 were elevated by ectopic FOXM1 expression in E6-knockdown GNM cells (Figure 5B, upper left panel). The representative invasiveness and sphere cells are shown in Figure 4B (lower panel). E6-mediated cell invasiveness due to increased FOXM1-mediated β-catenin nuclear translocation was observed in the increase of sphere formation efficacy in GNM cells (Figure 5B, upper right panel). Concomitantly, the Nanog, Oct4, and c-Myc proteins and their mRNA expressions were dose-dependently decreased by FOXM1 inhibitor (thiostrepton) in E6-overexpressing OECM-1 cells (Figure 5C). Similar observations to those seen in GNM oral cancer cells were also seen in TL-1 lung cancer cells (Figure S1). These results clearly indicate that the increased nuclear translocation of β-catenin due to E6-induced FOXM1 expression through the MZF1/NKX2-1 axis is responsible for invasiveness and stemness in HPV-positive oral and lung cancer cells.
Figure 5.
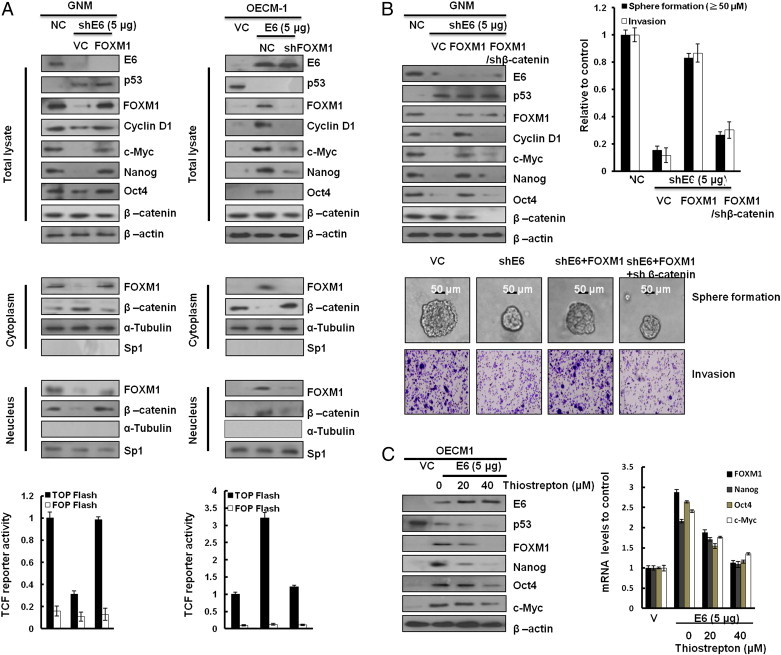
E6-induced FOXM1 expression promotes cell invasiveness and stemness through activating β-catenin/TCF signaling pathway in OCSCC cells. (A) Western blot analysis of the total levels of E6, p53, FOXM1, cyclin D1, c-Myc, Nanog, Oct4, and β-catenin; cytoplasmic and nuclear levels of β-catenin and FOXM1 in GNM and OECM-1 cells that were transfected with a vector (control, NC, or VC), FOXM1, shFOXM1, E6, or shE6 plasmids, respectively. β-Actin, α-tubulin, and SP1 were used as the protein loading controls of whole-cell extracts, as well as cytoplasmic and nuclear proteins. TOPFlash and FOPFlash activity levels were determined through luciferase reporter activity assay. FOPFlash was used as a negative control. β-Gal served as an internal control. Values are given as means ± SEMs for triplicate samples. (B) Western blot analysis of the total levels of E6, p53, FOXM1, cyclin D1, c-Myc, Nanog, Oct4, and β-catenin in GNM cells that were, respectively, transfected with a vector (control, NC, or VC), shE6, FOXM1, and shβ-catenin plasmids. The sphere formation and invasive cells were photographed (bottom) and quantified (upper right) The scale bar represents a length of 50 μm. (D) Western blot analysis of the total levels of E6, p53, FOXM1, cyclin D1, c-Myc, Nanog, Oct4, and β-catenin in OECM-1–E6 cells on treatment with thiostrepton at various dosages. Quantitative real-time PCR (right) was used to analyze the levels of FOXM1, Nanog, Oct4, and c-Myc mRNA expression. The data shown are the means ± SDs of three independent experiments.
E6-Induced FOXM1 Expression Is Responsible for HPV-Mediated Xenograft Metastatic Lung Tumor Formation in Nude Mice
We examined whether E6-induced FOXM1 expression could be responsible for HPV-infected tumor progression in nude mice. Four nude mice in each group were injected with E6-knockdown GNM or TL-1, FOXM1-knockdown GNM or TL-1, or ectopic FOXM1 expression in E6-knockdown GNM or TL-1 stable clones. The expression of E6 and FOXM1 in E6-knockdown, FOXM1-knockdown, and E6/FOXM1-knockdown cells and the further ectopic FOXM1 expression in E6-knockdown stable clones were confirmed by Western blot analysis (Figure 6A, upper panel). The change of FOXM1-mediated downstream genes Nanog, Oct4, and c-Myc in GNM and TL-1 cells subjected to different treatments was further evaluated by real-time polymerase chain reaction (PCR), indicating that these gene expression levels in both cells were markedly decreased by E6 knockdown, FOXM1 knockdown, and thiostrepton treatment. However, the decrease of these three gene expressions by E6 knockdown in both cells was reversed by ectopic FOXM1 expression. These results suggest that the expression of Nanog, Oct4, and c-Myc elevated by E6-mediated FOXM1 may be responsible for cell invasiveness and stemness in E6-positive oral and lung cancer cells.
Figure 6.
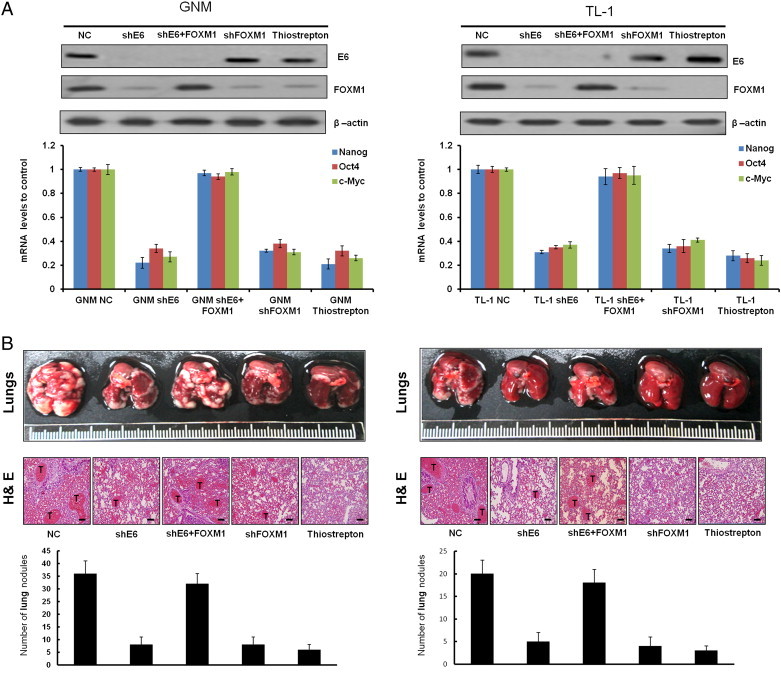
E6-induced FOXM1 expression is responsible for HPV-mediated metastatic lung tumor formation in nude mice. An in vivo metastasis assay was conducted by injecting nude mice with TL-1 (shGFP) and GNM (shGFP), a stable GNM (shE6) and TL-1 (shE6) clone (1 × 106 cells per mouse), or PBS through the tail vein. The mice were sacrificed, and the lungs were excised on day 42. Representative lungs bearing metastatic xenograft tumors were examined by hematoxylin and eosin staining, indicated with a “T”. The scale bar represents a length of 100 μm (right). Data are presented as means ± SEMs; data were compared between groups using the t test, and *P < .05 was considered to be statistically significant (the comparator was the control).
We next examined whether E6-mediated FOXM1 could promote tumor progression and metastasis in nude mice. The representative lung tumor nodules in the nude mice from each group are shown in Figure 6 (upper panel), and these tumors were confirmed by hematoxylin and eosin staining (Figure 6, middle panel). The lung tumor nodules in nude mice injected with E6-knockdown GNM or TL-1 stable clones decreased markedly compared with those injected with their NC cells (Figure 6, lower panel). The decrease in lung tumor nodules was observed in nude mice injected with FOXM1-knockdown GNM or TL-1 stable clones and in nude mice injected with NC cells treated with FOXM1 inhibitor—thiostrepton. Intriguingly, the decrease in lung tumor nodules was restored in nude mice injected with E6-knockdown GNM or TL-1 stable clones with ectopic FOXM1 expression. These results clearly indicate that E6-induced FOXM1 expression is responsible for HPV-mediated xenograft metastatic lung tumor formation.
HPV-Positive Patients with High-FOXM1 Tumors Exhibit the Worst Overall Survival and Relapse-Free Survival among the Four Subgroups
The data of HPV 16/18 DNA in OCSCC and NSCLC tumors were collected from previous studies [5], [6], [7], [10]. The presence of HPV DNA determined by nested PCR was further confirmed by in situ hybridization or p16 immunostaining [5], [6], [7], [10]. The immunohistochemical data showed that FOXM1 expression was positively correlated with NKX2-1 expression in oral and lung tumors (Tables 1 and S1). The positive association of HPV infection with FOXM1 expression was also observed in lung tumors (P < .001); however, this association between HPV and FOXM1 was only marginally observed in oral tumors; it did not reach statistical significance (P = .101). The representative FOXM1 and NKX2-1 immunostaining results in serial paraffin sections of tumors are shown in Figure S2. A Cox regression analysis showed that oral and lung cancer patients with high-FOXM1 tumors had shorter overall survival (OS) and shorter times to tumor recurrence than oral and lung cancer patients with low-FOXM1 tumors (Table 2). In this study population, the prognostic value of HPV infection for OS and relapse-free survival (RFS) was not observed in oral and lung cancers, but the prognostic value of HPV for RFS was seen in lung cancer patients. When oral and lung cancer patients were divided into four subgroups using two parameters, HPV and FOXM1, the highest hazard ratio (HR) value was observed in HPV-positive oral and lung cancer patients with high-FOXM1 tumors (Table 2). These results suggest that the prognostic significance of FOXM1 expression for OS and RFS was more pronounced in HPV-positive oral and lung cancer patients.
Table 1.
Relationship of NKX2-1 and FOXM1 Expression with HPV 16/18 Infection in Patients with Cancer
| Variables | Oral Cancer (n = 110) |
Variables | Lung Cancer (n = 117) |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | NKX2-1⁎ |
FOXM1⁎ |
No. | NKX2-1⁎ |
FOXM1⁎ |
||||||||||
| Low | High | P | Low | High | P | Low | High | P | Low | High | P | ||||
| HPV 16/18 infection | HPV 16/18 infection | ||||||||||||||
| No | 51 | 40 | 11 | .003 | 33 | 18 | .101 | No | 55 | 35 | 20 | .007 | 34 | 21 | < .001 |
| Yes | 59 | 30 | 29 | 29 | 30 | Yes | 62 | 24 | 38 | 12 | 50 | ||||
| NKX2-1⁎ | NKX2-1⁎ | ||||||||||||||
| Low | 70 | 49 | 21 | < .001 | Low | 59 | 29 | 30 | .028 | ||||||
| High | 40 | 13 | 27 | High | 58 | 17 | 41 | ||||||||
P value was obtained from Chi-square test.
The expressions of NKX2-1 and FOXM1 were evaluated by immunohistochemistry as the text described.
Table 2.
Cox Regression Analysis of HPV 16/18 DNA and FOXM1 Expression with OS and RFS of Patients with Cancer.
| Variables | Oral Cancer (n = 110) |
Variables | Lung Cancer (n = 117) |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | OS |
RFS |
No. | OS |
RFS |
||||||||||
| Median Survival (Months) | HR (95% CI)⁎ | P | Median Survival (Months) | HR (95% CI)⁎ | P | Median Survival (Months) | HR (95% CI)⁎ | P | Median Survival (Months) | HR (95% CI)⁎ | P | ||||
| HPV 16/18 DNA | HPV 16/18 DNA | ||||||||||||||
| Negative | 51 | 33.3 | 1 | 25.5 | 1 | No | 55 | 30.7 | 1 | 25.5 | 1 | ||||
| Positive | 59 | 23.1 | 0.92 (0.49-1.74) | .810 | 19.9 | 1.40 (0.78-2.51) | .259 | Yes | 62 | 21.5 | 1.43 (0.88-2.34) | .148 | 20.7 | 1.85 (1.12-3.03) | .015 |
| FOXM1† | FOXM1† | ||||||||||||||
| Low | 62 | 32.5 | 1 | 25.5 | 1 | Low | 46 | 35.3 | 1 | 30.3 | 1 | ||||
| High | 48 | 22.2 | 4.70 (2.50-8.87) | < .001 | 16.6 | 2.84 (1.60-4.95) | < .001 | High | 71 | 18.2 | 1.74 (1.06-2.86) | .030 | 13.6 | 1.97 (1.23-3.14) | .004 |
| HPV 16/18 DNA†/FOXM1 | HPV 16/18 DNA/FOXM1 protein† | ||||||||||||||
| Negative/low | 33 | 25.6 | 1 | 25.5 | 1 | Negative/low | 34 | 35.3 | 1 | 33.3 | 1 | ||||
| Positive/low | 29 | 42.3 | 0.16 (0.04-0.70) | .015 | 29.5 | 0.57 (0.20-1.64) | .299 | Positive/low | 12 | 39.4 | 0.45 (0.13-1.59) | .214 | 29.7 | 0.53 (0.18-1.53) | .241 |
| Negative/high | 18 | 38.8 | 2.73 (1.19-6.29) | .018 | 35.2 | 2.56 (01.17-5.64) | .019 | Negative/high | 21 | 30.9 | 0.94 (0.43-2.03) | .870 | 15.4 | 1.06 (0.50-2.26) | .880 |
| Positive/high | 30 | 15.1 | 3.71 (1.46-9.41) | .006 | 20.0 | 3.43 (1.47-8.00) | .004 | Positive/high | 50 | 17.8 | 1.76 (1.02-3.05) | .043 | 12.9 | 2.35 (1.37-4.03) | .002 |
CI, confidence interval.
HR adjusted for age, gender, smoke, tumor type, and stage.
HPV 16/18 DNA in tumors was detected by nested PCR, and the data were obtained from our previous reports.
Discussion
In the present study, we provided the evidence to demonstrate that FOXM1 upregulated by E6-mediated NKX2-1 was responsible for tumor progression and metastasis in HPV-associated cervical, oral cavity, and lung cancers. Importantly, a similar mechanistic action shown in the three types of HPV-positive cancer cells appear to support that FOXM1 may play a crucial role in HPV-associated tumorigenesis. Moreover, the findings from the cell model were further confirmed in xenograft tumors in nude mice. Therefore, we suggest that FOXM1 might be potentially targeted to suppress tumor progression and metastasis and, in turn, to improve outcomes in patients with HPV 16/18 infection. The association of HPV infection with cervical and oral cavity cancers has been documented [1], [2], [3], [4]; however, the association between HPV infection and lung cancer is still debated [25], [26], [27]. The involvement of HPV infection in Taiwanese lung tumorigenesis has been extensively studied [5], [6], [7], although the negative correlation between HPV infection and lung cancer has been reported elsewhere [25], [26], [27]. Therefore, the geographic variation has been considered to explain the difference in the association between HPV infection and lung cancer.
The involvement of FOXM1 expression in HPV-associated HNSCC and cervical tumor progression has been reported [11], [12], [13], [14], [15]; however, the mechanistic action of FOXM1 expression in HPV-associated tumor progression remains unclear. It is expected that FOXM1 expression suppressed by wild-type p53 may be de-repressed by E6-degraded p53. In the present study, we provide evidence that E6-mediated MZF1/NKX2-1 axis plays a crucial role in the up-regulation of FOXM1 transcription (Figure 2, Figure 3), which is in contrast to previous reports that showed FOXM1 up-regulation by E2F1 released by Rb phosphorylation through p53 inactivation [16], [17]. Up-regulation of FOXM1 by E6 through the MZF1/NKX2-1 axis could explain why FOXM1 expression was higher in HPV-positive cells than in HPV-negative cancer cells.
FOXM1 has been shown to promote tumor metastasis in human hepatocellular carcinoma, glioma, and pancreatic cancer through increased lysyl oxidase, NEDD4-1 and caveolin-1 expression [28], [29]. However, we did not observe FOXM1-mediated lysyl oxidase, NEDD4-1, and caveolin-1 being involved in the E6/MZF1/NKX2-1/FOXM1 axis–mediated invasiveness in GNM and TL-1 cells (Figure S3). Studies indicated that β-catenin was associated with lymph node metastasis in cervical cancer [30] and the increased expression of β-catenin indicated an increased risk of tumor local recurrence compared to the low expression of β-catenin in HNSCC [31]. Recently, nuclear β-catenin accumulation was associated with increased Nanog expression and predicted poor prognoses in NSCLC [32]. Therefore, β-catenin nuclear translocation has been shown to be linked with tumor metastasis and stemness in cases of cervical, HNSCC, and NSCLC. However, the molecular action of β-catenin nuclear translocation in HPV-associated OCSCC and NSCLC is still unclear. To the best of our knowledge, this is the first time to demonstrate that E6-induced FOXM1 expression through the MZF1/NKX2-1 axis promotes β-catenin nuclear translocation and, in turn, enhances cell invasiveness and stemness in HPV-positive OCSCC and NSCLC (Figures 4 and S1).
In summary, we provided evidence to demonstrate that the up-regulation of FOXM1 through the E6/MZF1/NKX2-1 axis may be responsible for HPV-infected tumor progression, poor survival, and faster tumor recurrence in HPV-positive OCSCC and NSCLC patients. Therefore, FOXM1 expression could feasibly predict outcomes in HPV-positive OCSCC and NSCLC patients. We suggest that the FOXM1 inhibitor thiostrepton might be useful in suppressing tumor aggressiveness and consequently improving outcomes in HPV-positive patients with tumors with high-expressing FOXM1.
Footnotes
This article refers to supplementary materials, which are designated by Table S1 and Figures S1 to S3 and are available online at www.neoplasia.com.
Grant support: This work was jointly supported by grants from the National Health Research Institute (NHRI96-TD-G-111-006 and NHRI97-TD-G-111-006) and the National Science Council (NSC-96-2628-B-040-002-MY3 and DOH100-TD-C-111-005) of Taiwan, ROC. Disclosure of potential conflicts of interest: No potential conflicts of interest were disclosed. Authors’ contributions: Conception and design—P.-M.C., Y.-C.W., C.-Y.C., and H.L. Development of methodology—P.-M.C., Y.-W.C., and H.L. Acquisition of data (provided animals, acquired and managed patients, provided facilities, and so on)—P.-M.C., Y.-C.W., T.-C.W., Y.-W.C., C.-Y.C., and H.L. Analysis and interpretation of data (e.g., statistical analysis, biostatistics, and computational analysis)—P.-M.C., Y.-C.W., T.-C.W., Y.-W.C., C.-Y.C., and H.L. Writing, review, and/or revision of the manuscript—P.-M.C., Y.-C.W., T.-C.W., Y.-W.C., C.-Y.C., and H.L. Administrative, technical, or material support (i.e., reporting or organizing data and constructing databases)—P.-M.C., Y.-C.W., T.-C.W., Y.-W.C., C.-Y.C., and H.L.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.neo.2014.09.010.
Appendix A. Supplementary data
Figure S1. E6-induced FOXM1 expression promotes cell invasiveness and stemness through activating β-catenin/TCF signaling pathway in lung cancer cells. (A) Western blot analysis of total levels of E6, p53, FOXM1, cyclin D1, c-Myc, Nanog, Oct4, and β-catenin; cytoplasmic and nuclear levels of β-catenin and FOXM1 in TL-1 and A549 cells that were transfected with a vector (control, NC, or VC), FOXM1, shFOXM1, E6, or shE6 plasmids, respectively. β-Actin, α-tubulin, and SP1 were used as the protein loading controls of whole-cell extracts, as well as cytoplasmic and nuclear proteins. TOPFlash and FOPFlash activity levels were determined through luciferase reporter activity assay. FOPFlash was used as a negative control. β-Gal served as an internal control. Values are given as means ± SEMs for triplicate samples. (B) Western blot analysis of the total levels of E6, p53, FOXM1, cyclin D1, c-Myc, Nanog, Oct4, and β-catenin in GNM cells that were, respectively, transfected with a vector (control, NC, or VC), shE6, FOXM1, and shβ-catenin plasmids. The sphere formation and invasive cells were photographed (bottom) and quantified (upper right) The scale bar represents a length of 50 μm. (C) Western blot analysis of the total levels of E6, p53, FOXM1, cyclin D1, c-Myc, Nanog, Oct4, and β-catenin in OECM-1–E6 cells on treatment with thiostrepton at various dosages. Quantitative real-time PCR (right) was used to analyze the levels of FOXM1, Nanog, Oct4, and c-Myc mRNA expression. The data shown are the means ± SDs of three independent experiments.
Figure S2. The representative immunostaining results of tumors in paraffin sections from OCSCC and NSCLC patients with or without HPV 16/18 infection. (A) HPV-negative OCSCC tumors (upper panel) and HPV-positive OCSCC tumors (lower panel). (B) HPV-negative NSCLC tumors (upper panel) and HPV-positive NSCLC tumors (lower panel).
Figure S3. LOX and caveolin-1 are not responsible for FOXM1-induced invasiveness in GNM and TL-1 cells. E6-knockdown GNM and TL-1 cells were transfected with FOXM1 expression vector and then co-transfected with LOX or caveolin-1 shRNA. The expression of E6, p53, FOXM1, LOX, and caveolin-1 was evaluated by Western blot analysis in both E6-knockdown cells with FOXM1 overexpression and LOX or caveolin-1 silencing. Boyden chamber assay was performed to determine the change of invasion capability in both cells with these treatments.
Table S1. Relationship of NKX2-1 and FOXM1 Expression with HPV 16/18 Infection in Patients with Cancer
References
- 1.Rositch A.F., Koshiol J., Hudgens M.G., Razzaghi H., Backes D.M., Pimenta J.M., Franco E.L., Poole C., Smith J.S. Patterns of persistent genital human papillomavirus infection among women worldwide: a literature review and meta-analysis. Int J Cancer. 2013;133:1271–1285. doi: 10.1002/ijc.27828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schlecht N.F., Platt R.W., Duarte-Franco E., Costa M.C., Sobrinho J.P., Prado J.C., Ferenczy A., Rohan T.E., Villa L.L., Franco E.L. Human papillomavirus infection and time to progression and regression of cervical intraepithelial neoplasia. J Natl Cancer Inst. 2003;95:1336–1343. doi: 10.1093/jnci/djg037. [DOI] [PubMed] [Google Scholar]
- 3.Anantharaman D., Gheit T., Waterboer T., Abedi-Ardekani B., Carreira C., McKay-Chopin S., Gaborieau V., Marron M., Lagiou P., Ahrens W. Human papillomavirus infections and upper aero-digestive tract cancers: the ARCAGE study. J Natl Cancer Inst. 2013;105:536–545. doi: 10.1093/jnci/djt053. [DOI] [PubMed] [Google Scholar]
- 4.Gillison M.L., Shah K.V. Role of mucosal human papillomavirus in nongenital cancers. J Natl Cancer Inst Monogr. 2003;31:57–65. doi: 10.1093/oxfordjournals.jncimonographs.a003484. [DOI] [PubMed] [Google Scholar]
- 5.Cheng Y.W., Chiou H.L., Sheu G.T., Hsieh L.L., Chen J.T., Chen C.Y., Su J.M., Lee H. The association of human papillomavirus 16/18 infection with lung cancer among nonsmoking Taiwanese women. Cancer Res. 2001;61:2799–2803. [PubMed] [Google Scholar]
- 6.Cheng Y.W., Wu M.F., Wang J., Yeh K.T., Goan Y.G., Chiou H.L., Chen C.Y., Lee H. Human papillomavirus 16/18 E6 oncoprotein is expressed in lung cancer and related with p53 inactivation. Cancer Res. 2007;67:10686–10693. doi: 10.1158/0008-5472.CAN-07-1461. [DOI] [PubMed] [Google Scholar]
- 7.Fei Y., Yang J., Hsieh W.C., Wu J.Y., Wu T.C., Goan Y.G., Lee H., Cheng Y.W. Different human papillomavirus 16/18 infection in Chinese non-small cell lung cancer patients living in Wuhan, China. Jpn J Clin Oncol. 2006;36:274–279. doi: 10.1093/jjco/hyl017. [DOI] [PubMed] [Google Scholar]
- 8.Koshiol J., Rotunno M., Gillison M.L., Van Doorn L.J., Chaturvedi A.K., Tarantini L., Song H., Quint W.G., Struijk L., Goldstein A.M. Assessment of human papillomavirus in lung tumor tissue. J Natl Cancer Inst. 2011;103:501–507. doi: 10.1093/jnci/djr003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moody C.A., Laimins L.A. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10:550–560. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- 10.Tung M.C., Wu H.H., Cheng Y.W., Wang L., Chen C.Y., Yeh S.D., Wu T.C., Lee H. Association of epidermal growth factor receptor mutations with human papillomavirus 16/18 E6 oncoprotein expression in non-small cell lung cancer. Cancer. 2013;119:3367–3376. doi: 10.1002/cncr.28220. [DOI] [PubMed] [Google Scholar]
- 11.Waseem A., Ali M., Odell E.W., Fortune F., Teh M.T. Downstream targets of FOXM1: CEP55 and HELLS are cancer progression markers of head and neck squamous cell carcinoma. Oral Oncol. 2010;46:536–542. doi: 10.1016/j.oraloncology.2010.03.022. [DOI] [PubMed] [Google Scholar]
- 12.Gemenetzidis E., Bose A., Riaz A.M., Chaplin T., Young B.D., Ali M., Sugden D., Thurlow J.K., Cheong S.C., Teo S.H. FOXM1 upregulation is an early event in human squamous cell carcinoma and it is enhanced by nicotine during malignant transformation. PLoS One. 2009;4:e4849. doi: 10.1371/journal.pone.0004849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang D.K., Son C.H., Lee S.K., Choi P.J., Lee K.E., Roh M.S. Forkhead box M1 expression in pulmonary squamous cell carcinoma: correlation with clinicopathologic features and its prognostic significance. Hum Pathol. 2009;40:464–470. doi: 10.1016/j.humpath.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 14.Xu N., Jia D., Chen W., Wang H., Liu F., Ge H., Zhu X., Song Y., Zhang X., Zhang D. FoxM1 is associated with poor prognosis of non-small cell lung cancer patients through promoting tumor metastasis. PLoS One. 2013;8:e59412. doi: 10.1371/journal.pone.0059412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan D.W., Yu S.Y., Chiu P.M., Yao K.M., Liu V.W., Cheung A.N., Ngan H.Y. Over-expression of FOXM1 transcription factor is associated with cervical cancer progression and pathogenesis. J Pathol. 2008;215:245–252. doi: 10.1002/path.2355. [DOI] [PubMed] [Google Scholar]
- 16.Barsotti A.M., Prives C. Pro-proliferative FoxM1 is a target of p53-mediated repression. Oncogene. 2009;28:4295–4305. doi: 10.1038/onc.2009.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Millour J., de Olano N., Horimoto Y., Monteiro L.J., Langer J.K., Aligue R., Hajji N., Lam E.W. ATM and p53 regulate FOXM1 expression via E2F in breast cancer epirubicin treatment and resistance. Mol Cancer Ther. 2011;10:1046–1058. doi: 10.1158/1535-7163.MCT-11-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lüscher-Firzlaff J.M., Westendorf J.M., Zwicker J., Burkhardt H., Henriksson M., Müller R., Pirollet F., Lüscher B. Interaction of the fork head domain transcription factor MPP2 with the human papilloma virus 16 E7 protein: enhancement of transformation and transactivation. Oncogene. 1999;18:5620–5630. doi: 10.1038/sj.onc.1202967. [DOI] [PubMed] [Google Scholar]
- 19.Chuang C.Y., Sung W.W., Wang L., Lin W.L., Yeh K.T., Su M.C., Hsin C.H., Lee S.Y., Wu B.C., Cheng Y.W. Differential impact of IL-10 expression on survival and relapse between HPV16-positive and -negative oral squamous cell carcinomas. PLoS One. 2012;7:e47541. doi: 10.1371/journal.pone.0047541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sung W.W., Wang Y.C., Lin P.L., Cheng Y.W., Chen C.Y., Wu T.C., Lee H. IL-10 promotes tumor aggressiveness via upregulation of CIP2A transcription in lung adenocarcinoma. Clin Cancer Res. 2013;19:4092–4103. doi: 10.1158/1078-0432.CCR-12-3439. [DOI] [PubMed] [Google Scholar]
- 21.Chang J.T. An economic and efficient method of RNAi vector constructions. Anal Biochem. 2004;334:199–200. doi: 10.1016/j.ab.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 22.Zhang N., Wei P., Gong A., Chiu W.T., Lee H.T., Colman H., Huang H., Xue J., Liu M., Wang Y. FoxM1 promotes β-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell. 2011;20:427–442. doi: 10.1016/j.ccr.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gong A., Huang S. FoxM1 and Wnt/β-catenin signaling in glioma stem cells. Cancer Res. 2012;72:5658–5662. doi: 10.1158/0008-5472.CAN-12-0953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park H.J., Gusarova G., Wang Z., Carr J.R., Li J., Kim K.H., Qiu J., Park Y.D., Williamson P.R., Hay N. Deregulation of FoxM1b leads to tumour metastasis. EMBO Mol Med. 2011;3:21–34. doi: 10.1002/emmm.201000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y.J., Tsai Y.C., Chen Y.C., Christiani D.C. Human papilloma virus and female lung adenocarcinoma. Semin Oncol. 2009;36:542–552. doi: 10.1053/j.seminoncol.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 26.Anantharaman D., Gheit T., Waterboer T., Halec G., Carreira C., Abedi-Ardekani B., McKay-Chopin S., Zaridze D., Mukeria A., Szeszenia-Dabrowska N. No causal association identified for human papillomavirus infections in lung cancer. Cancer Res. 2014;74:3525–3534. doi: 10.1158/0008-5472.CAN-13-3548. [DOI] [PubMed] [Google Scholar]
- 27.Ragin C., Obikoya-Malomo M., Kim S., Chen Z., Flores-Obando R., Gibbs D., Koriyama C., Aguayo F., Koshiol J., Caporaso N.E. HPV-associated lung cancers: an international pooled analysis. Carcinogenesis. 2014;35:1267–1275. doi: 10.1093/carcin/bgu038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dai B., Pieper R.O., Li D., Wei P., Liu M., Woo S.Y., Aldape K.D., Sawaya R., Xie K., Huang S. FoxM1B regulates NEDD4-1 expression, leading to cellular transformation and full malignant phenotype in immortalized human astrocytes. Cancer Res. 2010;70:2951–2961. doi: 10.1158/0008-5472.CAN-09-3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang C., Qiu Z., Wang L., Peng Z., Jia Z., Logsdon C.D., Le X., Wei D., Huang S., Xie K. A novel FoxM1-caveolin signaling pathway promotes pancreatic cancer invasion and metastasis. Cancer Res. 2012;72:655–665. doi: 10.1158/0008-5472.CAN-11-3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noordhuis M.G., Fehrmann R.S., Wisman G.B., Nijhuis E.R., van Zanden J.J., Moerland P.D., Loren Ver, van Themaat E., Volders H.H., Kok M. Involvement of the TGF-β and β-catenin pathways in pelvic lymph node metastasis in early-stage cervical cancer. Clin Cancer Res. 2011;17:1317–1330. doi: 10.1158/1078-0432.CCR-10-2320. [DOI] [PubMed] [Google Scholar]
- 31.Andrews NA, Jones AS, Helliwell TR, Kinsella AR. Expression of the E-cadherin-catenin cell adhesion complex in primary squamous cell carcinomas of the head and neck and their nodal metastases. Br J Cancer. 1997;75:1474–1480. doi: 10.1038/bjc.1997.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li X.Q., Yang X.L., Zhang G., Wu S.P., Deng X.B., Xiao S.J., Liu Q.Z., Yao K.T., Xiao G.H. Nuclear β-catenin accumulation is associated with increased expression of Nanog protein and predicts poor prognosis of non-small cell lung cancer. J Transl Med. 2013;11:114. doi: 10.1186/1479-5876-11-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. E6-induced FOXM1 expression promotes cell invasiveness and stemness through activating β-catenin/TCF signaling pathway in lung cancer cells. (A) Western blot analysis of total levels of E6, p53, FOXM1, cyclin D1, c-Myc, Nanog, Oct4, and β-catenin; cytoplasmic and nuclear levels of β-catenin and FOXM1 in TL-1 and A549 cells that were transfected with a vector (control, NC, or VC), FOXM1, shFOXM1, E6, or shE6 plasmids, respectively. β-Actin, α-tubulin, and SP1 were used as the protein loading controls of whole-cell extracts, as well as cytoplasmic and nuclear proteins. TOPFlash and FOPFlash activity levels were determined through luciferase reporter activity assay. FOPFlash was used as a negative control. β-Gal served as an internal control. Values are given as means ± SEMs for triplicate samples. (B) Western blot analysis of the total levels of E6, p53, FOXM1, cyclin D1, c-Myc, Nanog, Oct4, and β-catenin in GNM cells that were, respectively, transfected with a vector (control, NC, or VC), shE6, FOXM1, and shβ-catenin plasmids. The sphere formation and invasive cells were photographed (bottom) and quantified (upper right) The scale bar represents a length of 50 μm. (C) Western blot analysis of the total levels of E6, p53, FOXM1, cyclin D1, c-Myc, Nanog, Oct4, and β-catenin in OECM-1–E6 cells on treatment with thiostrepton at various dosages. Quantitative real-time PCR (right) was used to analyze the levels of FOXM1, Nanog, Oct4, and c-Myc mRNA expression. The data shown are the means ± SDs of three independent experiments.
Figure S2. The representative immunostaining results of tumors in paraffin sections from OCSCC and NSCLC patients with or without HPV 16/18 infection. (A) HPV-negative OCSCC tumors (upper panel) and HPV-positive OCSCC tumors (lower panel). (B) HPV-negative NSCLC tumors (upper panel) and HPV-positive NSCLC tumors (lower panel).
Figure S3. LOX and caveolin-1 are not responsible for FOXM1-induced invasiveness in GNM and TL-1 cells. E6-knockdown GNM and TL-1 cells were transfected with FOXM1 expression vector and then co-transfected with LOX or caveolin-1 shRNA. The expression of E6, p53, FOXM1, LOX, and caveolin-1 was evaluated by Western blot analysis in both E6-knockdown cells with FOXM1 overexpression and LOX or caveolin-1 silencing. Boyden chamber assay was performed to determine the change of invasion capability in both cells with these treatments.
Table S1. Relationship of NKX2-1 and FOXM1 Expression with HPV 16/18 Infection in Patients with Cancer