

SUMMARY
Dopaminergic neurons in the ventral tegmental area (VTA) are well known for their role in mediating the positive reinforcing effects of drugs of abuse. Here, we identify in rodents and humans a population of VTA dopamine neurons co-expressing corticotropin releasing factor (CRF). We provide further evidence in rodents that chronic nicotine exposure upregulates CRF mRNA in dopaminergic neurons of the posterior VTA, activates local CRF1 receptors, and blocks nicotine-induced activation of transient GABAergic input to dopaminergic neurons. Local downregulation of CRF mRNA and specific pharmacological blockade of CRF1 receptors in the VTA reversed the effect of nicotine on GABAergic input to dopaminergic neurons, prevented the aversive effects of nicotine withdrawal, and limited the escalation of nicotine intake. These results link the brain reward and stress systems within the same brain region in signaling the negative motivational effects of nicotine withdrawal.
INTRODUCTION
Drug addiction has been hypothesized to be driven by two mechanisms; reduction of the activity of the brain reward system1 and enhanced function of the anti-reward brain stress system2, concepts known as within- and between-system neuroadaptations, respectively3. Prominent downregulation of the mesolimbic dopamine (DA) reward system originating in the ventral tegmental area (VTA) and upregulation of the corticotropin-releasing factor (CRF) brain stress system originating in the extended amygdala have been observed in rodents, nonhuman primates, and humans during abstinence from drugs of abuse, including tobacco4,5.
The VTA is a critical region for nicotine dependence6, and several groups have examined the mechanisms behind DA and CRF neuroadaptations7–13. However, most of these studies were performed in nondependent animals, and these studies postulated that VTA CRF is released from axons that originate in the forebrain and not from local CRF neurons7–13. Thus, how the VTA DA and CRF systems interact in nicotine dependence and withdrawal is essentially unknown.
Here, we identify in rodents and humans a novel population of CRF neurons in the VTA and demonstrate that recruitment of these CRF neurons in the VTA after chronic nicotine contributes to a within-system neuroadaptation of DA neurons to mediate the negative motivational state elicited by nicotine withdrawal.
RESULTS
Nicotine dependence upregulates CRF mRNA in the pVTA
To test whether nicotine dependence upregulates CRF in the brain stress and reward systems, we first measured CRF mRNA in two key regions of the CRF brain stress system, the paraventricular nucleus of the hypothalamus (PVN) and central nucleus of the amygdala (CeA), as well as in the VTA, using quantitative real-time polymerase chain reaction (RT-PCR). Groups of mice were made nicotine-dependent by chronic exposure to nicotine delivered by osmotic minipumps (7 mg/kg/d)14,15. Brain punches of the PVN, CeA, and VTA (Fig. 1a) were sampled in saline-treated mice, dependent mice with nicotine minipumps, or 8 h after removal of the minipump (withdrawn mice)14. We consistently detected low levels of CRF mRNA in the VTA in saline-treated mice. CRF mRNA levels were 7–15 times lower in the VTA than in the CeA and PVN (F2,33=4.9, p=0.014, Fig. S1), in accordance with the fact that the CeA and PVN contain large populations of cell bodies that express CRF mRNA, whereas similar neurons have not been reported in the VTA16 until now. Chronic exposure to nicotine increased CRF mRNA levels in the VTA in both the dependent and withdrawn groups of mice (F2,100 = 2.7, p = 0.034), without altering CRF expression in the PVN or CeA (Fig. 1b). Considering that CRF neurons in the CeA and PVN project to and synapse with both DA and γ-aminobutyric acid (GABA) neurons in the VTA17, and that CRF release in the VTA is potentiated after repeated but not acute cocaine exposure8, the increase in CRF mRNA in the VTA observed herein could originate from either axonal transport of CRF mRNA to the VTA or the synthesis of CRF mRNA in local VTA neurons.
Fig. 1. Nicotine dependence increases CRF mRNA levels in the VTA.

a, Location of the brain tissue samples in the CeA, VTA, and PVN. b, CRF mRNA levels (ΔCt) in the CeA, VTA, and PVN relative to the housekeeping gene GAPDH normalized to saline in mice chronically exposed to saline (Sal) or nicotine (Nic) or withdrawn from chronic nicotine (WD) as measured by RT-PCR (n = 13–14 per group). A significant change in CRF mRNA was observed in the VTA but not CeA or PVN in both the Nic and WD groups (*p < 0.05, vs. Sal). Data represent mean ± SEM.
Identification of a novel population of CRF neurons in the pVTA
To test the hypothesis that CRF mRNA is synthesized locally in the VTA, we performed in situ hybridization (ISH) for CRF mRNA in a separate cohort of mice that were drug-naive or treated with acute nicotine (1.5 mg/kg), chronic nicotine (7 mg/kg/day for 12 days), or chronic nicotine and withdrawal (8 h). A significant population of CRF neurons with dense CRF mRNA in cell bodies could be detected bilaterally in the VTA in all groups, with the majority of neurons localized in the posterior VTA (pVTA), dorsal to the interpeduncular nucleus (IPN) (Fig. 2). No significant difference in the number of neurons was observed between groups (F24,180=0.82, p=0.71, Fig. S2), demonstrating that the increase in CRF mRNA was attributable to an increase in the production of CRF within neurons and not to an increase in the number of CRF neurons. To confirm the specificity of the CRF mRNA ISH, we performed urocortin mRNA ISH on adjacent sections. Urocortin is a structurally related peptide of the CRF family with significant affinity for the CRF2 receptor18. No urocortin-positive neurons were observed in the VTA (Fig. S3), whereas numerous urocortin-positive neurons were observed in the Edinger-Westphal nucleus (Fig. S3), an area that is known to contain a large population of urocortin neurons18. Complementary to these results, a recent study demonstrated that nicotine selectively activates dopaminergic neurons in the pVTA but not anterior VTA (aVTA), suggesting that CRF-expressing neurons in the pVTA may be dopaminergic19. To test this hypothesis, we then examined co-expression of CRF in VTA DA neurons in both mice and humans.
Fig. 2. Identification of VTA CRF neurons in mice.

a, Number of CRF neurons per section in the VTA (bregma range: −2.92 to −3.88) in mice (n = 24). Data represent mean ± SEM. b, Representative CRF mRNA in situ hybridization sections of the anterior (aVTA), posterior (pVTA), and tail (tVTA) of the VTA in naive mice. White arrows point to CRF neurons. c, Corresponding diagram from the Paxinos and Watson atlas. Scale bar = 200 μm
CRF mRNA is expressed in DA neurons
We performed double labeling of CRF mRNA and DA neurons using CRF mRNA radioactive (Fig. 3a), and fluorescent ISH coupled with tyrosine hydroxylase (TH) immunohistochemistry (Fig. 3c, d) in mice. CRF mRNA-positive neurons were located bilaterally in the VTA in TH-enriched regions (Fig. 3b), and 95.2 ± 4.8% of them co-expressed TH (Fig. 3c, d). To further confirm the existence of VTA CRF/DA neurons and test their relevance to humans, we performed CRF chromogenic ISH coupled with TH immunohistochemistry in VTA samples from three non-smoker humans without any psychiatric disorders (see Supplementary Table 1 for details). Of 600 CRF neurons, we found that 97.8 ± 0.7% of CRF neurons were colocalized with DA neurons (Fig. 4), confirming the existence of VTA CRF/DA neurons in both rodents and humans.
Fig. 3. Double labeling of CRF/DA neurons using CRF in situ hybridization and TH immunohistochemistry in mice.
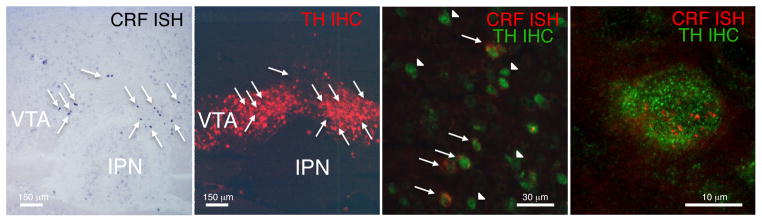
a, CRF mRNA radioactive in situ hybridization (black) demonstrating CRF-positive cell bodies (arrows) in the pVTA. b, TH immunohistochemistry (red) on an alternate section of the pVTA showing that dopaminergic cell bodies were located bilaterally in the pVTA in TH-immunoreactive areas. c, Double fluorescent labeling of CRF mRNA (red) and TH protein (green) in the pVTA at high magnification. Fluorescent ISH is less sensitive than radioactive or chromogenic ISH; thus, a limited number of neurons expressing CRF mRNA could be identified in the pVTA in nicotine-dependent mice using this procedure. However, it was used to achieve high-resolution imaging and assess the cellular colocalization of CRF mRNA with TH. Of 18 neurons that unambiguously expressed CRF mRNA, 16 were TH-positive (CRF+/TH+ neuron: arrow, CRF−/TH+ neuron: arrowhead). d, Confocal image of a single VTA neuron co-expressing TH and CRF mRNA. This experiment was repeated on 13 sections from three mice.
Fig. 4. Double labeling of CRF/DA neurons using CRF in situ hybridization and TH immunohistochemistry in humans.

Single TH immunohistochemistry (a, d, g, light brown) and CRF ISH (b, e, h, blue) performed on different but adjacent sections (distance: 80 μm) of the VTA (human sample HSB3750). Notice the similar distribution of TH (white arrow head) and CRF (black arrowhead) within the VTA (use blood vessel * as landmark). Double TH immunochemistry and CRF ISH (c, f, i) on the same section (adjacent to the other two sections; note blood vessel *). White arrows point to colocalized TH/CRF neurons. Very few neurons were single-labeled as TH-only (white arrow head) and CRF-only (black arrowhead). This experiment was repeated on brain samples from three human subjects.
Withdrawal-induced CRF peptide depletion in the pVTA and IPN
Increased CRF release during drug withdrawal is associated with decreased immunodensity of CRF peptide in neuropils. The decrease in CRF immunodensity reflects CRF depletion from synaptic vesicles subsequent to a local increase in CRF release20,21. Thus, we used CRF immunohistochemistry to examine whether the recruitment of CRF neurons in the pVTA is also associated with a local decrease in CRF peptide density in neuropils during withdrawal from chronic nicotine. Densitometry analysis revealed that both nicotine dependence and withdrawal from chronic nicotine decreased CRF peptide density compared with saline-treated mice in the pVTA (F2,31 = 4.40, p = 0.02; Fig. S4a), interpeduncular nucleus (IPN; F2,49 = 4.24, p = 0.020; Fig S4d), and CeA (F2,67 = 3.15, p = 0.049; Fig. S4c) but not aVTA (F2,23 = 0.03, p = 0.97; Fig. S4a) or PVN (F2,21 = 0.75, p = 0.48; Fig. S4b), demonstrating the specificity of these effects. The combined observations that chronic nicotine exposure increases CRF mRNA expression and decreases CRF peptide density in the pVTA and IPN suggest that CRF/DA neurons in the pVTA may release CRF locally in the pVTA and IPN during nicotine dependence. However, the possibility that CRF may also be released from extrinsic projection neurons, such as CRF neurons in the CeA, cannot be ruled out because decreased immunodensity was also observed in the CeA (Fig. S4c). The decreased CRF immunodensity in the CeA replicates results obtained with alcohol-dependent rats21 and extends this work to nicotine-dependent and -withdrawn mice.
pVTA CRF mRNA downregulation prevents nicotine dependence-induced dysregulation of GABA-DA synapses
The downregulation of VTA DA neuron activity is a key feature of addiction, and recent results have shown that nicotine-induced GABA currents are critical for the phasic activation of dopamine neurons22. To test whether CRF mRNA-expressing neurons in the pVTA play a role in the synaptic control of DA neurons, we tested the causal relationship between downregulation of CRF mRNA in the pVTA of nicotine-dependent and -withdrawn mice and the effect of nicotine on GABAergic currents in DA neurons. Local downregulation of CRF mRNA was performed using an adeno-associated viral vector that encodes a short-hairpin RNA targeting CRF mRNA (AAV2-shCRF) or a non-targeting sequence (AAV2-shSCR; Fig. S5a, b). The AAV2-shCRF vector decreased the amount of CRF mRNA by 88.0 ± 6.2% as measured by RT-PCR on VTA punches (t10 = 11.12, p = 0.0001, n = 6 mice/condition). Some neurons exhibited an even stronger downregulation, having no detectable CRF mRNA remaining after treatment with the silencing vector, such that the total number of CRF-positive neurons decreased by 26% (Fig. S5c, d).
Whole-cell voltage-clamp recordings of pharmacologically isolated GABAA receptor-mediated spontaneous inhibitory postsynaptic currents (sIPSCs) were first performed in putative pVTA DA neurons from naive mice. Dopamine neurons were identified at the time of recording by previously published functional criteria23,24 (Fig. S6), and a subset of neurons was collected after recording for TH expression using single-cell RT-PCR. To examine the effects of nicotine dependence on changes in GABAergic transmission in pVTA DA neurons with and without downregulation of CRF mRNA, neurons infected with either AAV2-shSCR or AAV2-shCRF were identified during withdrawal (6–8h) by the presence of green fluorescent protein (GFP) in the cell body (Fig. 5c). In naive mice, acute application of nicotine (1 μM) onto pVTA DA neurons produced a transient, significant increase in sIPSC frequency from 1.2 ± 0.2 Hz to 1.9 ± 0.3 Hz (t7 = 5.048, p = 0.0015, paired t-test; Fig. 5a, b; 172.9 ± 18.5% of control, t7 = 3.93, p = 0.0057, one-sample t-test; Fig. 5f), consistent with previous reports25,26. Nicotine also increased sIPSC frequency in pVTA DA neurons from mice implanted with saline minipumps and infected with AAV2-shSCR or AAV2-shCRF (176.9 ± 18.2% of control; t11 = 4.217, p = 0.0014 and 171.5 ± 16.7% of control; t11 = 4.248, p = 0.0013, respectively; Fig 5f). In contrast, in pVTA DA neurons from nicotine-dependent mice injected with AAV2-shSCR, acute application of nicotine (1 μM) produced no change in sIPSC frequency (97.7 ± 1.8% of control; t10 = 1.228, p = 0.2474; Fig. 5d, f), which was significantly different from the effects observed in naïve mice or mice implanted with saline minipumps (one-way ANOVA, F4,48= 4.704, p = 0.0028). However, during acute application of nicotine (1 μM) in pVTA DA neurons from nicotine-dependent mice injected with AAV2-shCRF, the nicotine-induced increase in sIPSC frequency was restored (171.2 ± 17.3 % of control; t9 = 4.108, p = 0.0026, one-sample t-test; Fig. 5e, f), which was significantly different from that observed in pVTA DA neurons from nicotine-dependent mice injected with AAV2-shSCR (unpaired t-test; t19 = 4.426, p = 0.0003), but not different from that observed in naive mice. To confirm the identity of the putative DA neurons, we performed single-cell RT-PCR after recordings to detect the presence of TH. The majority (13/18, 72%) of the VTA cells examined by single-cell PCR were TH-positive, and the electrophysiological results were similar when only TH-positive neurons were included (Fig. S7). Collectively, these data demonstrate that nicotine-induced activation of GABA neurons, a key mechanism required for DA neuron firing22, is deficient in nicotine-dependent mice, and that downregulation of CRF mRNA in the VTA can reverse the effect of nicotine dependence.
Fig. 5. Nicotine dependence causes dysregulation of GABA-DA synapses, which is reversed by downregulation of CRF mRNA.

a, Representative voltage-clamp recording of sIPSCs in a pVTA DA neuron from a naive mouse before and during the application of nicotine (1 μM). b, Box-and-whisker plot of average sIPSC frequency before and during nicotine application (n = 8 cells from three mice). c, 60× magnification photomicrograph of a pVTA DA neuron from a nicotine-dependent mouse using fluorescent optics to illustrate GFP expression (left panel) and infrared differential interference contrast (IR-DIC) optics during recording (right panel, * pipette). d, Representative voltage-clamp recording of sIPSCs in a pVTA DA neuron from a nicotine-dependent mouse injected with AAV2-shSCR before and during application of nicotine (1 μM). e, Representative voltage-clamp recording of sIPSCs in a pVTA DA neuron from a nicotine-dependent mouse injected with AAV2-shCRF before and during application of nicotine (1 μM). f, Summary of average change in sIPSC frequency produced by nicotine (*p < 0.05) in pVTA DA neurons from naive mice (n = 8 cells from three mice), naive mice injected with either AAV2-shSCR (n = 12 cells from three mice), or AAV2-shCRF (n = 12 cells from three mice) and nicotine-dependent mice injected with either AAV2-shSCR (n = 11 cells from four mice) or AAV2-shCRF (n = 10 cells from four mice; #p = 0.0003, vs. nicotine-dependent AAV2-shSCR). Data represent mean ± SEM.
pVTA CRF mRNA downregulation prevents the aversive effects of nicotine withdrawal
To test whether CRF mRNA-expressing neurons in the pVTA play a role in the motivational effects of nicotine dependence, we examined the causal relationship between downregulation of CRF mRNA in the pVTA of nicotine-dependent and -withdrawn mice and the expression of conditioned place aversion to nicotine withdrawal and anxiety-like behavior (Fig. 6a). In the place conditioning paradigm, a two-way ANOVA revealed a significant interaction between AAV2 injection and nicotine history (F1,40 = 5.80, p = 0.021; Fig. 6b). Nicotine-dependent and -withdrawn mice infused with the AAV2-shSCR control vector in the pVTA showed an aversive motivational response to the withdrawal-paired environment (p = 0.032), which was not observed in mice infused with the AAV2-shCRF vector (p = 0.6392). Mice that were injected with the AAV2-shCRF vector outside the pVTA (mostly located dorsal (red nucleus) and lateral (substantia nigra) to the pVTA) showed an aversive response to nicotine withdrawal similar to control mice (unpaired t-test t16 = 0.0255, p = 0.798; Fig S8), demonstrating the anatomical specificity of this effect. Furthermore, nondependent mice given either the AAV2-shSCR or AAV2-shCRF vector in the pVTA and an acute aversive injection of nicotine (1.75 mg/kg)14,15 showed an aversive response to the nicotine-paired environment (Fig. 6b), demonstrating that the lack of aversion to withdrawal in dependent mice infused with the silencing vector was not attributable to a general impairment of conditioned place aversion, but was specific to nicotine withdrawal in dependent mice. Altogether, these results suggest that CRF mRNA in the pVTA does not mediate a general aversive response to acute nicotine but specifically mediates aversion to nicotine withdrawal in dependent subjects. These results establish a causal relationship between the recruitment of CRF mRNA-expressing neurons in the pVTA during withdrawal from chronic nicotine and the aversive motivational response to nicotine withdrawal.
Fig. 6. Viral vector-mediated downregulation of CRF mRNA in the VTA prevents the aversive motivational response to nicotine withdrawal.

a, Timeline of experiment. b, Difference score (s) during testing of conditioned place aversion to nicotine withdrawal in nicotine-dependent and -withdrawn groups or nondependent groups given acute nicotine after AAV2-shSCR or AAV2-shCRF treatment (n = 11 mice per group). Mice infused with the silencing vector did not show the conditioned aversive motivational response (a negative difference score) to withdrawal from chronic nicotine (*p < 0.05) that was observed in mice given the control vector. Nondependent mice given AAV2-shSCR or AAV2-shCRF showed an aversive response to acute nicotine. Data represent mean ± SEM. The inset shows the viral vector injection site. c, Box-and-whisker plot showing time spent in the central area of the open field in nicotine-dependent and -withdrawn mice injected with AAV2-shSCR (n = 8) or AAV2-shCRF in the VTA (n = 8; *p < 0.05). Pictures below are example traces showing activity patterns in the open field of a mouse injected with the corresponding vector. See Fig. S5 for details on the viral vector.
Activation of the brain CRF-CRF1 receptor system is associated with increased anxiety-like behavior in humans and animals and is hypothesized to be responsible for the negative emotional state after protracted abstinence27. To test this hypothesis, we measured open field activity to evaluate anxiety-like behavior during protracted abstinence (3–4 weeks). Mice injected with the AAV2-shSCR vector spent significantly less time in the central area of the open field than mice injected with the AAV2-shCRF vector (unpaired t-test t14 = 2.84, p = 0.013; Fig. 6c), an effect that was not attributable to a decrease in locomotion (AAV2-shSCR: 2181 ± 215 cm; AAV2-shCRF: 2307 ± 146 cm; t14 = 0.49, p = 0.64). These results demonstrate that upregulation of CRF mRNA in the pVTA is required for the anxiogenic-like effects of protracted nicotine abstinence.
pVTA CRF mRNA downregulation decreases abstinence-induced escalation of nicotine intake
To test whether CRF mRNA-expressing neurons in the pVTA play a role in the motivation to take nicotine during abstinence, we infused the same AAV2-shCRF/shSCR vectors in the pVTA of rats, and measured nicotine self-administration 4 weeks later. In rats given short access (1 h/day) to nicotine self-administration for 10 days, downregulation of CRF mRNA in the VTA did not affect nicotine intake (unpaired t-test; t17 = 0.59, p = 0.28; Fig. 7). We then gave the rats long access (21 h/day) to nicotine self-administration and measured nicotine intake after 7 days of long access and after 48 h of abstinence27–29. Infusion of AAV2-shCRF decreased nicotine intake in long access rats (unpaired t-test t14 = 2.32, p = 0.018), and prevented abstinence-induced increases in nicotine intake, whereas the rats that received AAV2-shSCR showed a significant increase in nicotine intake after 48 h of abstinence (F1,14 = 4.91, p = 0.044; Fig. 7). These results demonstrate that pVTA CRF mRNA downregulation decreases excessive nicotine intake and is particularly effective at decreasing the abstinence-induced escalation of nicotine intake.
Fig. 7. Viral vector-mediated downregulation of CRF mRNA in the VTA decreases long-access nicotine self-administration and prevents abstinence-induced escalation of nicotine intake.
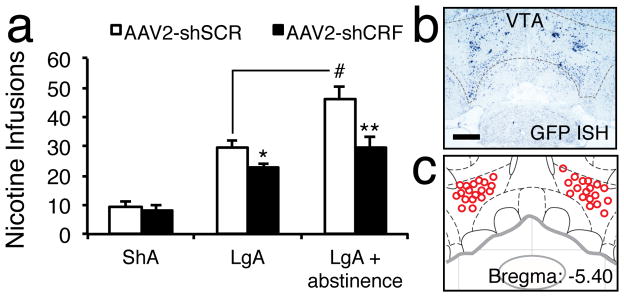
a, Number of nicotine self-administrations in rats injected with AAV2-shSCR or AAV2-shCRF and given short access (1 h/day for 10 days; ShA-AAV2-shCRF, n = 9; ShA-AAV2-shSCR, n = 9), long access (21 h/day for 7 days; LgA-AAV2-shCRF, n = 8; LgA-AAV2-shSCR, n = 7), or long access after 48 h of abstinence (LgA + abstinence; n = 11) to nicotine. AAV2-shCRF reduced the amount of nicotine infusions in the LgA group (*p < 0.05, vs. AAV2-shSCR) and LgA + abstinence group (**p < 0.001, vs. AAV2-shSCR). Comparisons of the AAV2-shSCR LgA groups before and after abstinence with regard to the number of infusions after abstinence demonstrate the abstinence-induced escalation of nicotine intake (#p < 0.05). Data represent mean ± SEM. b. Representative section of AAV2-shCRF using GFP ISH (dark blue). c, Viral vector injection site.
Blocking CRF1 receptors prevents the aversive effect of nicotine withdrawal
The present results revealed a depletion of CRF peptide in the pVTA during nicotine withdrawal, hypothesized to reflect a local increase in CRF release20,21. To further test this hypothesis, we examined whether the activation of CRF1 receptors in the pVTA is necessary for the conditioned aversive motivational response to a withdrawal-paired environment shown by nicotine-dependent and -withdrawn mice14,15. We administered the CRF1 receptor antagonist N,N-bis(2-methoxyethyl)-3-(4-methoxy-2-methylphenyl)-2,5-di-methyl-pyrazolo[1,5-a]pyrimidin-7-amine (MPZP; 20 mg/kg, s.c.)27 prior to place conditioning in nondependent mice given acute nicotine, nicotine-dependent mice with minipumps (7 mg/kg/day), or mice in spontaneous withdrawal (8 h) from chronic nicotine (Fig. 8a). A two-way ANOVA showed a significant effect of systemic MPZP treatment (F3,103 = 20.51, p < 0.0001; Fig 8b). Withdrawn mice pretreated with vehicle showed a conditioned place aversion to a withdrawal-paired environment that was blocked by MPZP (p = 0.0043), demonstrating that activation of CRF1 receptors during withdrawal is required for the aversive motivational response to nicotine withdrawal. Nicotine-dependent mice that were not in withdrawal and pretreated with saline showed a conditioned place preference for the nicotine-paired environment that was not blocked by MPZP pretreatment (p = 0.9085). These results suggest that the activation of CRF1 receptors is not required for the rewarding motivational response to chronic nicotine in dependent mice. To further evaluate the specificity of this effect, we tested the effect of CRF1 receptor antagonism on the conditioned avoidance response to an acute aversive dose of nicotine14. Nondependent mice given acute nicotine and pretreated with saline showed a conditioned place aversion to the nicotine-paired environment that was not blocked by MPZP pretreatment (p = 0.9228) similar to the results obtained using viral vector-mediated CRF mRNA silencing. Mice treated with chronic saline and pretreated with vehicle or MPZP showed no motivational response to a novel environment or to MPZP (p = 0.8697), confirming the specificity of these results. These results demonstrate that activation of CRF1 receptors is selectively required to mediate the aversive motivational response to nicotine withdrawal in dependent mice but not the aversive (in nondependent mice) or rewarding (in dependent mice) motivational effects of nicotine.
Fig. 8. CRF1 receptor antagonism prevents the aversive motivational response to withdrawal from chronic nicotine.

a, Timeline of experiment. b, Nicotine-dependent and -withdrawn mice pretreated with vehicle (n = 13) showed an aversion to the withdrawal-paired environment that was blocked by pretreatment with the CRF1 receptor antagonist MPZP (n = 14; *p < 0.05). Nicotine-dependent mice that were not experiencing withdrawal and were pretreated with vehicle (n = 15) showed a preference for the nicotine-paired environment that was not blocked by MPZP pretreatment (n = 15). Nondependent mice given acute nicotine and pretreated with vehicle (n = 16) showed an aversion to the nicotine-paired environment that was not blocked by MPZP pretreatment (n = 15). Mice with saline minipumps that were pretreated with vehicle (n = 11) or MPZP (n = 12) showed no motivational response to a novel environment or to MPZP. Data represent mean ± SEM. c, Schematic of the aVTA showing placements of the intra-aVTA cannulae. d. Schematic of the pVTA showing placements of the intra-pVTA cannulae. e, Nicotine-dependent and -withdrawn mice implanted with cannulae in the pVTA that were given vehicle (n = 11) showed an aversive motivational response to the withdrawal-paired environment that was blocked in mice that received intra-pVTA CRF1 receptor antagonist MPZP (n = 10; *p < 0.05). Dependent and withdrawn mice implanted with aVTA cannulae that were given vehicle (n = 10) or MPZP (n = 12) showed nicotine withdrawal aversions. Nondependent mice with pVTA cannulae that were pretreated with vehicle (n = 8) or MPZP (n = 10) and acute nicotine showed an aversive motivational response to the nicotine-paired environment. Control mice implanted with saline minipumps and pVTA cannulae that received vehicle (n = 12) or MPZP (n = 11) showed no motivational response to a novel environment or to MPZP. Data represent mean ± SEM.
To test whether activation of CRF1 receptors specifically in the pVTA mediates the conditioned aversive motivational response to nicotine withdrawal, we measured place conditioning in separate groups of mice after infusion, prior to conditioning, of MPZP (0.14 ng/0.3 μl) or vehicle in the aVTA (Fig. 8c) and pVTA (Fig. 8d). A two-way ANOVA revealed a significant interaction between treatment and nicotine history (F3,76 = 3.351, p = 0.0233; Fig. 8e). Nicotine-dependent mice that were in withdrawal and pretreated with vehicle showed an aversive response to the withdrawal-paired environment that was blocked in withdrawn mice pretreated with intra-pVTA MPZP (p = 0.0063). However, the ability of MPZP to block nicotine withdrawal aversions was not observed when the CRF antagonist was infused into the aVTA (p = 0.7108). These results demonstrate that CRF1 receptor activation, specifically in the pVTA, is necessary for the acquisition of the aversive motivational response to nicotine withdrawal in dependent mice. Similar to mice that received systemic MPZP or the AAV2-shCRF, mice in a nondependent motivational state that received intra-pVTA MPZP or vehicle prior to acute nicotine administration showed a conditioned avoidance to the acute nicotine-paired environment (p = 0.7323). Intra-VTA MPZP had no motivational effects on its own, such that mice implanted with saline minipumps showed neither a preference nor aversion to a MPZP-paired environment (p = 0.8996).
DISCUSSION
This report identifies a novel population of DA neurons in the VTA that co-express CRF in both rodents and humans. Chronic exposure to nicotine increased the levels of CRF mRNA in the pVTA associated with a depletion of CRF peptide in VTA and IPN neuropils and blocked nicotine-induced activation of GABAergic input onto DA neurons. Downregulation of CRF mRNA in the pVTA restored GABAergic control over DA neurons and prevented the motivational effects of nicotine withdrawal as measured by conditioned place aversion, anxiety-like behavior, and escalated nicotine self-administration. Pharmacological testing further demonstrated that the motivational effects of nicotine withdrawal were mediated by activation of CRF1 receptors in the pVTA.
The detection of CRF neurons in the brain can be very challenging. To confirm the existence of this novel population of CRF neurons in the VTA, we utilized a variety of complementary approaches: RT-PCR, radioactive and chromogenic ISH, immunohistochemistry, and fluorescent ISH combined with immunohistochemistry. These techniques revealed direct evidence of CRF neurons in the VTA in rodents and humans. To further confirm the existence of CRF neurons in the VTA, we used several controls, including RT-PCR of housekeeping genes, CRF immunodensity in the aVTA, and ISH of urocortin, a member of the CRF family that is structurally related to CRF. Confirming the specificity of the present results, housekeeping genes were not upregulated, CRF immunodensity was altered in the pVTA and IPN but not aVTA, and urocortin-positive neurons were not observed in the pVTA.
TH immunohistochemistry combined with CRF ISH clearly showed that the majority of CRF-positive neurons (95% in mice and 98% in humans) were colocalized with TH, a marker of DA neurons. CRF neurons were distributed bilaterally in the VTA, with the highest number observed in the pVTA in the region dorsal to the IPN. This increase in CRF mRNA was associated with the depletion of CRF in IPN and pVTA neuropils (but not aVTA), suggesting that the increase in CRF mRNA in the VTA was associated with increased CRF release in the pVTA and IPN. Virally-mediated CRF mRNA downregulation and pharmacological blockade of CRF1 receptors in the pVTA confirmed the physiological role of increased VTA CRF mRNA, CRF release, and CRF1 receptor activation in nicotine-dependent mice.
These results suggest that VTA CRF neurons may release CRF through axons in the IPN, and both axons and dendrites in the pVTA to act as autocrine and paracrine-like signals30. Indeed, unlike classic neurotransmitters, neuropeptides are not only found in axons but are also found abundantly in dendrites31. CRF specifically is found in dendrites associated with endomembranes and large dense-core vesicles32, and CRF immunoreactivity has been observed both in axon terminals and dendrites in the VTA17. Many groups have attempted to measure CRF in the VTA in rats but failed because of the low concentration, high degradation rate, and low stability and recovery of CRF using microdialysis. The only report of CRF microdialysis in the VTA has not been replicated, and involved intense footshock stress33. New techniques are being developed based on cell-based neurotransmitter fluorescent engineered reporters to reliably measure physiological CRF levels, but these methods are still in their initial stages of development.
The decreased CRF immunodensity in the CeA is in accordance with previous reports showing increased CRF release27 and reduction of negative emotional states after blockade of CRF1 receptors in the CeA34. The lack of increased CRF mRNA in the CeA suggests that the VTA and CeA CRF neurons may be upregulated at different time points. Recent reports showing increased CRF mRNA in the CeA 16–24 h into withdrawal35,36 may explain why an increase was not observed 8 h into withdrawal in the present study. Taken together, these studies demonstrate that blocking the CRF-CRF1 system in the CeA or VTA is sufficient to prevent the negative emotional states of nicotine withdrawal.
In addition to the upregulation of CRF mRNA in VTA DA neurons, chronic nicotine dysregulated GABA-DA synapses, demonstrated by the loss of inhibitory responses (sIPSCs) to acute nicotine in VTA DA neurons in dependent mice. Viral-mediated downregulation of CRF mRNA in the pVTA abolished the effect of chronic nicotine on GABA-DA synapses and prevented the motivational effect of withdrawal as measured by conditioned place aversion, anxiety-like behavior, and escalated nicotine self-administration after abstinence. These findings may explain why nicotine dependence produces in vivo decreases in tonic dopaminergic activity in the VTA15 and DA release in the nucleus accumbens37, whereas acute CRF1 receptor antagonism increases tonic DA activity38. These findings also may provide a mechanism for observed decreases in accumbal DA release in response to rewards after CRF administration in the VTA9, the decreased phasic dopamine release observed in cocaine-dependent rats39, and the decreased prefrontal dopamine metabolism after intra-VTA CRF administration40. Indeed, while pharmacological and optogenetic activation of VTA GABA neurons produces robust inhibition of DA neurons41,42, transient physiological activation of GABAergic input on DA neurons is required for DA firing22 because transient GABAergic inputs promote rebound firing in DA neurons. Altogether, these results demonstrate that the activation of a CRF-CRF1 system in the pVTA may dysregulate transient GABAergic inputs that are critical for dopaminergic firing. Similar effects of CRF on GABAergic input have been observed in the CeA43.
Increasing evidence suggests that a neurobiological switch occurs in the VTA during the transition from nondependent to drug-dependent motivational states15,44–46, suggesting that the effect of CRF1 receptor activation in dependent vs. non-dependent animals may be opposite. Our results showed a specific role for CRF in the pVTA in mediating the aversive response to nicotine withdrawal in dependent mice, but not to acute nicotine in nondependent mice. The specific effect on nicotine intake in LgA but not ShA rats is consistent with this hypothesis, demonstrating that the recruitment of VTA CRF/DA neurons significantly contributes to the development of nicotine dependence. The present results do not exclude the possibility that the expression of CRF in DA neurons alters plasticity in non-DA neurons and circuits in the VTA that may also alter DA function and have motivational significance46,47. However, taken with previous results demonstrating that nicotine withdrawal is signaled by a specific pattern of tonic VTA dopaminergic activity15, and activation of CRF1 receptors can mediate changes in VTA DA population activity38, the present results suggest that chronic nicotine leads to an upregulation of CRF in pVTA DA neurons that is released onto VTA CRF1 receptors to mediate the aversive motivational effects of nicotine withdrawal. Future studies are necessary to directly test this hypothesis for nicotine and other drugs of abuse.
The transition to drug dependence and drug addiction has been hypothesized to be driven and maintained by two relatively independent systems: downregulation of DA function in the VTA, a within-system neuroadaptation that leads to a reward deficit disorder48, and upregulation of CRF function, a between-system neuroadaptation that leads to a stress surfeit disorder49. Here, we identify a novel population of CRF neurons in the core of the brain incentive salience/reward system, in DA neurons of the pVTA, and show that the upregulation of CRF mRNA and activation of CRF1 receptors locally in the pVTA occur after the induction of nicotine dependence and are required for the aversive motivational effects of nicotine withdrawal. This within-system neuroadaptation links the brain reward and stress systems within the same neurobiological substrate, providing evidence that DA and CRF interact within the VTA to mediate the aversive motivational effects of nicotine withdrawal in dependent subjects.
ONLINE METHODS
Animals
All animal use procedures were approved by the University of Toronto Animal Care Committee in accordance with the Canadian Council on Animal Care guidelines and The Scripps Research Institute Institutional Animal Care and Use Committee and were in accordance with National Institutes of Health guidelines. Adult male C57BL/6 mice (2–4 months) (Charles River, Montreal, Canada, or Hollister, CA, USA) were housed in a temperature-controlled room with lights on from 7:00 AM to 7:00 PM. Adult male Wistar rats (2–4 months) (Charles River, Hollister, CA, USA) were housed in a temperature-controlled room with lights on from 8:00 PM to 8:00 AM. Animals were grouped 2–4 per cages. Behavioral testing was performed during the dark cycle.
Drugs
Nicotine hydrogen tartrate salt (Sigma-Aldrich, Ontario, Canada) was dissolved in saline, pH 7.0 ± 0.4, and administered via osmotic minipumps (chronic nicotine, 7 mg/kg/day, minipump model 1002, Alzet, Cupertino, CA, USA) or subcutaneous injection (acute nicotine, 1.75 mg/kg). Nicotine-dependent and -withdrawn mice had their minipumps removed 8 h prior to experimentation at a time that corresponded to peak motivational withdrawal14. The CRF1 receptor antagonist MPZP was synthesized at The Scripps Research Institute (Richardson et al., 2008), dissolved in (2-hydroxypropyl)-beta-cyclodextrin (HBC), and administered subcutaneously 20 min prior to conditioning or at a concentration of 0.14 μg/0.3 μl over 10 min for intra-VTA infusions.
Viral vector production
shRNA-encoding AAV2 vectors that target the CRF transcript were generated using the same procedure as described by Darcq et al. (2011). A shRNA sequence (shCRF, sense strand 5′-GGATCTCACCTTCCACCTTCT-3′) predicted to have high silencing efficiency was selected using Block-iT RNAi Designer (Life Technologies, Carlsbad, CA, USA). A “universal scramble” shRNA with no homology to any transcripts was used as a control (shSCR, sense strand 5′-GCGCTTAGCTGTAGGATTC-3′). An AAV2 shuttle plasmid that encodes shCRF or shSCR downstream of the mU6 promoter and enhanced green fluorescent protein (EGFP) under the control of the cytomegalovirus (CMV) promoter 3′-flanked by a β-globin intron was generated using Invitrogen Gateway technology. Helper-free AAV2 particles were produced by the triple transfection of AAV-293 cells (Agilent Technologies, Santa Clara, CA, USA) with the AAV2 shuttle plasmid described above, a plasmid that contains AAV2 rep and cap genes, and a plasmid that encodes the adenovirus helper functions. Two days later, cells were collected and lysed by three freeze-thaw cycles, treated with benzonase, and clarified by centrifugation. Viral vectors were purified by iodixanol gradient ultracentrifugation (Zolotukhin et al., 2002), followed by dialysis and concentration against Dulbecco phosphate-buffered saline (PBS) using centrifugal filter units (Millipore, Billerica, MA, USA). Genomic units (GU) were quantified by RT-PCR. The titers were 4.5–6.6 × 1011 GU/ml.
Viral vector and drug infusion
Mice and rats were anesthetized (1–5% isoflurane in oxygen mixture) and placed in a Kopf stereotaxic instrument. Double cannulae (Plastics One, Roanoke, VA, USA) were inserted bilaterally above the left and right VTA. The coordinates used were relative to bregma and from skull surface. Mouse coordinates: aVTA (anterior/posterior [AP], −3.3 mm; dorsal/ventral [DV], −4.4 mm; medial/lateral [ML], ±0.5 mm), pVTA (AP, −3.6 mm; DV, −4.4 mm; ML, ±0.5 mm). Each rat received two injections of the virus directed at different AP levels of the VTA. Rat coordinates: AP, −5.4 and −5.8 mm; ML, ±0.7 mm; DV, −8.2 mm. Each mouse received 0.75 μl/side and each rat received 1 μl/side of either the AAV2-shCRF or AAV2-shSCR at an infusion rate of 0.1 μl/min using injectors that protruded 1 mm beyond the cannula (for mice) or 2 mm beyond the cannula (for rats). The injectors were left in place for 10 additional minutes to ensure adequate diffusion of the solution. Infusions were delivered via polyethylene tubing (PE 20) that was connected to a Hamilton 10 μl syringe.
For intra-VTA MPZP infusion, dummy cannulae were secured in the injection cannulae by dust caps, and the animals were allowed to recover for 1 week prior to drug infusion and conditioning. On conditioning days, the dust cap and dummy cannulae were removed and replaced with injector cannulae. Mice were lightly restrained and infused with 0.14 μg/0.3 μl MPZP or 20% HBC (control solution) over 1 min. After the behavioral experiments were completed, the mice were sacrificed and perfused, and cannula placement was verified by Cresyl violet staining. Sixteen of 100 mice were excluded due to incorrect cannula placements.
Real-time RT-PCR
RNA was isolated using a Qiagen RNeasy extraction kit with DNAase to remove genomic DNA contamination, and a specified amount of cDNA was reverse-transcribed using Superscript III (Invitrogen, Foster City, CA, USA). Quantitative RT-PCR was performed using Taqman Gene Expression Assays for CRF (Crh, Mm01293920_s1) in a 7900HT Fast RT-PCR System (both from Life Technologies, Carlsbad, CA, USA). Quantification was performed using the ΔCt method with GAPDH as an endogenous control. For single-cell RT-PCR analysis, single cells were harvested from AAV2-shCRF- or AAV2-shSCR-injected brain slices by aspiration of neuronal cytoplasm immediately after electrophysiological recordings and processed with the Picopure RNA isolation kit (Life Technologies, Carlsbad, CA, USA) for RNA isolation and reverse transcription (RT) using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA). The cDNA obtained from single neurons was subjected to preamplification for 20 cycles, followed by RT-PCR to detect the expression of TH with forward (5-ACTGCTTCTCAACCACATCTT-3) and reverse (5-GGGTAGAATACAGCATGAAGGG-3) primers using the SSO Advanced SYBR Green Supermix (Bio-Rad, Hercules, CA, USA) according to manufacturer’s protocol.
Radioactive in situ hybridization
The mice were anesthetized with chloral hydrate (350 mg/kg, i.p.) and perfused via the ascending aorta with 0.9% saline followed by ice-cold 4% paraformaldehyde in 0.1 M borate buffer, pH 9.5. The brains were removed, postfixed for 3 h, and cryoprotected in 20% sucrose in 0.1 M phosphate buffer overnight at 4°C. Five one-in-four series of 30 μm-thick frozen coronal sections were cut, collected, and stored in 30% ethylene glycol and 20% glycerol in 0.1 M phosphate buffer at −20°C until processing. In situ hybridization was performed using 35S-labeled sense (control) and antisense cRNA probes labeled to similar specific activities using a full-length (1.2 kb) probe for mRNA that encodes CRF (1.2 kb; Dr. K. Mayo, Northwestern University, Evanston, IL, USA). Sections were mounted on Superfrost plus slides and dried under vacuum overnight. They were postfixed with 10% formalin for 30 min at room temperature, digested with 10 μg/ml proteinase K for 15 min at 37°C, and acetylated for 10 min. The probes were labeled to specific activities of 1–3 × 109 dpm/μg and applied to the slides at concentrations of ~107 cpm/ml overnight at 56°C in a solution that contained 50% formamide, 0.3 M NaCl, 10 mM Tris, 1 mM EDTA, 0.05% tRNA, 10 mM dithiothreitol, 1× Denhardt’s solution, and 10% dextran sulfate, after which they were treated with 20 μg/ml of ribonuclease A for 30 min at 37°C and washed in 15 mM NaCl/1.5 mM sodium citrate with 50% formamide at 70°C. The slides were then dehydrated and exposed to X-ray film (Kodak Biomax MR, Eastman Kodak, Rochester, NY, USA) for 18 h. They were coated with Kodak NTB-2 liquid emulsion and exposed at 4°C for 3–4 weeks as determined by the strength of the signal on the film. The slides were developed with Kodak D-19 and fixed with Kodak rapid fixer. One series of sections that adjoined those used for analysis was stained with thionin to facilitate the accurate localization of hybridization signals. The experimenters were blind to the experimental history of the animals.
Densitometry
The semiquantitative densitometric analysis of hybridization signals for CRF mRNA was performed on emulsion-dipped slides. Photomicrographs were captured using a Leica light microscope with a Hamamatsu Orca charge-coupled device camera through OpenLab software (version 3.1.5) and analyzed using ImageJ software. The optical densities of hybridization signals were determined under dark-field illumination at 400× magnification with a circular region of interest (ROI) with a 20 μm diameter that was placed over individual neurons. The size of the ROI was chosen on the basis of the average diameter of TH-immunoreactive neurons in the pVTA. The sections were analyzed at regular 120 μm intervals across the pVTA. Optical densities were corrected for the average background signal that was determined by sampling 20 cell-sized areas per section in non-signal areas adjacent to the pVTA. Optical density values are expressed in gray scale values of 1 to 256, corresponding to a gradation from low to high absorbance, respectively. Neurons on both sides of the brain were pooled for analysis to calculate the animal mean. Animal means were then grouped according to virus treatment, averaged, and statistically analyzed. The experimenters were blind to the experimental history of the animals during densitometry analysis.
Immunohistochemistry
Mice were anesthetized with 3% halothane, pre-perfused transcardially with a solution of 0.5 ml heparin/100 ml saline for 1–2 min, and then perfused with a solution of 4% paraformaldehyde in 0.1M phosphate buffer (PB), pH 7. The experimenters were blind to the drug history of the animals. The brains were removed from the skull, post-fixed at 4°C in the perfusate solution for 6–18 h, rinsed in several changes of PBS that contained 20% sucrose, and stored at 4°C in fresh 20% sucrose/PBS that contained 0.1% sodium azide. Coronal cryostat sections (40 μm) were obtained using a Reichert Jung cryostat. The sections were collected in strict anatomical order in a one-in-four series and stored at 4°C in PBS 0.1% azide prior to processing. The sections were incubated free-floating with shaking in multi-well plates, and all incubations were performed at room temperature unless otherwise specified. The samples were incubated for 20 min in 1% hydrogen peroxide/PBS to quench endogenous peroxidases, rinsed several times in PBS, and exposed to a blocking solution that contained PBS/Triton-X100 (0.3%), 1 mg/ml bovine serum albumin (BSA), and 5% normal donkey serum (Jackson Immuno Research, West Grove, PA, USA) for a minimum of 60 min. The sections were incubated overnight at 4°C in anti-CRF purified goat polyclonal antibody (sc-1761, Santa Cruz Biotechnology, Santa Cruz, CA, USA) diluted 1:200 in PBS, 0.5% Tween 20, and 5% normal donkey serum. Control sections were incubated in the antibody diluent. Following three rinses of 10 min each in PBS, the sections were incubated in Vector ImmPRESS Goat (Vector Labs, Burlingame, CA, USA) for 1 h, rinsed in PBS as above, and reacted with a DAB Substrate Kit (Vector Labs). The sections were monitored under a microscope to determine the optimal reaction time. The reaction was stopped in PBS. The sections were mounted on coated slides, air dried, dehydrated through a series of ethanol and xylene, and coverslipped with Permount. All brightfield photographs for analysis were taken with a Q Imaging Retiga 2000R color digital camera mounted on a Zeiss Axiophot microscope.
Combined TH immunohistochemistry and CRF fluorescent in situ hybridization
Mouse brains were snap-frozen using isopentane. Twenty-micrometer cryostat sections were mounted onto Superfrost Plus slides. A digoxigenin (DIG)-labeled CRF riboprobe was synthesized using a commercial kit (Roche, Indianapolis, IN, USA) from a plasmid containing full-length rat CRF cDNA (kind donation from Dr. K. Mayo, Northwestern University, Evanston, IL, USA). The sections were post-fixed in 4% formaldehyde for 1 min. Following PBS washes, proteins were acetylated in 0.1 M triethanolamine, pH 8.0, and 0.2% acetic acid. Following washes in 2× saline sodium citrate buffer, the sections were dehydrated in a graded ethanol-chloroform series. Pre-hybridization and hybridization were then performed at 70°C in a buffer containing 50% formamide, 2× SSC, 5× Denhardt’s, 0.5 mg/ml sheared salmon sperm DNA, and 0.25 mg/ml yeast RNA. The probe was diluted in the hybridization buffer (800 ng/ml) and incubated overnight on slides. Post-hybridization washes were performed in 50% formamide, 2× SSC, and 0.1% Tween-20. The slides were then blocked for 1 h and incubated with anti-DIG antibody conjugated to alkaline phosphatase (11093274910, 1:1000; Roche) and anti-TH antibody (AB152, 1:500; Millipore, Billerica, MA, USA) overnight at 4°C in TNT buffer (0.1 M Tris, pH 7.5, 0.15 M NaCl, 0.1% Tween-20) containing 1% blocking reagent (Roche). A donkey anti-rabbit secondary antibody conjugated to Alexa Fluor 488 (A-21206, 1:200, 2 h; Life Technologies, Carlsbad, CA, USA) was used to reveal the TH signal. Following TNT washes and incubation in 0.1 M Tris-HCl, pH 8, 0.1 M NaCl, 0.01 M MgCl2, HNPP combined with Fast Red TR (Roche) was then used to detect alkaline phosphatase. To enhance the signal, fresh substrate was applied three times for 30 min, and the slides were rinsed in TNT in-between. The slides were washed, air dried, and coverslipped with Vectashield HardSet-DAPI (Vector Laboratories, Burlingame, CA, USA). Images were taken using either epifluorescence (Zeiss Axiophot) or confocal microscopy (LaserSharp 2000, version 5.2; 488, 568, and 647 nm emission wavelengths; Bio-Rad, Hercules, CA, USA).
Human Brain samples (TH immunohistochemistry and CRF chromogenic in situ hybridization)
Human brain samples (snap-frozen and stored at −80 degrees) were obtained from the UCLA Brain Bank. For chromogenic in situ hybridization, brain sections were processed as described above for fluorescent in situ hybridization, but slides were incubated with NBT-BCIP (Sigma) as an alkaline phosphatase substrate at room temperature overnight in the dark. Sections were then rinsed in PBS Tween-20, and air-dried.
For double-labeling with TH, sections were then fixed in 4% paraformaldehyde/PBS for 15 min, rinsed in PBS, quenched for 20 min in 1% hydrogen peroxide/PBS, rinsed in PBS and blocked for 1 h at room temperature in PBS/TX100 (0.3%), BSA (1 mg/ml) and 5% normal donkey serum (Jackson). Sections were incubated in rabbit polyclonal TH antibody (Rabbit Millipore AB152) and diluted 1:1000 in PBS, 0.5% Tween 20, and 5% normal donkey serum overnight at room temperature in a humidity chamber. Following PBS rinses, the sections were incubated in Rabbit ImmPRESS (Vector Labs) for 1–2 h at room temperature. Following PBS rinses, the sections were incubated in either Vector ImmPACT Red (Vector Labs) or DAB substrate (Vector Labs). The reaction was stopped in PBS rinses and the sections were air-dried and coverslipped with DPX.
Brain slice preparation (electrophysiology)
Slices that contained the VTA were prepared as described previously24 from 20 adult male C57BL/6 mice subjected to brief anesthesia (3–5% isoflurane) followed by rapid decapitation and removal of the brain to an ice-cold high-sucrose solution (pH 7.3–7.4) that contained 206.0in mM sucrose, 2.5 mM KCl, 0.5 mM CaCl2, 7.0 mM MgCl2, 1.2 mM NaH2PO4, 26 mM NaHCO3, 5.0 mM glucose, and 5 mM HEPES. The brains were cut into transverse sections (300 μm) on a vibrating microtome (Leica VT1000S, Leica Microsystems, Buffalo Grove, IL, USA) and placed in an oxygenated (95% O2/5% CO2) artificial cerebrospinal fluid (aCSF) solution composed of the following: 120 mM NaCl, 2.5 mM KCl, 5 mM EGTA, 2.0 mM CaCl2, 1.0 mM MgCl2, 1.2 mM NaH2PO4, 26 mM NaHCO3, 1.75 mM glucose, and 5 mM HEPES. The slices were incubated in this solution for 30 min at 35–37°C, followed by 30 min equilibration at room temperature (21–22°C). Following equilibration, a single slice was transferred to a recording chamber mounted on the stage of an upright microscope (Olympus BX50WI).
Electrophysiological recording
Neurons were visualized on an upright microscope (Olympus BX50WI) with infrared differential interference contrast (IR-DIC) optics and an EXi Aqua camera (QImaging, Surrey, BC, Canada). To avoid photolytic damage, initial exposure to episcopic fluorescent illumination was brief (< 2 s). Fluorescence was detected using an X-Cite 120Q fluorescent illumination system (Lumen Dynamics, Mississauga, Ontario, Canada), and images were captured using QCapture software (QImaging). In all cases, the experimenter was blind to the nature of the viral vector injected into the mouse prior to experimental recording and data analysis. Whole-cell (voltage- and current-clamp) recordings were made with patch pipettes (3–5 MΩ; Warner Instruments) coupled to a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA, USA), low-pass filtered at 2–5 kHz, digitized (Digidata 1440A; Axon Instruments), and stored on a computer using pClamp 10 software (Axon Instruments). Series resistance was typically < 10 MΩ and continuously monitored with a hyperpolarizing 10 mV pulse. The intracellular solution used for recordings was composed of 145in mM KCl, 5 mM EGTA, 5 mM MgCl2, 10 mM HEPES, 2 mM Na-ATP, and 0.2 mM Na-GTP. To isolate only the inhibitory currents mediated by GABAA receptors, recordings (Vhold= −60mV) were performed in the presence of the glutamate receptor blockers 6,7-dinitroquinoxaline-2,3-dione (DNQX; 20 μM) and DL-2-amino-5-phosphonovalerate (AP-5; 50 μM) and the GABAB receptor antagonist CGP55845A (1 μM). Due to rapidly desensitizing effects, nicotine was bath applied for 5–7 min, but the period of analysis was restricted to 2 min after full wash-in. The frequency, amplitude, and decay of spontaneous inhibitory postsynaptic currents (sIPSCs) were analyzed and visually confirmed using semi-automated, threshold-based, mini-detection software (Mini Analysis, Synaptosoft Inc.). We determined averages of IPSC characteristics from baseline and experimental drug conditions containing a minimum of 60 events (time period of analysis varied as a product of individual event frequency), and we determined decay kinetics using exponential curve fittings, which are reported as decay time (ms). All of the detected events were used for event frequency analysis, but superimposed events were eliminated for amplitude and decay kinetic analysis. Action potential characteristics were evaluated using threshold-based event detection analysis in Clampfit 10.2 (Molecular Devices).
Place conditioning
The place conditioning apparatus was obtained from Med Associates (SOF-700RA-25 Two Chamber Place Preference Apparatus; St. Albans, VT, USA). One environment was black with a metal rod floor, and the other was white with a wire mesh floor. An intermediate gray area housed a removable partition. Each cage was cleaned between animals, and each group was fully counterbalanced. During preference testing, the dividing partition was removed, and the mice were given free access to both environments. A single 10 min preference test session was performed 5 days after the last conditioning day. All place conditioning and testing were performed between 10:00 AM and 6:00 PM.
The nicotine-dependent and -withdrawn groups of mice were conditioned according to modified place conditioning procedures as described previously14. Conditioning occurred only during withdrawal from chronic nicotine so that the motivational effects of withdrawal but not the direct effects of chronic nicotine were paired with the place conditioning environment. Eight hours after minipump removal, when the mouse was experiencing motivational withdrawal from chronic nicotine14, it was subcutaneously or intracranially pretreated with vehicle (HBC) or MPZP and confined to one of the conditioning environments for 1 h. The difference score for each animal was calculated by subtracting the time spent in the non-paired environment from the time spent in the withdrawal-paired environment during preference testing.
For nicotine-dependent mice (not withdrawn), conditioning occurred only during exposure to chronic nicotine so that the motivational effects of chronic nicotine in a dependent animal were paired with the place conditioning environment. The mice were subcutaneously pretreated with vehicle (20% HBC) or MPZP (20 mg/kg) and confined to one of the conditioning environments for 1 h. The difference score for each animal was calculated by subtracting the time spent in the non-paired environment from the time spent in the nicotine-paired environment during preference testing.
For the acute nicotine experiments, previously drug-naive mice were subcutaneously or intracranially pretreated with 20% HBC or MPZP, given a subcutaneous injection of 1.75 mg/kg nicotine or saline, and immediately confined to one of the conditioning environments for 1 h. The next day, the mouse was pretreated again with HBC or MPZP, given acute saline or nicotine, and confined to the other environment. The difference score for each animal was calculated by subtracting the time spent in the saline-paired environment from the time spent in the nicotine-paired environment. In all cases, the experimenter was blind to the previous drug and/or viral vector treatment.
Open field testing
Mice were placed in the center of a gray box measuring 41 × 41 × 38 cm for 5 min. The room was dark with the open field testing box illuminated by a soft red light. Locomotor activity and time spent in the center square area of the box were recorded by a video camera and calculated by the monitoring program (Ethovision XT, Noldus; Leesburg, VA, USA). The experimenter was blind to the mouse’s previous drug and viral vector history. The test box was cleaned with 70% alcohol between each mouse.
Nicotine self-administration
Two weeks following infusion, rats were implanted with intravenous catheters in the jugular vein under isoflurane anesthesia as described previously29. Detailed procedures for the escalation of nicotine self-administration have been described previously29. Briefly, three weeks following the injection of viral vectors in the VTA, rats were trained to nosepoke for food and water within their self-administration chambers in 21-h sessions but were not food-trained to respond for the lever that was associated with nicotine delivery. Subsequently, the active and inactive levers were extended, and the rats were allowed to self-administer nicotine (0.03251658240 or 0.0 mg/kg per 100251658240 μl/1251658240 s, free base, FR1, timeout 20 s) for 10 days (1 h per day). The rats were then allowed daily long access (LgA; 21 h) nicotine/saline self-administration sessions for 7 consecutive days, followed by 48 h of abstinence and another LgA session to measure abstinence-induced escalation of nicotine intake.
Statistical analysis
The behavioral data were analyzed with Statistica software using a one- or two-way ANOVA or Student’s t-test, where appropriate. In all cases, a normality test and an equal variance test were performed before the ANOVA to ensure its validity. For electrophysiology, statistical analysis was performed using Prism 5.02 software (GraphPad, San Diego, CA). Groups were analyzed for independent significance using a one-sample t-test, compared using an unpaired t-test for comparisons between two groups, and a one-way ANOVA for comparisons between three or more groups. All of the experiments and analyses were performed by experimenters who were blind to the treatments, and the animals’ order of treatment was randomized. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous publications13,25,37,39. Different numbers of animals between groups and between the beginning and end of the study are a result of the loss of data due to improper cannula placement, improper brain perfusion, damaged brain samples, or computer failure during testing. For all of the analyses, Bonferroni post hoc tests were used when appropriate. All of the data are expressed as mean ± standard “A supplementary methods checklist is available”.
Supplementary Material
Characteristics of human brain samples. Note that all subjects were selected to be non-smokers without any documented psychiatric disorder (based on DSM-IV) to compare the identification of CRF neurons in the VTA with the results obtained in naive mice.
Fig S1. CRF mRNA levels in the CeA, VTA, and PVN. CRF mRNA levels in the CeA, VTA, and PVN relative to the housekeeping gene GAPDH in mice chronically exposed to saline, measured by RT-PCR (n = 13 or 14 per group). Lower levels of CRF mRNA were observed in the VTA compared with the CeA and PVN (*p < 0.05). Data represent mean ± SEM.
Fig S2. Number of CRF mRNA-positive neurons in the VTA. Number of CRF neurons per section in the VTA (bregma range: −3.28 to −3.88) in naive, nondependent + acute nicotine (1.5 mg/kg), nicotine-dependent (12 days with minipumps), and nicotine-dependent and withdrawn (8 h) groups of mice (n = 6 per group). No difference was observed between groups. Data in Fig. 2 in the manuscript represent combined data from the four groups. Data represent mean ± SEM.
Fig S3. Urocortin in situ hybridization in the VTA and Edinger-Westphal nucleus. Notice the lack of urocortin neurons in the VTA and the prominent population of urocortin-positive neurons in the Edinger-Westphal nucleus. This result was repeated in six naive and six nicotine-dependent mice. Urocortin neurons were never observed in the VTA.
Fig S4. CRF immunohistochemistry. a–d. Quantification of immunodensity in the aVTA and pVTA (a), PVN (b), CeA (c) and IPN (d) in nondependent mice treated with saline (Sal), nicotine-dependent mice (Nic), and nicotine-dependent and -withdrawn mice (WD; n = 12–26 mice per group). Data represent mean ± SEM. e. Example of CRF-immunostained neuropils in the CeA showing CRF varicosities in a transversal fiber. This experiment was performed with one cohort.
Fig S5. Validation of the AAV2-shCRF viral vector. a, Timeline of viral vector infusion and behavioral experiments in mice. b, DNA construct used to produce vectors for CRF silencing (AAV2-shCRF) and control vectors (AAV2-shSCR). ITR, inverted terminal repeat; CMV, cytomegalovirus; EGFP, enhanced green fluorescent protein; hGH polyA, human growth hormone polyadenylation signal. c, Representative images of VTA CRF mRNA-containing cells taken at 20× magnification. d, Number of CRF mRNA-containing cells per section. Quantification of CRF-positive neurons restricted to GFP-positive areas in the pVTA revealed significant downregulation (−26%) of the number of CRF mRNA-containing cells in AAV2-shCRF mice compared with AAV2-shSCR mice (t12 = 2.7, p = 0.020). Data represent mean ± SEM. AAV2-shCRF also decreased the total number of CRF neurons in the entire VTA (including the region without GFP) by 18% (t12 = 2.4, p = 0.033). Notably, the mice were sacrificed approximately 4 weeks into withdrawal after the end of behavioral testing, demonstrating that the upregulation of CRF mRNA in nicotine-withdrawn mice and CRF silencing by the viral vector were both long-lasting. This experiment was performed with one cohort.
Fig S6. Electrophysiological recording of VTA DA neurons. a, Representative trace of a recorded VTA DA neuron. b, Definition of DA neuron action potential characteristics. c, Characterization of VTA dopamine neurons from naive (n = 8 cells), nicotine-dependent AAV2-shSCR (n = 11 cells), and nicotine-dependent AAV2-shCRF (n = 12 cells) mice (n = 3 naive, n = 4 nicotine-dependent AAV2-shSCR, n = 4 nicotine-dependent AAV2-shCRF). This experiment was performed with one cohort.
Fig. S7. Comparison of confirmed TH-positive VTA cells with all cells examined. a, Box-and-whisker plot showing the effects of acute nicotine in naive mice on sIPSC frequency in all VTA cells examined (left; *p = 0.0015, paired t-test) compared with TH+-only VTA cells (right; *p = 0.0051). The total number of cells for each condition is indicated in parentheses. b, Average change in sIPSC frequency in all VTA cells examined (left; *p = 0.0026, one-sample t-test; #p = 0.0003, unpaired t-test) compared with TH+-only VTA cells (right; *p = 0.0128, one-sample t-test; #p = 0.0005, unpaired t-test). Exact t and p values are noted under each condition. The total number of cells for each condition are indicated in parentheses. Data represent mean ± SEM.
Fig S8. Anatomical specificity of AAV2-shCRF VTA injections. Box-and-whisker plot showing that mice with AAV2-shCRF injections outside of the VTA (n = 7) showed an aversive motivational response to nicotine withdrawal that was not significantly different from mice injected with AAV2-shSCR (n = 11).
Acknowledgments
This work was supported by the Canadian Institutes of Health Research, National Institute on Drug Abuse (DA023597, DA035371), National Institute on Alcohol Abuse and Alcoholism (AA021491, AA015566, F32 AA020430, AA006420, AA016658, and INIA AA013498), Tobacco-Related Disease Research Program (12RT-0099), National Institute of Diabetes and Digestive and Kidney Diseases (DK026741), and the Clayton Medical Research Foundation
The authors thank Molly Brennan, Brenda Takabe, Carlos Arias, and the University of Toronto Division of Comparative Medicine staff for technical assistance and Michael Arends for editorial assistance. The authors would also like to thank Dr Nagra and James Riehl and the UCLA Brain bank (The Human Brain and Spinal Fluid Ressource Center) for providing the human samples.
Footnotes
AUTHOR CONTRIBUTIONS
TEG and OG designed the experiments. TEG, HVP, MC, and GMB performed minipump, cannulation, and viral vector surgeries. MR and MAH performed the electrophysiology experiments. TEG performed place conditioning and open field testing. AC performed self-administration experiments. CC, LAT, and PES performed ISH. CC and EC performed double ISH and immunohistochemistry. TEG, VC, PPS, ART and LC performed RT-PCR. JF and EC performed immunohistochemistry. CC, PK, and BLK supplied viral vectors. TEG and OG analyzed the data. TEG, CC, GFK, DVDK, and OG wrote the paper. All of the authors discussed the results and read the paper.
The authors declare no conflict of interest.
REFERENCE LIST
- 1.Volkow ND, Fowler JS, Wang GJ, Baler R, Telang F. Imaging dopamine’s role in drug abuse and addiction. Neuropharmacology. 2009;56 (Suppl 1):3–8. doi: 10.1016/j.neuropharm.2008.05.022. Epub 2008 Jun 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koob GF, Le Moal M. Addiction and the brain antireward system. Annu Rev Psychol. 2008;59:29–53. doi: 10.1146/annurev.psych.59.103006.093548. [DOI] [PubMed] [Google Scholar]
- 3.Koob GF, Bloom FE. Cellular and molecular mechanisms of drug dependence. Science. 1988 Nov 4;242(4879):715–23. doi: 10.1126/science.2903550. [DOI] [PubMed] [Google Scholar]
- 4.Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacol. 2010;35(1):217–38. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sarnyai Z, Shaham Y, Heinrichs SC. The role of corticotropin-releasing factor in drug addiction. Pharmacol Rev. 2001 Jun;53(2):209–43. [PubMed] [Google Scholar]
- 6.Picciotto MR, Kenny PJ. Molecular mechanisms underlying behaviors related to nicotine addiction. Cold Spring Harb Perspect Med. 2013 Jan 1;3(1):a012112. doi: 10.1101/cshperspect.a012112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boyson CO, Miguel TT, Quadros IM, Debold JF, Miczek KA. Prevention of social stress-escalated cocaine self-administration by CRF-R1 antagonist in the rat VTA. Psychopharmacology (Berl) 2011 Nov;218(1):257–69. doi: 10.1007/s00213-011-2266-8. Epub 2011 Apr 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hahn J, Hopf FW, Bonci A. Chronic cocaine enhances corticotropin-releasing factor-dependent potentiation of excitatory transmission in ventral tegmental area dopamine neurons. J Neurosci. 2009;29:6535–44. doi: 10.1523/JNEUROSCI.4773-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wanat MJ, Bonci A, Phillips PE. CRF acts in the midbrain to attenuate accumbens dopamine release to rewards but not their predictors. Nat Neurosci. 2013 Apr;16(4):383–5. doi: 10.1038/nn.3335. Epub 2013 Feb 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang B, You ZB, Rice KC, Wise RA. Stress-induced relapse to cocaine seeking: roles for the CRF(2) receptor and CRF-binding protein in the ventral tegmental area of the rat. Psychopharmacol. 2007;193(2):283–94. doi: 10.1007/s00213-007-0782-3. [DOI] [PubMed] [Google Scholar]
- 11.Wang HL, Morales M. Corticotropin-releasing factor binding protein within the ventral tegmental area is expressed in a subset of dopaminergic neurons. J Comp Neurol. 2008;509:302–18. doi: 10.1002/cne.21751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blacktop JM, Seubert C, Baker DA, Ferda N, Lee G, Graf EN, Mantsch JR. Augmented cocaine seeking in response to stress or CRF delivered into the ventral tegmental area following long-access self-administration is mediated by CRF receptor type 1 but not CRF receptor type 2. J Neurosci. 2011 Aug 3;31(31):11396–403. doi: 10.1523/JNEUROSCI.1393-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vranjkovic O, Gasser PJ, Gerndt CH, Baker DA, Mantsch JR. Stress-Induced Cocaine Seeking Requires a Beta-2 Adrenergic Receptor-Regulated Pathway from the Ventral Bed Nucleus of the Stria Terminalis That Regulates CRF Actions in the Ventral Tegmental Area. J Neurosci. 2014 Sep 10;34(37):12504–14. doi: 10.1523/JNEUROSCI.0680-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grieder TE, Sellings LH, Vargas-Perez H, Ting-A-Kee R, Siu EC, Tyndale RF, van der Kooy D. Dopaminergic signaling mediates the motivational response underlying the opponent motivational process to chronic but not acute nicotine. Neuropsychopharmacol. 2010;135:943–54. doi: 10.1038/npp.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grieder TE, George O, Tan H, George SR, Le Foll B, Laviolette SR, van der Kooy D. Phasic D1 and tonic D2 dopamine receptor signaling double dissociate the motivational effects of acute nicotine and chronic nicotine withdrawal. Proc Natl Acad Sci USA. 2012;109(8):3101–6. doi: 10.1073/pnas.1114422109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swanson LW, Sawchenko PE, Rivier J, Vale WW. Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology. 1983;36(3):165–86. doi: 10.1159/000123454. [DOI] [PubMed] [Google Scholar]
- 17.Tagliaferro P, Morales M. Synapses between corticotropin-releasing factor-containing axon terminals and dopaminergic neurons in the ventral tegmental area are predominantly glutamatergic. J Comp Neurol. 2008;506:616–26. doi: 10.1002/cne.21576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kozicz T, Bittencourt JC, May PJ, Reiner A, Gamlin PD, Palkovits M, Horn AK, Toledo CA, Ryabinin AE. The Edinger-Westphal nucleus: a historical, structural, and functional perspective on a dichotomous terminology. J Comp Neurol. 2011 Jun 1;519(8):1413–34. doi: 10.1002/cne.22580. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao-Shea R, Liu L, Soll LG, Improgo MR, Meyers EE, McIntosh JM, Grady SR, Marks MJ, Gardner PD, Tapper AR. Nicotine-mediated activation of dopaminergic neurons in distinct regions of the ventral tegmental area. Neuropsychopharmacol. 2011;36:1021–32. doi: 10.1038/npp.2010.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merlo Pich E, Lorang M, Yeganeh M, Rodriguez de Fonseca F, Raber J, Koob GF, Weiss F. Increase of extracellular corticotropin-releasing factor-like immunoreactivity levels in the amygdala of awake rats during restraint stress and ethanol withdrawal as measured by microdialysis. J Neurosci. 1995;15:5439–47. doi: 10.1523/JNEUROSCI.15-08-05439.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Funk CK, O’Dell LE, Crawford EF, Koob GF. Corticotropin-releasing factor within the central nucleus of the amygdala mediates enhanced ethanol self-administration in withdrawn, ethanol-dependent rats. J Neurosci. 2006;26:11324–32. doi: 10.1523/JNEUROSCI.3096-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tolu S, Eddine R, Marti F, David V, Graupner M, Pons S, Baudonnat M, Husson M, Besson M, Reperant C, Zemdegs J, Pagès C, Hay YA, Lambolez B, Caboche J, Gutkin B, Gardier AM, Changeux JP, Faure P, Maskos U. Co-activation of VTA DA and GABA neurons mediates nicotine reinforcement. Mol Psychiatry. 2013 Mar;18(3):382–93. doi: 10.1038/mp.2012.83. Epub 2012 Jul 3. [DOI] [PubMed] [Google Scholar]
- 23.Hnasko TS, Hjelmstad GO, Fields HL, Edwards RH. Ventral tegmental area glutamate neurons: electrophysiological properties and projections. J Neurosci. 2012 Oct 24;32(43):15076–85. doi: 10.1523/JNEUROSCI.3128-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nimitvilai S, Arora DS, McElvain MA, Brodie MS. Reversal of inhibition of putative dopaminergic neurons of the ventral tegmental area: interaction of GABA(B) and D2 receptors. Neuroscience. 2012 Dec 13;226:29–39. doi: 10.1016/j.neuroscience.2012.08.045. Epub 2012 Sep 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doyon WM, Dong Y, Ostroumov A, Thomas AM, Zhang TA, Dani JA. Nicotine decreases ethanol-induced dopamine signaling and increases self-administration via stress hormones. Neuron. 2013 Aug 7;79(3):530–40. doi: 10.1016/j.neuron.2013.06.006. Epub 2013 Jul 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mansvelder HD, Keath JR, McGehee DS. Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron. 2002 Mar 14;33(6):905–19. doi: 10.1016/s0896-6273(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 27.George O, Ghozland S, Azar MR, Cottone P, Zorrilla EP, Parsons LH, O’Dell LE, Richardson HN, Koob GF. CRF–CRF1 system activation mediates withdrawal-induced increases in nicotine self-administration in nicotine-dependent rats. Proc Natl Acad Sci USA. 2007;104:17198–203. doi: 10.1073/pnas.0707585104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohen A, Treweek J, Edwards S, Leão RM, Schulteis G, Koob GF, George O. Extended access to nicotine leads to a CRF(1) receptor dependent increase in anxiety-like behavior and hyperalgesia in rats. Addict Biol. 2013 Jul 22; doi: 10.1111/adb.12077. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cohen A, Koob GF, George O. Robust escalation of nicotine intake with extended access to nicotine self-administration and intermittent periods of abstinence. Neuropsychopharmacology. 2012 Aug;37(9):2153–60. doi: 10.1038/npp.2012.67. Epub 2012 May 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ludwig M, Leng G. Dendritic peptide release and peptide-dependent behaviours. Nat Rev Neurosci. 2006 Feb;7(2):126–36. doi: 10.1038/nrn1845. Review. [DOI] [PubMed] [Google Scholar]
- 31.Son SJ, Filosa JA, Potapenko ES, Biancardi VC, Zheng H, Patel KP, Tobin VA, Ludwig M, Stern JE. Dendritic peptide release mediates interpopulation crosstalk between neurosecretory and preautonomic networks. Neuron. 2013 Jun 19;78(6):1036–49. doi: 10.1016/j.neuron.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Treweek JB, Jaferi A, Colago EE, Zhou P, Pickel VM. Electron microscopic localization of corticotropin-releasing factor (CRF) and CRF receptor in rat and mouse central nucleus of the amygdala. J Comp Neurol. 2009 Jan 20;512(3):323–35. doi: 10.1002/cne.21884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang B, Shaham Y, Zitzman D, Azari S, Wise RA, You ZB. Cocaine experience establishes control of midbrain glutamate and dopamine by corticotropin-releasing factor: a role in stress-induced relapse to drug seeking. J Neurosci. 2005 Jun 1;25(22):5389–96. doi: 10.1523/JNEUROSCI.0955-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marcinkiewcz CA, Prado MM, Isaac SK, Marshall A, Rylkova D, Bruijnzeel AW. Corticotropin-releasing factor within the central nucleus of the amygdala and the nucleus accumbens shell mediates the negative affective state of nicotine withdrawal in rats. Neuropsychopharmacology. 2009 Jun;34(7):1743–52. doi: 10.1038/npp.2008.231. Epub 2009 Jan 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baiamonte BA, Valenza M, Roltsch EA, Whitaker AM, Baynes BB, Sabino V, Gilpin NW. Nicotine dependence produces hyperalgesia: role of corticotropin-releasing factor-1 receptors (CRF1Rs) in the central amygdala (CeA) Neuropharmacology. 2014 Feb;77:217–23. doi: 10.1016/j.neuropharm.2013.09.025. Epub 2013 Oct 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Torres OV, Gentil LG, Natividad LA, Carcoba LM, O’Dell LE. Behavioral, Biochemical, and Molecular Indices of Stress are Enhanced in Female Versus Male Rats Experiencing Nicotine Withdrawal. Front Psychiatry. 2013 May 20;4:38. doi: 10.3389/fpsyt.2013.00038. eCollection 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang L, Dong Y, Doyon WM, Dani JA. Withdrawal from chronic nicotine exposure alters dopamine signaling dynamics in the nucleus accumbens. Biol Psychiatry. 2012 Feb 1;71(3):184–91. doi: 10.1016/j.biopsych.2011.07.024. Epub 2011 Aug 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lodge DJ, Grace AA. Acute and chronic corticotropin-releasing factor 1 receptor blockade inhibits cocaine-induced dopamine release: correlation with dopamine neuron activity. J Pharmacol Exp Ther. 2005;314:201–6. doi: 10.1124/jpet.105.084913. [DOI] [PubMed] [Google Scholar]
- 39.Willuhn I, Burgeno LM, Groblewski PA, Phillips PE. Excessive cocaine use results from decreased phasic dopamine signaling in the striatum. Nat Neurosci. 2014 May;17(5):704–9. doi: 10.1038/nn.3694. Epub 2014 Apr 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kalivas PW, Duffy P, Latimer LG. Neurochemical and behavioral effects of corticotropin-releasing factor in the ventral tegmental area of the rat. J Pharmacol Exp Ther. 1987 Sep;242(3):757–63. [PubMed] [Google Scholar]
- 41.van Zessen R, Phillips JL, Budygin EA, Stuber GD. Activation of VTA GABA neurons disrupts reward consumption. Neuron. 2012 Mar 22;73(6):1184–94. doi: 10.1016/j.neuron.2012.02.016. Epub 2012 Mar 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tan KR, Yvon C, Turiault M, Mirzabekov JJ, Doehner J, Labouèbe G, Deisseroth K, Tye KM, Lüscher C. GABA neurons of the VTA drive conditioned place aversion. Neuron. 2012 Mar 22;73(6):1173–83. doi: 10.1016/j.neuron.2012.02.015. Epub 2012 Mar 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roberto M, Cruz MT, Gilpin NW, Sabino V, Schweitzer P, Bajo M, Cottone P, Madamba SG, Stouffer DG, Zorrilla EP, Koob GF, Siggins GR, Parsons LH. Corticotropin releasing factor-induced amygdala gamma-aminobutyric Acid release plays a key role in alcohol dependence. Biol Psychiatry. 2010 May 1;67(9):831–9. doi: 10.1016/j.biopsych.2009.11.007. Epub 2010 Jan 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nader K, van der Kooy D. Deprivation state switches the neurobiological substrates mediating opiate reward in the ventral tegmental area. J Neurosci. 1997;17:383–90. doi: 10.1523/JNEUROSCI.17-01-00383.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vargas-Perez H, Ting-A Kee R, Walton CH, Hansen DM, Razavi R, Clarke L, Bufalino MR, Allison DW, Steffensen SC, van der Kooy D. Ventral tegmental area BDNF induces an opiate-dependent like reward state in naive rats. Science. 2009;324:1732–4. doi: 10.1126/science.1168501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Laviolette SR, Alexson TO, van der Kooy D. Lesions of the tegmental pedunculopontine nucleus block the rewarding effects and reveal the aversive effects of nicotine in the ventral tegmental area. J Neurosci. 2002;22(19):8653–60. doi: 10.1523/JNEUROSCI.22-19-08653.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Steffensen SC, Stobbs SH, Colago EE, Lee RS, Koob GF, Gallegos RA, Henriksen SJ. Contingent and non-contingent effects of heroin on mu-opioid receptor-containing ventral tegmental area GABA neurons. Exp Neurol. 2006;202(1):139–51. doi: 10.1016/j.expneurol.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 48.Volkow ND, Fowler JS, Wang GJ, Swanson JM, Telang F. Dopamine in drug abuse and addiction: results of imaging studies and treatment implications. Arch Neurol. 2007;64(11):1575–9. doi: 10.1001/archneur.64.11.1575. [DOI] [PubMed] [Google Scholar]
- 49.Koob GF, Buck CL, Cohen A, Edwards S, Park PE, Schlosburg JE, Schmeichel B, Vendruscolo LF, Wade CL, Whitfield TW, Jr, George O. Addiction as a stress surfeit disorder. Neuropharmacology. 2014 Jan;76(Pt B):370–82. doi: 10.1016/j.neuropharm.2013.05.024. Epub 2013 Jun 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Characteristics of human brain samples. Note that all subjects were selected to be non-smokers without any documented psychiatric disorder (based on DSM-IV) to compare the identification of CRF neurons in the VTA with the results obtained in naive mice.
Fig S1. CRF mRNA levels in the CeA, VTA, and PVN. CRF mRNA levels in the CeA, VTA, and PVN relative to the housekeeping gene GAPDH in mice chronically exposed to saline, measured by RT-PCR (n = 13 or 14 per group). Lower levels of CRF mRNA were observed in the VTA compared with the CeA and PVN (*p < 0.05). Data represent mean ± SEM.
Fig S2. Number of CRF mRNA-positive neurons in the VTA. Number of CRF neurons per section in the VTA (bregma range: −3.28 to −3.88) in naive, nondependent + acute nicotine (1.5 mg/kg), nicotine-dependent (12 days with minipumps), and nicotine-dependent and withdrawn (8 h) groups of mice (n = 6 per group). No difference was observed between groups. Data in Fig. 2 in the manuscript represent combined data from the four groups. Data represent mean ± SEM.
Fig S3. Urocortin in situ hybridization in the VTA and Edinger-Westphal nucleus. Notice the lack of urocortin neurons in the VTA and the prominent population of urocortin-positive neurons in the Edinger-Westphal nucleus. This result was repeated in six naive and six nicotine-dependent mice. Urocortin neurons were never observed in the VTA.
Fig S4. CRF immunohistochemistry. a–d. Quantification of immunodensity in the aVTA and pVTA (a), PVN (b), CeA (c) and IPN (d) in nondependent mice treated with saline (Sal), nicotine-dependent mice (Nic), and nicotine-dependent and -withdrawn mice (WD; n = 12–26 mice per group). Data represent mean ± SEM. e. Example of CRF-immunostained neuropils in the CeA showing CRF varicosities in a transversal fiber. This experiment was performed with one cohort.
Fig S5. Validation of the AAV2-shCRF viral vector. a, Timeline of viral vector infusion and behavioral experiments in mice. b, DNA construct used to produce vectors for CRF silencing (AAV2-shCRF) and control vectors (AAV2-shSCR). ITR, inverted terminal repeat; CMV, cytomegalovirus; EGFP, enhanced green fluorescent protein; hGH polyA, human growth hormone polyadenylation signal. c, Representative images of VTA CRF mRNA-containing cells taken at 20× magnification. d, Number of CRF mRNA-containing cells per section. Quantification of CRF-positive neurons restricted to GFP-positive areas in the pVTA revealed significant downregulation (−26%) of the number of CRF mRNA-containing cells in AAV2-shCRF mice compared with AAV2-shSCR mice (t12 = 2.7, p = 0.020). Data represent mean ± SEM. AAV2-shCRF also decreased the total number of CRF neurons in the entire VTA (including the region without GFP) by 18% (t12 = 2.4, p = 0.033). Notably, the mice were sacrificed approximately 4 weeks into withdrawal after the end of behavioral testing, demonstrating that the upregulation of CRF mRNA in nicotine-withdrawn mice and CRF silencing by the viral vector were both long-lasting. This experiment was performed with one cohort.
Fig S6. Electrophysiological recording of VTA DA neurons. a, Representative trace of a recorded VTA DA neuron. b, Definition of DA neuron action potential characteristics. c, Characterization of VTA dopamine neurons from naive (n = 8 cells), nicotine-dependent AAV2-shSCR (n = 11 cells), and nicotine-dependent AAV2-shCRF (n = 12 cells) mice (n = 3 naive, n = 4 nicotine-dependent AAV2-shSCR, n = 4 nicotine-dependent AAV2-shCRF). This experiment was performed with one cohort.
Fig. S7. Comparison of confirmed TH-positive VTA cells with all cells examined. a, Box-and-whisker plot showing the effects of acute nicotine in naive mice on sIPSC frequency in all VTA cells examined (left; *p = 0.0015, paired t-test) compared with TH+-only VTA cells (right; *p = 0.0051). The total number of cells for each condition is indicated in parentheses. b, Average change in sIPSC frequency in all VTA cells examined (left; *p = 0.0026, one-sample t-test; #p = 0.0003, unpaired t-test) compared with TH+-only VTA cells (right; *p = 0.0128, one-sample t-test; #p = 0.0005, unpaired t-test). Exact t and p values are noted under each condition. The total number of cells for each condition are indicated in parentheses. Data represent mean ± SEM.
Fig S8. Anatomical specificity of AAV2-shCRF VTA injections. Box-and-whisker plot showing that mice with AAV2-shCRF injections outside of the VTA (n = 7) showed an aversive motivational response to nicotine withdrawal that was not significantly different from mice injected with AAV2-shSCR (n = 11).