

Abstract
Diabetes is linked to increased inflammation and susceptibility to certain infectious diseases including tuberculosis (TB). We previously reported that aerosol TB in mice with chronic (≥12 wk) hyperglycemia features increased bacterial load, over-production of several cytokines and increased immune pathology compared to normoglycemic controls. A similar phenotype exists in human diabetics with TB. The mechanisms of increased T cell activation in diabetes are unknown. In the current study, we tested the hypothesis that hyperglycemia modifies the intrinsic responsiveness of naïve T cells to TCR stimulation. Purified T cells from chronically hyperglycemic (HG) mice produced higher levels of Th1, Th2 and Th17 cytokines and proliferated more than T cells from normoglycemic controls after anti-CD3e or antigen stimulation. In this way, naïve T cells from HG mice resembled antigen-experienced cells although CD44 expression was not increased. Chromatin decondensation, another characteristic of antigen-experienced T cells, was increased in naïve T cells from HG mice. That phenotype depended on expression of the receptor for advanced glycation end products (RAGE) and could be reversed by inhibiting p38 MAPK. Chromatin decondensation and hyperresponsiveness to TCR stimulation persisted following transfer of T cells from HG mice into normoglycemic mice. We propose that chronic hyperglycemia causes RAGE-mediated epigenetic modification of naïve T cells leading to p38 MAPK-dependent chromatin decondensation. This pre-activation state facilitates transcription factor access to DNA, increasing cytokine production and proliferation following TCR stimulation. This mechanism may contribute to pathological inflammation associated with diabetes and might offer a novel therapeutic target.
Keywords: diabetes, hyperglycemia, T cells, adaptive immune response, chromatin decondensation
Introduction
Diabetes mellitus is characterized by hyperglycemia due to insulin deficiency caused by autoimmune destruction of pancreatic β cells (type 1 diabetes) or the combination of insulin resistance and relative insulin deficiency related to obesity, physical inactivity and inflammation (type 2diabetes). The incidence of type 2 diabetes has markedly increased in recent decades, mainly in developing countries; the International Diabetes Federation reported that ~80% of diabetic patients live in low or middle-income countries (1). Both types of diabetes are associated with complications stemming from microvascular and macrovascular pathology that is driven primarily by chronically elevated blood glucose (2). It is also recognized that defects in protective immunity occur as a complication of diabetes, notably increasing susceptibility and contributing to adverse outcomes of infection with certain human pathogens including but not limited to Klebsiellapneumoniae, Burkholderiapseudomallei and Mycobacterium tuberculosis (Mtb) (3).
We previously reported that a low aerosol dose of Mtb in chronically hyperglycemic (HG) mice was associated with higher bacterial burden, increased lung immune pathology and higher levels of proinflammatory cytokines compared to euglycemic mice with TB (4). An association between type 2 diabetes and high levels of Th1 and Th2 cytokines has also been described in patients with TB (5–7). Diabetes is associated with a delayed innate response to inhaled bacilli in mice, leads to delayed priming and expression of adaptive immunity and consequently higher lung bacterial burden (8). While higher antigen load may drive cytokine over-expression in diabetic hosts, we speculated that this hyper-inflammation could also result from impaired immune regulation as a complication of hyperglycemia.
In the current study we investigated the effects of hyperglycemia on T cell responses to TCR stimulation in the absence of infection. Our data show that T cells from HG mice have enhanced proliferation and cytokine production in response to stimulation with anti-CD3e mAb or antigen. Naïve T cells from HG mice behave functionally like antigen-experienced T cells despite having similar CD44 expression as euglycemic controls. We found that naïve T cells from chronically HG mice have a significantly higher frequency of decondensed nuclei, as occurs normally after primary activation on initial encounter with antigen. This pre-activation effect in HG mice depends on expression of the receptor for advanced glycation end products (RAGE), presumably in response to endogenous ligands upregulated in diabetic hosts including high mobility group box 1 (HMGB1) and S100 proteins (9,10) in addition to glycated proteins. This T cell phenotype, which is maintained after adoptive transfer into euglycemic hosts, may be a contributing factor in the pathological inflammation characteristic of TB and a wide range of other infectious and non-infectious complications of diabetes.
Material and Methods
Mice
Age matched (6 to 8 wk old) male C57BL/6 mice were obtained from Jackson Laboratory (Bar Harbor, ME), male C57BL/6 OT-II mice were a kind gift from Kenneth Rock (University of Massachusetts Medical School, UMMS) and RAGE−/− mice were donated by MedImmune, LLC (Gaithersburg, MD). Mice were housed in the Animal Medicine facility at UMMS where experiments were performed under protocols approved by the Institutional Animal Care and Use Committee and Institutional Biosafety Committee.
Mice were treated with 150 mg/kg body weight of streptozotocin (STZ, Sigma-Aldrich, St Louis, MO) by i.p. injection dissolved in phosphate citrate buffer (pH 4.5). All mice were at least 8 wk old with minimum weight of 25 g when treated with STZ. Blood glucose measurements were performed with a BD Logic glucometer (Becton Dickinson, Franklin Lakes, NJ) 10 d after STZ treatment and prior to experiment. Mice were considered hyperglycemic if their blood glucose was > 300 mg/dL and euglycemic or control when blood glucose was < 200 mg/dL. Urine ketones were tested by dip stick (LW Scientific Inc., Lawrenceville, GA); mice with diabetic ketoacidosis were excluded from the study. All mice were HG for 12 wk before starting the experiment.
Cell preparation
Splenocyte and lymphocyte isolation
Splenocytes were isolated by mechanical disruption of the spleen and passed through a 40 µm strainer. Red blood cells were lysed, splenocytes were washed and resuspended in RPMI. Lymphocytes were isolated from the inguinal, axillary and cervical lymph nodes. Lymph nodes were minced and incubated with 150 U/ml collagenase type IV and 60 U/ml DNAse in PBS with 5% FBS at 37°C under mild shaking for 40 min. After digestion, cells were passed through a 40 µm strainer, washed with media and pooled together with the splenocytes.
T cells separation
Total T cells, CD4+ or CD8+ T cells were isolated using Dynabeads® Untouched Mouse T cells (Life Technologies, Grand Island, NY) according to manufacturer’s guidelines. Briefly, pooled splenocytes and lymphocytes were resuspended in PBS with 0.1% BSA and 2 mM EDTA and incubated with FBS and the antibody mix at 4°C for 20 min. After the incubation, cells were washed and resuspended in the buffer with the magnetic beads. Cells were incubated at RT under mild shaking for 15 min and were separated with a magnet. T cell purity was determined by flow cytometry (LSRII, BD Biosciences, San Jose, CA) with anti-CD3 (145-2C11), anti-CD4 (GK1.5) and anti-CD8 (53-6.7) antibodies (eBiosciences, Inc., San Diego, CA).
Non-T cell separation
Non T-cells were isolated using Dynabeads® Mouse pan T (Thy1.2) (Life Technologies) by positive selection of T cells by magnetic beads following the manufacturer’s instructions. Pooled splenocytes and lymphocytes were resuspended in PBS with 0.1% BSA and 2 mM EDTA and incubated with the magnetic beads with gentle tilting and rotation at RT for 20 min. Cells were separated via magnet isolation and purity was checked by flow cytometry with anti-CD3, anti-CD4 and anti-CD8 antibodies.
Thymocytes isolation
Thymocytes were isolated by mechanical disruption of the thymus. Double negative (CD4−CD8−), double positive (CD4+CD8+), CD4+ and CD8+ populations were sorted after staining with the specific mAb and using BD FACS Aria cell sorter (BD Biosciences).
Cytokine Detection
Production of cytokines (IFN-γ, TNF-α, IL-12p40, IL-2, IL-10, IL-13, IL-17 and GM-CSF) was determined by multiplex ELISA in accordance with the manufacturer’s guidelines (R&D Systems, Minneapolis, MN) in media from pooled or individual samples of splenocytes from control or HG mice. Media was taken after 24, 48 and 72 h of in vitro stimulation with plate bound anti-CD3e (2.5 µg/ml). Absorbance was analyzed with a Multiskan Ascent microplate spectophotometer (Thermo Fisher Scientific, Waltham, MA).
Intracellular staining for IFN-γ was performed in 2×106 splenocytes 48 h after in vitro stimulation with plate-bound anti-CD3e (2.5 µg/ml). IL-2 (20 ng/ml) was included during the stimulation time and brefeldin A (1:1000 dilution, Biolegend, San Diego, CA) and anti-CD28 (2 µg/ml) were added 4 h before starting the staining. Cells were incubated with 1:50 dilution of Fc-block (BD Biosciences) for 10 min and then with the surface mAb anti-CD4 and anti-CD8 for 20 min. After washing the cells with PBS with 10% FBS and 0.05% sodium azide, cells were fixed and permeabilized with BD Cytofix/Cytoperm solution (BD Biosciencences) for 30 min at 4°C. After washing twice with Perm/Wash Buffer (BD Biosciences), cells were incubated with anti-IFN-γ (XMG1.2, Biolegend) for 30 min at 4°C and data was aquired on an LSRII (BD Biosciences). Results were analyzed using FlowJo v.7 (Tree Star, Ashland, OR). Cells were gated based on FSC/SSC for lymphocytes and FSC-A/FSC-H for singlets.
CFSE labeling
Cells were labeled with 1 µM CFSE (Vybrant® CFDA SE Cell Tracer Kit, Life Technologies) for in vitro experiments or 2 µM for in vivo experiments in PBS at 37°C for 10 min. RPMI with 10% FBS was added after the labeling and the cells were washed twice. Labeling efficiency was determined by both fluorescent microscopy and flow cytometry.
In vitro culture stimulation by anti-CD3e or OVA
96-well round bottom plates were coated with 2.5 µg/mL anti-CD3e mAb o/n at 4°C. Wells were washed twice with PBS and 0.15×106 cells per well were plated for the procedures. Supernatants and cells were taken after 24, 48 and 72 h of stimulation. CD4+ and CD8+ T cells proliferation were analyzed by CFSE dilution by flow cytometry after 48 and 72 h.
OVA stimulation was performed by adding 100 µg/ml of soluble OVA (Sigma-Aldrich) to 0.15×106 OT-II T cells. Cells were taken after 62 and 84 h of stimulation and proliferation of CD4+ T cells was assessed by CFSE dilution by flow cytometry.
In vitro crisscross culture
30% of negatively isolated CFSE labeled T cells from control or HG OT-II mice were incubated with 70% non-T cells from control or HG C57BL/6 mice in a 96-well round bottom plate. Stimulation with OVA was performed for 87 h and proliferation of CD4+ or CD8+T cells was analyzed by CFSE dilution by flow cytometry.
In vivo adoptive transfer
Pooled splenocytes and lymphocytes from control or HG OT-II mice were CFSE labeled and 20×106 cells were transferred to control or HG C57BL/6 mice via tail vein injection. After 24 h, 10 µg OVA in yeast β-glucan particles (YGP; provided by Dr. Gary Ostroff at UMMS) were delivered to the lungs by intra-tracheal instillation. Thoracic lymph nodes were harvested after 62 hand proliferation of CD4+ T cells was assessed by flow cytometry and CFSE dilution.
Flow Cytometry
Approximately, 1×106 cells were used for flow cytometry staining for surface markers as described above. The following mAb were used: anti-CD3, anti-CD4, anti-CD8, anti-CD44 (IM7), anti-CD62L (MEL-14), anti-CD19 (eBio1D3), anti-CD11b (N418) and anti-CD11c (M1/70). Regulatory T cells were stained using mouse Foxp3 buffer set (BD Biosciences) following manufacturer’s guidelines. Briefly, 2×106 cells were stained for surface markers CD4 and CD25 (PC61) as previously described. Cells were washed and fixed for 30 min at 4°C. Following several washes with the permeabilization buffer cells were incubated in this same buffer for 30 min at 37°C. The ab anti-Foxp3 (FJK-16s) was used to stain the cells for 20 min at RT. Data was acquired on an LSRII and analyzed with FlowJo v.7. Cells were gated based on FSC/SSC for lymphocytes and FSC-A/FSC-H for singlets.
Immunoblotting
Total protein was obtained from isolated T cells from control and HG mice and quantified by BCA assay kit (Pierce Biotechnology, Rockford, IL). Approximately, 20 µg of total protein were SDS-PAGE separated and transferred to a PVDF membrane (Bio-Rad Laboratories, Hercules, CA). After blocking the membrane with 5% BSA in TBST for 1 h, it was incubated with antibodies rabbit anti-mouse for P-p38 (Thr180/Tyr182), total p38, P-ERK1/2 (Thr202/Tyr204), ERK1/2, P-Akt (Ser473), Akt, P-FoxO3a (Thr32) and FoxO3a (Cell signaling Technology Inc., Danvers, MA) o/n under mild shaking at 4°C. The membrane was washed with TBST and incubated with the secondary antibody goat anti-rabbit Ig-G horseradish peroxidase-conjugated (Santa Cruz Biotechnology, Inc., Dallas, TX) for 1 h. After several washes with TBST, the membrane was incubated with SuperSignal West Pico chemiluminiscence substrate (Pierce Biotechnology) for 5 min, film was exposed and developed. Bands were quantified by ImageJ 1.46r (NIH, Bethesda, MD).
T cell and thymocytechromatin decondensation analysis
Control and HG mice T cells were isolated from pooled splenocytes and lymphocytes and then CD25+ T cells were depleted by magnetic bead separation (MiltenyBiotec Inc., Auburn, CA). The remaining cells were fixed with 4% paraformaldehye for 10 min at RT or o/n at 4°C. Four populations of thymocytes from control and HG mice (CD4+, CD8+, double-positive and double-negative) were fixed and individually sorted using a FACS Aria II cell sorter (BD Biosciences). T cells and thymocytes were adhered to slides by cytocentrifugation, washed with PBS and mounted with ProLong® Gold antifade reagent with DAPI (Life Technologies). Images were taken on a fluorescent microscope Nikon Eclipse E400. Chromatin decondensation was assessed by measuring the diameter of the nucleus using ImageJ. Percentages of T cells with nuclear diameter >6 µm and of thymocytes with diameter >6.5 µm were calculated.
Bone marrow chimeras
Bone marrow cells were isolated from control CD45.1 or HG CD45.2 mice. T cells were depleted or not from the bone marrow by CD90.2 (Thy1.2) microbeads (MiltenyBiotec Inc.). Chimeras of bone marrow from control and HG mice were transferred via tail vein into lethally irradiated CD90.1 C57/BL6 mice. After 7 wk, pooled splenocytes and lymphocytes from control or HG donor mice were stained with anti-CD90.1, anti-CD90.2, anti-CD3, anti-CD25, anti-CD19, anti-CD45.1 and anti-CD45.2 and sorted gating on CD90.2+ and then CD3+CD19−CD25− for the donor cells and then CD45.1+ for control and CD45.2+ for HG cells. T cells were fixed, adhered to slides by cytocentrifugation and mounted with ProLong® Gold antifade reagent with DAPI. Images were taken on a fluorescent microscope and the percentage of cells with the nucleus diameter >6 µm was analyzed for each population.
Statistics
Statistical analysis was performed using Sigma Plot 11.0 and Graph Pad Prism 6.0 and variance test was performed before the statistical analysis. When normality was confirmed, data were analyzed by Student’s t test or One Way ANOVA. Non-parametric data were analyzed by the Mann-Whitney U-test. The coefficient of correlation for DAPI fluorescent intensity and nucleus diameter in T cells was calculated by Pearson Product Moment Correlation. p values <0.05 were considered statistically significant.
Results
Hyperglycemia is associated with increased cytokine production by T cells
Hyperglycemic mice with TB over-express a broad range of Th1, Th2 and Th17 cytokines (4) and a similarly augmented cytokine response by diabetic humans with TB has been reported (5–7). To investigate whether the cytokine over-expression phenotype is restricted to antigen-specific responses in HG mice or if it is a generalized phenomenon, splenocytes from uninfected euglycemic control mice or HG mice were stimulated ex vivo with plate-bound anti-CD3e. Multiplex analysis of pooled samples demonstrated greater production of Th1, Th2 and Th17 cytokines from the HG group (Fig. 1A–C). Parallel samples from individual mice were tested by ELISA and confirmed the relative over-expression of IFN-γ, IL-10 and IL-17 by anti-CD3e stimulated splenocytes from HG mice (Fig. 1D). Basal cytokine expression in unstimulated splenocytes was below the limit of detection in samples from control and HG mice. Further confirmation of cytokine production by CD4+or CD8+T cells was done for IFN-γ by intracellular staining (Fig. 1E). Despite a trend for an increase in the percentage of CD4+ IFN-γ+T cells from HG mice, only the mean fluorescent intensity (MFI) was significantly higher in HG mice. There was no change in the percentage of IFN-γ+cells among CD8+ T cells but the MFI of IFN-γ staining was higher in the HG mice (Supplemental Fig. 1A).
Figure 1. Cytokine production by plate-bound anti-CD3e stimulated splenocytes from control and HG mice.

Media from cultures of splenocytes isolated from control or HG mice was collected after 24, 48 and 72 h of plate-bound anti-CD3e stimulation. A, Pooled samples from 3 individual cultures were analyzed for Th1 cytokines IFN-γ, TNF-α, IL-12p40 and IL-2 by multiplex ELISA. B, Pooled samples from 3 individual cultures were analyzed for Th2 cytokines IL-10 and IL-13 by multiplex ELISA. C, Pooled samples from 3 individual cultures were analyzed for Th17 cytokines IL-17 and GMCSF by multiplex ELISA. D, IFN-γ, IL-10 and IL-17 production were analyzed in media from individual cell cultures by ELISA. Line represents the mean of the values (n=5). E, Representative histogram of IFN-γ staining (left), percentage of IFN-γ+ cells (center) and mean fluorescent intensity (MFI) (right) in CD4+ T cells stimulated with plate-bound anti-CD3e (2.5 µg/ml) for 48 h. Statistical differences were analyzed by Student’s t test for each time point, *p<0.05, **p<0.01.
Hyperglycemia is associated with increase in CD4+ T cell proliferation
To determine whether the effects of hyperglycemia on T cell activation were limited to cytokine expression or extended to cell cycle regulation, splenocytes or lymphocytes from control or HG mice were isolated and labelled with CSFE as described in Materials and Methods. Cells from control and HG OT-II mice were stimulated in vitro with OVA for 62 and 84 h (Fig. 2A). The proportion of CD4+ T cells with CSFE dilution was significantly higher in the HG group. Similar results were obtained using cells from wildtype C57BL/6 control or HG mice stimulated with plate-bound anti-CD3e for 48 and 72 h (Supplemental Fig. 1B). Furthermore, the accelerated proliferative response of HG CD4+ T cells to anti-CD3e stimulation was not shared by CD8+ T cells (Supplemental Fig. 1C). The hyper-proliferative phenotype was observed only with T cells from mice that were hyperglycemic for ≥12 wk; it was not detected using cells from mice that were hyperglycemic for ≤4 wk (Supplemental Fig. 1D). The requirement for prolonged hyperglycemia is consistent with mechanism of other diabetic complications and mirrors the acquisition of TB susceptibility in diabetic mice (4).
Figure 2. In vitro and in vivo T cell proliferation in OT-II control and HG mice recognizing OVA.

A, Representative histograms (left) of CD4+ splenocytes and lymphocytes proliferation from control or HG mice stimulated with 100 µg/ml OVA in vitro for84 h. Percentage of CD4+ T cells proliferation from splenocytes (n=4–5) or lymphocytes (n=2–4) in control or HG mice stimulated with 100 µg/ml OVA in vitro for62 h and 84 h (right graphs). Proliferation of CD4+ T cells was detected by flow cytometry and CFSE dilution. B, Splenocytes depleted of T cells by negative selection with magnetic beads (non-T cells) from control or HG mice were used as APC. T cells from euglycemic OT-II mice or HG OT-II mice were isolated by negative selection by magnetic beads and labeled with CFSE. Purity of T cells was >75% and of non-T cells >97% after isolation. In vitro cultures of 30% T cells and 70% non-T cells were stimulated with 100 µg/ml OVA in vitro for 87 h (n=3). Proliferation of CD4+ T cells was detected by flow cytometry and CFSE dilution. Two-way ANOVA showed no statistically significant interaction between the source of T cells and of APC. C, Pooled splenocytes and lymphocytes from euglycemic OT-II mice (n=4) or HG OT-II mice (n=7–8) were labeled with CFSE and adoptively transferred to euglycemic control or HG mice. Beta-glucan particles loaded with OVA were delivered to the lungs by tracheal instillation followed by harvest of the thoracic lymph node 62 h later. Lymphocytes were isolated and proliferation of CD4+ T cells was detected by flow cytometry and CFSE dilution. All experiments were repeated at least twice. Statistical differences were analyzed by Student’s t test and two-way ANOVA followed by Bonferroni’s post analysis test, *p<0.05, **p<0.01.
While we focused on effects of hyperglycemia on intrinsic T cell functions, a concurrent alteration of APC function might also occur in the context of diabetes. To address that question we measured the proliferation of OVA-stimulated OT-II T cells co-incubated with autologous APC from control or HG mice. Increased proliferation of HG compared to control CD4+ T cells was observed in cultures with HG or control APC, indicated that the phenotype of accelerated proliferation could not be attributed to an effect of diabetes on APC function (Fig. 2B). Indeed, the proliferation of control T cells in the presence of HG APC was slightly lower than that seen in the presence of control APC.
To further investigate the impact of diabetes on APC function and to validate the physiological relevance of results from in vitro culture conditions, we performed a reciprocal adoptive transfer experiment between control and HG OT-II mice. Following transfer of CFSE-labelled donor T cells, recipient mice were challenged by tracheal instillation of OVA packaged in β-glucan particles. Lung-draining lymph nodes were harvested 62 h later and proliferation of transferred CD4+ T cells was assessed by flow cytometry (Fig. 2C). Using this in vivo system, we observed that the hyper-proliferative phenotype of HG T cells was maintained following transfer to a euglycemic host, supporting our interpretation to our in vitro data. Furthermore, the accelerated proliferation of HG T cells was reduced upon transfer to diabetic hosts. Thus, chronic hyperglycemia appeared to exert opposing effects on T cells and at least certain APC populations in vivo. Alternatively, the in vivo results might be explained by increased migration of activated, proliferating T cells from the spleen and lymph node. In either case, the in vivo data were consistent with the increased proliferative response of HG T cells observed in vitro.
T cell and APC populations in control and hyperglycemic mice
Naïve T cells (CD44−CD62Lhi) become primed during antigen presentation by DC and they proliferate to become effector cells, while ~10% survive to become memory T cells (CD44+) with self-renewal capacity (11). Memory T cells have a reduced threshold for proliferation and cytokine production within several hours of TCR re-stimulation. The hyper-proliferative phenotype we observed in vitro and in vivo would be predicted to lead to an increased proportion of antigen-experienced T cells in diabetic hosts. In fact, we found a similar basal percentage of naïve and memory T cells in control and HG mice (Fig. 3A). We also compared APC from these two groups, finding no impact of chronic hyperglycemia on the relative populations of CD19+, CD11c+ and CD11b+ cells (Fig. 3B). Therefore, it appeared unlikely that the observed functional differences in T cell activation were the result of a basal increase in the population of antigen-experienced T cells or a shift in steady state of APC populations in chronically HG mice. This increased proliferation of HG T cells could also be due to a defective population of regulatory T cells. We analyzed the proportion of regulatory T cells (CD4+Foxp3+CD25+) in control and HG mice (Fig. 3C), finding an increased percentage of these cells in HG mice.
Figure 3. Characterization of CD4+ and CD8+ T cells and APC phenotype and regulatory T cells in control and HG mice.

Splenocytes and lymphocytes were isolated from control or HG mice and stained for flow cytometry for the markers CD4, CD8, CD44, CD62L, CD19, CD11b, CD11c, CD25 and Foxp3. A, Percentage of naïve T cells, CD44−CD62Lhigh, and memory T cells, CD44+ (n=4). B, Percentage of other cell populations in spleen and lymph nodes (n=5). C, Percentage of regulatory T cells (CD4+CD25+Foxp3+). All experiments were repeated at least three times. Statistical differences were analyzed by Student’s t test.
Increased MAPK pathway activation in T cells from hyperglycemic mice
The breadth of the activation responses observed in T cells from HG mice suggested the involvement of one or more downstream regulators of TCR signaling. Candidates for such regulators include the MAPK family pathways (12, 13). We therefore compared basal, PMA- and anti-CD3e/CD28-stimulated p38 and ERK phosphorylation in purified T cells from control or HG mice (Fig. 4). Under basal conditions, p38 phosphorylation was significantly greater in T cells from HG mice whereas no difference in basal ERK phosphorylation was identified in T cells from control and HG mice. Following stimulation with PMA both p38 and ERK phosphorylation were significantly elevated in the HG group. When cells were stimulated with anti-CD3e/CD28, only p38 presented significantly higher activation in T cells from HG mice. This up-regulation of the MAPK pathway in T cells from HG mice could explain the augmented cytokine production and proliferation phenotype, confirmed by the reduction of cytokine levels with p38 inhibitor (Supplemental Fig. 2A) in splenocytes stimulated with plate-bound anti-CD3e for 48 h. Since differential activation was identified using PMA stimulation which bypasses the TCR, it suggests that hyper-activation phenotype in diabetes is not due to any perturbation of the cell surface receptor components.
Figure 4. MAPK activation in control and HG mice.

MAPK phosphorylation in T cells isolated by negative T cell selection by magnetic beads from control or HG mice, unstimulated or stimulated with 40 nM PMA or anti-CD3e/CD28 (2.5 µg/ml) for 15 min. A, p38 phosphorylation and B, ERK1/2 phosphorylation, mean ± SD (n=6–8). All experiments were repeated at least twice. Statistical differences were analyzed by Student’s t test, **p<0.01, ***p<0.001.
Stimulation of the TCR recruits and activates PI3K that modulates the activation of Akt and leads to the downstream inactivation of Forkhead box transcription factor family O (FOXO) transcription factors or mammalian target of rapamycin complex 1 (mTORC1) activation regulating T cell proliferation, growth and glycolytic metabolism (14). The increased proliferation observed in T cells from HG mice could reflect differential regulation in the Akt/mTOR pathway as well. Increased Akt activation in renal tissue from acute HG mice has been described () but this has not been previously examined in T cells. In contrast to the effects of diabetes on the kidney, we found no difference in basal Akt or FOXO3 phosphorylation between control and HG mice (Supplemental Fig. 2B). These results rule out Akt signaling as a mechanism contributing to the hyperproliferative phenotype of T cells from HG mice.
Hyperglycemia induces chromatin decondensation in T cells
Naïve T cells are characterized by a quiescent state, G0 phase, in which nuclear chromatin is condensed, leaving many gene promoters inaccessible by the transcription factors. When T cells are stimulated via TCR, chromatin is decondensed and the cells become larger and develop proliferative capacity (16). Although the proportion of antigen-experienced CD44+ T cells was not increased in HG mice, their enhanced activation response to TCR stimulation was reminiscent of previously activated T cells. We therefore compared the status of chromatin condensation/decondensation in T cells of HG and control mice. Cytospin preps of CD25− T cells were stained with DAPI and examined by fluorescent video microscopy (Fig. 5A) to measure the distribution of nuclear diameters grouped in 12 bins (Fig. 5B). Using a cutoff point of 6 µm, which reflects the upper limit of nuclear diameters of mature T cells with visibly condensed chromatin, we determined that a higher proportion of T cells from HG mice had decondensed nuclei (30.3 ± 6.9%) as compared to controls (10.8 ± 1.7%; Fig. 5C). As expected, stimulation with PMA for 5 h orplate-bound anti-CD3e for 24 h increased the percentage of T cells with nuclear diameter >6 µm and abrogated the differences between control and HG mice observed in unstimulated T cells (Fig. 5D). The enlarged nuclei also had lower fluorescence intensity, and there was an inverse correlation between the nucleus diameter and fluorescence signal (coefficient of correlation R=−0.81, p<0.0001) (Fig. 5E), confirming that the chromatin is decondensed as described by Rawlings et al. (16). By isolating CD4+ and CD8+ T cells we determined that only CD4+ T cells from HG mice presented an increased proportion of cells with nuclear diameter >6 µm (Supplemental Fig. 2C).
Figure 5. Chromatin decondensation in T cells from control and HG mice.

T cells from control or HG mice were isolated by negative selection with magnetic beads. CD25+ cells were depleted by magnetic beads and the remaining cells were DAPI stained. T cell purity was analyzed by flow cytometry staining and >90% of the cells were CD4+ or CD8+ and CD25−. The diameter of the nucleus was measured in individual cells by fluorescence microscopy. A, Representative images of T cells stained with DAPI from control and HG mice, scale bar indicates 10 µm. B, Nucleus diameter measurements grouped by size in the indicated bins, expressed as the % of all T cells, mean ± SD (n=5–6). C, Percentage of T cells with nuclei >6 µm in diameter (n=5–6). D, Percentage of T cells stimulated or not with 40 nM PMA or plate-bound anti-CD3e for 5 and 24 h, respectively, with nuclei >6 µm in diameter (n=4–6). E, Correlation of Pearson between DAPI fluorescence intensity and nucleus diameter in T cells from a representative control mouse stimulated with anti-CD3e for 24 h. F, Percentage of T cells with nuclei >6 µm in diameter following treatment with 1 µM SB203580 (p38 inhibitor) or 1 µM U0126 (ERK inhibitor) for 5 h (n=3–5). G, Percentage of T cells with nuclei >6 µm in diameter following stimulation with 40 nM PMA and treatment with 1 µM SB203580 or 1 µM U0126 for 5 h (n=3–5). All experiments were repeated at least three times. Statistical differences were analyzed by Student’s t test, *p<0.05, **p<0.01, ***p<0.001.
Activation of PKC modulates histone acetylation and therefore chromatin decondensation (17). Since the biochemical mechanisms of diabetic complications include the activation of certain PKC isoforms (2) we tested the effect of PKC inhibitors on nuclear condensation/decondensation in unstimulated T cells from HG and control mice. Cells were treated with two doses of the PKC inhibitor Gö6983: 7 nM for inhibition of PKC-α and β and 60 nM for inhibition of PKC-ζ. Neither of these treatments changed the percentage of T cells with decondensed chromatin from control or HG mice (Supplemental Fig. 2D). The Ca2+-calcineurin pathway is involved in T cell proliferation and effector functions (18) and also in the maintenance of anergy (19). Treatment with cyclosporine A to inhibit calcineurin did not change the difference in basal nuclear diameter of untreated T cells from HG mice (Supplemental Fig. 2E) indicating that the effect of the diabetic environment on chromatin decondensation is not calcineurin-dependent.
Since we observed that hyperglycemia was associated with p38 MAPK activation under basal conditions and activation of both p38 and ERK1/2 following stimulation with PMA, we tested the possible involvement of these MAPKs in T cell chromatin decondensation. Treating unstimulated T cells with the p38 inhibitor SB203580 reduced the proportion of cells with nuclear diameter >6 µm in the HG mice, eliminating the difference between the control and HG groups (Fig. 5F). In contrast, the ERK inhibitor U0126 did not reduce the proportion of T cells with decondensed nuclei in the HG group. Chromatin decondensation following TCR activation with anti-CD3ewas slightly inhibited by both inhibitors in control T cells (Fig. 5G). These results indicated that increased basal activation of p38 is required to maintain the elevated proportion of T cells with decondensed nuclei from HG mice and the TCR signaling in both control and HG mice. Increase CD44 expression follows TCR stimulation and is characteristic of memory T cells. We stimulated HG and control T cells with plate-bound anti-CD3e and observed similar increase in the proportion of CD44+ cells (Supplemental Fig. 2F). Surface expression of CD44 on activated T cells was not inhibited by co-treatment with SB203580 or U0126. These results indicate that p38 and ERK are not required for induction of CD44 following TCR stimulation. Since chromatin decondensation was dependent on p38 activation, we conclude that discrete signal pathways regulate chromatin decondensation and CD44 expression in T cells.
The effect of hyperglycemia on chromatin decondensation in the thymus
Chromatin condensation in T cells occurs during thymic development in the transition from double-negative to double-positive thymocytes (16). We considered whether chromatin decondensation of mature T cells in HG mice reflected changes exerted during thymocyte development. Double-negative, double-positive and single-positive thymocytes from control and HG mice were purified and stained with DAPI to assess nuclear diameter. There was no difference in the proportion of double-negative and double-positive thymocytes with nuclear diameter >6 µm between control and HG mice (Supplemental Fig. 3A). Consistent with normal trends in thymocyte development, the proportion of cells with decondensed nuclei was lower in double-positive compared to double-negative cells. In marked contrast to results obtained with mature splenic and lymph node T cells, the nuclear diameter of single-positive thymocytes was significantly lower in HG mice compared to controls (Supplemental Fig. 3A). The total number of the different cell populations in the thymus was not altered in HG mice (Supplemental Fig. 3B) and the total cell number in thymus was similar in the two groups (Supplemental Fig. 3C). These data suggest that the diabetic environment does influence the state of chromatin condensation in single-positive thymocytes but in a way opposite to that seen in mature peripheral T cells. It remains to be determined whether these two phenomena are linked or discrete effects.
The effects of hyperglycemia on T cell function do not involve bone marrow progenitors
Epigenetic mechanisms may drive some complications of diabetes and lymphocytes have been identified as targets for such chromatin modification (20, 21). Epigenetic effects of chronic hyperglycemia are thought to be responsible for the phenomenon of metabolic memory in diabetes in which complications progress after normalization of plasma glucose (22). We therefore questioned whether chromatin modification of lymphoid precursors in bone marrow was responsible for the phenotypic effects we observed in mature peripheral T cells from HG mice. To test that hypothesis we created bone marrow chimeras that were lethally irradiated, control mice were reconstituted with bone marrow progenitors transferred from HG and control donor mice. After 7 wk, CD25− T cells were isolated from chimeric mice and stained with DAPI to assess nuclear diameter. In this experiment, a significantly higher proportion of T cells from mice reconstituted by HG donors exhibited chromatin decondensation (Fig.6A, 6B). This indicated that bone marrow progenitors from HG donors retained the chromatin decondensation phenotype despite differentiation and maturation in a euglycemic environment. Adult bone marrow contains hematopoietic stems cells that can give rise to all blood cells types, Thy1− common lymphoid progenitors with restricted lymphoid reconstitution capacity, and Thy1+ prothymocytes that are periodically mobilized from the bone marrow and imported to the thymus (23, 24). Bone marrow also contains a fraction of mature T cells that migrate to the bone marrow after priming and are capable of mounting secondary response (25). To assess the stage of T cell development where the effects of hyperglycemia are mediated we reconstituted euglycemic recipient mice with bone marrow from HG or control donor mice that was depleted of Thy1.2+ cells prior to transfer. Depletion of Thy1.2+ donor cells abrogated the difference in mature T cell nuclear diameter between groups (Fig. 6C). If epigenetic reprogramming is responsible for the observed mature T cell phenotype, this must occur at a developmental stage later than common lymphoid progenitor cells. Since bone marrow containing Thy1.2+ cells reconstituted the mature T cell chromatin decondensation phenotype of donor HG mice, this could have reflected modification of prothymocytes or resident mature T cells. Regardless of their identity, these transferred cells retained an increased proportion with chromatin decondensation even in a euglycemic environment, along with the hyper-proliferative phenotype demonstrated in the experiments shown in Figure 2C.
Figure 6. Chromatin decondensation in T cells from lethally irradiated control mice reconstituted with bone marrow from control or HG mice.
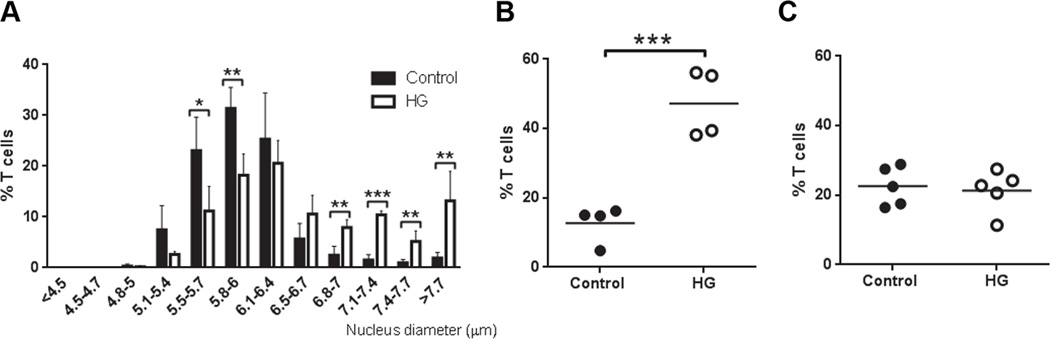
Seven weeks after bone marrow transfer, T cells were isolated from recipients by negative selection using magnetic beads. CD25+ cells were depleted by magnetic selection and the remaining T cells were DAPI stained for measurement of nuclear diameter. A, Nucleus diameter measurements grouped by size in the indicated bins, expressed as the % of all T cells, mean ± SD (n=4). B, Percentage of T cells with nuclei >6 µm in diameter, isolated from chimeric mice reconstituted with whole bone marrow from control or HG donor mice (n=4). C, Percentage of T cells with nuclei >6 µm in diameter, isolated from chimeric mice reconstituted with bone marrow depleted of Thy1.2+ cells prior to transfer (n=4). All experiments were repeated at least twice. Statistical differences were analyzed by Student’s t test, *p<0.05, **p<0.01, ***p<0.001.
Chromatin decondensation in T cells from HG mice depends on RAGE expression
Endogenous ligands for RAGE, including S100 proteins, HMGB1 and glycated proteins are elevated in hyperglycemic hosts and pathological RAGE signaling has been implicated in diabetic complications (26). To investigate the possible involvement of RAGE signaling in the T cell chromatin decondensation and activation phenotypes linked to chronic hyperglycemia, we prepared T cells from control RAGE−/− mice and HG RAGE−/− mice 12 wk after the induction of diabetes with STZ. The absence of RAGE expression abrogated in the effects of chronic hyperglycemia on chromatin decondensation in resting T cells (Fig. 7A) and reduced the ~2-fold increase in basal p38 MAPK phosphorylation that we previously identified in wildtype HG mice (Fig. 4A) to ~0.5-fold (Fig. 7B). RAGE deficiency did not inhibit the increased phosphorylation of p38 or ERK stimulated by PMA treatment of RAGE−/− control or HG T cells (Fig. 7B), nor did it influence Akt phosphorylation (Supplemental Fig. 4A). To further confirm that chromatin decondensation is dependent on RAGE, we incubated CD4+ T cells with high glucose media (30 mM D-glucose) or media containing 100 ng/ml S100A1 for 5 h and analyzed the nucleus diameter (Supplemental Fig. 4B, 4C). Only cells treated with S100A1 had a significant higher proportion of cells with nuclear diameter >6 µm. Cytokine production was analyzed in plate-bound anti-CD3e stimulated splenocytes from RAGE control and HG mice by ELISA as an evaluation of the final result of the signaling (Fig. 7C). As expected, no differences between control and HG RAGE−/− mice were observed. These data implicate the RAGE pathway in the basal increasedp38 MAPK signaling, chromatin decondensation and cytokine production that occurs in chronically hyperglycemic hosts.
Figure 7. Chromatin decondensation, MAPK phosphorylation and cytokine production in T cells from control and HG RAGE−/− mice.

A, Percentage of T cells with nuclei >6 µm in diameter (n=5) from control or HG WT or RAGE−/− mice unstimulated (left graph) or stimulated with 40 nM PMA (right graph) for 5 h. B, p38 (left graph) and ERK1/2 (right graph) phosphorylation in T cells from control or HG RAGE−/− mice, unstimulated or stimulated with 40 nM PMA for 15 min (n=5). C, Production of IFN-γ, IL-10 and IL-17 in splenocytes from control or HG RAGE−/− mice stimulated with plate-bound anti-CD3e for 24, 48 and 72 h. All experiments were repeated at least twice. Statistical differences were analyzed by Student’s t test, *p<0.05, ***p<0.001.
Discussion
We previously reported that mice with chronic but not acute STZ-induced diabetes have increased susceptibility to Mtb characterized by higher plateau lung bacterial load and increased immune pathology (4). This phenotype mimics at least some manifestations of TB disease in diabetic people (27). Susceptibility in mice is at least partly attributable to delayed recruitment of naïve myeloid cells into alveoli harboring resident macrophages initially infected by inhaled Mtb (8). This defect of innate immunity results in delayed initiation of the adaptive immune response that is required to restrict bacterial replication. The subsequently increased inflammatory response in the lung might simply result from a higher load of pathogen-associated molecular patterns and antigen but a diabetes-associated impairment of immune regulation could also be a contributing factor. Diabetes is associated with chronic inflammation and its delayed resolution in non-infectious inflammatory conditions (28, 29). Patients with metabolic syndrome, with a major risk factor for type 2 diabetes, have increased serum concentrations of both Th1 and Th2 cytokines that correlate positively with fasting glucose levels (30).
We studied naïve T cells isolated from control and HG mice in order to investigate whether diabetes causes an intrinsic perturbation of T cell activation in the absence of infection. Our data show that T cells from mice with chronic (≥12 wk) but not acute (≤4 wk) hyperglycemia express a broad range of cytokines at higher levels and proliferate more than T cells from control mice after TCR stimulation in vitro and in vivo. In this respect, naïve T cells from HG mice behave like antigen-experienced T cells despite no increase of CD44 expression. Developing T cells normally undergo nuclear condensation in the transition from double to single positive in the thymus. Compacted nuclear chromatin acts as a barrier to inadvertent T cell activation by limiting access of transcription factors to binding sites on DNA. Priming of naïve T cells by DC that present high affinity antigen to the TCR in the context of costimulatory molecules overcomes this barrier by triggering chromatin decondensation. This allows transcription factors such as Stat5to bind promoters involved in T cell proliferation and cytokine expression (15). Our data show that naïve T cells in diabetic hosts are pre-activated via RAGE signaling and have an exaggerated response to initial TCR stimulation. This dysregulated response might contribute to excessive immune pathology in the context of chronic infections like TB.
Several biochemical mechanisms for diabetic complications have been identified including increased polyol pathway flux, increased hexosamine pathway flux, increased protein glycation that alters function and produces ligands for RAGE, and PKC activation resulting from increased diacylglycerol synthesis and increased RAGE signaling (2). All these pathways lie downstream of a common initiating factor: hyperglycemia-driven mitochondrial overproduction of superoxide. We found that RAGE deficiency reversed the pre-activated phenotype of T cells in HG mice, linking that phenotype to an established diabetic complication pathway. Hyperglycemia promotes increased expression of RAGE and increased levels of its endogenous ligands S100 calgranulins and HMGB1 that bind with higher affinity than AGEs and are likely the most physiologically relevant ligands even in diabetic hosts (9, 10). We confirmed in vitro that S100A1, and not high glucose media, was enough to increase the percentage of T cells with the nucleus decondensed.
Ligand binding to RAGE activates several signaling pathways including NF-kB, focal adhesion kinase, PI3/Akt, Rho GTPase, Jak/STAT, Src kinases and MAPK (31–35). Histone acteyltransferases such as p300 are activated through some of these pathways, including p38 MAPK, and in turn are linked to chromatin decondensation (36–40). We found that treating unstimulated T cells from HG mice with SB203580 (p38 inhibitor) reduced the proportion of cells with decondensed chromatin to approximately the level present in control mice, indicating a role for p38 MAPK in this process. Nuclear size in T cells from control mice was not further reduced by p38 inhibition. It appears that p38 was involved in the basal chromatin decondensation of HG T cells but our results from adoptive transfer and radiation chimera experiments with euglycemic hosts suggest that the increased basal p38 activation in HG T cells does not depend on continuous RAGE stimulation but instead reflects a persistent alteration of cell function suggestive of epigenetic remodeling. Increased nuclear decondensation was only observed in CD4+ T cells from HG mice and this correlated with increased proliferation. This specific pre-activation of CD4+ T cells by hyperglycemia might be explained by a different role of p38 in T cell subsets; proliferation in CD4+ T cells versus apoptosis in CD8+ T cells (41).
A growing body of evidence indicates that epigenetics is an important mechanism of diabetic complications and metabolic memory. This latter concept refers to the progression of diabetic complications at the tissue level despite improved glycemic control. It is a well-recognized phenomenon in human diabetes and in animal models (22). The persistence of chromatin decondensation in unstimulated T cells derived from HG donor mice for up to 7 wk after transfer into euglycemic hosts is consistent with an epigenetic effect. Our data suggest that this occurs at a stage of T cell development later than the common lymphoid progenitor. We found that chronic hyperglycemia led to an increased prevalence of nuclear condensation in single-positive thymocytes while the opposite was observed in peripheral blood T cells. It is presently unknown whether these distinct perturbations of chromatin structure are linked to single underlying mechanism or if they are discrete effects that occur in the diabetic environment. The lack of a hyper-responsive T cell phenotype in mice that were hyperglycemic for <4 wk argues strongly against this being an artifact of the single dose of STZ given to induce diabetes.
Primary activation of naïve T cells is associated with chromatin decondensation and increased surface expression of CD44 that is maintained on memory T cells. Memory T cells mount a faster and quantitatively stronger response to TCR re-stimulation that occurs in naïve T cells. CD44 contributes to this augmented response by clustering TCR/CD3 complexes, by providing its associated Src family kinases Fyn and Lck, and by recruiting additional membrane receptors (42). Since the regulatory T cell population was not defective in HG mice we questioned whether the augmented response of T cells from HG mice reflected an increase of memory T cells (CD44+) in the diabetic host environment but found no evidence for this. The increased proportion of T cells with decondensed chromatin in HG mice was not matched by any change in the proportion of memory T cells compared to controls. This suggests that distinct pathways downstream of TCR regulate CD44 expression and chromatin decondensation. That conclusion is further supported by our finding that the marked increase of CD44 expression in cells stimulated by plate-bound anti-CD3e was not modulated by the p38 inhibitor SB203580 which was found to reverse chromatin decondensation in T cells from HG mice. We also found no evidence that diabetes repressed CD44 expression that was equally induced by anti-CD3e stimulation of T cells from control or HG mice (Supplemental Fig. 2F). Our data suggest that prolonged hyperglycemia expands a population of pre-activated T cells independent of TCR signaling or CD44, which function like antigen-experienced cells.
In summary, we found that naïve T cells from chronically HG mice mount a stronger response to TCR stimulation than T cells from control mice. In this way, these naïve cells act like antigen-experienced T cells yet they lack the increased expression of CD44 that characterizes memory T cell generated after normal immune priming. The breadth of cytokines expressed, spanning Th1, Th2 and Th17, suggests that hyperglycemia exerts a global effect on T cells rather than one limited to an isolated signaling pathway. We propose that chromatin decondensation, which is increased in T cells from HG mice, enables this widespread activation response by facilitating transcription factors access to their cognate binding sites in promoters of genes linked to cytokine expression and proliferation. Increased chromatin decondensation in HG mice depends on RAGE expression and is maintained by increased basal activation of p38 MAPK. The persistence of this phenotype for weeks after T cell transfer from a diabetic to a euglycemic host suggests an effect of epigenetic remodeling linked to RAGE signaling. This model is consistent with evidence that RAGE expression is increased in T cells from people with diabetes and that diabetes promotes epigenetic remodeling in lymphocytes (20, 22, 26, 43). An expanded population of pre-activated T cells in diabetic hosts may be a contributing factor to the increased immune pathology characteristic of TB and other infectious and non-infectious inflammatory conditions linked to diabetes. Our data support a model for complications driven by hyperglycemia, which is common to type 1 and type 2 diabetes (2). Our study did not address glucose-independent complication mechanisms that might be unique to type 2 diabetes and insulin resistance (44). However, hyperglycemia and hyperlipidemia are both associated with oxidative stress which may be a proximal common factor driving complications (44). A more complete understanding of T cell pre-activation as a novel complication of hyperglycemia may reveal targets for host-directed therapies to limit morbidity and mortality from infectious and non-infectious inflammatory disorders in people with diabetes.
Supplementary Material
Acknowledgements
We thank Dr. Kenneth Rock for the OT-II mice, MedImmune for the RAGE−/− mice and Dr. Gary Ostroff for β-glucan particles.
This work was supported by National Institutes of Health grant RO1 HL018849 (to H.K.).
Abbreviations used in this article
- FOXO
Forkhead box transcription factor family O
- HG
hyperglycemic
- HMGB1
high mobility group box 1
- Mtb
Mycobacterium tuberculosis
- mTORC1
mammalian target of rapamycin complex 1
- RAGE
receptor for advanced glycation end products
- TB
tuberculosis
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3:e442. doi: 10.1371/journal.pmed.0030442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 3.Koh GC, Peacock SJ, van der Poll T, Wiersinga WJ. The impact of diabetes on the pathogenesis of sepsis. Eur. J. Clin. Microbiol. Infect. Dis. 2012;31:379–388. doi: 10.1007/s10096-011-1337-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martens GW, Arikan MC, Lee J, Ren F, Greiner D, Kornfeld H. Tuberculosis susceptibility of diabetic mice. Am. J. Resp. Cell Molec. Biol. 2007;37:518–524. doi: 10.1165/rcmb.2006-0478OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar NP, Sridhar R, Banurekha VV, Jawahar MS, Fay MP, Nutman TB, Babu S. Type 2 diabetes mellitus coincident with pulmonary tuberculosis is associated with heightened systemic type 1, type 17, and other proinflammatory cytokines. Ann. Am. Thorac. Soc. 2013;10:441–449. doi: 10.1513/AnnalsATS.201305-112OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar NP, Sridhar R, Banurekha VV, Jawahar MS, Nutman TB, Babu S. Expansion of pathogen-specific T-helper 1 and T-helper 17 cells in pulmonary tuberculosis with coincident type 2 diabetes mellitus. J. Infect. Dis. 2013;208:739–748. doi: 10.1093/infdis/jit241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Restrepo BI, Fisher-Hoch SP, Pino PA, Salinas A, Rahbar MH, Mora F, Cortes-Penfield N, McCormick JB. Tuberculosis in poorly controlled type 2 diabetes: altered cytokine expression in peripheral white blood cells. Clin. Infect. Dis. 2008;47:634–641. doi: 10.1086/590565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vallerskog T, Martens GW, Kornfeld H. Diabetic mice display a delayed adaptive immune response to Mycobacterium tuberculosis. J. Immunol. 2010;184:6275–6282. doi: 10.4049/jimmunol.1000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Volz HC, Seidel C, Laohachewin D, Kaya Z, Muller OJ, Pleger ST, Lasitschka F, Bianchi ME, Remppis A, Bierhaus A, Katus HA, Andrassy M. HMGB1: the missing link between diabetes mellitus and heart failure. Basic Res. Cardiol. 2010;105:805–820. doi: 10.1007/s00395-010-0114-3. [DOI] [PubMed] [Google Scholar]
- 10.Yao D, Brownlee M. Hyperglycemia-induced reactive oxygen species increase expression of the receptor for advanced glycation end products (RAGE) and RAGE ligands. Diabetes. 2010;59:249–255. doi: 10.2337/db09-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pepper M, Jenkins MK. Origins of CD4(+) effector and central memory T cells. Nat. Immunol. 2011;12:467–471. doi: 10.1038/ni.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rudd CE. MAPK p38: alternative and nonstressful in T cells. Nat. Immunol. 2005;6:368–370. doi: 10.1038/ni0405-368. [DOI] [PubMed] [Google Scholar]
- 13.Jirmanova L, Giardino Torchia ML, Sarma ND, Mittelstadt PR, Ashwell JD. Lack of the T cell-specific alternative p38 activation pathway reduces autoimmunity and inflammation. Blood. 2011;118:3280–3289. doi: 10.1182/blood-2011-01-333039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Newton RH, Turka LA. Regulation of T cell homeostasis and responses by pten. Front. Immunol. 2012;3:151. doi: 10.3389/fimmu.2012.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Landau D, Eshet R, Troib A, Gurman Y, Chen Y, Rabkin R, Segev Y. Increased renal Akt/mTOR and MAPK signaling in type I diabetes in the absence of IGF type 1 receptor activation. Endocrine. 2009;36:126–134. doi: 10.1007/s12020-009-9190-2. [DOI] [PubMed] [Google Scholar]
- 16.Rawlings JS, Gatzka M, Thomas PG, Ihle JN. Chromatin condensation via the condensin II complex is required for peripheral T-cell quiescence. EMBO J. 2011;30:263–276. doi: 10.1038/emboj.2010.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clarke DL, Sutcliffe A, Deacon K, Bradbury D, Corbett L, Knox AJ. PKCbetaII augments NF-kappaB-dependent transcription at the CCL11 promoter via p300/CBP-associated factor recruitment and histone H4 acetylation. J. Immunol. 2008;181:3503–3514. doi: 10.4049/jimmunol.181.5.3503. [DOI] [PubMed] [Google Scholar]
- 18.Gorentla BK, Zhong XP. T cell Receptor Signal Transduction in T lymphocytes. J. Clin. Cell. Immunol. 2012;2012:5. doi: 10.4172/2155-9899.S12-005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heissmeyer V, Macian F, Im SH, Varma R, Feske S, Venuprasad K, Gu H, Liu YC, Dustin ML, Rao A. Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat. Immunol. 2004;5:255–265. doi: 10.1038/ni1047. [DOI] [PubMed] [Google Scholar]
- 20.Miao F, Smith DD, Zhang L, Min A, Feng W, Natarajan R. Lymphocytes from patients with type 1 diabetes display a distinct profile of chromatin histone H3 lysine 9 dimethylation: an epigenetic study in diabetes. Diabetes. 2008;57:3189–3198. doi: 10.2337/db08-0645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Villeneuve LM, Natarajan R. The role of epigenetics in the pathology of diabetic complications. Am. J. Physiol. Renal Physiol. 2010;299:F14–F25. doi: 10.1152/ajprenal.00200.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ceriello A, Ihnat MA, Thorpe JE. Clinical review 2: The "metabolic memory": is more than just tight glucose control necessary to prevent diabetic complications? J. Clin. Endocrinol. Metab. 2009;94:410–415. doi: 10.1210/jc.2008-1824. [DOI] [PubMed] [Google Scholar]
- 23.Donskoy E, Foss D, Goldschneider I. Gated importation of prothymocytes by adult mouse thymus is coordinated with their periodic mobilization from bone marrow. J. Immunol. 2003;171:3568–3575. doi: 10.4049/jimmunol.171.7.3568. [DOI] [PubMed] [Google Scholar]
- 24.Kondo M, Weissman IL, Akashi K. Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell. 1997;91:661–672. doi: 10.1016/s0092-8674(00)80453-5. [DOI] [PubMed] [Google Scholar]
- 25.Di Rosa F, Pabst R. The bone marrow: a nest for migratory memory T cells. Trends Immunol. 2005;26:360–366. doi: 10.1016/j.it.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 26.Yamamoto Y, Yamamoto H. RAGE-Mediated Inflammation, Type 2 Diabetes, and Diabetic Vascular Complication. Front. Endocrinol. 2013;4:105. doi: 10.3389/fendo.2013.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinez N, Kornfeld H. Diabetes and immunity to tuberculosis. Eur. J. Immunol. 2014;44:617–626. doi: 10.1002/eji.201344301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nguyen DV, Shaw LC, Grant MB. Inflammation in the pathogenesis of microvascular complications in diabetes. Front. Endocrinol. 2012;3:170. doi: 10.3389/fendo.2012.00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krisp C, Jacobsen F, McKay MJ, Molloy MP, Steinstraesser L, Wolters DA. Proteome analysis reveals antiangiogenic environments in chronic wounds of diabetes mellitus type 2 patients. Proteomics. 2013;13:2670–2681. doi: 10.1002/pmic.201200502. [DOI] [PubMed] [Google Scholar]
- 30.Surendar J, Mohan V, Rao MM, Babu S, Aravindhan V. Increased levels of both Th1 and Th2 cytokines in subjects with metabolic syndrome (CURES-103) Diabetes Technol. Ther. 2011;13:477–482. doi: 10.1089/dia.2010.0178. [DOI] [PubMed] [Google Scholar]
- 31.Fuentes MK, Nigavekar SS, Arumugam T, Logsdon CD, Schmidt AM, Park JC, Huang EH. RAGE activation by S100P in colon cancer stimulates growth, migration, and cell signaling pathways. Dis. Colon Rectum. 2007;50:1230–1240. doi: 10.1007/s10350-006-0850-5. [DOI] [PubMed] [Google Scholar]
- 32.Reddy MA, Li SL, Sahar S, Kim YS, Xu ZG, Lanting L, Natarajan R. Key role of Src kinase in S100B-induced activation of the receptor for advanced glycation end products in vascular smooth muscle cells. J. Biol. Chem. 2006;281:13685–13693. doi: 10.1074/jbc.M511425200. [DOI] [PubMed] [Google Scholar]
- 33.Fang F, Lue LF, Yan S, Xu H, Luddy JS, Chen D, Walker DG, Stern DM, Yan S, Schmidt AM, Chen JX, Yan SS. RAGE-dependent signaling in microglia contributes to neuroinflammation, A beta accumulation, and impaired learning/memory in a mouse model of Alzheimer's disease. FASEB J. 2010;24:1043–1055. doi: 10.1096/fj.09-139634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu X, Chen H, Zhu X, Ma Y, Liu Q, Xue Y, Chu H, Wu W, Wang J, Zou H. S100A9 promotes human lung fibroblast cells activation through receptor for advanced glycation end-product-mediated extracellular-regulated kinase 1/2, mitogen-activated protein-kinase and nuclear factor-kappaB-dependent pathways. Clin. Exp. Immunol. 2013;173:523–535. doi: 10.1111/cei.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Y, Liang C, Liu X, Liao B, Pan X, Ren Y, Fan M, Li M, He Z, Wu J, Wu Z. AGEs increased migration and inflammatory responses of adventitial fibroblasts via RAGE, MAPK and NF-kappaB pathways. Atherosclerosis. 2010;208:34–42. doi: 10.1016/j.atherosclerosis.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 36.Sparvero LJ, Asafu-Adjei D, Kang R, Tang D, Amin N, Im J, Rutledge R, Lin B, Amoscato AA, Zeh HJ, Lotze MT. RAGE (Receptor for Advanced Glycation Endproducts), RAGE ligands, and their role in cancer and inflammation. J. Transl. Med. 2009;7:17. doi: 10.1186/1479-5876-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang QE, Han C, Zhao R, Wani G, Zhu Q, Gong L, Battu A, Racoma I, Sharma N, Wani AA. p38 MAPK- and Akt-mediated p300 phosphorylation regulates its degradation to facilitate nucleotide excision repair. Nucleic Acids Res. 2013;41:1722–1733. doi: 10.1093/nar/gks1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sheppard KA, Rose DW, Haque ZK, Kurokawa R, McInerney E, Westin S, Thanos D, Rosenfeld MG, Glass CK, Collins T. Transcriptional activation by NF-kappaB requires multiple coactivators. Molec. Cell. Biol. 1999;19:6367–6378. doi: 10.1128/mcb.19.9.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Szerlong HJ, Prenni JE, Nyborg JK, Hansen JC. Activator-dependent p300 acetylation of chromatin in vitro: enhancement of transcription by disruption of repressive nucleosome-nucleosome interactions. J. Biol. Chem. 2010;285:31954–31964. doi: 10.1074/jbc.M110.148718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rainger JK, Bhatia S, Bengani H, Gautier P, Rainger J, Pearson M, Ansari M, Crow J, Mehendale F, Palinkasova B, Dixon MJ, Thompson PJ, Matarin M, Sisodiya SM, Kleinjan DA, Fitzpatrick DR. Disruption of SATB2 or its long-range cis-regulation by SOX9 causes a syndromic form of Pierre Robin sequence. Human Molec. Genet. 2014;23:2569–2579. doi: 10.1093/hmg/ddt647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Merritt C, Enslen H, Diehl N, Conze D, Davis RJ, Rincon M. Activation of p38 mitogen-activated protein kinase in vivo selectively induces apoptosis of CD8+ but not CD4+ T cells. Mol. Cell Biol. 2000;20:936–946. doi: 10.1128/mcb.20.3.936-946.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Foger N, Marhaba R, Zoller M. CD44 supports T cell proliferation and apoptosis by apposition of protein kinases. Eur. J. Immunol. 2000;30:2888–2899. doi: 10.1002/1521-4141(200010)30:10<2888::AID-IMMU2888>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 43.Akirav EM, Preston-Hurlburt P, Garyu J, Henegariu O, Clynes R, Schmidt AM, Herold KC. RAGE expression in human T cells: a link between environmental factors and adaptive immune responses. PloS One. 2012;7:e34698. doi: 10.1371/journal.pone.0034698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Giacco G, Brownlee M. Oxidative stress and diabetic complications. Circ. Res. 2010;107:1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.