

Abstract
In Pseudomonas aeruginosa quorum sensing (QS) activates the production of virulence factors, playing a critical role in pathogenesis. Multiple negative regulators modulate the timing and the extent of the QS response either in the pre-quorum or post-quorum phases of growth. This regulation likely increases P. aeruginosa phenotypic plasticity and population fitness, facilitating colonization of challenging environments such as higher organisms. Accordingly, in addition to the factors required for QS signals synthesis and response, also QS regulators have been proposed as targets for anti-virulence therapies. However, while it is known that P. aeruginosa mutants impaired in QS are attenuated in their pathogenic potential, the effect of mutations causing a dysregulated timing and/or magnitude of the QS response has been poorly investigated so far in animal models of infection. In order to investigate the impact of QS dysregulation on P. aeruginosa pathogenesis in a murine model of lung infection, the QteE and RsaL proteins have been selected as representatives of negative regulators controlling P. aeruginosa QS in the pre- and post-quorum periods, respectively. Results showed that the qteE mutation does not affect P. aeruginosa lethality and ability to establish chronic infection in mice, despite causing a premature QS response and enhanced virulence factors production in test tube cultures compared to the wild type. Conversely, the post-quorum dysregulation caused by the rsaL mutation hampers the establishment of P. aeruginosa chronic lung infection in mice without affecting the mortality rate. On the whole, this study contributes to a better understanding of the impact of QS regulation on P. aeruginosa phenotypic plasticity during the infection process. Possible fallouts of these findings in the anti-virulence therapy field are also discussed.
Introduction
Quorum sensing (QS) is an intercellular communication process based on the synthesis and secretion of signal molecules that bind to cognate receptors. The signal-activated receptors trigger the expression of target genes. Since the concentration of signal molecules is proportional to cell density, QS coordinates gene expression when the bacterial population reaches a critical threshold level. The population density at which gene expression is triggered is called the “quorum”, while the phase before expression is called the “pre-quorum” period [1]–[3].
QS processes are widespread in the bacterial world and they are studied with particular intensity in Pseudomonas aeruginosa. This bacterium is one of the most dreaded Gram-negative pathogens in developed countries, being responsible for both community- and hospital-acquired infections. In addition, P. aeruginosa chronic lung infection is the major cause of death in patients with cystic fibrosis (CF), a genetic disease affecting about 1/3,000 newborns in the Caucasian population. P. aeruginosa infections are difficult to eradicate as a consequence of intrinsic antibiotic resistance and growth in bacterial communities referred to as biofilms [4], [5]. Since in P. aeruginosa QS plays a critical role in the production of virulence factors and in biofilm formation, it is considered a very promising target for the development of anti-virulence drugs [6]–[9].
P. aeruginosa has at least three QS systems based on the production, secretion and perception of distinct signals: N-3-oxo-dodecanoyl-homoserine lactone (3OC12-HSL), N-butyryl-homoserine lactone (C4-HSL), and molecules belonging to the 2-alkyl-4-quinolones (AQs) family. The signal molecule 3OC12-HSL is required for optimal production of the other QS signals, though this hierarchy is dependent upon growth conditions [3], [10]–[13]. 3OC12-HSL is produced by the synthase LasI, encoded by the lasI gene, and perceived by the signal receptor LasR, encoded by lasR (Fig. 1). The LasR/3OC12-HSL complex activates the transcription of hundreds of genes, including: i) the lasI gene, thus generating the autoinduction circuit typical of most QS systems; ii) the genes required for C4-HSL and AQs synthesis and reception; iii) many genes involved in virulence factors production and biofilm formation [3], [10], [11].
Figure 1. Schematic representation of QteE- and RsaL-dependent regulation of the P. aeruginosa las QS system.

In the pre-quorum period, QteE binds to the LasR receptor and prevents the binding of the LasR-3OC12-HSL complex to the rsaL-lasI bidirectional promoter [20], hence delaying the onset of the QS response. Once the quorum has been reached, the LasR/3OC12-HSL complex triggers the transcription of both rsaL and lasI genes. The consequent increase of 3OC12-HSL levels, and thus of activated LasR, generates a positive feedback loop also responsible for the increase of RsaL levels. RsaL binding to the rsaL-lasI bidirectional promoter represses the expression of both rsaL and lasI genes, thus counteracting the positive feedback loop. This circuit provides 3OC12-HSL homeostasis [24]. Solid arrows represent positive control; T-shaped lines represent negative control; dashed arrows indicate information flow; curved arrows represent the transcription start points of the indicated genes.
In P. aeruginosa the autoinduction circuit is coupled to multiple negative regulatory mechanisms that modulate the timing and the extent of the QS response, hence limiting the metabolic burden for virulence factors production when unnecessary. Overall, this fine-tuning is believed to increase P. aeruginosa phenotypic plasticity and population fitness, ultimately facilitating colonization of challenging environments such as higher organisms' tissues [3], [14]–[17].
Multiple negative modulators delaying the onset of the QS response by restraining the expression of the lasI gene in the pre-quorum period have been identified so far [18]–[21]. As an example, QteE decreases LasR stability at low cell density. Accordingly, in a qteE mutant the transcription of both lasI and the 3OC12-HSL-dependent gene lasB, encoding the secreted virulence factor elastase, is prematurely activated (Fig. 1) [17], [20]. Moreover, the overexpression of qteE causes a strong reduction of virulence in potato and fruit fly models of infection [22], suggesting that QteE might limit P. aeruginosa virulence also in mammals. However, artificial gene overexpression could produce aberrant effects, and the impact of qteE mutation on 3OC12-HSL and virulence factor production, and on the overall pathogenic potential of P. aeruginosa in either test tube cultures or animal models of infections is still unexplored.
After the QS threshold has been reached, in the post-quorum growth phase, additional regulatory factors may intervene, modulating the overall levels of signal molecule produced by the bacterial population. The RsaL protein, a repressor of lasI transcription encoded by the rsaL gene, is the best characterized post-quorum regulator in P. aeruginosa. Since the LasR/3OC12-HSL complex activates both rsaL and lasI transcription, it generates a homeostatic regulatory loop that allows a population of P. aeruginosa cells to avoid 3OC12-HSL accumulation in the post-quorum period (Fig. 1) [23]–[25]. The effect of the rsaL mutation on 3OC12-HSL and virulence factor production in test tube cultures and in an insect infection model has been thoroughly investigated in a previous study [23], [26]. Briefly, it has been shown that rsaL mutation leads to enhanced motility and secreted virulence factors production such as pyocyanin and elastase, resulting in a hypervirulent phenotype in the Galleria mellonella model of systemic (acute) infection. On the other hand, the rsaL mutant strain displays increased sensitivity to different antibiotics and reduced ability to form biofilm [26].
P. aeruginosa mutants impaired in QS are less pathogenic in various animal models of infection, especially in those developed to investigate the acute infection process [27]. However, despite P. aeruginosa biofilm-based chronic infections have higher social and economic costs in developed countries [4], [28], studies on mammalian models of chronic infection have received less attention so far, likely due to the complexity to establish long-term chronic infection in animal models [27], [29], [30].
Given the importance of QS and phenotypic plasticity in P. aeruginosa pathogenicity, it is believed that pre- and post-quorum control of QS response could play a key role in the establishment and persistence of the infection [15], [16]. Accordingly, ancillary regulators controlling the timing and extent of the QS response have been proposed as targets for the development of anti-virulence therapies against P. aeruginosa [31]. However, to the best of our knowledge, the effect of mutations causing a dysregulated timing and/or magnitude of the QS response on P. aeruginosa virulence has never been investigated in mammalian models of chronic infection.
The general objective of this study has been to investigate the impact of QS dysregulation in P. aeruginosa pathogenesis, by selecting QteE and RsaL as representatives of two negative regulators that control the las QS system in the pre- and post-quorum periods, respectively. As summarized above, the effect of the rsaL mutation in test tube cultures has been thoroughly characterized by our group in previous studies, while the effect of this mutation in animal infection models has not been investigated so far [23], [24], [26]. On the other hand, very little is known about the effect of the qteE mutation on P. aeruginosa 3OC12-HSL production and virulence, both in vitro and in vivo. For this reason, the first part of this study has been focused on the characterization of the qteE mutant virulence-related phenotypes in test-tube cultures. Next, the effect of both qteE and rsaL mutations on P. aeruginosa pathogenesis has been investigated in a murine model of chronic infection. Results showed that a mutation in qteE causes a premature QS response and hyperproduction of virulence factors in P. aeruginosa cultures. However, the anticipation of the QS response in the pre-quorum period due to the qteE mutation does not affect P. aeruginosa pathogenicity, while the post-quorum dysregulation caused by the rsaL mutation hampers the establishment of chronic lung infection. Overall these findings contribute to fill-in the current gap of knowledge about the relevance of QS modulation in P. aeruginosa pathogenesis, and stimulate a re-discussion of the overall role played by QS during the infection process.
Results and Discussion
Phenotypic characterization of the P. aeruginosa qteE mutant
The effect of qteE mutation on P. aeruginosa 3OC12-HSL-dependent response was determined along growth by comparing the levels of this signal molecule and of selected QS-dependent virulence factors in wild type and in qteE cultures carrying either the empty vector pBBR1MCS-5 or its derivative plasmid (named pQteE) expressing the qteE gene.
As shown in Figure 2A, the qteE mutant produced detectable levels of 3OC12-HSL earlier than the wild type strain, reaching a 3OC12-HSL concentration about 6-fold higher at A600≈1. Interestingly, 3OC12-HSL levels measured in the qteE and in the wild type strains plateaued at the same level in the post-quorum phase of growth (A600≈2). This trend of 3OC12-HSL production in the qteE mutant is also consistent with previous western hybridization experiments showing that the positive effect of the qteE mutation on LasR protein stability is restricted to the pre-quorum period [20]. Conversely, as previously shown [24], the rsaL mutant disclosed normal 3OC12-HSL production in the pre-quorum period, while this mutant produced higher 3OC12-HSL levels than the wild type strain after the QS threshold has been reached (A600>1.8; Fig. 2A).
Figure 2. Effect of QS dysregulation caused by qteE mutation on P. aeruginosa virulence-related phenotypes.
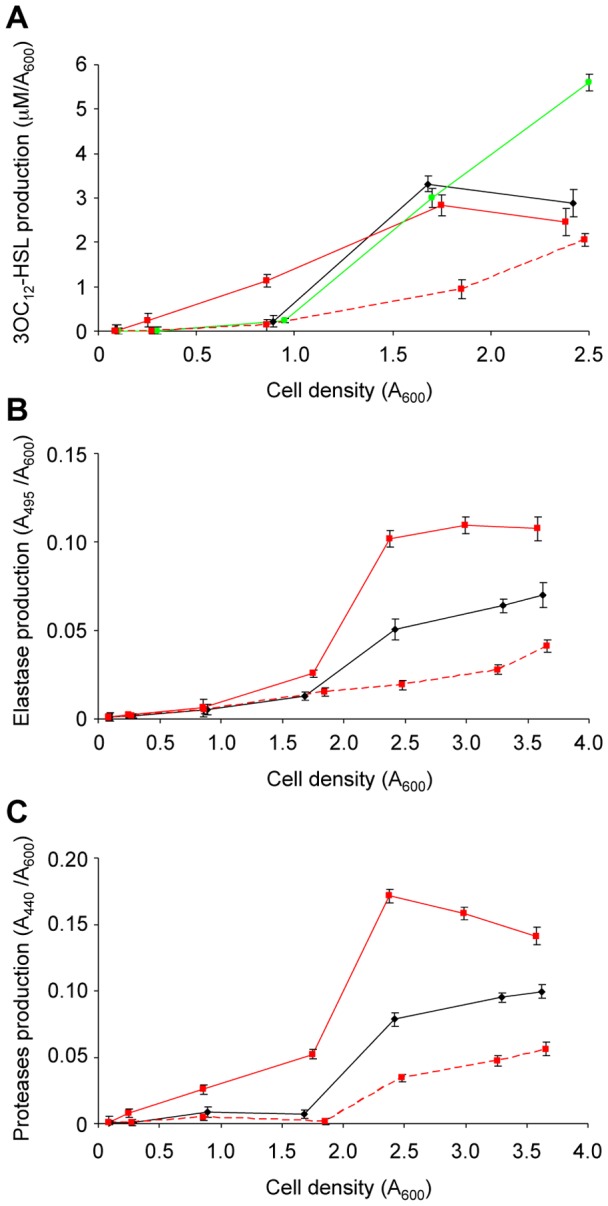
Levels of (A) 3OC12-HSL, (B) elastase, (C) proteases produced along growth by P. aeruginosa wild type (black lines), qteE (red lines) and rsaL strains (green line) carrying the pBBR1MCS-5 empty vector, or by the qteE strain carrying the pQteE plasmid (pBBR1MCS-5-derived) for the expression of qteE (dashed red line). Values are the means (± standard deviations) of at least three independent experiments.
In agreement with the precocious synthesis of 3OC12-HSL, the qteE mutant also anticipated the production of elastase (Fig. 2B) and protease (Fig. 2C). Differently from 3OC12-HSL levels, it seems that the anticipated expression of proteases and elastase levels in the qteE mutant causes accumulation of these secreted factors also in the post-quorum period (compare panels A, B and C of Fig. 2). The homeostatic control of 3OC12-HSL levels in the post-quorum period is likely due to specific mechanisms that do not affect proteases and elastase production, including the transcriptional repression exerted by RsaL on lasI, and the activity of the acyl-HSL degrading enzymes produced by P. aeruginosa [15], [23]–[25], [32]–[34]. Also the biosynthesis of the cytotoxic secondary metabolite pyocyanin is activated by the LasR/3OC12-HSL complex, though it starts later during the growth with respect to proteases and elastase biosynthesis [35]. Interestingly, when the wild type and qteE mutant cultures reached an A600≈3.5, the supernatants of the qteE mutant contained high pyocyanin levels, while this virulence factor was almost undetectable in the wild type strain (Fig. 3).
Figure 3. Effect of QS dysregulation caused by qteE mutation on pyocyanin production in P. aeruginosa.
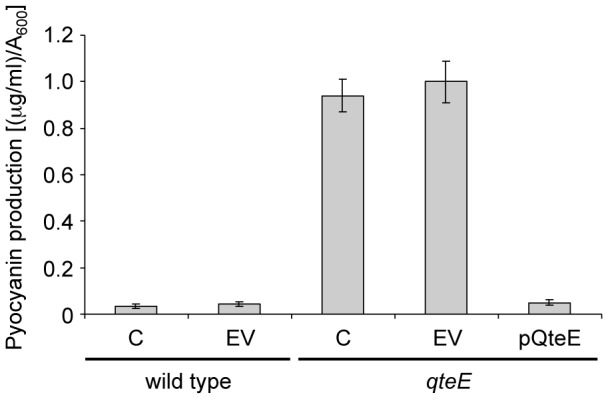
Levels of pyocyanin measured in cell-free supernatants from cultures of the indicated strains grown till A600≈3.5. C, no plasmid; EV, pBBR1MCS-5 empty vector; pQteE, pBBR1MCS-5 derivative plasmid for qteE expression.
The growth curve of the qteE mutant was similar to those of the rsaL mutant and wild type strains and was not affected by the presence of the pBBR1MCS-5 vector, ruling out the possibility that differences in the growth rates could account for the diverse phenotypes described above (Fig. S1 in File S1). Moreover, complementation of the qteE mutant strain with the pQteE plasmid caused a repression of both 3OC12-HSL, proteases, elastase, and pyocyanin production, ruling out any involvement of possible artefacts caused by qteE mutagenesis on the tested phenotypes (Figs. 2 and 3). We also verified that the pBBR1MCS-5 empty vector did not affect the levels of 3OC12-HSL, proteases, elastase and pyocyanin either in the wild type or in the qteE mutant per se (Fig. S2 in File S1 and Fig. 3).
Overall, the above results are in agreement with previous studies showing that the lasI and lasB promoters are prematurely activated in the qteE mutant with respect to the wild type [17], [20], and that QteE overexpression causes repression of elastase and pyocyanin production in P. aeruginosa [20], [22]. Notably, our results demonstrate that in the qteE mutant the production of 3OC12-HSL and of main QS-controlled virulence factors is anticipated, and that these exo-products accumulate along growth at higher levels with respect to the wild type strain, supporting the hypothesis that qteE may play a role in P. aeruginosa pathogenesis.
Effect of QS dysregulation on P. aeruginosa pathogenicity in a murine model of infection
A bacterial population can adopt distinct behaviours in acute and chronic infections, and distinctive phenotypes characterizing these two processes are often inversely regulated. An acute infection is rapid, systemic and carried out by a planktonic bacterial community expressing high levels of virulence factors. Conversely, in a chronic infection bacterial proliferation is limited to a specific host tissue (e.g., in the CF lung or in association with medical devices), and bacteria can persist in the host for extended periods of time, adopting a slow-growing sessile lifestyle (biofilm). In the biofilm mode of growth bacteria are more resistant to the host immune system and prolonged antibiotic therapies, and they produce limited amount of virulence factors despite high cell density [29], [36].
In order to test the effect of pre-quorum and post-quorum dysregulation of the lasI gene in vivo, the virulence of P. aeruginosa PAO1 wild type and of its isogenic qteE and rsaL mutant strains was compared by using the “agar beads” murine model of lung infection [37]–[39]. In this model, P. aeruginosa cells embedded in agar beads are inoculated into the murine lungs, where replicate in microaerobic/anaerobic conditions in the form of microcolonies, similarly to the growth in the mucus of CF patients [40]. Mice dying within three days from the challenge are killed by a systemic (acute) infection. The surviving mice are sacrificed after 14 days from the challenge and bacterial load is evaluated in their lungs: the subset of survived mice containing P. aeruginosa (CFU ≥1000) in their lungs are considered chronically infected, while the others are considered cleared from the infection [37]–[39]. This murine model of infection has been previously used to test the virulence of a P. aeruginosa PAO1 QS mutant impaired in both 3OC12-HSL and C4-HSL production, showing that the QS mutant had strongly attenuated ability to cause chronic infection with respect to the wild type, while the two strains showed similar ability to kill mice within three days from the challenge [41]. In addition, this infection model has been used to validate the anti-virulence activity of anti-Pseudomonas compounds, including QS inhibitors [7], [42].
When mice were challenged with the P. aeruginosa PAO1 wild type strain used in this study, 54% of mortality (20/37 mice) was observed in the first three days, while 76.5% of the survived mice (13/17 mice) were chronically infected after 14 days from the challenge (Fig. 4). Surprisingly, the ability of qteE mutant and of the wild type strains to cause both mice mortality and chronic infection, in the survived mice, did not show statistically significant differences (Fig. 4). Hence, despite the production of virulence factors regulated by QS is anticipated and increased in the qteE mutant in vitro, inactivation of this gene has no effect on P. aeruginosa ability to cause infection, at least in this model system.
Figure 4. Effect of QS dysregulation caused by qteE and rsaL mutations on P. aeruginosa pathogenesis in mice.
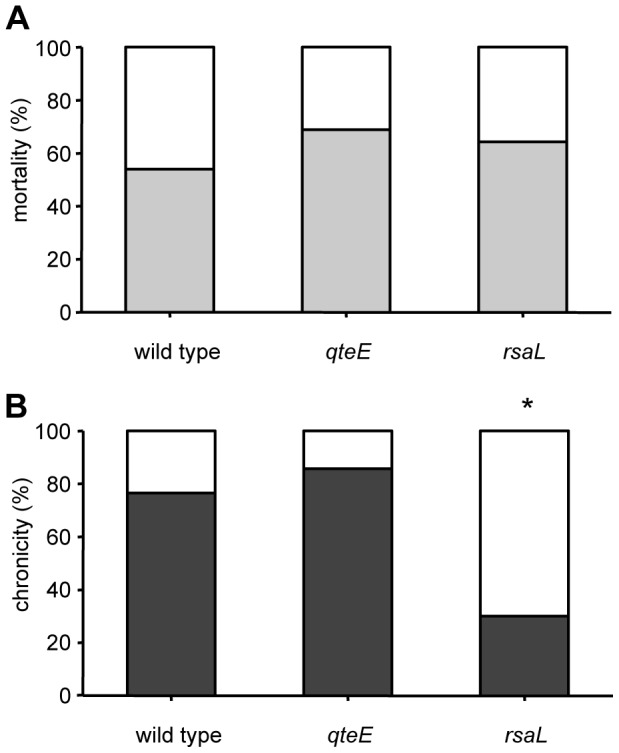
C57Bl/6 mice were infected with the indicated strains embedded in agar beads. (A) Mice mortality induced by bacteremia (light grey) and survival (white) were evaluated on challenged mice. (B) Clearance (white) and capacity to establish chronic airways infection (dark grey) were determined on surviving mice after 14 days from challenge. The results are averages of three independent experiments. Statistical significance is indicated by an asterisk comparing P. aeruginosa wild type versus qteE or rsaL strains (p<0.05).
Concerning the rsaL mutant, the ability of this strain to kill the mice by systemic infection (64.3%; 18/28 mice) did not show significant difference with respect to the wild type. This result was surprising, because the LD50 of the rsaL mutant is seven fold lower with respect to the wild type strain in the G. mellonella systemic infection model [26]. Although previous studies testing the virulence of QS defective or attenuated mutants showed a good correlation of the results between G. mellonella and murine infection models [43], it seems that this correlation is missing for mutants with dysregulated QS response.
Interestingly, only 30% (3/10 mice) of the survived mice developed a chronic infection after 14 days from the challenge with the rsaL strain, a percentage significantly lower than that observed with the wild type strain (76.5%; Fig. 4). Hence, the rsaL mutation decreased P. aeruginosa ability to cause chronic infection. This result strongly supports the hypothesis that the RsaL-mediated post-quorum homeostatic regulation of QS plays a positive role in the establishment of chronic lung infection in mice.
The surviving mice sacrificed 14 days from the challenge were either totally cleared (no P. aeruginosa cells in the lungs) or contained similar bacterial loads (>1000 CFU/lungs), independent from the P. aeruginosa strain used for the challenge (Fig. S3 in File S1). This result is overall in line with previous studies using this infection model [38].
Conclusions
Since the discovery that P. aeruginosa virulence genes expression is QS-dependent and that QS mutants have attenuated pathogenicity in animal models of infection, researchers have tried to explain why QS favours the infection. An early and still in vogue theory is that the QS-control of virulence factors avoids the stimulation of the host immune response at early infection stages, when the size of the bacterial population is small [1]. Another hypothesis, not excluding the former one, is that QS could be important to save energy from unprofitable exoproducts production in environments with high mass transfer, allowing their synthesis only if bacteria are within a low diffusion rate environment, such as an infected tissue [44]. However, considering the key role played by the QS circuitry in P. aeruginosa central and secondary metabolism, and its poorly understood links with other cellular regulatory networks, it seems quite an hard task to find an univocal and simple explanation for the role played by QS in the infection.
P. aeruginosa has evolved as a tough versatile organism, able to thrive in a wide range of environmental niches rather than as a specialized pathogen. In accordance with its phenotypic plasticity, P. aeruginosa can cause a range of different acute and chronic infections in almost all areas of the human body [4], [16], [36], implying that the relevance of the different factors affecting the timing and extent of the QS response in vivo could be dependent upon the kind of infection.
In the lung infection model used in this study, the similar pathogenic behaviour of the P. aeruginosa wild type and qteE mutant indicates that the restrained production of 3OC12-HSL and expression of virulence genes in the pre-quorum period does not favour P. aeruginosa ability in either mice killing or in establishing a chronic lung infection. This finding argues against the hypothesis that delaying virulence factors production until cells amass to a certain density could favour the establishment of the infection [1], [20].
A relevant finding of this study is that the post-quorum homeostatic regulation of QS exerted by RsaL favours the establishment of the chronic P. aeruginosa infection. The importance of this factor also in the human chronic infection is supported by studies on P. aeruginosa strains isolated from the chronically infected lungs of CF patients. During the course of this infection, that can last for decades, the P. aeruginosa population that initially settles in the lung is subject to a microevolution process leading to the emergence of mutants with phenotypic traits unusual in the environmental strains, including loss of motility, increased ability to form biofilm, increased antibiotic resistance, reduced production of secreted virulence factors [45]. It is striking how these phenotypes inversely correlate with those disclosed by the rsaL mutant [26]. Accordingly, in the CF chronic lung infection there is a positive selection for cells expressing high levels of RsaL [46], [47].
Since rsaL transcription is strongly dependent upon LasR, this factor should not be expressed in a lasR mutant. Hence it is interesting to discuss our results by considering that lasR mutants are frequently isolated from the lungs of CF patients. It is still under debate whether these mutants arise because they are social cheaters gaining a growth advantage by utilizing “public goods” (i.e., virulence factors) produced by neighbour wild type cells, rather than producing their own [45], [48], [49], or whether they are better adapted than the wild type to the peculiar environment of the CF lung [50], [51]. Overall, it is still unclear whether and how the emergence of lasR mutants could contribute to the CF lung decline. However, a recent work showed that lasR mutants were able to produce very high levels of pyocyanin under the slow-growing conditions typical of the chronic infection, while wild type cells did not [13]. Moreover, in co-cultivation experiments, the lasR mutant was able to cooperate with the wild type for pyocyanin production [13]. Pyocyanin overproduction in the lasR mutant is due to the loss of repression normally exerted by RsaL on phenazine biosynthetic genes, because RsaL itself is not expressed as a consequence of lasR mutation [13]. However, mutations in rsaL are not commonly isolated in CF clinical samples, suggesting that the constitutive expression of QS regulated factors caused by this mutation is unfavourable in the CF lung environment, and that a mutation in the rsaL gene can be tolerated only when associated to the lack of expression of the entire LasR regulon.
It has been proposed that targeting the function and the cellular levels of the regulatory factors that modulate the QS pre-quorum and post-quorum response could be a strategy to inhibit P. aeruginosa virulence [31]. Though the hypervirulent phenotype disclosed by the rsaL mutant in vitro and in the G. mellonella infection model might cause some concern, our results indicate that a compound targeting RsaL could reduce the ability of P. aeruginosa to establish a chronic infection. Moreover, since the rsaL mutant is also less resistant to antibiotics, with respect to the wild type [26], such compound could synergize with drugs currently used in the CF therapy.
In conclusion, our results contribute to a better understanding of the QS regulatory factors involved in the establishment of the chronic infection caused by P. aeruginosa, indicate the RsaL homeostatic regulator of QS as a promising target for drugs specific against this kind of infection, and highlight the importance of carrying out further studies about the role played by QS modulation in mammalian infection models.
Materials and Methods
Bacterial strains and culture conditions
Pseudomonas aeruginosa wild type, substrain PAO1-UW, and its qteE and rsaL mutant derivatives were supplied by The University of Washington Genome Center (www.genome.washington.edu/UWGC/pseudomonas) [52]. Escherichia coli DH5α [53] was used for cloning purposes. Bacterial strains were grown at 37°C in Luria-Bertani broth (LB) [54] with 200 r.p.m shaking; 20 mg/L and 100 mg/L Gentamicin was added to the E. coli and P. aeruginosa strains for plasmid maintenance, respectively.
Plasmids construction
A DNA region of 1,280 bp encompassing the entire qteE gene, including its native promoter region, was amplified from the PAO1-UW genome by PCR and cloned into the KpnI-HindIII sites of the pBBR1MCS-5 vector [55], generating the pQteE plasmid. The PCR was performed with the following Forward and Reverse oligonucleotides: 5′-CGGGGTACCGAGGACTACCAGAAAGCCC-3′, and 5′-ATAAAGCTTTCAGGCCAGCCCATAGCT-3′; the KpnI and HindIII restriction sites introduced in the oligonucleotides are underlined.
Phenotypic assays
Plasmids pBBR1MCS-5 and pQteE were inserted in the PAO1-UW strain and in the PAO1-UW qteE mutant by conjugation, as previously described [56]. Strains were grown 16 hours at 37°C in LB supplemented with 100 µg/ml Gentamicin. For the phenotypic assays, cultures were diluted to an A600 of 0.02 in LB and incubated at 37°C with 200 r.p.m shaking. Cell-free supernatants were collected every hour after 3 hours of incubation. The concentration of 3OC12-HSL, proteases, elastase and pyocyanin in the cell-free supernatants were measured as previously described [24], [57]–[60].
The average measurements and relative standard deviations were calculated from three independent experiments.
Mouse model of P. aeruginosa lung infection
C57Bl/6 male mice (20–22 gr) were purchased by Charles River Laboratories (Calco, Italy). All mice were maintained under specific pathogen-free conditions in sterile cages which were put into a ventilated isolator. Fluorescent lights were cycled 12 hours on/12 hours off, and ambient temperature (23±1°C) and relative humidity (40–60%) were regulated. The P. aeruginosa agar-beads mouse model was used [37]. PAO1-UW wild type strain and the isogenic qteE and rsaL mutant strains were used for inclusion in the agar beads, as previously described [38], [39], [61]. Briefly, mice were anesthetized with 2.5% avertin (2,2,2-tribromethanol, 97%; Sigma Aldrich) in 0.9% NaCl, intubated with a 22-gauge venous catheter and inoculated with P. aeruginosa 2×106 CFU. Animals were observed twice a day and those showing more than 25% of body weight loss and had evidence of severe clinical disease, such as scruffy coat, inactivity, loss of appetite, poor locomotion, or painful posture, were sacrificed before the termination of the experiments with an overdose of carbon dioxide. Fourteen days after infection, mice were sacrificed by CO2 administration and murine lungs were excised, homogenized and plated onto Trypticase Soy Agar plates for CFU counting. Recovery of ≥1,000 CFU from lung cultures was indicative of chronic infection [37], [38]. The results are averages of at least three independent experiments. Overall, 37, 45 and 28 mice were infected with the P. aeruginosa wild type, the qteE mutant or the rsaL mutant strains, respectively (10-12 mice for each experimental group, for experiment).
Statistical analysis was performed using Fisher's exact test (two-tailed) for categorical variables. Differences were considered statistically significant at p value <0.05.
Ethics statement
Animal studies were carried out according to protocols approved by the IRCCS - San Raffaele Scientific Institute (Milan, Italy) Institutional Animal Care and Use Committee (IACUC), and in strict accordance with the Italian Ministry of Health guidelines for the use and care of experimental animals.
Supporting Information
This file contains Figures S1-S3. Figure S1, Growth curve of P. aeruginosa PAO1 wild type, qteE and rsaL strains. Figure S2, The presence of pBBR1MCS-5 vector does not affect virulence-related phenotypes in P. aeruginosa. Figure S3, Bacterial load in the lungs of mice infected with P. aeruginosa.
(PDF)
Acknowledgments
This work is dedicated to the memory of our dear colleague and mentor Prof. Gerd Doring. We also thank Marcella Facchini and Camilla Riva for assistance in experimental infections.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by grants from the Ministry of University and Research of Italy (Futuro in Ricerca 2010 - RBFR10LHD1; www.miur.it), and the Italian Cystic Fibrosis Research Foundation (FFC 14/2010 and FFC 13/2011; www.fibrosicisticaricerca.it). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Fuqua WC, Winans SC, Greenberg EP (1994) Quorum sensing in bacteria: the LuxR-LuxI family of cell density-responsive transcriptional regulators. J Bacteriol 176: 269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pesci EC, Iglewski BH (1999) Quorum sensing in Pseudomonas aeruginosa. In: Dunny GM, Winans SC, editors.Cell-cell signaling in bacteria.Washington D.C.: American Society for Microbiology. pp. 259–273. [Google Scholar]
- 3. Williams P, Cámara M (2009) Quorum sensing and environmental adaptation in Pseudomonas aeruginosa: a tale of regulatory networks and multifunctional signal molecules. Curr Opin Microbiol 12: 182–191. [DOI] [PubMed] [Google Scholar]
- 4. Driscoll JA, Brody SL, Kollef MH (2007) The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs 67: 351–368. [DOI] [PubMed] [Google Scholar]
- 5. Rosenthal VD, Bijie H, Maki DG, Mehta Y, Apisarnthanarak A, et al. (2012) International Nosocomial Infection Control Consortium (INICC) report, data summary of 36 countries, for 2004-2009. Am J Infect Control 40: 396–407. [DOI] [PubMed] [Google Scholar]
- 6. Rasko DA, Sperandio V (2010) Anti-virulence strategies to combat bacteria-mediated disease. Nat Rev Drug Discov 9: 117–128. [DOI] [PubMed] [Google Scholar]
- 7. Imperi F, Massai F, Facchini M, Frangipani E, Visaggio D, et al. (2013) Repurposing the antimycotic drug flucytosine for suppression of Pseudomonas aeruginosa pathogenicity. Proc Natl Acad Sci USA 110: 7458–7463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. LaSarre B, Federle MJ (2013) Exploiting quorum sensing to confuse bacterial pathogens. Microbiol Mol Biol Rev 77: 73–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rampioni G, Leoni L, Williams P (2014) The art of war against bacteria: a deception strategy targeting quorum sensing communication systems. Bioorg Chem doi:10.1016/j.bioorg.2014.04.005 [DOI] [PubMed] [Google Scholar]
- 10. Smith RS, Iglewski BH (2003) Pseudomonas aeruginosa quorum sensing as a potential antimicrobial target. J Clin Invest 112: 1460–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schuster M, Greenberg EP (2006) A network of networks: quorum-sensing gene regulation in Pseudomonas aeruginosa . Int J Med Microbiol 296: 73–81. [DOI] [PubMed] [Google Scholar]
- 12. Dekimpe V, Déziel E (2009) Revisiting the quorum-sensing hierarchy in Pseudomonas aeruginosa: the transcriptional regulator RhlR regulates LasR-specific factors. Microbiology 155: 712–723. [DOI] [PubMed] [Google Scholar]
- 13. Cabeen MT (2014) Stationary phase-specific virulence factor overproduction by a lasR mutant of Pseudomonas aeruginosa . PLoS One 9: e88743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mellbye B, Schuster M (2011) The sociomicrobiology of antivirulence drug resistance: a proof of concept. MBio 2: e00131–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Venturi V, Rampioni G, Pongor S, Leoni L (2011) The virtue of temperance: built-in negative regulators of quorum sensing in Pseudomonas . Mol Microbiol 82: 1060–1070. [DOI] [PubMed] [Google Scholar]
- 16. Brown SP, Cornforth DM, Mideo N (2012) Evolution of virulence in opportunistic pathogens: generalism, plasticity, and control. Trends Microbiol 20: 336–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gupta R, Schuster M (2013) Negative regulation of bacterial quorum sensing tunes public goods cooperation. ISME J 7: 2159–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chugani SA, Whiteley M, Lee KM, D′Argenio D, Manoil C, et al. (2001) QscR, a modulator of quorum-sensing signal synthesis and virulence in Pseudomonas aeruginosa . Proc Natl Acad Sci USA 98: 2752–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Diggle SP, Winzer K, Lazdunski A, Williams P, Cámara M (2002) Advancing the quorum in Pseudomonas aeruginosa: MvaT and the regulation of N-acylhomoserine lactone production and virulence gene expression. J Bacteriol 184: 2576–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Siehnel R, Traxler B, An DD, Parsek MR, Schaefer AL, et al. (2010) A unique regulator controls the activation threshold of quorum-regulated genes in Pseudomonas aeruginosa . Proc Natl Acad Sci USA 107: 7916–7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Seet Q, Zhang LH (2011) Anti-activator QslA defines the quorum sensing threshold and response in Pseudomonas aeruginosa . Mol Microbiol 80: 951–965. [DOI] [PubMed] [Google Scholar]
- 22. Liang H, Duan J, Sibley CD, Surette MG, Duan K (2011) Identification of mutants with altered phenazine production in Pseudomonas aeruginosa . J Med Microbiol 60: 22–34. [DOI] [PubMed] [Google Scholar]
- 23. Rampioni G, Bertani I, Zennaro E, Polticelli F, Venturi V, et al. (2006) The quorum-sensing negative regulator RsaL of Pseudomonas aeruginosa binds to the lasI promoter. J Bacteriol 188: 815–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rampioni G, Schuster M, Greenberg EP, Bertani I, Grasso M, et al. (2007) RsaL provides quorum sensing homeostasis and functions as a global regulator of gene expression in Pseudomonas aeruginosa . Mol Microbiol 66: 1557–1565. [DOI] [PubMed] [Google Scholar]
- 25. Rampioni G, Polticelli F, Bertani I, Righetti K, Venturi V, et al. (2007) The Pseudomonas quorum-sensing regulator RsaL belongs to the tetrahelical superclass of H-T-H proteins. J Bacteriol 189: 1922–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rampioni G, Schuster M, Greenberg EP, Zennaro E, Leoni L (2009) Contribution of the RsaL global regulator to Pseudomonas aeruginosa virulence and biofilm formation. FEMS Microbiol Lett 301: 210–217. [DOI] [PubMed] [Google Scholar]
- 27. Papaioannou E, Utari PD, Quax WJ (2013) Choosing an appropriate infection model to study quorum sensing inhibition in Pseudomonas infections. Int J Mol Sci 14: 19309–19340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.European Centre for Disease Prevention and Control (2013) Point prevalence survey of healthcare-associated infections and antimicrobial use in European acute care hospitals 2011–2012. Stockholm, Sweden. Catalogue Number: TQ-01-13-314-EN-C. 216 p.
- 29. Furukawa S, Kuchma SL, O'Toole GA (2006) Keeping their options open: acute versus persistent infections. J Bacteriol 188: 1211–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bragonzi A (2010) Murine models of acute and chronic lung infection with cystic fibrosis pathogens. Int J Med Microbiol 300: 584–593. [DOI] [PubMed] [Google Scholar]
- 31. Hirakawa H, Tomita H (2013) Interference of bacterial cell-to-cell communication: a new concept of antimicrobial chemotherapy breaks antibiotic resistance. Front Microbiol 4: 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang JJ, Petersen A, Whiteley M, Leadbetter JR (2006) Identification of QuiP, the product of gene PA1032, as the second acyl-homoserine lactone acylase of Pseudomonas aeruginosa PAO1. Appl Environ Microbiol 72: 1190–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sio CF, Otten LG, Cool RH, Diggle SP, Braun PG, et al. (2006) Quorum quenching by an N-acyl-homoserine lactone acylase from Pseudomonas aeruginosa PAO1. Infect Immun 74: 1673–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wahjudi M, Papaioannou E, Hendrawati O, van Assen AH, van Merkerk R, et al. (2011) PA0305 of Pseudomonas aeruginosa is a quorum quenching acylhomoserine lactone acylase belonging to the Ntn hydrolase superfamily. Microbiology 157: 2042–2055. [DOI] [PubMed] [Google Scholar]
- 35. Winstanley C, Fothergill JL (2009) The role of quorum sensing in chronic cystic fibrosis Pseudomonas aeruginosa infections. FEMS Microbiol Lett 290: 1–9. [DOI] [PubMed] [Google Scholar]
- 36. Coggan KA, Wolfgang MC (2012) Global regulatory pathways and cross-talk control Pseudomonas aeruginosa environmental lifestyle and virulence phenotype. Curr Issues Mol Biol 14: 47–70. [PubMed] [Google Scholar]
- 37. Cash HA, McCullough B, Johanson WG Jr, Bass JA (1979) A rat model of chronic respiratory infection with Pseudomonas aeruginosa . Am Rev Respir Dis 119: 453–459. [DOI] [PubMed] [Google Scholar]
- 38. Bragonzi A, Paroni M, Nonis A, Cramer N, Montanari S, et al. (2009) Pseudomonas aeruginosa microevolution during cystic fibrosis lung infection establishes clones with adapted virulence. Am J Respir Crit Care Med 180: 138–145. [DOI] [PubMed] [Google Scholar]
- 39. Facchini M, De Fino I, Riva C, Bragonzi A (2014) Long term chronic Pseudomonas aeruginosa airway infection in mice. J Vis Exp 17: 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Worlitzsch D, Tarran R, Ulrich M, Schwab U, Cekici A, et al. (2002) Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J Clin Invest 109: 317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wu H, Song Z, Givskov M, Doring G, Worlitzsch D, et al. (2001) Pseudomonas aeruginosa mutations in lasI and rhlI quorum sensing systems result in milder chronic lung infection. Microbiology 147: 1105–1113. [DOI] [PubMed] [Google Scholar]
- 42. Hentzer M, Wu H, Andersen JB, Riedel K, Rasmussen TB, et al. (2003) Attenuation of Pseudomonas aeruginosa virulence by quorum sensing inhibitors. EMBO J 22: 3803–3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jander G, Rahme LG, Ausubel FM (2000) Positive correlation between virulence of Pseudomonas aeruginosa mutants in mice and insects. J Bacteriol 182: 3843–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. West SA, Winzer K, Gardner A, Diggle SP (2012) Quorum sensing and the confusion about diffusion. Trends Microbiol 20: 586–594. [DOI] [PubMed] [Google Scholar]
- 45. Folkesson A, Jelsbak L, Yang L, Johansen HK, Ciofu O, et al. (2012) Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: an evolutionary perspective. Nat Rev Microbiol 10: 841–851. [DOI] [PubMed] [Google Scholar]
- 46. Son MS, Matthews WJ Jr, Kang Y, Nguyen DT, Hoang TT (2007) In vivo evidence of Pseudomonas aeruginosa nutrient acquisition and pathogenesis in the lungs of cystic fibrosis patients. Infect Immun 75: 5313–5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Starkey M, Hickman JH, Ma L, Zhang N, De Long S, et al. (2009) Pseudomonas aeruginosa rugose small-colony variants have adaptations that likely promote persistence in the cystic fibrosis lung. J Bacteriol 191: 3492–3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sandoz KM, Mitzimberg SM, Schuster M (2007) Social cheating in Pseudomonas aeruginosa quorum sensing. Proc Natl Acad Sci USA 104: 15876–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rumbaugh KP, Diggle SP, Watters CM, Ross-Gillespie A, Griffin AS, et al. (2009) Quorum sensing and the social evolution of bacterial virulence. Curr Biol 19: 341–345. [DOI] [PubMed] [Google Scholar]
- 50. D'Argenio DA, Wu M, Hoffman LR, Kulasekara HD, Déziel E, et al. (2007) Growth phenotypes of Pseudomonas aeruginosa lasR mutants adapted to the airways of cystic fibrosis patients. Mol Microbiol 64: 512–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Harrison F, Muruli A, Higgins S, Diggle SP (2014) Development of an ex vivo porcine lung model for studying growth, virulence, and signaling of Pseudomonas aeruginosa. Infect Immun. 82: 3312–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Winsor GL, Lam DK, Fleming L, Lo R, Whiteside MD, et al. (2011) Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res D596–600. [DOI] [PMC free article] [PubMed]
- 53. Grant SG, Jessee J, Bloom FR, Hanahan D (1990) Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc Natl Acad Sci USA 87: 4645–4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sambrook J, Fritsch EF, Maniatis T (1989)Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor NY: Cold Spring Harbor Laboratory Press.
- 55. Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, et al. (1995) Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166: 175–176. [DOI] [PubMed] [Google Scholar]
- 56. Figursky DH, Helinski DR (1979) Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc Natl Acad Sci USA 76: 1648–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Massai F, Imperi F, Quattrucci S, Zennaro E, Visca P, et al. (2011) A multitask biosensor for micro-volumetric detection of N-3-oxo-dodecanoyl-homoserine lactone quorum sensing signal. Biosens Bioelectron 26: 3444–3449. [DOI] [PubMed] [Google Scholar]
- 58. Ohman DE, Burns RP, Iglewski BH (1980) Corneal infections in mice with toxin A and elastase mutants of Pseudomonas aeruginosa . J Infect Dis 142: 547–555. [DOI] [PubMed] [Google Scholar]
- 59. Kessler E, Israel M, Landshman N, Chechick A, Blumberg S (1982) In vitro inhibition of Pseudomonas aeruginosa elastase by metal-chelating peptide derivatives Infect Immun. 38: 716–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Longo F, Rampioni G, Bondì R, Imperi F, Fimia GM, et al. (2013) A new transcriptional repressor of the Pseudomonas aeruginosa quorum sensing receptor gene lasR . PLoS One 8: e69554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bragonzi A, Worlitzsch D, Pier GB, Timpert P, Ulrich M, et al. (2005) Nonmucoid Pseudomonas aeruginosa expresses alginate in the lungs of patients with cystic fibrosis and in a mouse model. J Infect Dis 192: 410–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This file contains Figures S1-S3. Figure S1, Growth curve of P. aeruginosa PAO1 wild type, qteE and rsaL strains. Figure S2, The presence of pBBR1MCS-5 vector does not affect virulence-related phenotypes in P. aeruginosa. Figure S3, Bacterial load in the lungs of mice infected with P. aeruginosa.
(PDF)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.