

Abstract
Background and Objectives
Morquio A syndrome (mucopolysaccharidosis IVA; MPS IVA) is a lysosomal storage disorder caused by deficiency of N-acetylgalactosamine-6-sulfatase, an enzyme required for degradation of the glycosaminoglycan keratan sulfate. Enzyme replacement therapy with elosulfase alfa provides a potential therapy for Morquio A syndrome. We analyzed the pharmacokinetics and pharmacodynamics of elosulfase alfa in Morquio A patients from a phase III clinical trial.
Methods
In a randomized double-blind study, elosulfase alfa at 2.0 mg/kg was administrated weekly or every other week for 24 weeks. Pharmacokinetic parameters of elosulfase alfa were determined at weeks 0 and 22 by non-compartmental analysis. Safety was assessed throughout the study. The relationship of pharmacokinetic parameters to patient demographics, pharmacodynamic assessments, immunogenicity, and efficacy and safety outcomes were assessed graphically by treatment group.
Results
Elosulfase alfa exposure and half-life (t ½) increased for both dose regimens during the study. There appeared to be no consistent trend between drug clearance (CL) and patient’s sex, race, body weight, or age. All patients developed anti-drug antibodies, but no association was noted between total antibody titer and CL. In contrast, positive neutralizing antibody (NAb) status appeared to associate with decreased CL and prolonged t ½ for patients in the cohort dosed weekly. NAb may interfere with receptor-mediated cellular uptake and lead to increased circulation time of elosulfase alfa.
Conclusion
Despite the association between NAb and decreased drug clearance, neither dosing cohort showed associations between drug exposure and change in urinary keratan sulfate, 6-min walk test distances, or the occurrence of adverse events.
Electronic supplementary material
The online version of this article (doi:10.1007/s40262-014-0173-y) contains supplementary material, which is available to authorized users.
Key Points
| Elosulfase alfa is an enzyme replacement therapy for Morquio A syndrome that was evaluated in patients with Morquio A syndrome |
| Neutralizing anti-elosulfase alfa antibodies may interfere with elosulfase alfa uptake from plasma into cells |
| No associations were apparent between pharmacokinetic parameters and changes in urine keratan sulfate levels, the pharmacodynamic readout |
| Variability in elosulfase alfa exposure in plasma may not translate directly to pharmacodynamic, efficacy or safety outcomes in patients. |
Introduction
Morquio A syndrome (mucopolysaccharidosis IV A; MPS IVA; OMIM 253000) is an autosomal, recessive, lysosomal storage disease caused by a deficiency of N-acetylgalactosamine-6-sulfatase (GALNS; EC 3.1.6.4), an enzyme required for degradation of the glycosaminoglycans (GAGs) chondroitin-6-sulfate and keratan sulfate (KS). Morquio A syndrome is rare, with an incidence estimated to range from 1 in 76,000 to 1 in 640,000 live births in different populations [1–3].
Patients with Morquio A syndrome appear healthy at birth but develop multiple clinical signs owing to the accumulation of GAGs causing cellular disruptions in multiple cells, tissues, and organs. The progress of the disease and the severity of individual clinical manifestations can vary considerably between patients. Prominent skeletal and connective tissue abnormalities are common, with patients often manifesting dwarfism with short trunk and neck, spinal abnormalities, genu valgum, pectus carinatum, hip dysplasia, and joint instability and hypermobility as a result of ligamentous laxity and joint bone deformities [4]. Pulmonary and cardiac manifestations are also common, which can result in reduced endurance and impact both quality of life and mortality; death often results from cardiorespiratory or neurological complications [4]. Patients with slowly progressing disease rarely survive their sixth decade, and patients with rapidly progressing disease often die in their second or third decade [4].
Enzyme replacement therapy (ERT) by intravenous infusion of recombinant human GALNS (elosulfase alfa; Vimizim™, BioMarin Pharmaceutical Inc., Novato, CA, USA) is approved by the US Food and Drug Administration for treatment of patients with Morquio A syndrome. Elosulfase alfa is expected to increase the degradation of accumulated KS and chondroitin-6-sulfate that cause the Morquio A clinical manifestations. The MOR-004 phase III study was designed to assess the safety and efficacy of two different elosulfase alfa dosing regimens, 2.0 mg/kg every other week (QOW) and 2.0 mg/kg weekly (QW), in comparison with placebo [5]. The elosulfase alfa dose level of 2.0 mg/kg was chosen based on prior in vitro analysis in a human cell culture model of Morquio A syndrome [6] and a prior phase 1/2 dose-escalation study in patients with Morquio A syndrome [7]. Results from the phase I–II study indicated that elosulfase alfa had a short plasma elimination half-life of about 40 min following repeat intravenous infusion and that increases in the area under the plasma concentration-time curve from time zero to the time of last measurable concentration (AUClast) and the maximum concentration in plasma (C max) were greater than dose proportional when the dose was increased from 1.0 to 2.0 mg/kg/week [8]. Here, we report the pharmacokinetic and pharmacodynamic results of elosulfase alfa in patients with Morquio A syndrome from the pivotal phase III trial.
Methods
Study Design and Patients
Data from the MOR-004 study served as the basis for this analysis [5]. In brief, MOR-004 was a phase III, randomized, double-blind, placebo-controlled study of elosulfase alfa in patients with Morquio A syndrome over 24 weeks, conducted at 33 study centers in 17 countries. The study protocol was approved by an Institutional Review Board, Independent Ethics committee, or Research Ethics Board at each participating clinical site. Each participant or his/her legally authorised representative provided written informed consent before entering the study. Patients, investigators, and site personnel were blinded to treatment assignments until the final analysis was complete.
All study participants had a documented clinical diagnosis of Morquio A syndrome based on clinical signs and symptoms, together with either a documented decrease in GALNS enzyme activity as assayed in fibroblasts or leukocytes or a genetic test confirming the Morquio A diagnosis [5].
A total of 176 patients entered the study, of whom 59 were randomized to placebo, 59 to receive elosulfase alfa 2.0 mg/kg every other week (QOW), and 58 to receive elosulfase alfa 2.0 mg/kg weekly (QW); one patient from the QW group discontinued the study. As the study was conducted in a blinded manner, samples for pharmacokinetic analysis were collected from 65 patients (17 receiving placebo, 24 receiving elosulfase alfa 2.0 mg/kg/QOW, and 24 receiving elosulfase alfa 2.0 mg/kg/QW); pharmacokinetic parameters were estimated only for the 48 patients receiving elosulfase alfa.
Administration of Elosulfase Alfa
To reduce the risk of potential infusion-associated reactions, patients were pretreated with an antihistamine at approximately 30–60 min prior to infusion. At the discretion of the investigator, patients with known risk factors for infusion-associated reactions could receive additional agents prior to infusion, such as a sedating antihistamine (e.g., diphenhydramine or chlorpheniramine), H2-receptor blockers, montelukast sodium, or steroids. Elosulfase alfa (2.0 mg/kg) or placebo solution was diluted with saline to a final volume of 250 mL, allowed to reach room temperature, and was administered intravenously. Patients randomized to the QOW group received infusions of elosulfase alfa or placebo on alternating weeks. Each infusion was administered over approximately 4 h.
Pharmacokinetic Analysis
Blood samples for pharmacokinetic analysis were drawn at weeks 0 and 22 from an IV line separate from that for infusion. Samples were drawn predose (within 15 min prior to dosing), 60 and 120 min after beginning infusion, within 5 min prior to ending the infusion, and then at 5, 15, 30, 60, 120, and 180 min postinfusion. Because of missing samples one patient was excluded from pharmacokinetic analysis, two patients (from QOW and QW cohorts) lacked pharmacokinetic values at week 22, and one patient (QW cohort) lacked pharmacokinetic values at week 0. Plasma elosulfase alfa concentrations were measured by a validated electrochemiluminescence (ECL) assay using a sandwich format (Supplementary Methods S1.1). The lower limit of quantitation for elosulfase alfa was 15.6 ng/mL. The in-study inter-assay accuracy (% relative error) and precision (% coefficient of variation, CV) did not exceed 12.0 % (BioMarin, unpublished results).
Pharmacokinetic parameters of elosulfase alfa were estimated based on the plasma concentration–time data using Phoenix WinNonlin 6.1 (Pharsight Corporation, Cary, NC, USA) by non-compartmental analysis. Because the elosulfase alfa mean elimination half-life (t ½) is short (mean t ½ <1 h) relative to its dosing interval (1 or 2 weeks), each infusion was treated as a single dose for pharmacokinetic analysis. Maximum concentration in plasma (C max) and the time of maximum concentration (t max) were recorded directly from the observed data. Other pharmacokinetic parameters estimated in this study include t ½, area under the plasma concentration–time curve from time 0 to the last measurable concentration, estimated by linear trapezoidal rule (AUClast), area under the plasma concentration–time curve extrapolated to infinity (AUC0–∞), clearance of the absorbed fraction (CL), volume of distribution based on terminal phase (V z), and volume of distribution at steady state (V ss).
Efficacy and Safety Evaluations
The primary endpoint of the study was the distance traveled during a 6-min walk test (6MWT), a test of endurance that was performed based on American Thoracic Society guidelines [9]. The secondary efficacy endpoints were the number of stairs climbed per minute in the 3-min stair climb test (3MSCT) and the urinary keratan sulfate (uKS) normalized to creatinine [5].
Safety was assessed by examining the incidence, severity, and relationship to the study drug of treatment-emergent adverse events (AEs) reported during the study [5]. Additionally, infusion-associated reactions, laboratory results, vital signs, physical examination results, and medication use were assessed. Electrocardiograms and echocardiograms were performed at baseline and at week 24. Immunogenicity was also assessed (see Sect. 2.6; unpublished data, BioMarin Pharmaceutical Inc). Potential hypersensitivity AEs were identified by using the standardized Medical Dictionary of Regulatory Activities (MedDRA, version 15.0) queries for broad anaphylactic reaction and broad angioedema [5]. Full safety data from this study were published elsewhere [5]; in brief, in the QW dosing cohort 22.4 % of patients had AEs that led to an elosulfase alfa infusion interruption or discontinuation that required medical intervention, but for both dosing groups no AEs led to permanent treatment discontinuation [5].
Pharmacodynamic Efficacy
Urinary keratan sulfate was quantitatively measured by keratanase II digestion followed by liquid chromatography and tandem mass spectrometry (LC-MS/MS) analysis of samples obtained from first morning voids on two separate days at baseline, and then at weeks 2 and 4, then every 4 weeks thereafter, as well as within 1 week of an early withdrawal. The measured concentrations of the two major KS disaccharides generated by digestion were summed and normalized to urine creatinine concentrations [5] (Supplementary Methods S1.2). Assay sensitivity was 0.104 μg/mL for each disaccharide and in-study between-run precision was less than 9 % CV [5].
Immunogenicity Analysis
Serum samples were collected at baseline, at weeks 2 and 4, and every 4 weeks thereafter, and within 1 week of an early-termination visit. Samples for immunogenicity testing were drawn prior to administration of elosulfase alfa or placebo. Total elosulfase alfa-specific antibody (TAb) and neutralizing elosulfase alfa-specific antibody (NAb) were evaluated by validated ECL bridging and fluorescence competition ELISA assays, respectively. NAb, which is a subset of TAb, was not assessed if TAb was negative.
The total antibody assay measures multiple anti-drug antibody isotypes in a single assay. TAb-positive samples were confirmed for elosulfase alfa specificity and serially diluted to obtain a titer (Supplementary Methods S1.3). The validated NAb assay detects antibodies capable of inhibiting elosulfase alfa from binding to plate-immobilized cation-independent mannose-6-phosphate receptor (CI-M6PR) and produces a qualitative assessment of positive or negative (Supplementary Methods S1.4). The in-study between-run precision was below 17 % (TAb) and 28 % (NAb) CV, consistent with assay validation performance (BioMarin unpublished results). As elosulfase alfa is active in the acidic environment of the lysosome and not the neutral pH of blood, no assay was developed to detect antibodies that inhibit enzymatic activity.
Statistical Analyses
Demographic characteristics were summarized for the pharmacokinetic population. Pharmacokinetic parameters were summarized descriptively for weeks 0 and 22 and by treatment group. As analyses of the pharmacokinetic population were not powered to assess statistical significance, the relationship of pharmacokinetic parameters to demographic characteristics, immunogenicity, pharmacodynamics, efficacy, and AE parameters were assessed graphically by treatment group.
Results
Patient Characteristics
In the MOR-004 trial, patients with Morquio A syndrome were randomized to receive placebo, elosulfase alfa 2.0 mg/kg/QW, or elosulfase alfa 2.0 mg/kg/QOW. The demographic characteristics of the pharmacokinetic analysis population were generally comparable between those receiving elosulfase alfa QW and QOW (Supplemental Table S1) with the exception of sex: the QW pharmacokinetic population was 60.9 % female (14/23) and the QOW pharmacokinetic population was 25.0 % female (6/24). Demographic characteristics and baseline characteristics for patients who were included in the pharmacokinetic analysis (Supplemental Table S1) were similar to those of the general study population [5].
Pharmacokinetics
Elosulfase alfa was administered as an infusion, typically over 4 h. The mean plasma concentration of elosulfase alfa during and after infusion is shown in Fig. 1, at the initial infusion (week 0) and at week 22 for both dosing cohorts. At both weeks 0 and 22 and in both dosing cohorts, elosulfase alfa was detectable in plasma at the first time point after beginning infusion (60 min) and reached maximum concentrations between 120 and 240 min. After 240 min, the typical endpoint for elosulfase alfa infusions, plasma elosulfase alfa concentration decreased with time. The pharmacokinetic parameters were comparable between the dosing cohorts at week 0 but differed at week 22 (Table 1). At week 0, the t ½ was ~7 min for both dosing cohorts, but at week 22 the QOW cohort had a t ½ of ~19 min and the QW cohort had a t ½ of ~36 min. For the QOW cohort, exposure increased by 76 or 84 %, as measured by AUClast or C max, respectively, between weeks 0 and 22, and for the QW cohort, exposure increased by 181 or 192 % (by AUClast or C max, respectively).
Fig. 1.
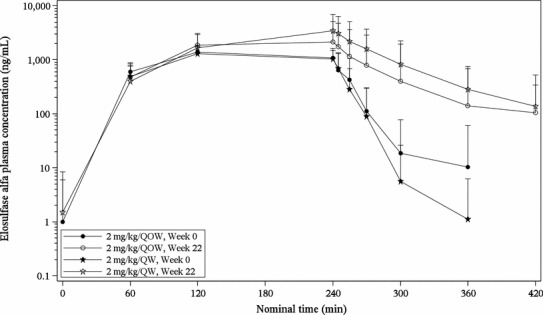
Mean plasma concentration profiles of elosulfase alfa during and after infusion. Mean plasma concentration of elosulfase alfa during and after infusion is displayed with standard error bars at nominal pharmacokinetic sampling time for weeks 0 and 22 and by treatment group. Time scale is nominal time, with 0 min the beginning of infusion and 240 min the typical infusion end point. QOW every other week, QW weekly
Table 1.
Pharmacokinetic parameters for elosulfase alfa 2.0 mg/kg/QW or 2/0 mg/kg/QOW in patients with Morquio A syndrome
| Parameter | Elosulfase alfa 2.0 mg/kg/QOW | Elosulfase alfa 2.0 mg/kg/QW | Ratio of Elosulfase alfa QOW/QWa (%) |
|---|---|---|---|
| Week 0 | |||
| n | 24 | 22 | |
| AUC 0–∞, ng·min/mL | 287,597 (96,432.1), 14 | 231,074 (103,207.4), 15 | 124.5 |
| AUClast, ng·min/mL | 248,720 (97,063.7), 24 | 237,884 (100,328.6), 22 | 104.6 |
| C max, ng/mL | 1,438 (435.3), 24 | 1,494 (534.1), 22 | 96.2 |
| CL, mL/min/kg | 7.54 (2.002), 14 | 10.04 (3.733), 15 | 75.1 |
| V ss, mL/kg | 219.42 (95.483), 12 | 395.74 (315.636), 14 | 55.4 |
| V z, mL/kg | 68.79 (34.008), 14 | 123.66 (144.115), 15 | 55.6 |
| t ½, min | 6.57 (3.110), 14 | 7.52 (5.484), 15 | 87.4 |
| t max, min | 150 (58.1), 24 | 172 (75.3), 22 | 87.2 |
| Week 22 | |||
| n | 23 | 22 | |
| AUC 0–∞, ng·min/mL | 463,460 (491,418.9), 19 | 619,080 (422,048.3), 20 | 74.9 |
| AUClast, ng·min/mL | 411,687 (420,279.7), 23 | 577,371 (416,316.6), 22 | 71.3 |
| Cmax, ng/mL | 2,616 (2,702.1), 23 | 4,036 (3,237.1), 22 | 64.8 |
| CL, mL/min/kg | 6.50 (2.942), 19 | 7.08 (12.997), 20 | 91.8 |
| V ss, mL/kg | 245.19 (273.145), 17 | 649.67 (1,841.703), 20 | 37.7 |
| V z, mL/kg | 120.11 (71.076), 19 | 299.52 (543.309), 20 | 40.1 |
| t ½, min | 19.25 (19.217), 19 | 35.86 (21.485), 20 | 53.7 |
| t max, min | 159 (60.6), 23 | 202 (90.8), 22 | 78.5 |
| Week 22/Week 0b (%) | |||
| n | 23 | 21 | |
| AUC 0–∞, ng·min/mL | 179.2 | 328.6 | |
| AUClast, ng·min/mL | 176.3 | 280.6 | |
| C max, ng/mL | 183.6 | 291.6 | |
| CL, mL/min/kg | 87.0 | 46.4 | |
| V ss, mL/kg | 127.0 | 188.9 | |
| V z, mL/kg | 147.0 | 246.0 | |
| t ½, min | 280.0 | 696.0 | |
| t max, min | 119.8 | 145.7 | |
Values are expressed as mean (SD), n unless otherwise indicated
For patients who have missing values of AUC0–∞, t ½, CL, V z and V ss, the parameters could not be estimated due to insufficient data in the terminal phase of the plasma profile. For patients who have missing values of V ss only, their V ss was not reported due to a negative value. Adjusting for infusion caused a negative MRTinf value. The V ss value was also negative because of the relationship: V ss = MRTinf·CL
AUC 0–∞ area under the plasma concentration–time curve from time zero to infinity, AUC last area under the plasma concentration–time curve from time zero to the time of last measurable concentration, CL total clearance of drug after intravenous administration, C max observed maximum plasma concentration, MRTinf mean residence time extrapolated to infinity, SD standard deviation, QOW every other week, QW weekly, V ss apparent volume of distribution at steady-state, V z apparent volume of distribution based upon the terminal phase, t ½ elimination half-life
a Ratio is ratio of means
b Only patients with pharmacokinetic data available for both visits are included
Pharmacokinetics and Patient Demographics
The relationship between the demographic characteristics of patients included in the pharmacokinetic analysis and the pharmacokinetic results were examined to determine if pharmacokinetics of elosulfase alfa were influenced by demographics. Male and female patients had comparable clearance of elosulfase alfa at both weeks 0 and 22 (Fig. 2). At week 0, white patients (n = 20) had higher elosulfase alfa clearance than non-white patients (n = 9; Fig. 2a), but this trend was not observed at week 22 (Fig. 2b, c). At week 0, patients with increased body weight and increased age showed decreased clearance of elosulfase alfa (Fig. 2a), but neither trend was observed at week 22 (Fig. 2b, c). The relationships between the pharmacokinetic parameters AUClast, AUC0–∞, C max, V ss, and t ½ and patient demographic characteristics were also examined and no consistent relationships were observed (data not shown).
Fig. 2.
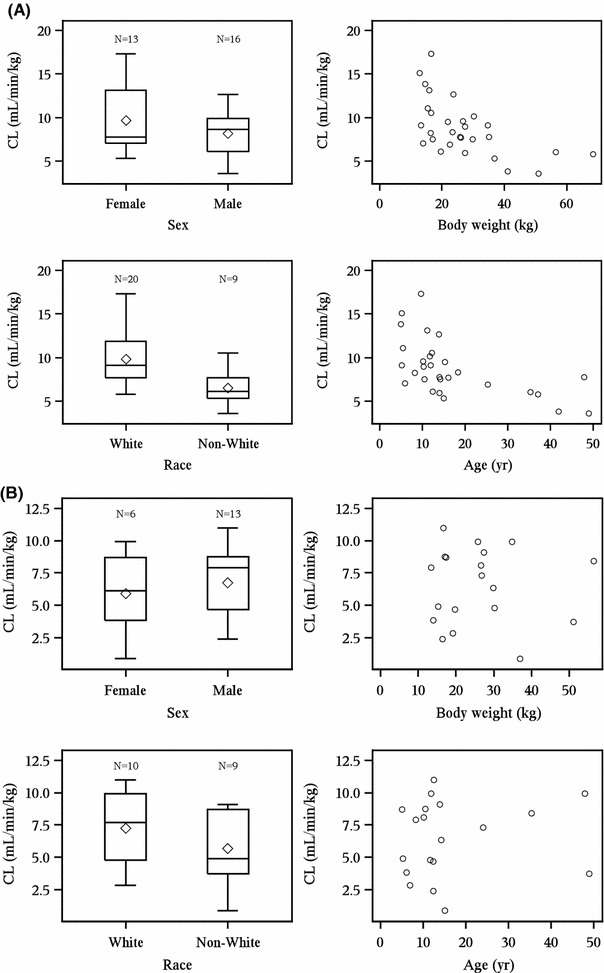
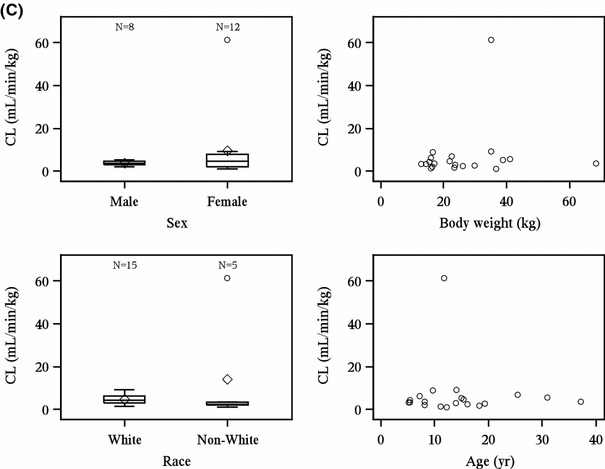
Patient demographics and pharmacokinetic results. Patient demographics and pharmacokinetic results at (a) week 0; (b) week 22, QOW dosing group; (c) week 22, QW dosing group. CL total clearance of drug after intravenous administration, QOW every other week, QW weekly. The bottom and top of whiskers represents the minimum and maximum values excluding outliers, which are plotted separately. The lower and upper ends of the box represent the first and third quartile, the bar within the box represents the median value and the diamond within the box represents the mean value
Immunogenicity and Pharmacokinetics
All patients treated with elosulfase alfa were positive for total antibody against elosulfase alfa (TAb) by week 24 (unpublished data, BioMarin Pharmaceutical Inc). Given the immunogenicity of elosulfase alfa, the relationship between immunogenicity and pharmacokinetic parameters was examined to determine if the pharmacokinetics of elosulfase alfa was influenced by development of anti-drug antibodies. TAb titers at week 24 were plotted against elosulfase alfa CL at week 22 (Fig. 3a) and no general association was observed between TAb titer and CL for either dosing group. Similarly, TAb titers at week 24 were plotted against elosulfase alfa mean t ½ at week 22, and no general association was observed between TAb titer and mean t ½ for either group (Fig. 3b).
Fig. 3.
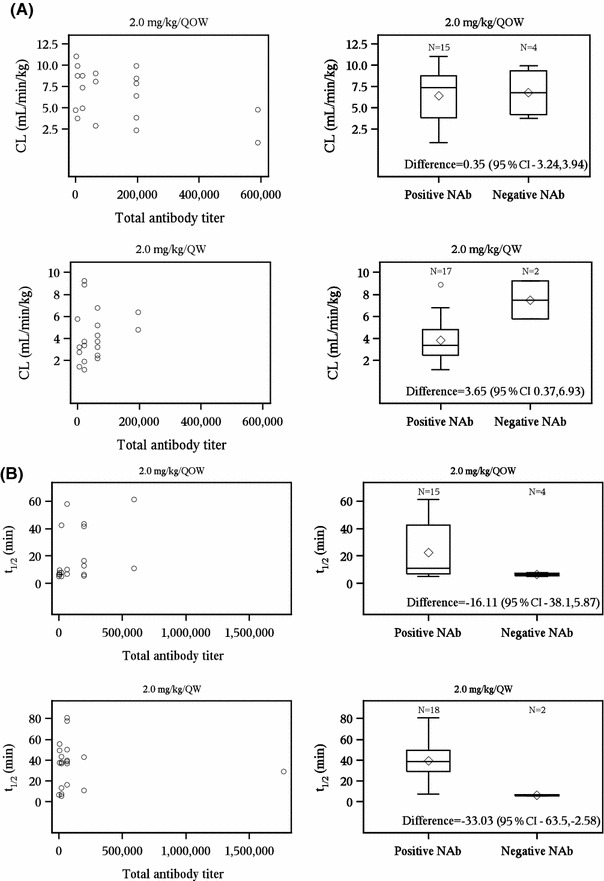
Association between elosulfase alfa pharmacokinetics and patient immunogenicity status. Pharmacokinetic values are from week 22 and patient immunogenicity status is from week 24. a Elosulfase alfa clearance and immunogenicity. b Elosulfase alfa plasma elimination half-life and immunogenicity. The bottom and top of whiskers represents the minimum and maximum values excluding outliers, which are plotted separately. The lower and upper ends of the box represent the 1st and 3rd quartile, the bar within the box represents the median value and the diamond within the box represents the mean value. CL total clearance of drug after intravenous administration, NAb neutralizing elosulfase alfa-specific antibodies that inhibit cellular receptor binding, TAb total anti-elosulfase alfa antibody, QOW every other week, QW every week
The mean elosulfase alfa CL at week 22 was compared between patients either positive or negative for NAb at week 24 (Fig. 3a). For patients in the QOW cohort, there was little difference in mean CL between NAb-negative patients (n = 4/19) and NAb-positive patients (n = 15/19; mean difference CL 0.35 mL/min/kg; 95 % CI −3.24 to 3.94), but for patients in the QW cohort, NAb-negative patients (n = 2/19) had higher mean CL than NAb-positive patients (n = 17/19; mean difference CL 3.65 mL/min/kg; 95 % CI 0.37–6.93). Similarly, when the mean elosulfase alfa t ½ was compared between NAb-negative and NAb-positive patients (Fig. 3b), in the QOW cohort, there was little difference in mean t ½ between NAb-negative patients (n = 4/19) and NAb-positive patients (n = 15/19; mean difference t ½ −16.11 min; 95 % CI −38.1 to 5.87 min), but in the QW cohort, NAb-negative patients (n = 2/20) had decreased mean t ½ compared with NAb-positive patients (n = 18/20; mean difference t ½ −33.03 min; 95 % CI −63.5 to −2.58 min).
Pharmacokinetics/Pharmacodynamics and Efficacy
The overall pharmacodynamic results of this study were reported elsewhere [5]; in brief, both QW and QOW dosing of elosulfase alfa led to reductions in uKS when compared with placebo. To evaluate whether the variability in patient pharmacodynamics was related to elosulfase alfa exposure (as measured either by AUClast or C max in plasma), levels of uKS from patients with Morquio A syndrome were assessed over time and compared with elosulfase alfa exposure (Fig. 4). No relationship between exposure to elosulfase alfa at week 22 and patients’ change in uKS between weeks 0 and 24 was observed.
Fig. 4.
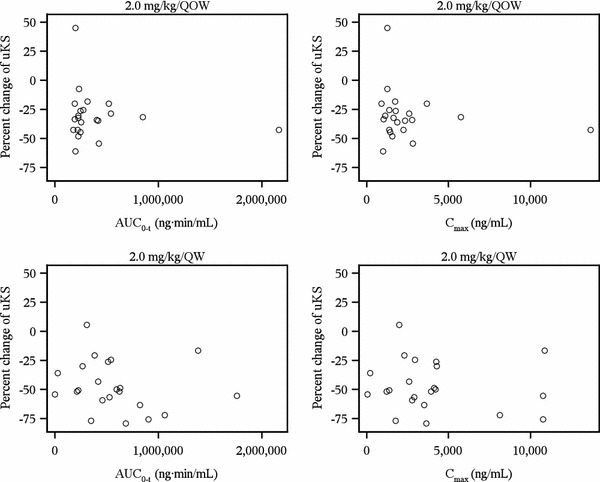
Elosulfase alfa exposure and percent change in uKS over 24 weeks. Pharmacokinetic values are from week 22. Percent change of uKS is the percent change in creatinine-normalized uKS levels between baseline and week 24. AUC last area under the plasma concentration-time curve from time zero to the time of last measurable concentration, C max observed maximum plasma concentration, qow every other week, uKS urinary keratan sulfate
The overall efficacy findings of this study were reported elsewhere [5]. In brief, QW dosing of elosulfase alfa led to statistically significant improvements in 6MWT distances vs. placebo but QOW dosing did not, and neither QW nor QOW dosing was associated with statistically significant improvements in 3MSCT results. Here, the relationship between patients’ exposures and efficacy outcomes was examined (Supplementary Figure S1, Supplementary Figure S2). For patients in both the QOW and QW cohorts, exposure at week 22 (as measured by either AUClast or C max in plasma) showed no evident association with changes in 6MWT or 3MSCT between weeks 0 and 24 (Supplementary Figure S1, Supplementary Figure S2).
Pharmacokinetics and Adverse Events
The occurrence of AEs [5] was analyzed against elosulfase alfa exposure (AUClast and C max); the results indicated no substantial differences between patients with and without AEs (Supplementary Figure S3). In addition, increase in elosulfase alfa exposure (AUClast and C max) was not associated with increase in occurrence of AEs.
Discussion
Morquio A syndrome is a progressive lysosomal storage disorder that results from deficiency of the lysosomal enzyme GALNS, causing broad morbidity and early mortality. Patients with Morquio A syndrome can present with differing disease manifestations and severity due to the heterogeneous nature of the disease [4, 10]. Currently, no treatment options are available that address the biological causes of Morquio A syndrome, but ERT with recombinant human GALNS enzyme (elosulfase alfa) may represent a possible treatment option. This study examined the pharmacokinetic and pharmacodynamic characteristics of elosulfase alfa 2.0 mg/kg dosing either weekly or every other week in patients with Morquio A syndrome during MOR-004, a 24-week, phase III, randomized, double-blind, placebo-controlled study conducted at 33 study centers in 17 countries.
Data from previous studies of elosulfase alfa support dosing at 2.0 mg/kg/QW. Prior studies demonstrated that, compared with elosulfase alfa 1.0, 2.0 mg/kg/QW yielded higher plasma concentrations of elosulfase alfa and sustained these concentrations above the Kuptake of the CI-M6PR for longer [6, 8]. A prior dose-escalation study showed elosulfase alfa dose-dependent declines in mean normalized uKS, with the greatest declines observed at 2.0 mg/kg QW, the highest dose tested [7]. The MOR-004 study demonstrated that, compared with placebo, elosulfase alfa 2.0 mg/kg/QW produced statistically significant differences in 6MWT distance and was associated with reductions in uKS, whereas the 2.0 mg/kg/QOW dose reduced uKS but was not associated with significant change in 6MWT distances [5]. These data support dosing elosulfase alfa at 2.0 mg/kg/QW and suggest that both dose and frequency of administration may impact outcomes and decreases in uKS.
Pharmacokinetic parameters were comparable between the two dosage regimens at week 0. Variability in elosulfase alfa pharmacokinetics among patients was not explained by patient demographics, as the demographics characteristics of patients did not appear to correlate with clearance of elosulfase alfa or other pharmacokinetic parameters. Although elosulfase alfa has a short plasma half-life, its intracellular half-life is estimated to be 5–7 days in human Morquio fibroblasts, which supports the QOW and QW dosing [11]. Pharmacokinetics of elosulfase alfa are time dependent, as demonstrated by increased plasma exposure, decreased CL, and prolonged half-life at week 22 compared with week 0. The time-dependent change in at least some of the pharmacokinetic parameters of elosulfase alfa are likely Nab related. All patients in this study developed antibodies against elosulfase alfa by week 16 of the trial. However, for both dosing groups, there were no apparent associations between total antibody titer and clearance of elosulfase alfa, nor were there associations between total antibody titer and the elosulfase alfa elimination half-life. The majority of patients developed neutralizing antibodies capable of inhibiting elosulfase alfa binding to the CI-M6PR in an in vitro assay. At each assessed time point, NAb positivity was slightly higher in the QW cohort than in the QOW cohort (Schweighardt et al., manuscript submitted for publication). At week 22 within the QW group, clear differences in elimination half-life and CL were noted between NAb-positive and NAb-negative patients. At week 22 within the QOW group, NAb-positive and NAb-negative patients had a clear difference in elimination half-life but the difference in CL was less pronounced, consistent with the small change in CL (14 %) observed from week 0 to week 22. For patients receiving weekly elosulfase alfa, only two patients were NAb negative at week 24. Both of these two patients tested positive for NAb during the study, but were no longer positive at week 24 when these analyses were performed. These patients demonstrated elosulfase alfa clearance at the upper range of the values exhibited in NAb-positive patients and elimination half-life at the lower end. Although the numbers of NAb-negative patients at week 24 were small (n = 2), these results are consistent with results observed in the independent elosulfase alfa clinical trial MOR-002, where again NAb positivity was associated with prolonged elimination half-life and decreased clearance of elosulfase alfa [8]. Importantly, these observed alterations in elosulfase alfa pharmacokinetics were not correlated with changes in efficacy or safety.
Despite variability in elosulfase alfa exposure between patients during the trial, there were no associations observed between exposure and efficacy, pharmacodynamics, and safety for either dosing group. The change in patients’ levels of uKS between weeks 0 and 24 was not correlated with elosulfase alfa clearance at week 22 for either dosing group. Similarly, changes in patients’ results for either the 6MWT or the 3MSCT between weeks 0 and 24 were not correlated with elosulfase alfa clearance at week 22 for either dosing group. Additionally, for patients in both dosing groups the occurrence of AEs was not correlated with elosulfase alfa exposure. These results indicate that the observed variability in elosulfase alfa exposure in plasma may not translate directly to pharmacodynamic, efficacy, or safety outcomes in patients, possibly because these plasma concentrations do not directly reflect the concentration where the drug is enzymatically active, at the likely site of action within the intracellular lysosomal compartment [6]. Although the 24-week duration of this study did not allow for the assessment of the long-term effects of elosulfase alfa, a long-term extension of this study (MOR-005; NCT01415427) and a patient registry will allow continued monitoring and assessment of the efficacy and safety of elosulfase alfa treatment in patients with Morquio A.
Compared with ERTs for other mucopolysaccharidosis diseases, such as laronidase for MPS I, idursulfase for MPS II, and galsulfase for MPS VI, the clearance of elosulfase alfa is about two- to threefold higher, which is in agreement with its two- to fourfold higher dose recommended for clinical use [12–16]. Enzyme replacement therapies for MPS diseases generally have a short plasma elimination half-life, from 26 min for galsulfase to 1.5 to 3.6 h for laronidase. However, their intracellular or tissue half-life is much longer, between 1 and 2 days in rat tissue for idursulfase and 5–7 days in human Morquio fibroblasts for elosulfase alfa [11]. The long intracellular or tissue half-life supports their weekly dosing recommended for clinical use as the site of action of these ERTs is within the intracellular lysosomal compartment.
Conclusion
This paper summarized pharmacokinetic characteristics and their relationship to the demographics, immunogenic responses, pharmacodynamics, efficacy, and safety of elosulfase alfa observed in patients with Morquio A syndrome in a phase III trial. Exposure was seen to increase over the 24-week study period in patients who received elosulfase alfa 2.0 mg/kg either QW or QOW. Variability in elosulfase alfa exposure in plasma appeared not to translate directly to pharmacodynamic, efficacy, or safety outcomes in patients, possibly because the drug’s site of action is within the intracellular lysosomal compartment. In combination with the primary efficacy and safety outcomes of this trial [5], these data support the dosing of elosulfase alfa at 2.0 mg/kg/QW in patients with Morquio A.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary material 3 (PS 173 kb) Supplementary Figure S1: Pharmacokinetic parameters and patient outcomes in the 6-min walk test. Pharamcokinetic values from week 22; change in distance achieved in 6MWT from baseline to week 24. AUC last area under the plasma concentration-time curve from time zero to the time of last measurable concentration, C max observed maximum plasma concentration, qow every other week
Supplementary material 4 (PS 178 kb) Supplementary Figure S2: Patients’ pharmacokinetic parameters and outcomes in the 3-min stair climb. Pharamcokinetic values from week 22; change in stairs climbed per minute in 3MSC from baseline to week 24. AUC last area under the plasma concentration-time curve from time zero to the time of last measurable concentration, 3MSC 3-min stair climb test, C max observed maximum plasma concentration, qow every other week
Supplementary material 5 (PS 268 kb) Supplementary Figure S3: Patients’ elosulfase alfa exposure and adverse events. Number of adverse events is the occurrence of adverse events (with incidences >10 % in the elosulfase alfa treatment group vs. placebo) per patient between baseline and week 24 compared with pharmacokinetic values from week 22. AE adverse events, AUC last area under the plasma concentration-time curve from time zero to the time of last measurable concentration, C max observed maximum plasma concentration, qow every other week
Acknowledgments
Medical writing support for this manuscript was furnished by Karl Zawadzki, PhD, all of which was funded by BioMarin Pharmaceuticals, Inc.
Conflict of interest
Yulan Qi, Donald G Musson, Becky Schweighardt, Troy Tompkins, Lynne Jesaitis, Adam J. Shaywitz, Ke Yang, and Charles A. O’Neill are employees of BioMarin Pharmaceutical.
Patient informed consent statement
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Each participant or his/her legally authorized representative provided written informed consent before entering the study in compliance with the applicable local regulations.
Details of the contributions of individual authors
All authors were involved in the analysis and interpretation of data, drafting of the manuscript, and critical revisions of the manuscript for important intellectual content.
References
- 1.Hendriksz CJ, Harmatz P, Beck M, Jones S, Wood T, Lachman R, Gravance CG, Orii T, Tomatsu S. Review of clinical presentation and diagnosis of Mucopolysaccharidosis IVA. Mol Genet Metab. 2013;110:54–64. doi: 10.1016/j.ymgme.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nelson J. Incidence of the mucopolysaccharidoses in Northern Ireland. Hum Genet. 1997;101:355–358. doi: 10.1007/s004390050641. [DOI] [PubMed] [Google Scholar]
- 3.Nelson J, Crowhurst J, Carey B, Greed L. Incidence of the mucopolysaccharidoses in Western Australia. Am J Med Genet A. 2003;123A:310–313. doi: 10.1002/ajmg.a.20314. [DOI] [PubMed] [Google Scholar]
- 4.Harmatz P, Mengel KE, Giugliani R, Valayannopoulos V, Lin SP, Parini R, Guffon N, Burton BK, Hendriksz CJ, Mitchell J, Martins A, Jones S, Guelbert N, Vellodi A, Hollak C, Slasor P, Decker C. The Morquio A Clinical Assessment Program: baseline results illustrating progressive, multisystemic clinical impairments in Morquio A subjects. Mol Genet Metab. 2013;109:54–61. doi: 10.1016/j.ymgme.2013.01.021. [DOI] [PubMed] [Google Scholar]
- 5.Hendriksz CJ, Burton BK, Fleming TR, Harmatz PR, Hughes D, Jones SA, Lin SP, Mengel E, Scarpa M, Valayannopoulos V, Giugliani R, STRIVE Investigators, Slasor P, Lounsbury D, Dummer W. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): a phase 3 randomised placebo-controlled study, J Inherit Metab Dis. 2014. (Epub ahead of print). [DOI] [PMC free article] [PubMed]
- 6.Dvorak-Ewell M, Wendt D, Hague C, Christianson T, Koppaka V, Crippen D, Kakkis E, Vellard M. Enzyme replacement in a human model of mucopolysaccharidosis IVA in vitro and its biodistribution in the cartilage of wild type mice. PLoS One. 2010;5:e12194. doi: 10.1371/journal.pone.0012194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hendriksz C, Vellodi A, Jones S, Takkele H, Lee S, Chesler S, Decker C. Long term outcomes of a phase 1/2, multicenter, open-label, dose-escalation study to evaluate the safety, tolerability, and efficacy of BMN 110 in patients with mucopolysaccharidosis IVA (Morquio A syndrome) Mol Genet Metab. 2012;105:S35. doi: 10.1016/j.ymgme.2011.11.076. [DOI] [Google Scholar]
- 8.Qi Y, Musson D, Martell L, Van Tuyl A, Decker C, Santos S, Garcia L, Lau K, Devereaux D, O’Neill C. Phase 1/2 pharmacokinetic studies of recombinant human N-acetylgalactosamine-6-sulfatase (rhGALNS) in MPS IVA patients. In: Poster presentation at the 2012 AAPS Annual Meeting and Exposition, October 14–17, 2012; Chicago. 2012; Poster T3338.
- 9.ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med. 2002;166:111–117. doi: 10.1164/ajrccm.166.1.at1102. [DOI] [PubMed] [Google Scholar]
- 10.Montaño AM, Tomatsu S, Gottesman GS, Smith M, Orii T. International Morquio A registry: clinical manifestation and natural course of Morquio A disease. J Inherit Metab Dis. 2007;30:165–174. doi: 10.1007/s10545-007-0529-7. [DOI] [PubMed] [Google Scholar]
- 11.BioMarin Pharmaceutical. Vimizim (elosulfase alfa) for the treatment of mucopolysaccharidosis type IV A (Morquio A syndrome). Briefing document for the endocrinologic and metabolic drugs advisory committee, FDA, http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/EndocrinologicandMetabolicDrugsAdvisoryCommittee/UCM375127.pdf. 19 Nov 2013, p. 53.
- 12.BioMarin Pharmaceutical. Naglazyme (galsulfase): highlights of prescribing information, Novato; 2013.
- 13.BioMarin Pharmaceutical, Genzyme Corporation, Aldurazyme (laronidase): Highlights of prescribing information, Novato and Cambridge; 2013.
- 14.Harmatz P, Ketteridge D, Giugliani R, Guffon N, Teles EL, Miranda MC, Yu ZF, Swiedler SJ, Hopwood JJ, MPS VI Study Group Direct comparison of measures of endurance, mobility, and joint function during enzyme-replacement therapy of mucopolysaccharidosis VI (Maroteaux–Lamy syndrome): results after 48 weeks in a phase 2 open-label clinical study of recombinant human N-acetylgalactosamine 4-sulfatase. Pediatrics. 2005;115:e681–e689. doi: 10.1542/peds.2004-1023. [DOI] [PubMed] [Google Scholar]
- 15.Kakkis ED, Matynia A, Jonas AJ, Neufeld EF. Overexpression of the human lysosomal enzyme alpha-l-iduronidase in Chinese hamster ovary cells. Protein Expr Purif. 1994;5:225–232. doi: 10.1006/prep.1994.1035. [DOI] [PubMed] [Google Scholar]
- 16.Shire Human Genetic Therapies, Elaprase (idursulfase): Highlights of prescribing information, Lexington; 2013.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material 3 (PS 173 kb) Supplementary Figure S1: Pharmacokinetic parameters and patient outcomes in the 6-min walk test. Pharamcokinetic values from week 22; change in distance achieved in 6MWT from baseline to week 24. AUC last area under the plasma concentration-time curve from time zero to the time of last measurable concentration, C max observed maximum plasma concentration, qow every other week
Supplementary material 4 (PS 178 kb) Supplementary Figure S2: Patients’ pharmacokinetic parameters and outcomes in the 3-min stair climb. Pharamcokinetic values from week 22; change in stairs climbed per minute in 3MSC from baseline to week 24. AUC last area under the plasma concentration-time curve from time zero to the time of last measurable concentration, 3MSC 3-min stair climb test, C max observed maximum plasma concentration, qow every other week
Supplementary material 5 (PS 268 kb) Supplementary Figure S3: Patients’ elosulfase alfa exposure and adverse events. Number of adverse events is the occurrence of adverse events (with incidences >10 % in the elosulfase alfa treatment group vs. placebo) per patient between baseline and week 24 compared with pharmacokinetic values from week 22. AE adverse events, AUC last area under the plasma concentration-time curve from time zero to the time of last measurable concentration, C max observed maximum plasma concentration, qow every other week