

Abstract
This article focuses on elucidating the key presentation features of neurotrophic ligands at polymer interfaces. Different biointerfacial configurations of the human neural cell adhesion molecule L1 were established on two-dimensional films and three-dimensional fibrous scaffolds of synthetic tyrosine-derived polycarbonate polymers and probed for surface concentrations, microscale organization, and effects on cultured primary neurons and neural stem cells. Underlying polymer substrates were modified with varying combinations of protein A and poly-d-lysine to modulate the immobilization and presentation of the Fc fusion fragment of the extracellular domain of L1 (L1-Fc). When presented as an oriented and multimeric configuration from protein A-pretreated polymers, L1-Fc significantly increased neurite outgrowth of rodent spinal cord neurons and cerebellar neurons as early as 24 h compared to the traditional presentation via adsorption onto surfaces treated with poly-d-lysine. Cultures of human neural progenitor cells screened on the L1-Fc/polymer biointerfaces showed significantly enhanced neuronal differentiation and neuritogenesis on all protein A oriented substrates. Notably, the highest degree of βIII-tubulin expression for cells in 3-D fibrous scaffolds were observed in protein A oriented substrates with PDL pretreatment, suggesting combined effects of cell attachment to polycationic charged substrates with subcellular topography along with L1-mediated adhesion mediating neuronal differentiation. Together, these findings highlight the promise of displays of multimeric neural adhesion ligands via biointerfacially engineered substrates to “cooperatively” enhance neuronal phenotypes on polymers of relevance to tissue engineering.
Introduction
Neurodegenerative diseases and traumatic injuries can cause irreversible damage to the central nervous system (CNS), resulting in a significant deficit in motor and sensory function due to a limited endogenous capacity for regeneration and targeted axonal regrowth [1]. An ideal strategy for CNS repair relies on promoting regrowth/sprouting, neuronal survival, synaptogenesis, and remyelination of host axons while stimulating transplanted exogenous neural cells to survive, migrate, and integrate within host tissue [2]. Engineered biomaterials involving specific neural cell adhesion ligands have been recently explored for their ability to support neurite outgrowth and neuronal differentiation relevant for CNS repair [3, 4]. However, the effective integration of neuronal bioactivity along with transplantable substrate designs remains a major challenge. Conventional bioactive or biomimetic substrates for neural engineering applications have presented peptides, extracellular matrix (ECM) proteins, growth factors, and cell adhesion proteins [5–9].
A key adhesion ligand for neuronal survival and differentiation is the neural cell adhesion molecule L1. L1 is a transmembrane glycoprotein of the immunoglobulin superfamily that also shares several binding domains with fibronectin [10]. L1 acts through homophilic (L1–L1) interactions, as well as heterophilic interactions [11–13] and plays a critical role in neural cell adhesion, [14–16] neurite fasciculation, neuronal protection [17, 18], synaptic plasticity, axonal outgrowth and adhesion, [19] subcellular synapse organization and cell migration [20, 21]. It is expressed on the cell surface of postmitotic neurons in the CNS and peripheral nervous system (PNS), as well as Schwann cells in the PNS [22]. L1 has also been shown to improve functional recovery and improved corticospinal tract regrowth following spinal cord injury in mice [23].
The role of L1 in neuronal differentiation of neural stem cells has also been investigated. Primary mouse neural stem cells demonstrated enhanced neuronal differentiation and decreased astrocyte differentiation when cultured on substrates pretreated with L1-Fc or fibroblasts engineered to express L1-Fc, compared to poly-d-lysine (PDL) and laminin-treated surfaces [24]. Additionally, L1-Fc treated surfaces primarily promoted GABAergic differentiation over other cell types, suggesting that L1 can influence neuronal subtype specification in the absence of growth factors [24]. In another study, L1-transfected mouse embryonic stem cells exhibited enhanced process extension and migration in comparison to non-transfected stem cells when injected into a spinal cord lesion site, which induced enhanced functional recovery [25]. Both studies demonstrate that L1 promotes neuronal versus astrocytic differentiation of stem cells.
Despite the widespread interest in the biological activity of L1, mechanisms and approaches for optimally presenting L1 from biomedically relevant materials for cell transplantation remain to be systematically examined. A variety of materials of natural and synthetic origin, have been previously shown to promote adhesion, proliferation, neurite extension, and neuronal differentiation of neural cells in vitro and in vivo [26–28]. Synthetic polymer-based biomaterial scaffolds have the added advantage of controlled chemistries and mechanical properties [29, 30], while enabling display or release of neurotrophic factors [31, 32]. Of the various scaffold configurations proposed to date [27, 28, 30], electrospun polymer substrates have exhibited excellent neurogenic properties, due to their high surface area and porosity, and fibrous ECM-like geometries [33, 34].
In this study, we combined both 2-D and 3-D substrate configurations fabricated from biodegradable synthetic polymers, with the goal of modulating controlled surface presentation of the neural adhesion molecule L1. Polymer films (2-D) and fibrous polymer scaffolds (3-D) were fabricated from tyrosine-derived polycarbonates, a combinatorial library of degradable polymers with tunable mechanical properties, surface properties, and degradation rates [35, 36]. Specifically, the base monomer poly(desaminotyrosyl tyrosine ethyl ester carbonate) [poly(DTE carbonate)] can be copolymerized with variable amounts of desaminotyrosyl tyrosine (DT) and/or poly(ethylene glycol) (PEG), resulting in polymers with similar structures but variable degradation rates and protein adsorption properties. We used three different approaches to establish biointerfaces of L1-Fc: (1) the conventional adsorption of L1-Fc onto substrates pretreated with PDL, (2) presentation of L1-Fc from substrates pretreated with PDL and protein A and (3) presentation of L1-Fc from substrates pretreated with protein A alone. We hypothesized that the presentation of L1-Fc via substrate-coated protein A would promote the outward display of surface-bound L1-Fc, while presenting L1-Fc in a more optimal, physiologically-relevant manner, compared to the random orientation of L1-Fc/PDL coated substrates. We report that multimeric presentation of L1-Fc via binding to protein A enhanced adhesion and neurite outgrowth in 2-D configurations and that this effect was also replicated in a 3-D configuration, consisting of aligned scaffolds of polymer fibers biofunctionalized with L1-Fc. This is also the first report of effects of substrate biofunctionalized L1 on human neural progenitor cell behavior.
Materials and Methods
Biointerfaces on Polymer Thin Films
A 1% (w/v) poly(DTE-co-10% DT-co-1% PEG1k carbonate) polymer solution was prepared by dissolving the polymer in tetrahydrofuran (THF) (Sigma-Aldrich, St. Louis, MO, USA) overnight at room temperature. The polymer solution was then filtered using a 0.45 μm Whatman pTFE filter and spin-coated onto 12 mm glass coverslips. The thin films were dried for at least 24 h under vacuum and UV sterilized for 15 min, prior to coating with proteins. The thin films were then coated with 100 μg/ml of PDL (70–150 kDa; Sigma-Aldrich) for 1 h at room temperature. After three washes with ultrapure water, the films were coated with 1.25 μg/ml of protein A (PA) (Invitrogen, Carlsbad, CA, USA) for 1 h at room temperature. Following three washes with ultrapure water, the films were coated with 2, 10, or 50 μg/ml of human L1-Fc (R&D Systems, Minneapolis, MN, USA) overnight at 4°C. L1-Fc is the fusion of the Fc region of human IgG1, which contains two disulfide bonds, to two extracellular domains of human L1. For controls, the thin films were kept hydrated with phosphate buffered saline (PBS, Lonza, Walkersville, MD, USA) overnight. The films were washed three times with PBS and the cells were seeded immediately. The resulting conditions were as follows: L1-Fc/PDL, L1-Fc/PA/PDL, and L1-Fc/PA and the corresponding controls.
Verification of L1-Fc Functionality
Enzyme-linked immunosorbent assay (ELISA) was used to determine the presence of L1-Fc on different substrates. Each substrate was prepared as described above. Next, the thin films were washed in PBS one time for 2 min and treated with a blocking solution composed of 1% bovine serum albumin (BSA, Sigma Aldrich, St. Louis, MO, USA), in PBS for 1.5 h at room temperature. After one wash for 2 min in PBS, the samples were incubated with a monoclonal mouse anti-L1 antibody, clone L1.1 (1 μg/ml) (Invitrogen), specific for the extracellular domain of human L1 for 1.5 h and washed three times, for 5 min each, with PBS containing 0.05% Tween-20 (PBST). The substrates were then treated with the secondary antibody, alkaline phosphatase conjugated-goat anti-mouse IgG antibody (Sigma-Aldrich) for 1.5 h at room temperature. Following three 5 min washes with PBST, the substrates were reacted with alkaline phosphatase yellow (pNPP) liquid substrate (Sigma-Aldrich) for up to 30 min in the dark at room temperature. The reaction was stopped with 3N NaOH and the substrates were read on an absorbance plate reader at 405 nm. The absorbance readings were used to assess the presence of L1-Fc on each substrate compared to the respective controls.
Quantification of Protein Layer on Polymer Thin Films
Deposition of polypeptide and protein layers on polymer thin films was measured using a quartz crystal microbalance with dissipation (QCM-D) (Qsense E4, Biolin Scientific, Linthicum Heights, MD, USA). Polymer thin film substrates using gold-coated sensor crystals (QSX301, Biolin Scientific, Linthicum Heights, MD, USA) were prepared as described above using spin coating except dioxane (Fisher Scientific, Pittsburgh, PA, USA) was used as the solvent. Prior to measurement, the polymer coated sensor crystals were equilibrated in PBS for at least 1 h at 37°C to establish a stable baseline. Subsequently, PDL (100 μg/ml), PA (20 μg/ml) and L1-Fc (10 μg/ml) were perfused through the flow system for 30 min at a flow rate of 24.2 μl/min. The deposition of each layer was followed by a PBS wash for at least 30 min. Control experiments were conducted replacing deposition of respective layer with PBS perfusion. The frequency and dissipation were measured during each experiment. For raw data analysis the fifth overtone (25 MHz) was used. Hydrated mass per surface area of L1-Fc was modeled from the frequency and dissipation of the fifth and seventh overtone according to the Voigt model (Q-tools software) [37]. From the information obtained from the Voigt model and the bulk L1-Fc concentration applied to the crystals, the percent of L1-Fc deposited onto each thin film was also determined.
AFM Analysis of Biointerfaces on Thin Films
Atomic force microscopy (AFM) was utilized to visualize L1-Fc spatial distribution and the thickness of each thin film. Each thin film was prepared as described above. Next, films were blocked in a blocking solution of 1% BSA in PBS for 30 min at room temperature. Following three washes with PBS, the substrates were treated with the L1.1 antibody (1 μg/ml) for 30 min. The substrates were washed three times with PBS and incubated with Nanogold® goat anti-mouse IgG antibody (1:40 dilution) (Nanoprobes, Yaphank, NY, USA) for 30 min. To amplify the gold signal, thin films were treated for 5 min with a silver enhancement kit, which would increase the gold particle size from 30 to 100 nm, depending on treatment time (LI Silver Enhancing Kit, Invitrogen). We chose to use the shortest treatment time possible as large variability in particle size has been shown to increase with time [38]. The samples were then washed with ultra-pure water and air dried in a desiccator overnight prior to AFM imaging. AFM height images in tapping mode were collected using a Multimode AFM having a Nanoscope-IIIa controller equipped with a J-type piezo scanner (Veeco Metrology Group, Santa Barbara, CA, USA).
Fibrous Polymer Scaffold Fabrication and Characterization
Fibrous scaffolds were fabricated by electrospinning poly(DTE-co-10% DT-co-1% PEG1k carbonate). An 18% (w/v) solution was prepared by dissolving the polymer in glacial acetic acid (Fisher) overnight at room temperature. Fibrous scaffolds were produced by flowing the polymer solution through an 18-gauge needle at a flow rate of 1 ml/h, controlled by a programmable syringe pump (KD Scientific, Holliston, MA, USA). A voltage of +24 kV was applied to the needle by a 30 kV dual polarity power supply (Gamma High Voltage Research, Inc, Ormond Beach, FL, USA) and the fibers were collected on a rapidly rotating mandrel spaced 18 cm from the needle. The scaffolds were allowed to dry under vacuum for at least 3 days and were UV sterilized for 30 min prior to protein treatment and cell culture.
Surface morphology of the electrospun scaffolds was observed on an AMRAY 1830 I scanning electron microscope (SEM). Samples for SEM imaging were dried under vacuum, and sputter-coated with gold–palladium [39]. Imaging was performed at 20 kV acceleration potential. Quantification of the fiber diameter distribution was based upon measurement of 100 individual fibers in 1,000× SEM images using NIH-ImageJ software (http://rsb.info.nih.gov/ij/).
Fiber alignment was quantified from SEM images using ImageJ, similar to a previously described method [40]. Briefly, ImageJ was used to measure the angle of alignment of each fiber in a given SEM image and the angle difference between each fiber and the mean fiber angle was calculated. This data is shown as a histogram of angle differences. The alignment of at least 100 fibers was measured.
Primary Neuronal Cell Cultures
Spinal cord neurons (SCNs) were obtained following a protocol that was adopted and modified from what was previously described, [41] using embryonic day 15 rat embryos obtained from pregnant Sprague–Dawley rats (Taconic, Germantown, NY, USA). The SCNs were cultured in serum free neurobasal medium (Invitrogen) supplemented with 1× B27 supplement (Invitrogen), 2 mM glutamine (Invitrogen) and 1% penicillin/streptomycin (Lonza) and maintained for 24 h.
Cerebellar neurons (CNs) were obtained from P8 Sprague–Dawley rat pups, following a protocol previously described [42]. The cells were cultured in serum free neurobasal-A medium (Gibco), supplemented with 1× B27 supplement, 25 mM KCl, 2 mM glutamine, and 1% penicillin/streptomycin.
Quantification of Neurite Outgrowth
For neurite outgrowth studies of SCNs and CNs, both cell types were plated at 2 × 104 cells/cm2 onto thin films or 3.1 × 104 cells/cm2 onto scaffolds pretreated with different L1-Fc presentations and controls. The cells were fixed and immunostained with mouse monoclonal βIII-tubulin (clone TUJ1, Covance, Berkeley, CA, USA) after 24 h in culture. For each condition, ten fields of view at 40× magnification were imaged using a Leica TCS.SP2 confocal microscope system (Leica Microscope, Exton, PA). For image analysis, total neurites were measured using NIH-ImageJ imaging software. Only neurites that were equal to or greater than the diameter of a cell body were measured. In the case of SCNs, the average length of the longest neurite was also measured for cells with multiple membrane processes.
Functional Blocking Assays of Cell Adhesion and Neurite Outgrowth
To determine if L1-Fc presentation affected the availability of different binding sites, we investigated how blocking interactions between substrate bound L1-Fc and cellular L1 or αvβ3 integrin receptors influenced neurite outgrowth and cell attachment. To block L1–L1 homophilic interactions, each substrate was treated with 50 μg/ml of anti-L1 antibody, clone L1.1 or mouse IgG1 isotype control (R&D Systems), for 1 h, prior to seeding SCNs. In order to competitively inhibit the interaction between L1 and αvβ3, SCNs were treated with various concentrations of cyclic RGD (cyclo-RGDfV) peptide (Peptides International, Louisville, KY, USA) for 1 h prior to plating the cells onto the different L1-presenting substrates. SCNs were cultured for 24 h, immunostained with βIII-tubulin, and analyzed for cell attachment and neurite outgrowth. For each metric, ten fields of view were taken for each condition and the number of nuclei for each view was counted or neurites were measured using NIH-ImageJ imaging software.
hESC-Derived Neural Progenitor Cell Culture and Differentiation
ENStem-A neural progenitor cells (NPCs) were purchased from Millipore (Millipore, Temecula, CA, USA). These NPCs were derived from NIH approved H9 human embryonic stem cells (hESCs). For cell expansion and maintenance, NPCs were cultured in 6 cm culture dishes coated with ¼x stock Matrigel (BD Biosciences, Franklin Lakes, NJ, USA) in ENStem-A neural expansion medium (Millipore) at 37°C in 5% CO2. Medium was changed every 2 days and cells were passaged, by mechanical dissociation, once they reached 90–95% confluence (every 4–5 days) at a ratio of 1:2 or 1:3.
Differentiation of NPCs was initiated once the cells reached about 85–90% confluence, by removing ENStem-A medium, washing two times with warm neurobasal medium, and replacing the medium with neural differentiation medium (NDM) composed of neurobasal medium, 1× B27 supplement, 2 mM glutamax (Invitrogen), 1% penicillin/streptomycin. After 4 days in NDM, the cells were passaged with Accutase (Invitrogen), plated on L1-Fc coated thin films and scaffolds at a density of 6.25 × 104 or 2 × 104 cells/cm2 respectively, and allowed to differentiate for 7 days, with medium changes every 2 days.
Immunocytochemistry of Neuronal Phenotypes
After 24 h (SCNs or CNs) or 7 days (NPCs), culture medium was removed and the cells were fixed in 4% paraformaldehyde for 20 min at room temperature and then washed three times with PBS. The cells were then permeabilized with 0.1% Triton-X, followed by three PBS washes. To prevent non-specific antibody binding, the samples were blocked with 5% normal goat serum (MP Biomedicals, Solon, OH, USA) in PBS (blocking buffer), for 1 h at room temperature. Primary antibodies were then diluted in the blocking buffer and applied to the samples overnight at 4°C. Following the removal of the primary antibody, the cells were washed three times, for 20 min each time, and the secondary antibodies, along with 1 μg/ml of Hoechst 33258 (Sigma-Aldrich) were diluted in blocking buffer and applied for 1 h at room temperature, followed by three, 20 min washes with PBS. Primary antibodies mouse anti-βIII-tubulin and mouse anti-nestin (IgG1) (Millipore) followed by isotype specific Alexa Fluor 488, 594, or 647 goat-anti mouse secondary antibodies (Invitrogen) were used for the studies.
Statistical Analysis
The data is expressed as mean ± the standard error of the mean. Variance of analysis using one-way ANOVA was used followed by post hoc means comparison with Tukey’s test. Differences between conditions were considered statistically significant with p values <0.05. All data represents three independent experiments performed in duplicate.
Results
Protein A Coated Polymer Films Support Increased Surface Adsorption of L1-Fc
The relative amount of the neuritogenesis-promoting domain exposed within L1-Fc adsorbed onto polymer thin films treated with PDL, PA/PDL, or PA was determined by ELISA (Fig. 1a). We examined three different concentrations (2, 10, and 50 μg/ml) to characterize L1-Fc epitope exposure on the surface using multimeric versus random modes of presentation. ELISA results showed increased and dose-dependent L1-Fc increased exposure of the neuritogenesis-promoting epitope on PA containing substrates compared to PDL controls. These results support a more outward display of L1 on PA-treated substrates, given the nature and location of antibody binding epitopes within the N-terminal, first and second Ig-like domains of the ectodomain of L1 [43].
Fig. 1.

Presentation of L1-Fc by protein A results in enhanced L1 adsorption and improved directed presentation of L1-Fc. a ELISA was used to determine the relative amounts of L1-Fc on the polymer thin films treated with and without PA. L1-Fc (2, 10, 50 μg/ml) was adsorbed on PDL with and without PA (PDL, PA/PDL) or PA alone (PA). b Representative QCM-D plots of frequency versus time: L1-Fc deposition on PDL (red); PA/PDL (blue), and PA (green). c Voigt mass of L1-Fc deposition modeled from QCM-D data subsequent to PDL, PA/PDL, or PA deposition. d The percent of L1-Fc adsorbed onto each surface during the QCM-D, from a bulk concentration of 10 μg/ml of L1-Fc. PDL poly-d-lysine. PA protein A (* denotes p < 0.05)
The amount of L1-Fc adsorbed on the polymer thin films was quantified using QCM-D. Representative QCM-D plots of frequency over time show subsequent frequency drops indicating deposition of PDL, PA, and L1-Fc, respectively (Fig. 1b). An average frequency change for the deposition of PDL of Δf = −9 ± 0.9 Hz was observed. The frequency change for the deposition of PA was higher for surfaces with prior PDL deposition with Δf = −15 ± 0.3 Hz as compared to Δf = 7 ± 4.1 Hz for PA on uncoated polymer surfaces. For both PDL and PA, only small changes of dissipation were observed with ΔD < 10. Deposition of L1-Fc subsequent to deposition of PA and PDL led to much larger changes of −70 ± 17.3 Hz and a dissipation change of ΔD = 8 ± 3.4. The hydrated mass per surface area of L1-Fc was calculated using the Voigt model for all conditions (Fig. 1c). The Voigt mass of L1-Fc was highest with m = 2.8 ± 0.84 μg/cm2 subsequent to coating the polymer surface with both PDL and PA. A slightly lower mass of L1-Fc was deposited on the polymer surface treated only with PA with m = 1.9 ± 0.56 μg/cm2. In comparison, the deposited mass of L1-Fc onto the exclusively PDL treated polymer surfaces was even lower with m = 0.8 ± 0.15 μg/cm2. The percent of L1-Fc deposited onto the surface from the added bulk ligand concentration is shown in (Fig. 1d). The results of the ELISA and QCM-D studies confirm that PA-based presentation of L1-Fc results in increased levels of L1-Fc adsorption onto the polymer thin films.
Spatial Distribution of L1-Fc when Presented on PDL and Protein A
We hypothesized that L1-Fc presentation was multimeric and oriented, in a specific manner, on PA coated polymer thin films (Fig. 2b) compared to a more randomly oriented presentation on PDL coated polymer films (Fig. 2a). AFM, using dry tapping mode, was used to quantify the spatial distribution, substrate thickness, and relative sizes of L1 complexes using gold-conjugated antibodies and silver counterstaining. L1-Fc adsorbed onto PDL resulted in complexes diffusely distributed on the thin films with a surface thickness of 23.5 ± 1.9 nm. L1-Fc presented from PDL and PA or PA alone also resulted in diffusely distributed L1-Fc protein complexes with surface thicknesses 23.5 ± 1.8 and 23.2 ± 1.5 nm respectively (Fig. 2c). While there was no difference in the thickness of each substrate, which was a result of adsorbed L1-Fc complexed with the primary and gold-conjugated-silver enhanced secondary antibodies, it was clear that PA-presented L1-Fc resulted in increased L1-Fc adsorbed to the surface, as is indicated by the increase in the density of the complexes in the two-dimensional images and three-dimensional images of substrate topography (Fig. 2c). We also measured the width of the protein complexes using the cross-sectional profiles of the complexes shown in the third column of Fig. 2c. Thin films of L1-Fc/PDL exhibited well-defined peaks indicative of less aggregation, whereas L1-Fc presented from PA/PDL or PA alone resulted in peaks that were wider and less defined, suggesting the close apposition of multiple complexes, and greater density that is consistent with increased surface concentrations of L1-Fc quantified via ELISA in Fig. 1a.
Fig. 2.
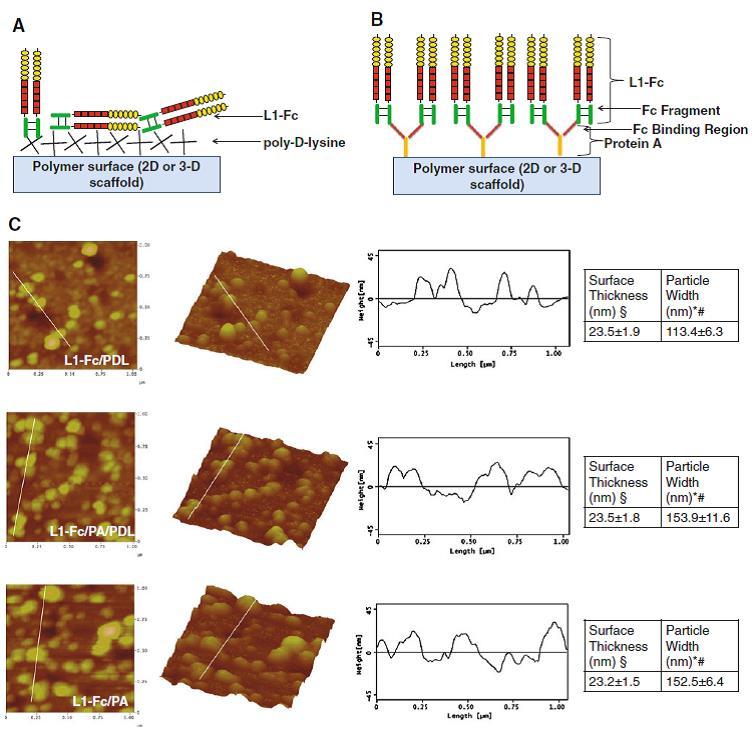
Differentially treated polymer films alter surface presentation of L1-Fc. a Schematic representation of how L1-Fc might randomly orient when coated onto a layer of PDL. b PA presentation will result in four L1-Fc molecules per PA molecule [68], enabling the multimeric, oriented presentation of L1-Fc. c AFM images (1 × 1 μm2, two and three dimensional) and cross-sectional profiles of the protein complexes, which correspond to the white lines drawn on the images, of thin polymer films coated with L1-Fc/PDL, L1-Fc/PA/PDL, or L1-Fc/PA. Thin films in which L1-Fc is presented from PA revealed increased L1-Fc adsorption as shown by the increase in protein complexes in the 2-D and 3-D images. PA presented L1-Fc resulted in cross-sectional profiles with wider peaks compared to L1-Fc adsorbed on PDL. A summary of the thickness of the thin films and the width of the protein complexes is shown in the tables to the far right. PDL poly-d-lysine, PA protein A (* denotes p < 0.05 compared to both PDL and PA/PDL controls, § denotes p < 0.05 compared to PA control, # denotes p < 0.05 compared to the L1-Fc/PDL condition)
Multimeric L1 Presentation Enhances Neurite Outgrowth of Cultured Neurons on 2-D Films
Spinal cord neurons were cultured on variable L1-Fc configurations for 24 h (Fig. 3a–c). At increasing concentrations of L1-Fc, L1-Fc elicited longer neurite extensions of SCNs when presented on films containing PA compared to thin films only coated with PDL (Fig. 3a). The average SCN neurite lengths on the L1-deficient controls PDL, PA/PDL, and PA averaged around 53 ± 2.4 μm (Fig. 3b) and were comparable to 2 μg/ml of L1-Fc presented from PDL and PA/PDL. In contrast, the earliest significant increase in neurite length (75.9 ± 8.4 μm) was elicited by 2 μg/ml of L1-Fc presented from PA alone, suggesting that PA enhances the efficacy of low concentrations of L1-Fc. Increased L1-Fc concentrations progressively elicited enhanced neurite extension. Both L1-Fc/PA/PDL and L1-Fc/PA promoted increased neurite lengths on 10 μg/ml (145.1 ± 12.1 and 154.8 ± 12.7 μm, respectively) and 50 μg/ml (186.4 ± 5.5 and 182.8 ± 13.4 μm, respectively) of L1-Fc in comparison to the L1-Fc/PDL substrates (73.4 ± 4.5 and 93.1 ± 10.3 μm). These results show that (1) PA presentation greatly improves L1-Fc mediated neurite outgrowth of SCNs and (2) PDL does not additionally contribute to enhanced cell attachment or extensions when L1-Fc is presented from PA.
Fig. 3.
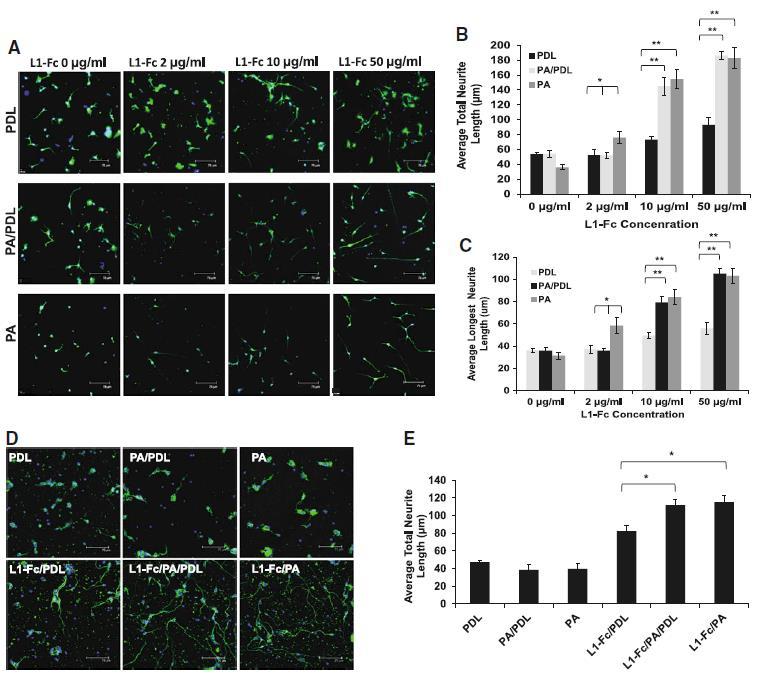
Protein A presentation of L1-Fc enhances neurite outgrowth in spinal cord and cerebellar neurons. a Spinal cord neurons (SCNs) were plated on L1-Fc adsorbed onto PDL (L1-Fc/PDL), a combination of PDL and PA (L1-Fc/PA/PDL), and PA alone (L1-Fc/PA), as well as control surfaces that did not contain L1-Fc. SCNs cultured for 24 h on L1-Fc/PA/PDL or L1-Fc/PA extended longer neurites compared to those on L1-Fc/PDL treated films. b Quantification of the average total neurite outgrowth indicated that on 10 or 50 μg/ml of L1-Fc, there was no significant difference in outgrowth between the two conditions in which L1-Fc was presented from protein A (with or without PDL). c Quantification of the average length of the longest neurite per cell. d Morphology of CNs that were seeded onto the same substrates listed above, where 10 μg/mL of L1-Fc was used. e Substrates in which L1-Fc was presented using PA resulted in enhanced neurite outgrowth of CNs compared to neurons cultured on L1-Fc presented from PDL. Scale bar 75 μm. PDL poly-d-lysine, PA protein A (* denotes p < 0.05, ** denotes p < 0.01)
Cell-L1 binding has been reported to be cell type specific [44]. Therefore, we also examined how a different source of neurons, namely CNs, responded to variable L1-Fc presentations after 24 h. Similar to the observation with SCNs, polymer films where L1-Fc was presented via PA/PDL or PA alone promoted enhanced neurite outgrowth of CNs (112 ± 5.8 and 115.3 ± 7.8 μm, respectively) when compared to L1-Fc coated onto PDL (82.4 ± 6.6 μm) (Fig. 3e). In addition, CNs grown on L1-Fc/PA extended thinner, finer neurites compared to thicker extension observed on L1-Fc/PA/PDL surfaces (Fig. 3d) suggesting that PDL may have a modest cooperative influence over neurite adhesion for cerebellar neurons.
Multimeric L1 Presentation Enhances Neurite Outgrowth of Cultured Neurons in 3-D, Fibrous Substrates
Next, neurite extension was examined on fibrous scaffolds fabricated from poly(DTE carbonate) polymer, combined with fiber-based multimeric L1-Fc presentation. Scaffolds with aligned fiber orientation, designed to promote directional neuronal cell growth [45, 46], were fabricated by electrospinning and visualized using SEM. The morphology of the aligned scaffolds is shown in Fig. 4a, where the average fiber diameter was found to be 1.25 ± 0.5 μm. The alignment quantification of the fibers demonstrates that the scaffolds have a high degree of fiber alignment, with the predominant fiber counts falling at an angle difference of 0°, which represents the fiber axis parallel to the reference line (Fig. 4b). All scaffolds treated with L1-Fc showed extensive neurite outgrowth along the direction of the aligned fibers (Fig. 4c). However, L1-Fc/PA/PDL and L1-Fc/PA promoted significantly longer neurite outgrowth (99.5 ± 5.2 and 103 ± 6.7 μm, respectively) compared to L1-Fc/PDL (71.8 ± 5.1 μm) and the controls (Fig. 4d).
Fig. 4.
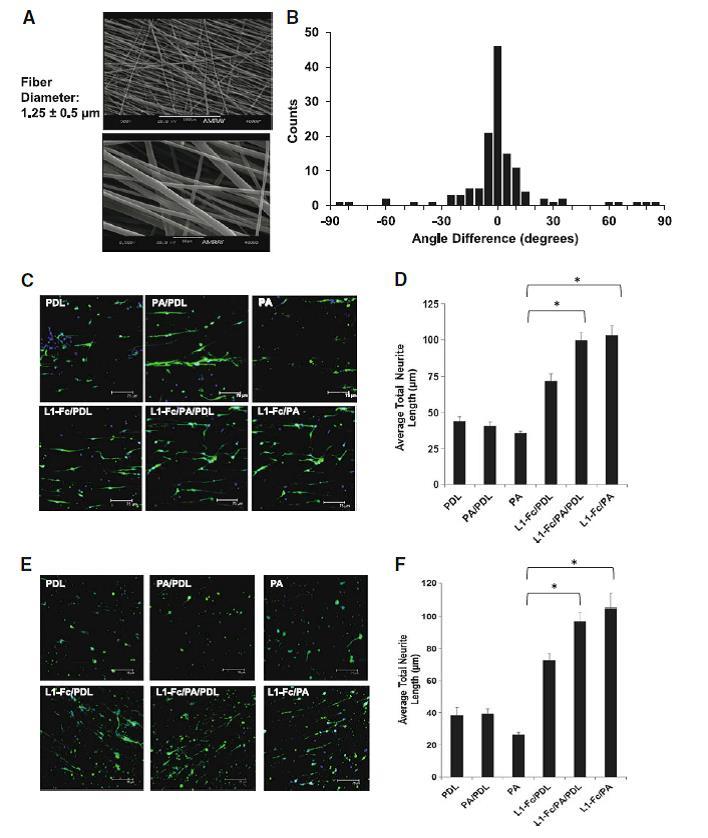
Protein A-presented L1-Fc increases neurite outgrowth in aligned, polymeric scaffolds. a Morphology and fiber diameter of aligned scaffolds that were fabricated by electrospinning. b Histogram depicting the degree of fiber alignment of the scaffolds. c Spinal cord neurons (SCNs) were plated onto scaffolds treated with L1-Fc presented from PDL, PA/PDL, or PA alone, as well as control surfaces that did not contain L1-Fc. PA presented L1-Fc results in longer neurite lengths when cultured on aligned scaffolds. Neurons cultured on 10 μg/ml L1-Fc passively adsorbed onto the fibers extended longer neurites than on the PDL control. The three-dimensional scaffold results are similar to those observed on the polymer thin films. d Quantification of the average total neurite lengths of SCNs cultured on the different L1-Fc presentations showed L1-Fc/PDL resulted in SCNs with shorter neurites compared to L1-Fc/PA/PDL and L1-Fc/PA, suggesting that the manner in which L1-Fc is presented on nanofibrous scaffolds influences its efficacy. (e, f) Cerebellar neurons (CNs) were plated onto scaffolds pretreated with different L1-Fc presentations and controls as mentioned above. PA presented L1-Fc resulted in CNs that were more spread and grew longer along the fibers compared to CNs cultured on L1-Fc adsorbed onto PDL. The three-dimensional scaffold results correlate to the results observed on the polymer thin films. Scale bar 75 μm. PDL poly-d-lysine, PA protein A (* denotes p < 0.05)
As observed with SCNs, all presentations of L1-Fc resulted in directional growth of CNs, but enhanced neuritogenesis was observed on L1-Fc/PA/PDL (97.04 ± 5.6 μm) or L1-Fc/PA (104.88 ± 9.1 μm) presenting scaffolds as well (Fig. 4e–f).
Role of L1–L1 Homophilic Cell-Substrate Adhesion Underlying Neurite Outgrowth on Multimeric L1 Films
To establish that the multimeric L1-Fc presenting films engendered L1 cell receptor-mediated adhesion, each surface was treated with a monoclonal antibody against L1 prior to plating SCNs. A significant reduction in neurite outgrowth of SCNs cultured on L1-Fc/PA/PDL and L1-Fc/PA treated surfaces in the presence of the anti-L1 antibody was observed, with a more pronounced inhibition seen in the L1-Fc/PA condition. Given that L1 multimeric exposure is differentially more amenable to inhibition on L1-Fc/PA substrates indicates a more potent role for L1–L1 adhesion underlying neurite outgrowth of SCNs cultured on L1-Fc/PA compared to L1-Fc/PDL (Fig. 5a). In the case of SCNs cultured on L1-Fc/PDL, neurite outgrowth was also reduced in the presence of the anti-L1 antibody, however not as markedly as on the other substrates (Fig. 5b), suggesting that the binding epitopes on L1 are not as accessible for SCN binding when L1-Fc is adsorbed onto PDL treated films. Thin films treated with the isotype control resulted in SCNs with a similar morphology to SCNs cultured on untreated thin films. Additionally, we examined specific binding of the Fc region of L1-Fc to protein A by blocking the Fc region of L1-Fc with an anti-Fc antibody. Neurite outgrowth was greatly reduced in SCNs cultured on L1-Fc/PA/PDL and L1-Fc/PA surfaces, but not on L1-Fc/PDL treated surfaces, suggesting that L1-Fc linkages with protein A are critical to the orientation and activity of L1 (data not shown).
Fig. 5.
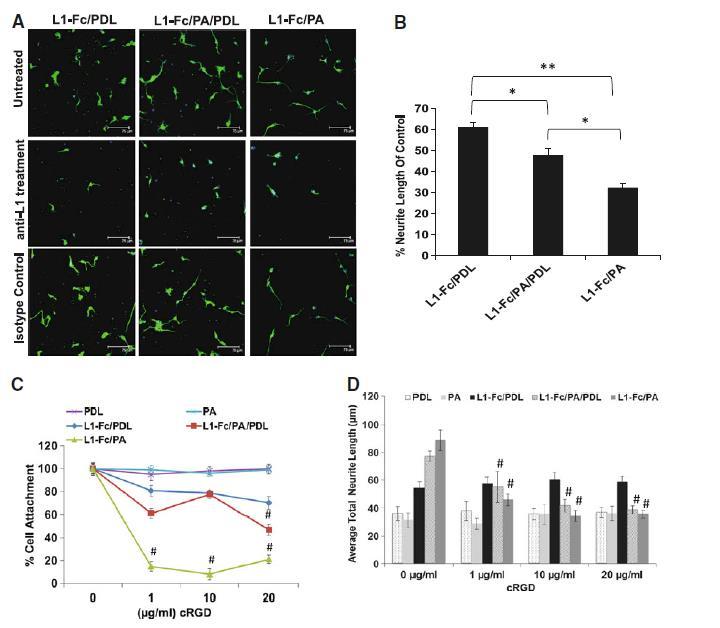
Anti-L1 antibody and cyclic RGD treatment reduces cell attachment and neurite outgrowth. a Reduced neurite outgrowth of SCNs was observed for each L1-Fc presentation (L1-Fc/PDL, L1-Fc/PA/PDL, and L1-Fc/PA) when the surface was pretreated with a monoclonal anti-L1 antibody. b SCNs cultured on L1-Fc/PA treated thin films exhibited a drastic decrease in neurite outgrowth compared to SCNs cultured on L1-Fc/PDL and L1-Fc/PA/PDL treated thin films when the anti-L1 antibody directed against the second Ig-like domain was added, suggesting that the epitope to which the L1 antibody binds is more exposed due to an outward display of L1-Fc presented from PA. c Dose-dependent reduction in cell attachment was observed in SCNs pretreated with cyclic RGD peptide prior to plating onto the different L1-Fc containing substrates. SCNs cultured on L1-Fc/PA treated thin films exhibited the greatest reduction in cell attachment indicating that L1-Fc presented from PA strongly interacts with αvβ3 integrins, which was interrupted by the presence of cyclic RGD peptide. Cell attachment on control substrates was not affected. d Similar to cell attachment, neurite outgrowth of SCNs was greatly diminished, in a dose-dependent manner, in the presence of cyclic RGD peptide. The greatest inhibition of neurite outgrowth, compared to untreated SCNs, was observed in SCNs cultured on L1-Fc/PA thin films, where as little as 1 μg/ml of cyclic RGD peptide inhibited cell attachment by 80%. Addition of 10 μg/ml of cyclic RGD peptide also resulted in a decrease in average neurite length of SCNs cultured on L1-Fc/PA/PDL coated thin films, although the effect was less prominent. The average neurite length of SCNs cultured on L1-Fc/PDL was not affected by any concentration of cyclic RGD, similar to the controls. Scalebar 75 μm. SCNs spinal cord neurons, PDL poly-d-lysine, PA protein A (* denotes p < 0.05, ** denotes p < 0.01)
Multimeric L1 Films Support RGD Peptide-Sensitive Integrin Mediated Cell Adhesion and Neurite Outgrowth
The L1 molecule contains one RGD sequence located within the sixth-Ig like domain, which has been reported to bind to integrins α5β1 and αvβ3 [47]. The interaction of αvβ3 and L1 has been reported to stimulate neurite outgrowth of the rat pheochromocytoma cell line, PC12, as well as chick dorsal root ganglion cells [48, 49]. We examined the possible interactions of αvβ3 cell adhesion receptors and L1-presentation and the resultant effects on cell responses. To this end, cyclic RGD peptides (cyclo-RGDfV) reported to bind αvβ3 with high affinity [50] and inhibit binding of L1 and αvβ3 [48], were added to SCNs at different concentrations, prior to cell plating onto the different substrates, and cell attachment and neurite outgrowth were examined. Attachment of SCNs cultured on L1-Fc/PA (Fig. 5c) was considerably reduced to 14.9 ± 3.2% in the presence of 1 μg/ml of cyclic RGD and further reduced to 8.2 ± 2.9% when treated with 10 μg/ml of cyclic RGD. Cell attachment was mildly affected, following cyclic RGD peptide treatment of SCNs cultured on L1-Fc/PDL with no significant decrease, compared to untreated cells. These results suggest that cell attachment on L1-Fc/PDL-based substrates is only partially dependent on L1-integrin binding. In contrast, on L1-Fc/PA substrates, αvβ3-L1 binding plays a major role in cell attachment. When PDL and PA were combined to present L1-Fc, only treatment with 20 μg/ml of cyclic RGD peptide resulted in a significant reduction of cell attachment.
Reduced neurite lengths were observed in SCNs pretreated with cyclic RGD peptides and the results were quantified (Fig. 5d). Neurite outgrowth was reduced by 51% from 88.7 ± 7.5 to 45 ± 4.2 μm in SCNs cultured on L1-Fc/PA coated substrates. A further reduction in neurite length was observed after treatment with 10 and 20 μg/ml of cyclic RGD peptide with no significant difference between the three concentrations of cyclic RGD peptide. In contrast, neurite outgrowth was not affected by cyclic RGD treatment in SCNs cultured on L1-Fc/PDL. In the case of SCNs cultured on L1-Fc/PA/PDL coated thin films, outgrowth was reduced by 28.5% from 77.2 ± 3.7 to 55 ± 11.1 μm when treated with 1 μg/ml of cyclic RGD peptide.
L1 Films Promote Neuronal Differentiation of Human NPCs but Oriented Presentation is Not Necessary in 2-D
Next, we examined the ability of L1-Fc to support neuronal differentiation and outgrowth of human embryonic stem cell derived-NPCs. We used laminin coated PDL substrates (LN/PDL) as the positive control. NPCs were cultured in NDM for 4 days, plated onto substrates treated with PDL, LN/PDL, L1-Fc/PDL, PA/PDL, and L1-Fc/PA/PDL and cultured an additional 7 days in NDM. In contrast to the primary neuronal cultures, L1-Fc/PA coated substrates were not included for the human NPC studies as NPCs failed to attach to this surface, suggesting the importance of cooperative presentation of PDL and multimeric presentation of L1 for these cells. After 7 days in culture, NPCs were fixed and immunostained for the neural stem cell marker nestin and the early neuronal marker βIII-tubulin, clone TUJ1 (Fig. 6a). A reduction in nestin expression and an increase in βIII-tubulin, along with neurite extensions, confirmed early differentiation into neurons. In order to quantify the degree of neuronal differentiation, the percent of nestin positive+ cells and βIII-tubulin+ cells was determined by taking the ratio of nestin+ or βIII-tubulin+ cells to the total number of cells, as indicated by Hoechst-stained nuclei. Both presentations of L1-Fc resulted in an increase in βIII-tubulin+ cells compared to the PDL and PA/PDL (Fig. 6b), suggesting that L1-Fc promoted neuronal differentiation of NPCs. However, there was no significant difference in the degree of neuronal differentiation elicited by the two different L1-Fc presentations in 2-D.
Fig. 6.
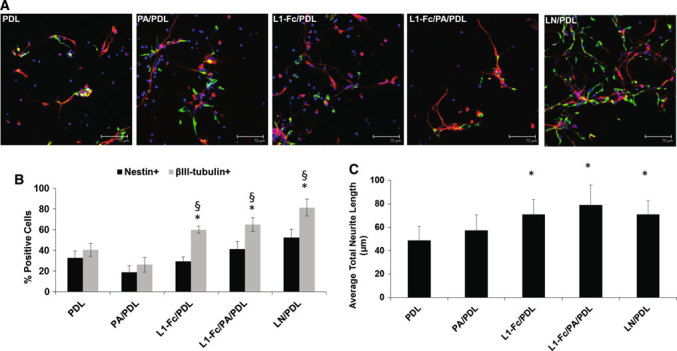
L1-Fc supports neuronal differentiation and neuritogenesis of neural progenitor cells. a NPCs were cultured on PDL, PA/PDL, L1-Fc/PDL, L1-Fc/PA/PDL, and LN/PDL for 7 days in neuronal differentiation media and then immunostained for the nestin (green) to identify neural stem/progenitor cells, βIII-tubulin (red) to identify neurons, and Hoechst 33258 (blue) to label the nuclei. After 7 days, NPCs differentiated toward the neuronal lineage, to some degree, when cultured on all substrates, as in indicated the βIII-tubulin+ cells. b Quantification of the percent of NPCs (nestin+) and neuronal cells (βIII-tubulin+) of NPCs after 7 days of differentiation. Both L1-Fc substrates promoted increased neuronal differentiation over controls and had a greater neuronal to neural progenitor cell population. c Average neurite lengths were quantified for NPCs after 7 days in culture. L1-Fc/PDL and L1-Fc/PA/PDL coated thin films promote a significant increase in neurite length compared to the PDL and PA/PDL controls. LN/PDL served as a positive control. Scalebar 75 μm. NPCs neural progenitor cells, PDL poly-d-lysine, PA protein A, LN laminin (* denotes p < 0.05 compared to PDL and PA/PDL controls, § denotes p < 0.05 comparing nestin+ cells to βIII-tubulin+ cells within each condition)
Additionally, neurite outgrowth was quantified for NPCs cultured on each substrate (Fig. 6c). L1-Fc/PDL and L1-Fc/PA/PDL coated thin films significantly promoted increased neurite outgrowth compared to PDL or PA/PDL controls. The average neurite lengths on L1-Fc/PDL and L1-Fc/PA/PDL were 71.1 ± 12.7 and 79.1 ± 16.5 μm, respectively, which was comparable to those elicited by LN/PDL coated controls (71.1 ± 11.8 μm). There was no significant difference in lengths of neurites observed on LN/PDL, L1-Fc/PDL, and L1-Fc/PA/PDL. Thus, while L1-Fc promoted neurite outgrowth comparable to that of laminin, the nature of L1-Fc presentation did not appear to sensitively influence the extent of neurite length of differentiating NPCs on 2-D films.
Oriented, Multimeric L1 Presentation Triggers Enhanced Neuronal Differentiation of Human NPCs in 3-D Fibrous Scaffolds
The human NPCs were cultured on aligned, fibrous scaffold pretreated with different L1-Fc presentations, laminin, and controls to investigate how the combination of topographical cues from the scaffold and L1-Fc affect neuronal differentiation and neurite outgrowth of NPCs. Scaffolds treated with L1-Fc, with or without PA, promoted directed neuritogenesis of NPCs, comparable to LN, as is indicated by the elongated processes that follow the direction of the aligned scaffolds (Fig. 7a). To determine the degree to which L1-Fc influenced neuronal differentiation of NPCs, the percent of nestin+ and βIII-tubulin+ cells were quantified by taking the ratio of nestin+ and βIII-tubulin+ cells to the total number of cells, as labeled by Hoechst (Fig. 7b). A smaller nestin+ population compared to βIII-tubulin population was observed on both L1-Fc presentations, similar to the LN/PDL treated scaffold, which served as a positive control. Additionally, both L1-Fc treated scaffolds yielded in an increase in neuronal differentiation compared to PDL and PA/PDL controls (Fig. 7b). Both presentations of L1-Fc from the fibrous scaffolds promoted increased neurite lengths compared to the controls. Notably, NPCs cultured on protein A-presented L1-Fc (L1-Fc/PA/PDL) exhibited an increase in βIII-tubulin+ cells (65.9 ± 5.4%) compared to those cultured on L1-Fc/PDL (48.1 ± 4.6%). Further, the average total neurite length of βIII-tubulin+ positive cells was quantified (Fig. 7c). The oriented display of L1-Fc, presented from PA/PDL, promoted increased neurite lengths over L1-Fc adsorbed onto PDL treated scaffolds. These results suggest that protein A presented L1-Fc has a greater influence on neuronal differentiation of NPCs when presented from aligned fibrous scaffolds compared to two-dimensional thin films.
Fig. 7.
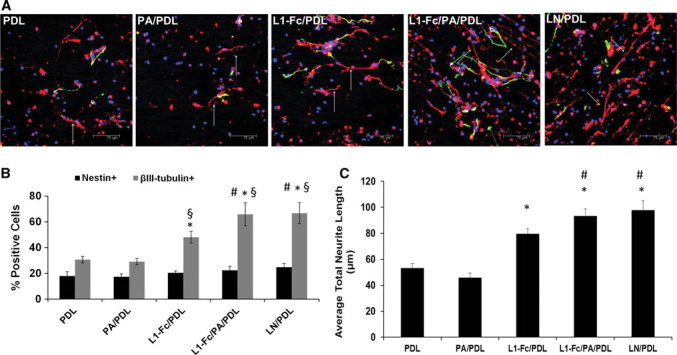
L1-Fc supports neuronal differentiation and neuritogenesis of neural progenitor cells within aligned scaffolds. a NPCs were cultured on aligned, fibrous scaffolds pretreated with L1-Fc presented from PDL or PA/PDL. After 7 days of differentiation, the NPCs were immunostained for the nestin (green) to identify neural progenitor cells, βIII-tubulin (red) to identify neurons, and Hoechst 33258 (blue) to label the nuclei. NPCs cultured on scaffolds treated with L1-Fc, with or without PA, extended neurites along the scaffold fibers. NPCs differentiated toward the neuronal lineage, to some degree, when cultured on all substrates, as in indicated the βIII-tubulin+ cells. b Quantification of the percent of NPCs (nestin+) and neuronal cells (βIII-tubulin+). Both L1-Fc substrates promoted increased neuronal differentiation over controls and had a greater neuronal to neural progenitor cell population. L1-Fc/PA/PDL scaffolds resulted in more βIII-tubulin+ cells compared to L1-Fc/PDL coated scaffolds. c Quantification of the average total neurite length. NPCs cultured on L1-Fc/PA/PDL treated scaffolds extended longer neurites compared to those cultured on L1-Fc/PDL. Scale bar 75 μm. NPCs neural progenitor cells, PDL poly-d-lysine, PA protein a, LN laminin (* denotes p < 0.05 compared to PDL and PA/PDL controls, § denotes p < 0.05 comparing nestin+ cells to βIII-tubulin+ cells within each condition, # denotes p < 0.05 compared to the L1-Fc/PDL condition)
Discussion
Biointerfacial display of neurotrophic factors has the potential to significantly influence neural cell behaviors relevant to CNS repair, including adhesion, differentiation, and neurite extension [51, 52]. In this study, we investigated multiple presentation schemes for the neural cell adhesion molecule L1 to elucidate the effects of presentation on primary neurons and human embryonic stem cell-derived NPCs on 2-D and 3-D polymer substrates. We found that protein A (PA)-mediated oriented and multimeric display of L1-Fc significantly enhanced neurite extension of primary spinal cord and cerebellar neurons relative to L1-Fc presented from PDL. We also report that the combination of multimeric L1-Fc presentation and fibrous geometries of 3-D scaffolds cooperatively enhanced neuronal differentiation of human NPCs.
The spatial mode of presentation of biochemical cues such as growth factors and adhesion ligands plays a critical role in determining their bioactivity and function. For example, immobilized interferon gamma can potentiate neuronal differentiation of rat NPCs compared to soluble interferon gamma [53]. Similarly, while L1 has been largely used as a soluble ligand [54, 55], L1 can also be anchored onto substrates as L1-Fc and presented via PA [56, 57], or used in a trimeric form, which was reported to result in increased PC12 cell neurite outgrowth relative to monomeric L1 [58]. Using a combination of PDL treatment to enable improved cell-polymer adhesion and L1-Fc/PA presentation, we report on the concentrations and biointerfacial configurations that promote and enhance L1 bioactivity. We also obtained two broad insights regarding L1-functionalized tyrosine-derived polycarbonate biomaterials: (a) PA/polymer interfaces can elicit multimeric and oriented presentation of L1 from such biomaterials. (b) The L1-biointerfaces can markedly modulate adhesion and neuronal activity of both primary neurons and human NPCs.
The use of potentially implantable and biodegradable polymer substrates in this study has implications for translating the insights from this study to in vivo models. While the biomaterials used in this study are biodegradable, the chosen composition shows minimal hydrolytic degradation over the time frame of our study [59]. Furthermore, tyrosine-derived polycarbonate biomaterials, with varying degradation rates, did not elicit any changes in cell behavior (proliferation and differentiation) after 14 days in culture [60]. L1-functionalized substrates can selectively promote neuronal cell attachment compared to astrocytes and fibroblasts [61] and thus be potentially advantageous over more generic cell adhesive treatments such as poly-lysine (PDL or PLL). The multimeric nature of PA-based L1-Fc presentation could be important for both promoting cell adhesion for a given concentration of L1 as well as enhancing neurite extension activity, as the second Ig-like domain of L1 regulates neurite outgrowth of neural retinal cells and directly mediates L1–L1 homophilic interactions [62, 63]. The marked inhibition of neurite outgrowth following antibody-based blocking studies of the L1-Fc/PA treated surfaces suggests that the second Ig-like domain of L1 is more exposed on PA-treated substrates compared to those based on PDL. Another domain within L1 is the RGD domain located within the sixth Ig-like domain. Our studies with cyclic RGD peptide pretreatment showed reduction in SCN attachment and neurite outgrowth when plated onto L1-Fc/PA/PDL and L1-Fc/PA treated thin films, suggesting that this domain was also exposed and available for αvβ3 integrin binding. Thus, we propose that the PA-based treatment of the polymers not only facilitated oriented display of L1-Fc, but also engendered a display of L1-Fc molecules in close proximity to one another, which may mirror the close apposition of highly organized and clustered L1 molecules on the cell surface [43, 64]. Using AFM imaging of substrate counterstained with L1 antibodies, we established that L1-Fc/PA substrates showed adsorbed L1-Fc. The cross-sectional profiles acquired from the AFM images of L1-Fc/PA/PDL and L1-Fc/PA substrates depicted wider, less defined peaks, which suggest that these protein complexes were more proximally associated compared to L1-Fc adsorbed on PDL, where the peaks were narrow and more defined. The results of the AFM images, in combination with the functional results, where neurons extended longer neurites on L1-Fc/PA treated substrates, indicate that the arrangement of PA-presented L1-Fc influences the efficacy of L1-Fc. The presentation of multimeric, closely associated L1-Fc, in combination with fibrous scaffolds could prove beneficial to the design of neurotrophic materials for neural tissue engineering.
Both spinal cord and cerebellar neurons displayed increased neurite lengths when cultured on PA-presented L1-Fc. One interesting difference between the behaviors of rodent neurons and human NPCs was evident through the polymer-treatment with PA and PDL. While the neuronal cultures showed robust adhesion and neurite extension even in the absence of PDL, a combination of PDL and PA was necessary for the NPC cultures suggesting that multimeric L1-based receptor-ligand binding is critical but not sufficient for the NPC cultures, requiring cooperative cell-substrate adhesion facilitated by polycationic PDL treatment.
The possible role of L1 presentation on neurogenesis in three-dimensional substrates is of great interest to scalable constructs for cell transplantation. We observed that fibrous, aligned polymer scaffolds treated with L1-Fc resulted in an increase in neuronal differentiation and neurite lengths compared to the scaffolds that were not treated with L1-Fc. However, in contrast to the two-dimensional thin films, NPCs cultured on L1-Fc/PA/PDL treated scaffolds had a larger neuronal population and enhanced neurite lengths relative to NPCs cultured on L1-Fc/PDL treated scaffolds. These results suggest that the NPC response to the different L1-Fc presentations on 2-D thin films and the 3-D fibrous scaffolds could be due to the combined effects of fiber topography and the outward display of L1-Fc. This suggests cooperative enhancement of L1 signaling on subcellular scales of substrate geometry, a phenomenon that deserves further mechanistic analysis. Scaffold topography has been shown to influence and enhance cellular responses, [65–67] as the 3-D architecture better emulates in vivo conditions compared to 2-D thin films.
Summary and Conclusions
We investigated biointerfaces incorporating L1-Fc on to polymer films and aligned, fibrous polymer scaffolds that would elicit multimeric, oriented presentation of L1-Fc. While L1-Fc has been shown to be effective in the treatment of nervous system injury models, we have demonstrated that the function of L1-Fc can be enhanced in vitro with the optimal substrate presentation using protein A. We showed that on two-dimensional films and aligned, fibrous scaffolds, treated with protein A-presented L1-Fc promotes enhanced neurite lengths of primary neuronal cells, as well as increased neuronal differentiation and neuritogenesis of human embryonic stem cell-derived NPCs. We combined this presentation of L1-Fc with poly(DTE carbonate), which can be fine-tuned for the desired stiffness, degradation rate, and protein adsorption, all of which are important factors to consider when designing implantable devices for nerve injury repair. The bioactive, three-dimensional substrates investigated in this study could prove as useful implantable devices for neural tissue engineering applications.
REFERENCES
- 1.Xu X, Warrington AE, Bieber AJ and Rodriguez M, CNS Drugs 25(7), 555 (2011) 10.2165/11587830-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thuret S, Moon LD and Gage FH, Nat Rev Neurosci 7(8), 628 (2006) 10.1038/nrn1955. [DOI] [PubMed] [Google Scholar]
- 3.Yue XS, Murakami Y, Tamai T, Nagaoka M, Cho CS, Ito Y and Akaike T, Biomaterials 31(20), 5287 (2010) 10.1016/j.biomaterials.2010.03.035. [DOI] [PubMed] [Google Scholar]
- 4.Niere M, Braun B, Gass R, Sturany S and Volkmer H, Biomaterials 27(18), 3432 (2006) 10.1016/j.biomaterials.2006.01.037. [DOI] [PubMed] [Google Scholar]
- 5.Cooke MJ, Phillips SR, Shah DS, Athey D, Lakey JH and Przyborski SA, Cytotechnology 56(2), 71 (2008) 10.1007/s10616-007-9119-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shin H, Jo S and Mikos AG, Biomaterials 24(24), 4353 (2003) 10.1016/S0142-9612(03)00339-9. [DOI] [PubMed] [Google Scholar]
- 7.Yu TT and Shoichet MS, Biomaterials 26(13), 1507 (2005) 10.1016/j.biomaterials.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 8.Silva GA, Czeisler C, Niece KL, Beniash E, Harrington DA, Kessler JA and Stupp SI, Science 303(5662), 1352 (2004) 10.1126/science.1093783. [DOI] [PubMed] [Google Scholar]
- 9.Cooke MJ, Zahir T, Phillips SR, Shah DS, Athey D, Lakey JH, Shoichet MS and Przyborski SA, J Biomed Mater Res A 93(3), 824 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Moos M, Tacke R, Scherer H, Teplow D, Fruh K and Schachner M, Nature 334(6184), 701 (1988) 10.1038/334701a0. [DOI] [PubMed] [Google Scholar]
- 11.Castellani V, Adv Exp Med Biol 515, 91 (2002) 10.1007/978-1-4615-0119-0_8. [DOI] [PubMed] [Google Scholar]
- 12.Felding-Habermann B, Silletti S, Mei F, Siu CH, Yip PM, Brooks PC, Cheresh DA, O’Toole TE, Ginsberg MH and Montgomery AM, J Cell Biol 139(6), 1567 (1997) 10.1083/jcb.139.6.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silletti S, Mei F, Sheppard D and Montgomery AM, J Cell Biol 149(7), 1485 (2000) 10.1083/jcb.149.7.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Appel F, Holm J, Conscience JF and Schachner M, J Neurosci 13(11), 4764 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maretzky T, Schulte M, Ludwig A, Rose-John S, Blobel C, Hartmann D, Altevogt P, Saftig P and Reiss K, Mol Cell Biol 25(20), 9040 (2005) 10.1128/MCB.25.20.9040-9053.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schultheis M, Diestel S and Schmitz B, Cell Mol Neurobiol 27(1), 11 (2007) 10.1007/s10571-006-9113-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loers G, Chen S, Grumet M and Schachner M, J Neurochem 92(6), 1463 (2005) 10.1111/j.1471-4159.2004.02983.x. [DOI] [PubMed] [Google Scholar]
- 18.Roonprapunt C, Huang W, Grill R, Friedlander D, Grumet M, Chen S, Schachner M and Young W, J Neurotrauma 20(9), 871 (2003) 10.1089/089771503322385809. [DOI] [PubMed] [Google Scholar]
- 19.Loers G and Schachner M, J Neurochem 101(4), 865 (2007) 10.1111/j.1471-4159.2006.04409.x. [DOI] [PubMed] [Google Scholar]
- 20.Huang ZJ, Nat Neurosci 9(2), 163 (2006) 10.1038/nn1638. [DOI] [PubMed] [Google Scholar]
- 21.Mehrke G, Jockusch H, Faissner A and Schachner M, Neurosci Lett 44(3), 235 (1984) 10.1016/0304-3940(84)90028-4. [DOI] [PubMed] [Google Scholar]
- 22.Maness PF and Schachner M, Nat Neurosci 10(1), 19 (2007) 10.1038/nn1827. [DOI] [PubMed] [Google Scholar]
- 23.Chen J, Wu J, Apostolova I, Skup M, Irintchev A, Kugler S and Schachner M, Brain 130(Pt 4), 954 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Dihne M, Bernreuther C, Sibbe M, Paulus W and Schachner M, J Neurosci 23(16), 6638 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen J, Bernreuther C, Dihne M and Schachner M, J Neurotrauma 22(8), 896 (2005) 10.1089/neu.2005.22.896. [DOI] [PubMed] [Google Scholar]
- 26.Cooke MJ, Vulic K and Shoichet MS, Soft Matter 6(20), 4988 (2010) 10.1039/c0sm00448k. [DOI] [Google Scholar]
- 27.Madigan NN, McMahon S, O’Brien T, Yaszemski MJ and Windebank AJ, Respir Physiol Neurobiol 169(2), 183 (2009) 10.1016/j.resp.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nomura H, Tator CH and Shoichet MS, J Neurotrauma 23(3–4), 496 (2006) 10.1089/neu.2006.23.496. [DOI] [PubMed] [Google Scholar]
- 29.Delcroix GJ, Schiller PC, Benoit JP and Montero-Menei CN, Biomaterials 31(8), 2105 (2010) 10.1016/j.biomaterials.2009.11.084. [DOI] [PubMed] [Google Scholar]
- 30.Subramanian A, Krishnan UM and Sethuraman S, J Biomed Sci 16, 108 (2009) 10.1186/1423-0127-16-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Horne MK, Nisbet DR, Forsythe JS and Parish CL, Stem Cells Dev 19(6), 843 (2010) 10.1089/scd.2009.0158. [DOI] [PubMed] [Google Scholar]
- 32.Koh HS, Yong T, Chan CK and Ramakrishna S, Biomaterials 29(26), 3574 (2008) 10.1016/j.biomaterials.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 33.Timnak A, Gharebaghi FY, Shariati RP, Bahrami SH, Javadian S, Emami Sh H and Shokrgozar MA, J Mater Sci Mater Med 22(6), 1555 (2011) 10.1007/s10856-011-4316-5. [DOI] [PubMed] [Google Scholar]
- 34.Griffin J, Delgado-Rivera R, Meiners S and Uhrich KE, J Biomed Mater Res A 97(3), 230 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ertel SI and Kohn J, J Biomed Mater Res 28(8), 919 (1994) 10.1002/jbm.820280811. [DOI] [PubMed] [Google Scholar]
- 36.Meechaisue C, Dubin R, Supaphol P, Hoven VP and Kohn J, J Biomater Sci Polym Ed 17(9), 1039 (2006) 10.1163/156856206778365988. [DOI] [PubMed] [Google Scholar]
- 37.Voinova MV, Rodahl M, Jonson M, Kasemo B (1999) Physica Scripta 59(5):391–396
- 38.Neagu C, Werf KO, Putman CA, Kraan YM, Grooth BG, Hulst NF and Greve J, J Struct Biol 112(1), 32 (1994) 10.1006/jsbi.1994.1004. [DOI] [PubMed] [Google Scholar]
- 39.Rossi MP, Xu J, Schwarzbauer J and Moghe PV, Biointerphases 5(4), 105 (2010) 10.1116/1.3507236. [DOI] [PubMed] [Google Scholar]
- 40.Wang HB, Mullins ME, Cregg JM, Hurtado A, Oudega M, Trombley MT and Gilbert RJ, J Neural Eng 6(1), 016001 (2009) 10.1088/1741-2560/6/1/016001. [DOI] [PubMed] [Google Scholar]
- 41.Jiang XY, Fu SL, Nie BM, Li Y, Lin L, Yin L, Wang YX, Lu PH and Xu XM, J Neurosci Methods 158(1), 13 (2006) 10.1016/j.jneumeth.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 42.Mercado ML, Nur-e-Kamal A, Liu HY, Gross SR, Movahed R and Meiners S, Neuroscience 24(1), 238 (2004) 10.1523/JNEUROSCI.4519-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dahlin-Huppe K, Berglund EO, Ranscht B and Stallcup WB, Mol Cell Neurosci 9(2), 144 (1997) 10.1006/mcne.1997.0608. [DOI] [PubMed] [Google Scholar]
- 44.Kadmon G and Altevogt P, Differentiation 61(3), 143 (1997) 10.1046/j.1432-0436.1997.6130143.x. [DOI] [PubMed] [Google Scholar]
- 45.Xie J, MacEwan MR, Li X, Sakiyama-Elbert SE and Xia Y, ACS Nano 3(5), 1151 (2009) 10.1021/nn900070z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xie J, Willerth SM, Li X, Macewan MR, Rader A, Sakiyama-Elbert SE and Xia Y, Biomaterials 30(3), 354 (2009) 10.1016/j.biomaterials.2008.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Montgomery AM, Becker JC, Siu CH, Lemmon VP, Cheresh DA, Pancook JD, Zhao X and Reisfeld RA, J Cell Biol 132(3), 475 (1996) 10.1083/jcb.132.3.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yip PM and Siu CH, J Neurochem 76(5), 1552 (2001) 10.1046/j.1471-4159.2001.00152.x. [DOI] [PubMed] [Google Scholar]
- 49.Yip PM, Zhao X, Montgomery AM and Siu CH, Mol Biol Cell 9(2), 277 (1998) 10.1091/mbc.9.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pfaff M, Tangemann K, Muller B, Gurrath M, Muller G, Kessler H, Timpl R and Engel J, J Biol Chem 269(32), 20233 (1994). [PubMed] [Google Scholar]
- 51.Stabenfeldt SE, Munglani G, Garcia AJ and LaPlaca MC, Tissue Eng Part A 16(12), 3747 (2010) 10.1089/ten.tea.2009.0837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Potter W, Kalil RE and Kao WJ, Front Biosci 13, 806 (2008) 10.2741/2721. [DOI] [PubMed] [Google Scholar]
- 53.Leipzig ND, Xu C, Zahir T and Shoichet MS, J Biomed Mater Res A 93(2), 625 (2010). [DOI] [PubMed] [Google Scholar]
- 54.Doherty P, Williams E and Walsh FS, Neuron 14(1), 57 (1995) 10.1016/0896-6273(95)90240-6. [DOI] [PubMed] [Google Scholar]
- 55.Lavdas AA, Efrose R, Douris V, Gaitanou M, Papastefanaki F, Swevers L, Thomaidou D, Iatrou K and Matsas R, J Neurochem 115(5), 1137 (2010) 10.1111/j.1471-4159.2010.07003.x. [DOI] [PubMed] [Google Scholar]
- 56.Oliva AA Jr, James CD, Kingman CE, Craighead HG and Banker GA, Neurochem Res 28(11), 1639 (2003) 10.1023/A:1026052820129. [DOI] [PubMed] [Google Scholar]
- 57.Shi P, Shen K and Kam LC, Dev Neurobiol 67(13), 1765 (2007) 10.1002/dneu.20553. [DOI] [PubMed] [Google Scholar]
- 58.Hall H, Bozic D, Fauser C and Engel J, J Neurochem 75(1), 336 (2000) 10.1046/j.1471-4159.2000.0750336.x. [DOI] [PubMed] [Google Scholar]
- 59.Magno MHR, Kim J, Srinivasan A, McBride S, Bolikal D, Darr A, Hollinger JO and Kohn J, J Mater Chem 20, 8885 (2010) 10.1039/c0jm00868k. [DOI] [Google Scholar]
- 60.Abramson SD (2002) Dissertation, Rutgers University and University of Medicine and Dentistry of New Jersey and Robert Wood Johnson Medical School, New Brunswick, USA
- 61.Webb K, Budko E, Neuberger TJ, Chen S, Schachner M and Tresco PA, Biomaterials 22(10), 1017 (2001) 10.1016/S0142-9612(00)00353-7. [DOI] [PubMed] [Google Scholar]
- 62.Zhao X and Siu CH, J Biol Chem 270(49), 29413 (1995) 10.1074/jbc.270.49.29413. [DOI] [PubMed] [Google Scholar]
- 63.Zhao X, Yip PM and Siu CH, J Neurochem 71(3), 960 (1998) 10.1046/j.1471-4159.1998.71030960.x. [DOI] [PubMed] [Google Scholar]
- 64.Hall H, Carbonetto S and Schachner M, J Neurochem 68(2), 544 (1997) 10.1046/j.1471-4159.1997.68020544.x. [DOI] [PubMed] [Google Scholar]
- 65.Levenberg S, Huang NF, Lavik E, Rogers AB, Itskovitz-Eldor J and Langer R, Proc Natl Acad Sci USA 100(22), 12741 (2003) 10.1073/pnas.1735463100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Smith LA, Liu X, Hu J and Ma PX, Biomaterials 31(21), 5526 (2010) 10.1016/j.biomaterials.2010.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shahbazi E, Kiani S, Gourabi H and Baharvand H, Tissue Eng Part A 17(23–24), 3021 (2011) 10.1089/ten.tea.2011.0121. [DOI] [PubMed] [Google Scholar]
- 68.Semler EJ, Dasgupta A and Moghe PV, Tissue Eng 11(5–6), 734 (2005) 10.1089/ten.2005.11.734. [DOI] [PubMed] [Google Scholar]