

Abstract
The ability of a retinoid X receptor (RXR) to heterodimerize with many nuclear receptors, including LXR, PPAR, NGF1B and RAR, underscores its pivotal role within the nuclear receptor superfamily. Among these heterodimers, PPAR:RXR is considered an important signalling mediator of both PPAR ligands, such as fatty acids, and 9-cis retinoic acid (9-cis RA), an RXR ligand. In contrast, the existence of an RXR/9-cis RA signalling pathway independent of PPAR or any other dimerization partner remains disputed. Using in vivo chromatin immunoprecipitation, we now show that RXR homodimers can selectively bind to functional PPREs and induce transactivation. At the molecular level, this pathway requires stabilization of the homodimer–DNA complexes through ligand-dependent interaction with the coactivator SRC1 or TIF2. This pathway operates both in the absence and in the presence of PPAR, as assessed in cells carrying inactivating mutations in PPAR genes and in wild-type cells. In addition, this signalling pathway via PPREs is fully functional and can rescue the severe hypothermia phenotype observed in fasted PPARα−/− mice. These observations have important pharmacological implications for the development of new rexinoid-based treatments.
Keywords: coactivators, homodimers, PPAR, rexinoid, RXR
Introduction
The family of nuclear hormone receptors plays a unique role in pathways involved in metabolic regulation by directly linking the presence or absence of signalling molecules to gene activation or repression. Within the nuclear receptor superfamily, a pivotal place is occupied by the retinoid X receptor (RXR). There are three RXR isotypes (RXRα, RXRβ and RXRγ), each encoded by a separate gene. All RXR isotypes bind with stereo-selectivity the vitamin A derivative 9-cis retinoic acid (RA). The physiological significance of the 9-cis RA signalling pathway is supported by the presence of 9-cis RA in the developing embryo and the identification of enzymes that contribute to its biosynthesis (Mertz et al, 1997; Romert et al, 1998). At the molecular level, several mechanisms are proposed for the biological action of 9-cis RA. First, 9-cis RA can bind to retinoic acid receptors (RARs) and activate the RAR:RXR heterodimer and the corresponding set of target genes. However, all-trans RA is a more abundant natural ligand for RAR. Second, in in vitro assays, 9-cis RA can induce the formation of RXR homodimers that bind to DR1 sequences (Zhang et al, 1992). It is so far unclear as to what extent this signalling mechanism is used in vivo, as few RXR-specific target genes have been identified. The third mechanism for 9-cis RA signalling is so far the best documented; it relies on the ability of RXR-containing heterodimers to mediate transcriptional activation in response to the RXR ligand (Mangelsdorf and Evans, 1995; Minucci and Ozato, 1996). Examples of such complexes include the heterodimers PPAR:RXR, LXR:RXR, NGF1B:RXR and also RAR:RXR.
Among the heterodimers that can convey a 9-cis RA response, PPAR:RXR has been extensively studied and is considered an important mediator of both 9-cis RA action and of signalling by fatty acids and their derivatives (Desvergne and Wahli, 1999). Most of the ligand-dependent interactions of nuclear receptors and cofactors have been assessed for each receptor independently, and analysed in solution. In this context, the ligand-dependent interaction of PPAR on the one hand and of RXR on the other hand with the cofactor SRC1 is indeed well documented. Thus, the ability of PPAR:RXR to become transcriptionally active in the presence of either a PPAR or an RXR ligand has been interpreted as the ability of either receptor to recruit coactivator(s) (DiRenzo et al, 1997).
It was recently shown that cofactors may interact differently with DNA-bound nuclear receptor heterodimers (Yang et al, 2000). To understand rexinoid action on genes controlled by a PPRE, we analysed coactivator recruitment by DNA-bound PPAR and RXR. Surprisingly, we found that DNA-bound PPAR:RXR does not interact with SRC1 in the presence of a PPAR ligand. In contrast, in the presence of an RXR agonist, SRC1 stabilizes the binding of a functional RXR homodimer to the PPRE. Using chromatin immunoprecipitation (ChIP) to assess complex formation both in primary keratinocyte cultures and in whole animal models, we show that RXR homodimers bind to PPREs via specific coactivator recruitment, irrespective of the presence of PPAR. Moreover, the resultant ternary complex is able to substitute functionally for PPAR:RXR and regulate complex metabolic pathways in vivo. Taken together, the data presented identify a new mechanism for RXR/9-cis RA signalling independent of a dimerization partner, in which the response element, through allosteric control of transcription factor/coactivator interactions, is able to discriminate functionally between competing signalling pathways.
Results
So far, the ligand-dependent interactions of PPAR, RXR or PPAR:RXR with coactivators have been assessed in solution and in the absence of DNA. As a first approach to determine how DNA-bound PPAR:RXR interacts with SRC1, we used electrophoretic mobility shift analysis (EMSA). As was shown previously (IJpenberg et al, 1997), recombinant PPARγ and RXR proteins form heterodimers that bind to the malic enzyme PPRE (MEp) (Figure 1A, lanes 2 and 3). Importantly, no recruitment of the SRC1 nuclear receptor interacting domain (SRC1(B); Jeannin et al, 1998) by DNA-bound PPAR:RXR is observed in the presence of the PPARγ ligand BRL49653 (Figure 1A, lanes 6 and 7). Similarly, no SRC1(B) recruitment was observed using PPARα and PPARβ with their appropriate ligands (data not shown). In contrast, in the presence of the RXR ligand 9-cis RA, a low-mobility complex binds to the PPRE, accompanied by a reduction in DNA-bound PPAR:RXR (Figure 1A, compare lanes 4, 6 and 8). In the presence of both PPAR and RXR ligands, a pattern similar to that of 9-cis RA alone is obtained (Figure 1A, compare lane 5 with 4, and lane 9 with 8). Remarkably, in the absence of PPAR (lanes 10–13), the same high-affinity low-mobility complex still binds to MEp in the presence of 9-cis RA. Furthermore, co-incubation with a polyclonal PPAR antibody (PPAR-Ab) that disrupts the PPAR:RXR interface reduces binding of PPAR:RXR but does not affect the complex formed in the presence of 9-cis RA and SRC1 (lanes 14–17). These results suggest that DNA-bound PPAR:RXR does not interact with SRC1 and that the observed ternary complex, comprising SRC1, may not contain PPAR. To assess directly the presence of SRC1 in the 9-cis RA-dependent ternary complex, DNA-dependent pull-down experiments were performed using either GST-SRC1(B) or full-length SRC1. Western blot analysis of the pulled-down protein complexes confirmed the tethering of SRC1 to MEp in the presence of RXR and its cognate ligand (Figure 1B, lanes 1–7, top two panels). As seen in the EMSA, the SRC1 pull-down was independent of the presence of PPAR (Figure 1B, lanes 8–13).
Figure 1.

Specific interaction of SRC1 with DNA-bound RXR:RXR complexes. (A) EMSA: Radiolabelled MEp was incubated with RXRα, PPARγ and SRC1(B), in the presence or absence of 1 μM 9-cis RA or 5 μM BRL49653, as indicated. The arrowheads indicate bound PPAR:RXR (grey), RXR:RXR (open) and ternary SRC1(B)-containing complex (black). Ab: polyclonal PPAR antibody; p.i.: preimmune serum. (B) Pull-down assays: Different combinations of RXRα, PPARγ, and either full-length SRC1 or the SRC1 nuclear receptor interacting domain (SRC1(B)) were incubated in 96-well plates coated with biotinylated DNA, either MEp-biotin or PDK1-biotin, in the presence of 40 μM 9-cis RA and/or 10 μM Rosiglitazone. After washing of the wells, retained proteins were analysed by Western blot with antibodies directed against GST (recognizing the GST-SRC1(B) or SRC1 (recognizing the full-length SRC1) as indicated. (C) EMSA: Analyses were performed as in (A), but in the absence of PPAR. DR1G: synthetic RXRE; MEp: functional PPRE from the malic enzyme promoter; AcoA: functional PPRE from the acyl-CoA oxidase promoter; HD: functional PPRE from the bifunctional enzyme promoter; MEd: nonfunctional DR1 element in the malic enzyme promoter.
Although in vitro assays have shown that 9-cis RA can induce the binding of RXR homodimers to DR1-like elements (Nakshatri and Chambon, 1994), little if any binding has been seen with natural PPREs so far. In contrast, our results strongly suggest that SRC1 triggers the formation of a ternary complex composed of RXR homodimers and SRC1, which binds to MEp with a high affinity in the presence of 9-cis RA. As a positive control for RXR homodimer binding, we used the synthetic rexinoid response element DR1G (Nakshatri and Chambon, 1994). As expected, RXR homodimers efficiently bind to DR1G in the presence of 9-cis RA even in the absence of SRC1 (Figure 1C). Upon addition of SRC1(B), an RXR:RXR:SRC1(B) complex is formed, which migrates exactly like the ternary complex bound to MEp. We next assessed the specificity of RXR homodimer binding by either DNA-dependent pull-down analysis or EMSA, and found that the functional PPREs from the PDK1 promoter (Figure 1B, lower two panels) and from the acyl-CoA oxidase (AcoA) and the bifunctional enzyme (HD) promoters (Figure 1C, lanes 5–8) all tethered SRC1 in the presence of 9-cis RA and RXR. In contrast, RXR homodimers do not bind to the nonfunctional malic enzyme distal DR1-like element (MEd) (IJpenberg et al, 1997) under any of the conditions tested (Figure 1C, lanes 9 and 10).
To further analyse RXR homodimer binding to the PPREs, EMSA was performed using the light-stable synthetic rexinoids LG100268 and LG100754. Whereas the first compound is a potent RXR pan-agonist, LG100754 has been characterized as an agonist of PPAR:RXR and RXR:VDR heterodimers, but an antagonist of RXR homodimers (Lala et al, 1996). Based on our results, we postulated that RXR:RXR would bind to PPREs only upon activation by LG100268, but not by LG100754. Indeed, the RXR:RXR/SRC1 ternary complex bound to the PPRE (MEp) as well as to the RXRE (DR1G) in the presence of either 9-cis RA or LG100268, but not in the presence of the RXR homodimer antagonist LG100754 (Figure 2A). These results were confirmed using full-length SRC1 in pull-down experiments (data not shown). Altogether, the results confirm that the ternary complexes binding to functional PPREs in response to RXR agonists are RXR homodimers stabilized by SRC1. As a first approach to assess the functionality of RXR homodimer binding to PPREs, we cotransfected NIH-3T3 cells with a reporter plasmid controlled by MEp (IJpenberg et al, 1997) and an RXR-encoding plasmid. As expected, reporter activity was induced in the presence of 9-cis RA (Figure 2B). Treatment of the cells with LG100268 equally resulted in induction of reporter gene expression. In contrast, treatment with LG100754 had no effect, in agreement with the in vitro binding assay. Despite the high expression levels of SRC1 in these cells (data not shown), coexpression of a dominant-negative form of SRC1 (SRCdn; Kurokawa et al, 1995) reduced the RXR-dependent response (Figure 2C; 9-cis RA). The PPAR:RXR-dependent response, on the other hand, is not or only weakly altered by SRCdn (Figure 2C; Wy and BRL, respectively). Altogether, the results show that RXR homodimer antagonists inhibit the formation of RXR:SRC1 ternary complexes. In contrast, RXR:RXR:SRC1 complexes are induced by RXR agonists and are able to mediate reporter gene induction when transfected into exogenous cells.
Figure 2.

RXR antagonists preclude RXR:RXR/SRC-1 complex formation. (A) EMSA: Radiolabelled MEp (left panel) or the synthetic RXRE DR1G (right panel) was incubated with RXRα and SRC1(B) in the presence or absence of 1 μM 9-cis RA, LG100268 or LG100754 as indicated. The open arrowhead indicates the RXR homodimer complex, and the black arrowhead indicates the ternary complex. (B) NIH-3T3 cells were transfected with the MEp-containing reporter gene, RXR encoding expression vector, and pCMV-β-galactosidase as internal control for transfection efficiency. Following transfection, the cells were cultivated for 48 h in the presence of 1 μM 9-cis RA, LG100268 or LG100754 as indicated, or with vehicle alone. CAT activity levels were standardized for β-galactosidase expression and expressed as fold induction with respect to the CAT activity obtained in the presence of the vehicle alone. (C) NIH-3T3 cells were transfected with the MEp-containing reporter gene and either an RXR- or a PPAR-encoding expression vector as indicated, together with pCMV-β-galactosidase as internal control for transfection efficiency. A vector expressing a dominant-negative form of SRC1 (SRCdn) was added at increasing doses, as indicated; total transfected DNA was maintained equal under each condition by use of the corresponding empty vector. Numbers indicate the relative proportions (μg) of transfected expression vectors. Following transfection, the cells were cultivated for 48 h in the absence or presence of either 1 μM 9-cis RA, 100 μM Wy14,643, 5 μM BRL49653 or vehicle alone. CAT activity levels were standardized for β-galactosidase expression and expressed as fold induction with respect to the activity observed in the presence of vehicle alone.
We next analysed the specificity of RXR homodimer/coactivator interaction by performing EMSA in the presence of the nuclear receptor interaction domains of TIF2 and p300. Similar to our observations with SRC1, TIF2 is able to stabilize the binding of 9-cis RA-activated RXR homodimers to MEp (Figure 3). In contrast, p300 is unable to interact with the DNA-bound RXR homodimer, but readily forms a complex with PPAR:RXR regardless of the presence of ligand. As p300 has been shown to interact with RXR in solution, this latter result further suggests an allosteric role for DNA, which affects nuclear receptor/coactivator interactions.
Figure 3.

Specificity of RXR/coactivator association. EMSA: Radiolabelled MEp was incubated with PPARγ and RXRα (A), or RXR alone (B) and the receptor interacting domains of either SRC1 (amino acids 1226–1441; S), TIF2 (amino acids 624–869; T) or p300 (amino acids 31–220; 300), in the presence or absence of 1 μM 9-cis RA or 5 μM BRL49643 as indicated. The open arrowhead indicates the RXR homodimer, and the black arrowhead indicates the ternary complex.
These results emphasize that, in vitro, RXR homodimer formation onto PPREs requires selective and ligand-dependent interaction with specific coactivators. We next addressed the in vivo relevance of this RXR homodimer signalling mechanism. To this end, we analysed the regulation of well-characterized PPAR target genes in vivo, in the presence and absence of PPARs (Figure 4A). By crossing PPARα−/− and PPARβ−/− mice (Peters et al, 1997; Tan et al, 2001), we obtained PPARα−/−β−/− pups from which we derived primary keratinocyte cultures. These cells can be considered as devoid of PPARs since PPARγ is undetectable in keratinocytes (Tan et al, 2001). As seen in Figure 4B, the PPRE reporter gene transfected in wild-type primary keratinocytes is activated to a similar extent by the PPAR ligand and by 9-cis RA. In PPARα−/−β−/− keratinocytes, regulation by 9-cis RA remains unaffected, whereas regulation by the PPAR ligand is lost. Finally, regulation by both ligands is lost in cells derived from pups with a keratinocyte-specific deletion of RXRα (Li et al, 2001). Subsequent ChIP analysis performed on the corresponding cell extracts demonstrates that in wild-type cells, both PPAR and RXR are able to bind to the transfected PPRE in the presence of either RXR or PPAR activator (Figure 4C). Importantly, and as proof of the concept, 9-cis RA still induces RXR binding to the reporter gene PPRE in the absence of PPAR. Using the same approach, identical complex formation is observed onto the endogenous PPRE of the PDK1 gene (Figure 4C), a newly characterized PPAR target gene (Di Poï et al, 2002). These results demonstrate that in vivo RXR homodimers can bind to functional PPREs, both endogenous and transfected elements, and mediate transactivation.
Figure 4.

In vivo cofactor-specific binding of RXR homodimers to PPREs. Experiments were performed using primary keratinocyte cultures derived from either wild-type mice (WT), compound knock-out mice for PPARα and PPARβ (PPARα−/−β−/−), and mice carrying a keratinocyte-specific deletion of RXRα (RXRα−/−). Primary cultures were transfected with a PPRE-luciferase reporter gene and treated with either PPAR ligands (P; a mixture of 10 μM Wy14,643 and 5 μM L1165041), RXR ligand (X; 0.5 μM 9-cis RA) or vehicle (V; DMSO), and subjected to analyses. (A) Schematic outline of the procedure. (B) Reporter gene induction in response to various ligands. Luciferase activity levels were standardized for β-galactosidase expression used as internal control of transfection efficiency, and expressed as the fold induction with respect to the activity observed with the control reporter gene. (C) PPAR and RXR binding to PPREs in vivo. Chromatin from the various primary keratinocyte cultures was immunoprecipitated with either a pan-PPAR antibody (PPAR-Ab), an RXR antibody (RXR-Ab) or preimmune serum (p.i.). Enrichment of either a DNA fragment encompassing the PPRE from the transfected reporter gene (rep PPRE), a PPRE-containing fragment from the endogenous PDK1 promoter (PDK1 PPRE) or a control DNA fragment from the PDK1 gene (PDK1 control) was evaluated by PCR. Aliquots of the extracts were also analysed before immunoprecipitation (input). (D) Ligand-dependent coactivator association in solution. Western blot analyses were performed on proteins after immunoprecipitation (IP) with either an antibody against PPAR (PPAR-Ab) or RXR (RXR-Ab). Proteins were separated by SDS–PAGE, blotted and probed with either SRC1 or TIF2 primary antibody as indicated. Bands were detected by chemiluminescence with horseradish peroxidase. The Western blot control with tubulin antibody was performed on cell lysates prior to immunoprecipitation. (E) SRC1 binds to DNA-bound RXR, in a 9-cis RA-dependent manner. ChIP of PPRE-containing genes was performed as described in (C), using anti-SRC1 or preimmune serum (p.i.).
We next examined RXR homodimer interaction with coactivators in vivo. To address this, we first performed Western blot analysis to evaluate the interaction between nuclear receptor and cofactor, either on DNA or in solution. Immunoprecipitation performed with an RXR antibody revealed that both SRC1 and TIF2 strongly interact with RXR in the presence of 9-cis RA (Figure 4D). Fainter but clearly positive signals are also seen in the extracts obtained with the PPAR antibody. To discriminate between interaction in solution and when bound to DNA, we next performed a ChIP using an SRC1 antibody. The results demonstrate that ligand-activated PPAR loses its ability to interact with SRC1 upon binding to the response element (compare Figure 4D and E). In contrast, SRC1 is specifically recruited to the PPRE by activated RXR, irrespective of the presence of PPAR (Figure 4E).
These results show that in the absence of PPAR, RXR homodimers can functionally substitute for PPARs by binding to PPREs as a complex with SRC1. It can be argued that the homodimer pathway uncovered herein may be an artifact of deleting PPARs. Therefore, to support the existence of a functionally relevant RXR homodimer pathway even in the presence of PPARs, we performed a sequential ChIP experiment using primary keratinocyte cultures derived from wild-type mice. Following treatment with 9-cis RA, we performed a first ChIP using either an SRC1 or a p300 antibody. Subsequently, the antibodies were dissociated from the complexes, and a second ChIP was performed with either a PPAR or an RXR antibody (Figure 5). If SRC1 is exclusively recruited by activated RXR homodimers, PCR amplification of the PDK1 gene should only be observed following sequential immunoprecipitation with SRC1 and RXR antibodies. Indeed, no amplification is observed after the second immunoprecipitation with the PPAR antibody, showing that the complex pulled down with the SRC1 antibody contains RXR but no PPAR (Figure 5A, compare lanes 3 and 4). In contrast, and in agreement with our results from Figure 3, the complex pulled down with the p300 antibody contains both PPAR and RXR (Figure 5A, lanes 6 and 7). The same results were obtained using BMS 649, a synthetic RXR specific ligand (Egea et al, 2002) (Figure 5B). This clearly shows that RXR homodimer signalling via PPREs occurs in vivo and requires SRC1, whereas DNA-bound PPAR:RXR does not interact with SRC1. To further demonstrate the functional significance of SRC1 for RXR homodimer signalling, primary keratinocytes from wild-type and SRC1−/− mice (Xu et al, 1998) were transfected with a PPRE reporter gene and treated with either PPAR- or RXR-specific ligands (Figure 5C). The RXR agonist-induced response in SRC1−/− cells was significantly reduced as compared to wild-type cells, whereas PPAR agonist-induced reporter gene activity remained unaffected. Thus, in the presence of DNA, coactivators constitute a molecular switch between two competing signalling pathways, PPAR:RXR and RXR homodimers.
Figure 5.
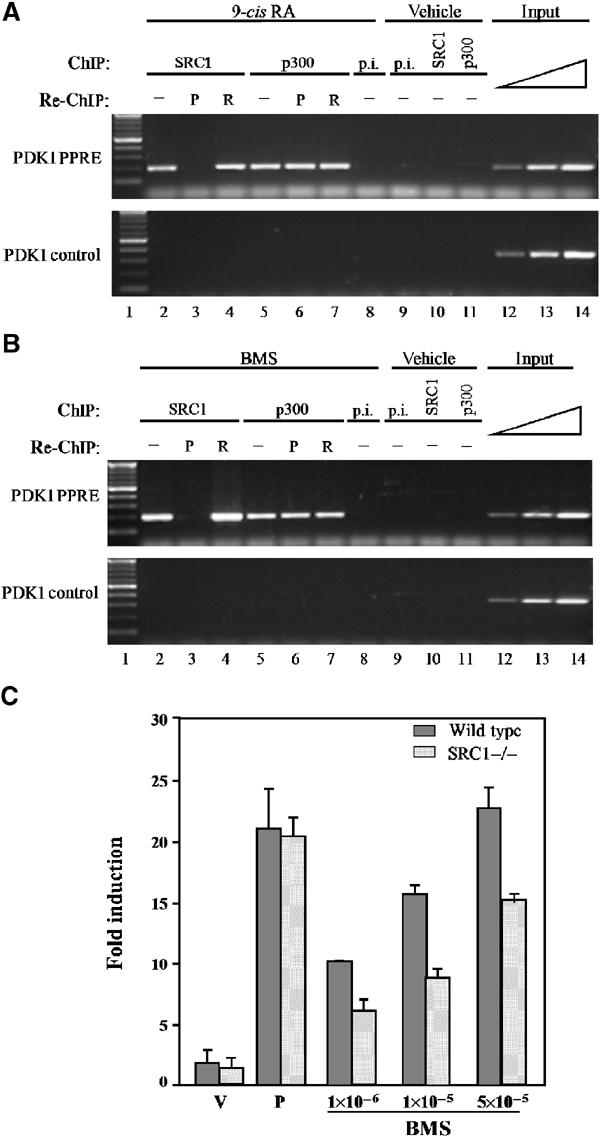
RXR:RXR/SRC1 ternary complexes bind to PPREs in the presence of PPAR. Primary keratinocytes from wild-type mice were treated with either 0.5 μM 9-cis RA (A), 1 μM BMS 649 (B) or vehicle. Following immunoprecipitation (ChIP) with either an SRC1-specific antibody (SRC1), a p300-specific antibody (p300) or preimmune serum (p.i.), the complexes were subjected to a second immunoprecipitation (re-ChIP) with either a PPAR- (P) or an RXR- (R) specific antibody. Enrichment of a PPRE-containing fragment from the endogenous PDK1 promoter (PDK1 PPRE) and a control DNA fragment from the PDK1 gene (PDK1 control) was evaluated by PCR. Aliquots of the extracts were also analysed before immunoprecipitation (input). (C) Primary keratinocytes from either wild-type (grey bars) or SRC1−/− (hatched bars) mice were transfected with a PPRE-luciferase reporter gene and treated with either vehicle (V; DMSO), PPAR ligands (P; a mixture of 10 μM Wy14,643 and 5 μM L1165041) or increasing doses of BMS as indicated. Luciferase activity levels were standardized for β-galactosidase expression used as internal control of transfection efficiency and expressed as the fold induction with respect to the activity observed with the activity of the reporter gene in the presence of vehicle alone.
The malic enzyme (ME) gene used throughout this study is abundantly expressed in the liver and is induced by both PPAR and RXR activators. Only low levels of SRC1 are expressed in the liver; however, TIF2 is abundantly expressed and interacts with both PPAR and RXR, at least in solution (Figure 6A and B). To further demonstrate the relevance of the RXR homodimer pathway in vivo, wild-type and PPARα−/− mice were treated for 5 days either with the PPARα ligand Wy14,643, or 9-cis RA. Analysis by ChIP of liver extracts from wild-type mice shows that RXR binds to MEp in the presence of either activator. In the absence of PPARα, RXR no longer binds to the PPRE upon treatment with the PPARα activator. However, in the presence of 9-cis RA, RXR still binds the PPRE and is able to induce the activity of the ME gene (Figure 6C, left and middle panels, and Figure 6D). Remarkably, ChIP using a TIF2 antibody results in an MEp enrichment, which is observed only in the presence of RXR and 9-cis RA and is independent of PPARα expression (Figure 6C, right panel). In addition to RXR homodimer binding, these observations could also reflect the binding to the PPRE by other RXR heterodimers. PPARβ, PPARγ and RAR are the only other known nuclear receptors that able to bind to DR1 elements as heterodimers with RXR. To rule out activation of these alternative pathways as a possible transactivation mechanism, PPARα−/− mice were administered specific activators for each of these putative partners. However, none of these activators was able to induce ME gene activity (Figure 6D, right panel).
Figure 6.

RXR homodimers are functional in vivo. (A) SRC1 and TIF2 expression in the liver. Western blot analysis of SRC-1 and TIF2 protein expression levels in a liver protein extract. For the purpose of comparison and control, a protein extract from pup skin was used. Equal loading was verified by β-tubulin levels. (B) PPAR and RXR interaction with TIF2 in solution. Wild-type (WT) and PPARα−/− (KO) mice were fed for 5 days with either Wy14,643 (50 mg/kg/day), 9-cis RA (30 mg/kg/day) or vehicle (solvent). Western blot analyses were performed on protein extracts from the liver and subjected to immunoprecipitation (IP) with either an antibody against PPAR (PPAR-Ab) or RXR (RXR-Ab). Proteins were separated by SDS–PAGE, blotted and probed with a TIF2 primary antibody. Bands were detected by chemiluminescence with horseradish peroxidase. The Western blot control with tubulin antibody was performed on cell lysates prior to immunoprecipitation. (C) ChIP of MEp with PPAR, RXR and TIF2 antibodies. Wild-type (WT) and PPARα−/− (KO) mice were fed for 5 days with either Wy14,643 (50 mg/kg/day), 9-cis RA (30 mg/kg/day) or vehicle (solvent). ChIP of liver extracts was performed with either a PPARα (PPAR-Ab), RXRα (RXR-Ab) or TIF2 (TIF2-Ab) antibody as indicated, and analysed by PCR for enrichment of the MEp element (top panels) or a control sequence (bottom panels). p.i.: preimmune serum. (D) Evaluation of ME gene activity in livers of mice treated with various ligands. Wild-type (WT) or PPARα KO (KO) mice were treated for 5 days with either a PPARα ligand (Wy: Wy14,643, 50 mg/kg/day), RXR ligand (9-cis RA, 30 mg/kg/day), PPARβ ligand (LD: L165041, 30 mg/kg/day), PPARγ ligand (Tro: troglitazone, 250 mg/kg/day), RAR ligand TTNPB (3 μg/kg/day) or vehicle (solvent, 0.5% carboxymethylcellulose), as indicated. Total liver RNA was isolated and expression levels of the ME and L27 genes were analysed by RNase protection assay using gene-specific probes. The indicated ratio of ME to L27 expression levels was determined for each of the samples. An arbitrary value of 1 was assigned to the value ratio obtained in mice treated with the solvent alone. (E) ChIP of the apoCIII DR1 element, in the liver. Wild-type (WT) and PPARα−/− (KO) mice were fed for 5 days with either Wy14,643 (50 mg/kg/day), 9-cis RA (30 mg/kg/day) or vehicle (solvent). ChIP of liver extracts was performed with either a PPARα (PPAR-Ab), RXRα (RXR-Ab) or TIF2 (TIF2-Ab) antibody as indicated, and analysed by PCR for enrichment of the apoCIII DR1 element (top panels) or a control sequence (bottom panels). p.i.: preimmune serum.
Having shown in vivo binding of RXR:RXR:SRC1 to the PDK1 PPRE in primary keratinocytes, as well as RXR:RXR:TIF2 to MEp in the liver, we extended our analysis of RXR homodimer signalling to the apoCIII gene. Intriguingly, in the liver, apoCIII gene expression is downregulated by fibrates in a PPARα-dependent manner (Peters et al, 1997), but upregulated by RXR-specific agonists (Vu-Dac et al, 1998). Thus, in vivo ChIP experiments, following the same experimental approach as described above, were performed using the DR1 element from the apoCIII promoter (Vu-Dac et al, 1998). As shown in Figure 6E, both RXR and TIF2 interact with the response element in a 9-cis RA-dependent manner, regardless of the presence or the absence of PPAR. Remarkably, PPAR:RXR heterodimers do not bind to this element in vivo, suggesting that negative regulation by fibrates occurs via an indirect mechanism. Alternatively, another as yet unidentified element in the apoCIII promoter could confer its PPAR-dependent repression.
Finally, to illustrate that activated RXR homodimers can functionally compensate for the absence of PPAR activity, we exposed PPARα−/− mice to an overnight fast, thus inducing severe hypothermia (Kersten et al, 1999). However, as shown in Figure 7, activation of RXR through administration of 9-cis RA for 5 days prior to fasting enabled the PPARα−/− mice to tolerate the fast and maintain their body temperature at a similar level as wild-type mice. Importantly, this final experiment shows that activated RXR homodimers are able to regulate complex metabolic pathways in vivo.
Figure 7.

Activation of the RXR homodimer signalling pathway prevents fasting-induced hypothermia. Following a 5-day treatment with 9-cis RA (30 mg/kg/day) or vehicle (V), wild-type (circle) and PPARα−/− (triangle) mice were subjected to an overnight fast. Each circle corresponds to the rectal temperature of an individual mouse. Rectal temperatures under fasting conditions are shown on the left, and rectal temperatures of similar groups of mice measured under nonfasting conditions are shown on the right.
Discussion
Taken together, our detailed analysis of rexinoid action via PPREs has brought up two important concepts with far-reaching pharmacological implications. Firstly, our demonstration that the ligand-dependent interaction with SRC1 or TIF2 is a key step in the binding of RXR:RXR to DR1 elements underscores a putative new role for coactivator molecules in stabilizing receptor binding under suboptimal conditions. Coactivator interaction may provide the means to overcome functional constraints, more particularly those imposed upon binding to DNA. At the same time, the allosteric effect of the DNA imposes selectivity in receptor/coactivator interactions (Zamir et al, 1997). Herein we show that PPRE-bound RXR homodimers selectively interact with TIF2 and SRC1, whereas within the context of the DNA-bound heterodimer, PPAR is unable to recruit SRC1. In agreement with our results, Yang et al (2000) previously reported the differential recruitment of DRIP205 and SRC1 by PPAR and RXR ligands respectively, but did not specifically demonstrate the (lack of) involvement of PPAR in the ternary SRC1 complex formed in the presence of 9-cis RA. A selective role of the DNA has also been reported for the SRC1 interaction with TR:RXR bound to mutated TREs (Takeshita et al, 1998). These results all point to the particular attention that should be given to the various levels of functional constraint when searching for selective nuclear receptor modulators. Indeed, whereas coactivators may interact with many transcription factors, selective recruitment of specific coactivators by a given receptor drives the formation of specific transcription complexes, which may result in different signalling cascades. In that respect, different nuclear receptor ligands could be considered as selective modulators recruiting specific cofactors (Sporn et al, 2001), leading to the activation of a specific subset of target genes. Clearly, a complete functional analysis of new modulators should not only take into account the complexity of the interactions between proteins, ligands and coactivators, but also include steric constraints imposed by DNA binding. Secondly, our results illustrate a novel mechanism for RXR homodimer signalling in vivo. Rather than just acting as a silent partner for other nuclear receptors, it is now quite well established that RXR can function as a bona fide receptor in vivo (Mascrez et al, 1998; Vivat-Hannah et al, 2003). However, in most reports, the existence of an RXR/9-cis RA signalling pathway independent of PPAR or any other RXR dimerization partners is either ignored or disputed (Germain et al, 2002). Indeed, our results provide an explanation for certain apparently contradictory observations, such as the reported downregulation of the apoCIII gene by fibrates in a PPARα-dependent manner (Peters et al, 1997), and its upregulation by RXR-specific agonists (Vu-Dac et al, 1998). It may also be the cause of the reported rexinoid-mediated activation of PPARα target genes in PPARα−/− mice (Ouamrane et al, 2003). Taken together, the physiological and pharmacological implications of this new signalling pathway are far-reaching, especially in view of the possible therapeutic use of RXR-specific activators.
Materials and methods
Reagents: 9-cis RA and TTNPB were from Sigma, troglitazone and Wy14,643 from CalBiochem and BRL49653 from Alexis. L165041 was synthesized in-house (Berger et al, 1999). SRC1 (sc-8995) and GRIP1/TIF2 (sc-8996) antibodies are from Santa Cruz Biotechnology; β-tubulin antibody is from Pharmingen and the GST antibody is from Amersham. The RXRα antibody was a kind gift from C Rochette-Egly and P Chambon. The rexinoids, LG and BMS were a kind gift from R Heyman (Ligand) and H Gronemeyer, respectively The PPARα antibody was developed in–house, whereas the panPPAR antibody used in EMSA and in ChIP is a kind gift from Dr M Dauca (Université de Nancy, France).
Animals: Mice were housed in a temperature-controlled room (23°C) on a 10-h dark, 14-h light cycle. PPARα−/− mice are a kind gift from F Gonzales (Lee et al, 2002). SRC1−/− mice are a kind gift from B O'Malley (Xu et al, 1998). Gavage with either Wy14,643 (50 mg/kg/day), 9-cis RA (30 mg/kg/day), L165041 (30 mg/kg/day), troglitazone (250 mg/kg/day), TTNPB (3 μg/kg/day) or vehicle (0.5% carboxymethylcellulose) for 5 days was performed on male mice weighing between 23 and 25 g. Rectal temperature was measured with a lubricated digital probe inserted 2 cm into the colon and held until a steady temperature was obtained (Ellab thermometer, Copenhagen).
Proteins: Proteins for EMSA as well as PPARγ and full-length SRC1 used in the DNA-dependent pull-down assays were obtained by in vitro TNT® coupled reticulocyte lysate system (Promega). Mouse RXRβ was prepared as a nuclear extract of Sf9 cells infected with recombinant baculovirus overexpressing recombinant H-2RIIBP (Marks et al, 1992). His-tagged mRXRα was produced in Sf9 cells and purified using a nickel column (Ni-NTA, Qiagen) (Dilworth et al, 1999). The nuclear receptor interaction domains of the human SRC1, mouse p300 and human TIF2 were expressed as GST fusion proteins in bacteria and purified as described by the manufacturer (Amersham Pharmacia Biotech). The SRC1 nuclear receptor interacting domain (SRC1(B); Jeannin et al, 1998) corresponds to amino acids 568–781 according to the SRC1 protein sequence as curated in GenBank under number NM_003743 (i.e. to amino acids 188–401 of the previous GenBank accession number AAC50305). pGEX2T-p300 (amino acids 39–220) was obtained from Dr M Leid. pGEX2T-TIF2 (amino acids 624–869) was derived from the pSG5-TIF2.5 clone obtained from Dr H Gronemeyer.
EMSA: EMSAs were performed according to classical procedures, as described before (IJpenberg et al, 1997).
Pull-down assays: DNA-dependent pull-down experiments were performed in 96-well plates (Streptawell, High bind, Roche) coated with biotinylated double-stranded oligonucleotide encompassing either the proximal PPRE of the malic enzyme promoter (MEp) (sense oligonucleotide: TTTTC CTGGG CATTC TGGGT CAAAG TTGAT CCCCT CCTG) or the PPRE of the PDK1 promoter (sense oligonucleotide: AGCAA GGGGC TGGGT GAGGA GAAAG GTGAC CCACA GAAG). Wells were washed three times with 300 μl of wash buffer (10 mM Tris pH 7.5, 1 mM EDTA, 1 M NaCl) and twice with 300 μl of incubation buffer (20 mM Hepes pH 7.5, 1 mM EDTA, 110 mM KCl, 10% glycerol, 0.1% Triton X-100). Different combinations of RXR (70 ng), PPAR (5 μl), full-length SRC-1 (15 μl), GST-SRC1(B) (1 μg), 9-cis RA (40 μM final concentration) and Rosiglitazone (10 μM final concentration) were mixed in incubation buffer supplemented with 0.01% BSA and protease inhibitors (Roche) in a total reaction volume of 50 μl, added to the wells and incubated in the dark for 2 h and 30 min at 4°C. Following incubation, the wells were washed four times with 300 μl of incubation buffer. A 50 μl portion of SDS–PAGE sample buffer was added to each well. The plate was covered with parafilm, heated for 5 min at 95°C and centrifuged for 1 min at 1000 rpm. Samples were analysed by Western blot using antibodies directed against either GST or SRC1.
Cell culture: Mouse primary keratinocytes were isolated, cultured and transfected as described in Tan et al (2001). The chart figuring the treatment and analyses to which the primary keratinocytes were subjected is shown in Figure 4A. NIH-3T3 cells were routinely cultured in DMEM supplemented with 10% fetal calf serum. Transfections were performed using Lipofectamine as instructed by the manufacturer (Gibco).
Western blot: Analyses were performed according to standard procedures, as described (Di Poï et al, 2002).
In vivo ChIP: After the indicated treatment, mice were killed by cervical dislocation. The liver was rapidly perfused with prewarmed (37°C) PBS for 5 min, followed by 0.2% collagenase (in PBS) for 10 min. The liver was diced, forced through a 60 μm stainless steel sieve and the hepatocytes were collected into DMEM containing 1% formaldehyde. After incubation at 37°C for 15 min, hepatocytes were pelleted and ChIP was performed using PPARα- and RXRα-specific antibodies as previously described. PCR was performed using primers flanking either the ME PPRE (−496 to −230) or an unrelated control sequence (−1958 to −1592). ChIP in primary keratinocytes in culture was performed as described by Di-Poï et al (2002).
Double chromatin immunoprecipitation (re-ChIP): Re-ChIP assays were performed with modifications of the procedure described by Di-Poï et al (2002). Briefly, chromatin was crosslinked using 1% formaldehyde for 10 min at 37°C and sonicated in lysis buffer (10 mM EDTA, 50 mM Tris–HCl, pH 8.0, 1% SDS, protease inhibitor cocktail (Roche Biochemicals, Mannheim, Germany)) to achieve crosslinked DNA of 200–600 bp in length. After centrifugation, 10% of the supernatant was used as input, and the remaining amount was subjected to the ChIP procedure. Complexes were eluted from the sepharose beads by sequential incubation with 1 bed volume of re-ChIP buffer (0.5 mM DTT, 1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris–HCl, pH 8.1), followed by 1 bed volume of Immunopure Gentle Ag/Ab Elution buffer (Pierce) containing 0.1 mM DTT (15 min per incubation step). The eluates were pooled and diluted 20-fold in ChIP dilution buffer (1 mM EDTA, 20 mM Tris–HCl, pH 8.1, 50 mM NaCl, 1% Triton X-100) and subjected to another ChIP procedure. PCR was performed using 5–10 μl of final re-ChIP eluate.
RNase protection assay (RPA): Gene-specific probes for mouse malic enzyme (nt 922–1273 from the ATG start codon) and L27 (nt 157–356) were subcloned into pGEM3Zf(+) (Promega). Antisense riboprobes were synthesized by in vitro transcription with Sp6 RNA polymerase. A ratio of 1:1 and 1:20 [α-32P]UTP to cold UTP was used for ME and L27, respectively. Total RNA was prepared from the liver of mice fed with the indicated agonists using the RNeasy minikit (Qiagen). RPA was carried out with 10 μg of total RNA as previously described (Tan et al, 2001).
Acknowledgments
We thank Marco Alvez for the synthesis of L165041, MG Parker, M Leid, H Gronemeyer, P Escher and E Jeannin for the kind gift of various plasmids, R Heyman (Ligand) and H Gronemeyer for providing the synthetic retinoids (LG) and BMS, respectively. We are also grateful to F Gonzalez for the gift of the PPARα null mice. This work was supported by the Etat de Vaud, the Human Frontier Science Program and the Swiss National Science Foundation.
References
- Berger J, Leibowitz MD, Doebber TW, Elbrecht A, Zhang B, Zhou G, Biswas C, Cullinan CA, Hayes NS, Li Y, Tanen M, Ventre J, Wu MS, Berger GD, Mosley R, Marquis R, Santini C, Sahoo SP, Tolman RL, Smith RG, Moller DE (1999) Novel peroxisome proliferator-activated receptor (PPAR) gamma and PPARdelta ligands produce distinct biological effects. J Biol Chem 274: 6718–6725 [DOI] [PubMed] [Google Scholar]
- Desvergne B, Wahli W (1999) Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev 20: 649–688 [DOI] [PubMed] [Google Scholar]
- Di-Poï N, Tan NS, Michalik L, Wahli W, Desvergne B (2002) Antiapoptotic role of PPARbeta in keratinocytes via transcriptional control of the Akt1 signaling pathway. Mol Cell 10: 721–733 [DOI] [PubMed] [Google Scholar]
- Dilworth FJ, Fromental-Ramain C, Remboutsika E, Benecke A, Chambon P (1999) Ligand-dependent activation of transcription in vitro by retinoic acid receptor alpha/retinoid X receptor alpha heterodimers that mimics transactivation by retinoids in vivo. Proc Natl Acad Sci USA 96: 1995–2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiRenzo J, Soderstrom M, Kurokawa R, Ogliastro MH, Ricote M, Ingrey S, Horlein A, Rosenfeld MG, Glass CK (1997) Peroxisome proliferator-activated receptors and retinoic acid receptors differentially control the interactions of retinoid X receptor heterodimers with ligands, coactivators, and corepressors. Mol Cell Biol 17: 2166–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egea PF, Mitschler A, Moras D (2002) Molecular recognition of agonist ligands by RXRs. Mol Endocrinol 16: 987–997 [DOI] [PubMed] [Google Scholar]
- Germain P, Iyer J, Zechel C, Gronemeyer H (2002) Co-regulator recruitment and the mechanism of retinoic acid receptor synergy. Nature 415: 187–192 [DOI] [PubMed] [Google Scholar]
- IJpenberg A, Jeannin E, Wahli W, Desvergne B (1997) Polarity and specific sequence requirements of peroxisome proliferator-activated receptor (PPAR)/retinoid X receptor heterodimer binding to DNA. A functional analysis of the malic enzyme gene PPAR response element. J Biol Chem 272: 20108–20117 [DOI] [PubMed] [Google Scholar]
- Jeannin E, Robyr D, Desvergne B (1998) Transcriptional regulatory patterns of the myelin basic protein and malic enzyme genes by the thyroid hormone receptors alpha1 and beta1. J Biol Chem 273: 24239–24248 [DOI] [PubMed] [Google Scholar]
- Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W (1999) Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J Clin Invest 103: 1489–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa R, Soderstrom M, Horlein A, Halachmi S, Brown M, Rosenfeld MG, Glass CK (1995) Polarity-specific activities of retinoic acid receptors determined by a co-repressor. Nature 377: 451–454 [DOI] [PubMed] [Google Scholar]
- Lala DS, Mukherjee R, Schulman IG, Koch SS, Dardashti LJ, Nadzan AM, Croston GE, Evans RM, Heyman RA (1996) Activation of specific RXR heterodimers by an antagonist of RXR homodimers. Nature 383: 450–453 [DOI] [PubMed] [Google Scholar]
- Lee Y, Yu X, Gonzales F, Mangelsdorf DJ, Wang MY, Richardson C, Witters LA, Unger RH (2002) PPAR{alpha} is necessary for the lipopenic action of hyperleptinemia on white adipose and liver tissue. Proc Natl Acad Sci USA 99: 11848–11853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Chiba H, Warot X, Messaddeq N, Gerard C, Chambon P, Metzger D (2001) RXR-alpha ablation in skin keratinocytes results in alopecia and epidermal alterations. Development 128: 675–688 [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Evans RM (1995) The RXR heterodimers and orphan receptors. Cell 83: 841–850 [DOI] [PubMed] [Google Scholar]
- Marks MS, Hallenbeck PL, Nagata T, Segars JH, Appella E, Nikodem VM, Ozato K (1992) H-2RIIBP (RXR beta) heterodimerization provides a mechanism for combinatorial diversity in the regulation of retinoic acid and thyroid hormone responsive genes. EMBO J 11: 1419–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascrez B, Mark M, Dierich A, Ghyselinck NB, Kastner P, Chambon P (1998) The RXRalpha ligand-dependent activation function 2 (AF-2) is important for mouse development. Development 125: 4691–4707 [DOI] [PubMed] [Google Scholar]
- Mertz JR, Shang E, Piantedosi R, Wei S, Wolgemuth DJ, Blaner WS (1997) Identification and characterization of a stereospecific human enzyme that catalyzes 9-cis-retinol oxidation. A possible role in 9-cis-retinoic acid formation. J Biol Chem 272: 11744–11749 [DOI] [PubMed] [Google Scholar]
- Minucci S, Ozato K (1996) Retinoid receptors in transcriptional regulation. Curr Opin Genet Dev 6: 567–574 [DOI] [PubMed] [Google Scholar]
- Nakshatri H, Chambon P (1994) The directly repeated RG(G/T)TCA motifs of the rat and mouse cellular retinol-binding protein II genes are promiscuous binding sites for RAR, RXR, HNF-4, and ARP-1 homo- and heterodimers. J Biol Chem 269: 890–902 [PubMed] [Google Scholar]
- Ouamrane L, Larrieu G, Gauthier B, Pineau T (2003) RXR activators molecular signalling: involvement of a PPARalpha-dependent pathway in the liver and kidney, evidence for an alternative pathway in the heart. Br J Pharmacol 138: 845–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM, Hennuyer N, Staels B, Fruchart JC, Fievet C, Gonzalez FJ, Auwerx J (1997) Alterations in lipoprotein metabolism in peroxisome proliferator-activated receptor alpha-deficient mice. J Biol Chem 272: 27307–27312 [DOI] [PubMed] [Google Scholar]
- Romert A, Tuvendal P, Simon A, Dencker L, Eriksson U (1998) The identification of a 9-cis retinol dehydrogenase in the mouse embryo reveals a pathway for synthesis of 9-cis retinoic acid. Proc Natl Acad Sci USA 95: 4404–4409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sporn MB, Suh N, Mangelsdorf DJ (2001) Prospects for prevention and treatment of cancer with selective PPARgamma modulators (SPARMs). Trends Mol Med 7: 395–400 [DOI] [PubMed] [Google Scholar]
- Takeshita A, Yen PM, Ikeda M, Cardona GR, Liu Y, Koibuchi N, Norwitz ER, Chin WW (1998) Thyroid hormone response elements differentially modulate the interactions of thyroid hormone receptors with two receptor binding domains in the steroid receptor coactivator-1. J Biol Chem 273: 21554–21562 [DOI] [PubMed] [Google Scholar]
- Tan NS, Michalik L, Noy N, Yasmin R, Pacot C, Heim M, Fluhmann B, Desvergne B, Wahli W (2001) Critical roles of PPAR beta/delta in keratinocyte response to inflammation. Genes Dev 15: 3263–3277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivat-Hannah V, Bourguet W, Gottardis M, Gronemeyer H (2003) Separation of retinoid X receptor homo- and heterodimerization functions. Mol Cell Biol 23: 7678–7688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu-Dac N, Gervois P, Torra IP, Fruchart JC, Kosykh V, Kooistra T, Princen HM, Dallongeville J, Staels B (1998) Retinoids increase human apo C-III expression at the transcriptional level via the retinoid X receptor. Contribution to the hypertriglyceridemic action of retinoids. J Clin Invest 102: 625–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Qiu Y, DeMayo FJ, Tsai SY, Tsai MJ, O'Malley BW (1998) Partial hormone resistance in mice with disruption of the steroid receptor coactivator-1 (SRC-1) gene. Science 279: 1922–1925 [DOI] [PubMed] [Google Scholar]
- Yang W, Rachez C, Freedman LP (2000) Discrete roles for peroxisome proliferator-activated receptor gamma and retinoid X receptor in recruiting nuclear receptor coactivators. Mol Cell Biol 20: 8008–8017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamir I, Zhang J, Lazar MA (1997) Stoichiometric and steric principles governing repression by nuclear hormone receptors. Genes Dev 11: 835–846 [DOI] [PubMed] [Google Scholar]
- Zhang XK, Lehmann J, Hoffmann B, Dawson MI, Cameron J, Graupner G, Hermann T, Tran P, Pfahl M (1992) Homodimer formation of retinoid X receptor induced by 9-cis retinoic acid. Nature 358: 587–591 [DOI] [PubMed] [Google Scholar]