

Lanthionine synthetase C-like 2 (LanCL2) is a novel regulator of Akt, promoting maximum Akt activation and cell survival in liver cells. LanCL2 regulates Akt activation by directly facilitating mTORC2 phosphorylation of Akt.
Abstract
The serine/threonine protein kinase Akt controls a wide range of biochemical and cellular processes under the modulation of a variety of regulators. In this study, we identify the lanthionine synthetase C–like 2 (LanCL2) protein as a positive regulator of Akt activation in human liver cells. LanCL2 knockdown dampens serum- and insulin-stimulated Akt phosphorylation, whereas LanCL2 overexpression enhances these processes. Neither insulin receptor phosphorylation nor the interaction between insulin receptor substrate and phosphatidylinositide 3-kinase (PI3K) is affected by LanCL2 knockdown. LanCL2 also does not function through PP2A, a phosphatase of Akt. Instead, LanCL2 directly interacts with Akt, with a preference for inactive Akt. Moreover, we show that LanCL2 also binds to the Akt kinase mTORC2, but not phosphoinositide-dependent kinase 1. Whereas LanCL2 is not required for the Akt-mTORC2 interaction, recombinant LanCL2 enhances Akt phosphorylation by target of rapamycin complex 2 (mTORC2) in vitro. Finally, consistent with a function of Akt in regulating cell survival, LanCL2 knockdown increases the rate of apoptosis, which is reversed by the expression of a constitutively active Akt. Taken together, our findings reveal LanCL2 as a novel regulator of Akt and suggest that LanCL2 facilitates optimal phosphorylation of Akt by mTORC2 via direct physical interactions with both the kinase and the substrate.
INTRODUCTION
The serine/threonine protein kinase Akt belongs to the protein kinase A, G, and C (AGC) family and plays a central role in a variety of cellular functions, including cell proliferation, cell survival, and glucose metabolism (Lawlor and Alessi, 2001). Deregulation of Akt activity is closely associated with several human diseases, such as cancer, diabetes, and cardiovascular and neurological diseases. Hyperactivation of Akt is one of the most common hallmarks in human malignancy, making Akt and its signaling pathways important therapeutic targets in cancer treatment (Bellacosa et al., 2005). On the other hand, Akt has an established function to suppress neuronal death (Datta et al., 1997; Dudek et al., 1997). Restoration of decreased activity of Akt represents an important approach in treating neurodegenerative diseases, including Parkinson's disease (Namikawa et al., 2000; Ries et al., 2006).
Akt can be activated by a wide range of extracellular signals, including many growth factors and cytokines. In the case of insulin signaling, insulin receptor is activated and recruits insulin receptor substrate (IRS) to the plasma membrane, where IRS is phosphorylated and in turn provides a docking site for the p85 regulatory subunit of phosphatidylinositide 3-kinase (PI3K). The interaction with IRS induces a conformational change in p85, which activates the catalytic subunit p110 of PI3K (Ruderman et al., 1990; Myers et al., 1992). Phosphatidylinositol (3,4,5)-trisphosphate, the product of PI3K, recruits phosphoinositide-dependent kinase 1 (PDK1) and Akt to the plasma membrane, where Akt is phosphorylated by PDK1 at Thr-308 in the T loop (Alessi et al., 1997). Akt is also phosphorylated in the C-terminal hydrophobic motif at Ser-473 by mammalian target of rapamycin complex 2 (mTORC2; Sarbassov et al., 2005). Dual phosphorylation of Akt results in its full activation. In addition, Akt activity is negatively regulated by protein phosphatases. The phosphatases responsible for dephosphorylating Thr-308 and Ser-473 sites belong to the protein phosphatase 2A (PP2A) and PP2C family, respectively (Gao et al., 2005).
The activity of endogenous Akt is also subject to context-dependent regulation/fine-tuning by many interacting proteins. In the past two decades, more than 20 of these proteins have been discovered, including both activators and inhibitors of Akt function (Franke, 2008). Many of these regulators do not have discernible enzymatic activity, and they regulate Akt function either by directly stimulating/inhibiting its kinase activity or affecting its subcellular localization or access to other regulators.
The eukaryotic lanthionine synthetase C (LanC)-like proteins are homologues of prokaryotic LanC, which is a cyclase involved in the biosynthesis of lantibiotics (Knerr and van der Donk, 2012). The human genome encodes three LanC-like proteins, LanCL1, 2, and 3, the functions of which are largely unknown. Human LanCL2 has been suggested to have a role in adriamycin sensitizing and the abscisic acid signaling pathway (Park and James, 2003; Landlinger et al., 2006; Sturla et al., 2009; Magnone et al., 2012). But the biochemical nature and mechanism of action of LanCL2 remain elusive. Here we investigated the potential function of LanCL2 in mammalian cells by examining its role in major signaling pathways. Our findings reveal LanCL2 to be a novel regulator of Akt activity and cell survival.
RESULTS
LanCL2 positively regulates Akt phosphorylation
To probe the function of LanCL2 in mammalian cells, we used lentivirus-delivered short hairpin RNA (shRNA) to knock down LanCL2 in multiple cell lines and examined the response of several major signaling pathways. LanCL2 knockdown suppressed serum-stimulated Akt phosphorylation on both Ser-473 and Thr-308 in human hepatocarcinoma HepG2 cells (Figure 1A), suggesting impaired Akt activation. This effect on Akt was further validated by decreased phosphorylation of GSK3β, a known substrate of Akt, in LanCL2 knockdown cells (Figure 1B). Insulin stimulation of Akt phosphorylation was similarly affected by LanCL2 knockdown, and two independent shRNAs elicited similar effects (Figure 1C), confirming the on-target specificity of the RNA interference (RNAi). The phosphorylation levels of two other kinases in the AGC family, S6K1 and cPKC, were not affected by LanCL2 knockdown, and neither was phosphorylation of Erk (Figure 1A). Hence, the effect of LanCL2 knockdown appears to be specific to Akt. Conversely, overexpression of FLAG-tagged LanCL2 modestly, but reproducibly, increased Akt phosphorylation (Figure 1D).
FIGURE 1:

LanCL2 is necessary for maximal Akt phosphorylation. All experiments were performed in HepG2 cells unless otherwise indicated, with cell lysates subjected to Western analysis. (A) Cells were infected with lentiviruses expressing LanCL2 shRNA (“L”) or a hairpin of scrambled sequence as control (“C”) for 1 d; this was followed by puromycin selection for 2 d. The cells were then either maintained in growth medium (“growth”), or serum starved overnight (“starved”) and restimulated with 10% FBS for 20 min (“stimulated”). (B) Cells were infected as in A and maintained in growth medium before lysis. (C) Cells were infected with lentiviruses expressing LanCL2 shRNAs (“L-1” and “L-2”), serum starved overnight, and then stimulated with 30 nM insulin for 20 min. (D) Cells were transfected with FLAG-tagged LanCL2 together with a plasmid carrying a puromycin-resistant gene and selected in puromycin for 3 d. (E) Three cell lines as indicated were treated as in A, serum starved overnight, and then stimulated with 10% FBS for 20 min. (F) Cells were infected with lentivirus expressing LanCL1 shRNA for 1 d; this was followed by puromycin selection for 2 d. The cells were then serum starved overnight and restimulated with 10% FBS for 20 min.
Interestingly, the effect of LanCL2 knockdown was observed only in HepG2 cells and not in several other human cell lines examined under the same conditions, including HEK293, HeLa, and MCF-7 (unpublished data). We therefore wondered whether this might be a liver cell–specific function of LanCL2 and set out to test this idea. We knocked down LanCL2 in three other liver cell lines—hepatocarcinoma Hep3B cells and SV40 large T antigen–immortalized Ea1C-35 and THLE-2 liver cells—and found that serum-stimulated Akt phosphorylation was impaired in all three cell lines (Figure 1E). Collectively these data suggest a positive role of LanCL2 in the regulation of Akt activation in liver cells. Of note, LanCL2 was found to be expressed in all cell lines we examined (Supplemental Figure S1), with some variation in expression levels that did not correlate with its knockdown effect on pAkt. Knockdown of LanCL1, which is also expressed in HepG2 cells, did not affect Akt phosphorylation (Figure 1F).
LanCL2 does not regulate Akt phosphorylation through the canonical insulin-signaling pathway or its phosphatase PP2A
In search of the mechanism by which LanCL2 regulates Akt phosphorylation, we first asked whether LanCL2 functions through the insulin receptor–IRS–PI3K pathway. As shown in Figure 2A, LanCL2 knockdown had no effect on the level of insulin receptor or its phosphorylation at the activation sites Tyr-1150/1151 in response to insulin. Moreover, the insulin-stimulated interaction between endogenous IRS1 and the p85 subunit of PI3K was also unaffected by LanCL2 knockdown (Figure 2B). Thus LanCL2 does not appear to regulate Akt phosphorylation through these canonical upstream components. We then investigated whether LanCL2 functions by regulating the phosphatase PP2A. As shown in Figure 2C, okadaic acid, a specific inhibitor of PP2A, did not rescue Akt phosphorylation at Thr-308 in LanCL2 knockdown cells, suggesting that LanCL2 does not function through inhibiting PP2A.
FIGURE 2:
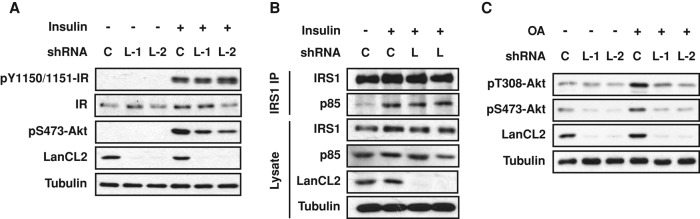
LanCL2 does not function through the insulin-IRS-PI3K pathway or PP2A. (A) HepG2 cells were infected with lentiviruses expressing LanCL2 shRNAs (“L-1” and “L-2”) or control shRNA (“C”) for 1 d; this was followed by puromycin selection for 2 d. The cells were then serum starved overnight and stimulated with 30 nM insulin for 20 min; this was followed by cell lysis and Western analysis. (B) Cells treated as in A were serum starved overnight and stimulated with 100 nM insulin for 10 min before lysis and immunoprecipitation (IP) of endogenous IRS1. (C) Cells were treated as in A, serum starved, and then stimulated with 10% FBS for 20 min with or without pretreatment with 1 μM okadaic acid (“OA”) for 15 min; this was followed by Western analysis.
LanCL2 interacts with Akt
With the known upstream regulators of Akt and its phosphatase PP2A ruled out as targets of LanCL2 regulation, we considered the possibility of a direct interaction between LanCL2 and Akt. This possibility was first tested in HEK293 cells as an exogenous system because of the ease of achieving higher transfection efficiency. Coimmunoprecipitation (co-IP) of transiently expressed FLAG-LanCL2 and endogenous Akt was observed (Figure 3A), and the reciprocal co-IP performed with hemagglutinin (HA)-Akt and FLAG-LanCL2 further confirmed this interaction (Figure 3B). Next we examined and confirmed this interaction in HepG2 cells by either pulling down endogenous Akt with His-LanCL2 purified from bacteria, or performing co-IP of transiently expressed FLAG-LanCL2 and HA-Akt (Figure 3, C and D). The interaction was also detected between purified His-LanCL2 and GST-Akt (Figure 3E), suggesting that it is direct and not mediated by other protein(s).
FIGURE 3:

LanCL2 interacts with Akt. (A) FLAG-tagged LanCL2 was transfected in HEK293 cells, and FLAG IP was carried out 24 h after transfection. Endogenous Akt was detected by Western blotting. (B) FLAG-tagged LanCL2 and HA-tagged Akt were coexpressed in HEK293 cells, and HA IP was carried out after 24 h; this was followed by Western analysis. (C and E) His pull-down assay was performed with purified His-LanCL2 protein and HepG2 cell lysates (C) or purified GST-Akt protein (E) and analyzed by Western blotting. (D) HepG2 cells were cotransfected with FLAG-LanCL2 and HA-Akt; this was followed by IP with FLAG antibody. (F) FLAG-tagged LanCL2 was coexpressed with different HA-tagged Akt deletion mutants and FLAG IP was carried out 48 h after transfection.
To map the LanCL2 interaction site on Akt, we generated various deletion mutants of Akt and examined their binding to LanCL2. As shown in Figure 3F, the Akt fragment aa 120–433, containing mostly the kinase domain, bound to LanCL2 to a similar extent as intact Akt. It is not feasible to map the Akt-interacting domain on LanCL2, because our preliminary data suggest a double seven-helix barrel-fold structure of LanCL2 similar to that of nisin cyclase (NisC) and LanCL1 (Li et al., 2006; Zhang et al., 2009), making it unlikely that any fragment of LanCL2 maintains its native fold.
Next we asked whether the interaction between LanCL2 and Akt was affected by the activation status of Akt. Interestingly, serum stimulation diminished Akt interaction with LanCL2, as shown in Figure 4A. Furthermore, LanCL2 displayed higher affinity for a kinase-inactive mutant of Akt (K179M; Franke et al., 1995) than for a constitutively active Akt (myr-Akt; Andjelkovic et al., 1997; Figure 4B). These observations suggested that LanCL2 might have a higher affinity for inactive Akt. Alternatively, the interaction might simply be influenced by the subcellular localization of Akt, as both serum stimulation and tagging by a myristoylation signal (myr-Akt) translocate Akt to the plasma membrane. To distinguish between the two possibilities, we compared Akt-K179M with wild-type Akt (Akt-WT) under serum stimulation, as Akt translocation is dependent on its PH domain but independent of its kinase activity. As shown in Figure 4C, LanCL2 displayed higher affinity for Akt-K179M than for Akt-WT. Taken together, these findings suggest that LanCL2 favors binding to inactive Akt.
FIGURE 4:
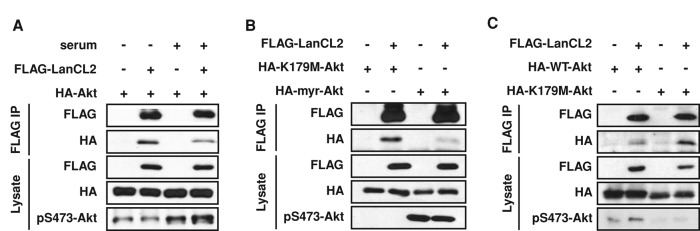
LanCL2 favors binding to inactive Akt. (A) HepG2 cells were cotransfected with FLAG-LanCL2 and HA-Akt for 24 h, serum starved overnight, and then stimulated with 10% FBS for 20 min before cell lysis and IP with anti-FLAG antibody. (B) HepG2 cells were cotransfected with FLAG-LanCL2 and myr-Akt or K179M-Akt for 24 h; this was followed by IP with anti-FLAG antibody. (C) HepG2 cells were cotransfected with FLAG-LanCL2 and HA-Akt-WT or HA-Akt-K179M, and treated as in A; this was followed by IP with anti-FLAG antibody.
LanCL2 enhances Akt phosphorylation by mTORC2
In light of the physical interaction between Akt and LanCL2 and the lack of any known catalytic activity for LanCL2, we set out to test a simple model in which LanCL2 facilitates Akt recruitment to its kinases. PDK1 and mTORC2 are known to phosphorylate Thr-308 and Ser-473, respectively (Alessi et al., 1997; Sarbassov et al., 2005), although other kinases may also be involved in a context-dependent manner. Indeed, we found that LanCL2 coimmunoprecipitated mTOR and rictor (core components of mTORC2; Figure 5A), but not PDK1 (unpublished data). While knockdown of rictor did not affect LanCL2-mTOR pull down, knockdown of mTOR clearly weakened LanCL2-rictor interaction (Figure 5, B and C), suggesting that mTOR, rather than rictor, may directly interact with LanCL2. Indeed, we also consistently found a small amount of raptor in the LanCL2 immunoprecipitates, as would be expected if LanCL2 interacted with mTOR.
FIGURE 5:

LanCL2 binds mTOR and facilitates Akt phosphorylation by mTORC2. (A) HepG2 cells were transfected with FLAG-tagged LanCL2, and FLAG IP was carried out 48 h after transfection. (B and C) HepG2 cells were infected with lentiviruses expressing a control shRNA (“C”), rictor shRNA (“ric”), or mTOR shRNA (“M”) for 2 d; this was followed by puromycin selection for 2 d. Cells were then transfected with FLAG-tagged LanCL2, and FLAG IP was carried out 48 h after transfection. (D) HepG2 cells were infected with lentiviruses expressing control shRNA or LanCL2 shRNA (“L”) and selected by puromycin. Cells were then transfected with HA-Akt, and HA IP was carried out 48 h after transfection. (E) Endogenous raptor and rictor were immunoprecipitated from HepG2 cells and subjected to mTORC1 and mTORC2 kinase assays, using GST-S6K1 and His-Akt as substrates, respectively. Phosphorylation at Thr-389-S6K1 or Ser-473-Akt was detected as a readout of the kinase activity. Purified His-LanCL2 (5 μg) was added before the kinase assay in the indicated samples. (F) HEK293 cells were transfected with FLAG-LanCL2 or an empty vector; this was followed by FLAG IP and Western analysis.
A straightforward scaffold model would predict the dependence of Akt-rictor interaction on LanCL2. However, that is not the case, as knockdown of LanCL2 had no detectable effect on the Akt-rictor interaction (Figure 5D). Nevertheless, this observation does not rule out the possibility that LanCL2 binding may be important for optimal catalytic activity of mTORC2 toward Akt. To test this model, we performed in vitro kinase assays with immunoprecipitated endogenous mTORC2 and recombinant Akt as the substrate, with and without the addition of His-LanCL2 purified from bacteria. As shown in Figure 5E, recombinant LanCL2 clearly enhanced Akt Ser-473 phosphorylation in vitro. Importantly, mTORC1 kinase activity toward S6K1 was not affected by recombinant LanCL2 (Figure 5E), which is consistent with our observation that LanCL2 knockdown did not affect S6K1 Thr-389 phosphorylation in cells (Figure 1A). These observations strongly suggest that LanCL2 specifically regulates Akt phosphorylation by mTORC2, even though LanCL2 may interact with both mTORC1 and mTORC2. The physical interaction between LanCL2 and Akt may have contributed to this specificity, as we did not detect any LanCL2-S6K1 interaction (Figure 5F).
The effect of LanCL2 on mTORC2 kinase activity toward Akt prompted us to examine another substrate of mTORC2, SGK1 (Garcia-Martinez and Alessi, 2008). Phosphorylation by mTORC2 activates SGK1, which in turn phosphorylates NDRG1 at Thr-346. The latter is widely used as the readout of SGK1 activity in cells (Murray et al., 2004). Interestingly, NDRG1 phosphorylation was suppressed by LanCL2 knockdown (Supplemental Figure S2), suggesting that LanCL2 may facilitate SGK1 phosphorylation by mTORC2 as well. However, we were not able to reliably assess whether LanCL2 interacted with SGK1 due to difficulty in detecting endogenous SGK1 with available antibodies.
LanCL2 promotes cell survival through Akt
Among a wide range of cellular functions controlled by Akt is cell survival. Activated Akt protects a variety of cell types from apoptosis (Duronio, 2008). We therefore wondered whether LanCL2 was involved in this cellular process because of its role in regulating Akt phosphorylation. Indeed, we observed cleavage of poly(ADP-ribose) polymerase (PARP), a marker of apoptosis, induced by LanCL2 knockdown in HepG2 cells (Figure 6A). Apoptosis induced by tumor necrosis factor α (TNFα)/cycloheximide (CHX) or adriamycin was also enhanced by LanCL2 knockdown (Figure 6A). DNA fragmentation, another important marker of programmed cell death, was also significantly increased in LanCL2 knockdown cells as measured by terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assays (Figure 6B). Importantly, expression of a constitutively active form of Akt (myr-Akt) effectively suppressed TNFα/CHX-induced apoptosis in LanCL2 knockdown cells (Figure 6C), suggesting that LanCL2 promotes cell survival through activation of Akt.
FIGURE 6:
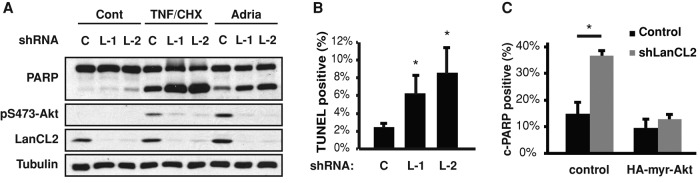
LanCL2 depletion sensitizes cells to apoptosis through down-regulating Akt phosphorylation. (A) HepG2 cells were infected with lentiviruses expressing LanCL2 shRNA (“L-1” and “L-2”) or control (“C”), selected with puromycin, and then treated with 10 ng/ml TNFα and 10 μg/ml CHX for 3.5 h or 5 μM adriamycin for 17 h; this was followed by Western analysis. (B) Cells treated with TNFα and CHX as in A were subjected to TUNEL assays. Data shown are the average of three independent experiments, and ∼30,000 cells were examined for TUNEL signals for each data point. (C) Lentivirus-infected and puromycin-selected cells were transfected with HA-myr-Akt or a control plasmid; this was followed by TNFα/CHX treatment as in A. The cells were fixed and immunostained for cleaved PARP and HA. The percentage of transfected cells positive for cleaved PARP was calculated. Data shown are the average of three independent experiments. A paired t test was performed to compare each data point with the control. *, p < 0.05.
DISCUSSION
In the current study, we have established LanCL2 as a novel regulator of Akt phosphorylation. LanCL2 interacts with Akt and promotes the maximal phosphorylation of Akt in response to mitogenic signals. We also show that LanCL2 binds mTORC2 and directly promotes the mTORC2 kinase activity toward Akt. Furthermore, we demonstrate the biological significance of this novel regulation by showing that LanCL2 promotes cell survival through Akt.
Our findings add another player to the growing list of modulators for Akt, a central regulator of myriad cellular and developmental processes. The observation that LanCL2 favors binding to inactive Akt is reminiscent of several other positive regulators of Akt, such as APPL (Adaptor protein containing PH domain, PTB domain and leucine zipper motif) and APE (Akt-phosphorylation enhancer) (Mitsuuchi et al., 1999; Anai et al., 2005). On activation, Akt undergoes a dramatic conformational change in the catalytic domain (Yang et al., 2002), where LanCL2 binds, which may lead to the dissociation of LanCL2. This binding preference for inactive Akt also implies that LanCL2 is involved in the activation of Akt rather than maintaining its phosphorylation or activity. Supporting this notion, we find that LanCL2 does not function through the phosphatase of Akt, PP2A, but instead acts directly to enhance Akt phosphorylation by mTORC2.
Enhancing Akt phosphorylation through its kinases is one of the reported mechanisms for Akt-interacting proteins. Freud-1/Aki1 and BSTA are two examples in this category, functioning as scaffold proteins to facilitate Akt-PDK1 and Akt-rictor binding, respectively (Nakamura et al., 2008; Yao et al., 2013). LanCL2, however, does not seem to be necessary for the tethering of Akt and mTORC2, despite its interaction with both proteins. Nevertheless, LanCL2 may contribute to optimal catalysis by orienting the substrate (Akt) for the kinase (mTORC2) or rendering Akt more accessible to mTORC2 for subsequent phosphorylation. In other words, LanCL2 may help to achieve a catalytically more effective interaction between Akt and mTORC2. This catalytic function of scaffold proteins has been proposed before. For instance, the yeast scaffold protein Ste5 is found to primarily function through tethering components of the mitogen-activated protein kinase (MAPK) pathway but it most likely also contributes to orient kinase/substrate for optimal catalysis (Park et al., 2003). Interestingly, NDRG1 phosphorylation is also suppressed in LanCL2 knockdown cells, indicating a dampened SGK1 activity. LanCL2 may facilitate SGK1 phosphorylation by mTORC2 in the same way as Akt. On the other hand, the absence of a LanCL2-S6K1 interaction coincides with the absence of a LanCL2 effect on mTORC1 kinase activity toward S6K1, corroborating our hypothesis that the physical interaction is necessary for LanCL2-mediated enhancement of substrate phosphorylation by mTOR complexes.
It is noteworthy that the phosphorylation of Akt on Thr-308 is also affected by LanCL2 knockdown, yet LanCL2 does not interact with PDK1. This is in agreement with previous reports that Akt phosphorylation on Thr-308 by PDK1 is dependent on the prior phosphorylation on Ser-473 (Scheid et al., 2002; Yang et al., 2006). Therefore, when Ser-473 phosphorylation is attenuated by LanCL2 knockdown, so is the phosphorylation on Thr-308.
LanCL2 was identified in a screen for genes whose down-regulation results in anticancer drug resistance and therefore was designated “testis-specific adriamycin sensitivity protein (TASP)” (GenBank accession no. AB035966). Two other reports also showed that LanCL2 overexpression in human epithelial cells of amniotic origin (UAC) and a human uterine sarcoma cell line (MES-SA) increased adriamycin-induced cell death rate (Erbay et al., 2003; Landlinger et al., 2006). Our observation that LanCL2 knockdown cells are more sensitive to adriamycin appears to contradict those earlier reports. However, it is important to note that the function of LanCL2 may be highly dependent on cell type and cellular context. Indeed, in another report using human gastric, pancreatic, colon, and breast cancer cell lines, the expression level of LanCL2 was not correlated with adriamycin sensitivity (Gyorffy et al., 2005). Of the various cell lines we have examined, inhibition of Akt activation by LanCL2 knockdown is only consistently observed in liver cells. While it remains to be determined what other types of cells may harbor the same mechanism, this novel function of LanCL2 is observed in several liver cell lines, including both hepatocarcinoma and noncancerous cells. It is not clear why LanCL2 regulates Akt only in liver cells, while its expression appears to be ubiquitous, but the stoichiometry of various components in the Akt pathway likely varies from tissue to tissue, which could determine the relative contribution of these regulators. As Akt is a well-established regulator of glucose metabolism and organ growth in the liver, future probing of the physiological functions of LanCL2 in those contexts is warranted.
MATERIALS AND METHODS
Antibodies and reagents
The antibodies used were obtained from the following sources. Anti-LanCL2 antibody was generated by Proteintech Group (Chicago, IL) using full-length recombinant LanCL2 protein as the antigen. Anti-LanCL1 antibody was from Abnova (Taipei, Taiwan). Anti-FLAG M2 was from Sigma-Aldrich (St. Louis, MO); anti-HA (16B12) from Covance (San Diego, CA), anti-HA (rabbit) from Promega (Madison, WI), anti-tubulin from Abcam (Cambridge, England), anti-pPKC from EMD Millipore (Billerica, MA), anti-His and anti-GST from Santa Cruz Biotechnology (Dallas, TX), anti-rictor and anti-raptor (for IP) from Bethyl Laboratories (Montgomery, TX), and all other antibodies from Cell Signaling Technology (Beverly, MA). Cobalt beads and anti-HA agarose beads were purchased from Thermo Fisher (Waltham, MA). TNFα was from Cell Signaling Technology. Adriamycin and okadaic acid were from LC laboratories (Woburn, MA). Fibronectin was from EMD Millipore. GST-Akt was from SignalChem (Richmond, BC, Canada). His-Akt was from Millipore. All other reagents were obtained from Sigma-Aldrich.
Plasmids
Human LanCL2 cDNA was subcloned in the p3×FLAG-CMV-14 vector (Sigma-Aldrich), with the 3×FLAG tag fused at the C-terminus of LanCL2. Human Akt1 cDNA was subcloned in the pCDNA3 vector (Invitrogen) with an HA tag at the N-terminus. pCMV6myristoylated-HA-Akt (HA-myr-Akt) was previously described (Erbay et al., 2003). pCDNA3HA-Akt1-K179M was subcloned from pLNCX-HA-Akt1-K179M (Addgene plasmid 9007; Ramaswamy et al., 1999).
Cell culture and transfection
All cell lines used in this study were obtained from the American Type Culture Collection (Manassas, VA). The following media were used to maintain various types of cells: DMEM/Ham's F-12 containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (pen/strep) for HepG2 cells, DMEM containing 10% FBS and 1% pen/strep for HEK293 cells, MEM containing 10% FBS and 1% pen/strep for Hep3B cells, MFE support medium (Xenotech) for Ea1C-35 cells, and BEGM (LONZA) supplemented with 70 ng/ml phosphoethanolamine and 5 ng/ml EGF for THLE-2 cells. All cells were maintained at 37°C with 5% CO2. Plasmid DNA transfection was performed using TransIT-LT1 (Mirus Bio) in HepG2 cells or Polyfect (Qiagen) in HEK293 cells, according to the manufacturers' instructions.
Lentivirus-mediated RNAi
LanCL1 and LanCL2 lentiviral shRNA constructs in the pLKO vector were obtained from Sigma-Aldrich (TRC library). Their clone IDs are LanCL2 #1 (L1), TRCN0000045403; LanCL2 #2 (L2), TRCN0000045406; LanCL1, TRCN0000011687. mTOR and rictor lentiviral shRNA constructs were from Addgene, as plasmid 1855 and plasmid 1853, respectively (Sarbassov et al., 2005). Lentivirus packaging was performed by cotransfecting pLKO-shRNA, pCMV-dR8.91, and pCMV-VSV-G into HEK293T cells using TransIT-LT1 at 0.5, 0.45, and 0.05 μg, respectively (for one well in a six-well plate). Media containing viruses were collected 48 h after transfection. HepG2 and other cells were infected with the viruses in the presence of 8 μg/ml polybrene for 24 h and were then subjected to selection with various concentrations of puromycin.
Western blotting
Cells were lysed in MIPT lysis buffer (50 mM Tris-Cl, pH 7.2, 137 mM NaCl, 25 mM NaF, 25 mM β-glycerophosphate, 2 mM EDTA, 2 mM ethylene glycol tetraacetic acid (EGTA), 10 mM sodium pyrophosphate, 0.3% Triton X-100, and 1× protease inhibitor cocktail [Sigma-Aldrich]) and microcentrifuged at 10,000 × g for 10 min at 4°C. The supernatant was mixed 1:1 with 2× SDS sample buffer and heated at 95°C for 5 min. Proteins were resolved on SDS–PAGE, transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore), and incubated with various antibodies following the manufacturers' recommendations. Detection of horseradish peroxidase–conjugated secondary antibodies was performed with Western Lightning Chemiluminescence Reagent Plus (Perkin Elmer), and images were developed on x-ray films.
Immunoprecipitation
Cells were lysed in MIPT lysis buffer or NP40-based lysis buffer (20 mM Tris-Cl, pH 7.5, 0.2% Nonidet P-40, 10% glycerol, 1 mM EDTA, 1.5 mM MgCl2, 137 mM NaCl, 50 mM NaF, 1 mM NaVO3, 12 mM β-glycerophosphate, 1× protease inhibitor cocktail [Sigma-Aldrich]) and microcentrifuged at 10,000 × g for 10 min at 4°C. The supernatant was incubated with anti-FLAG beads or anti-HA beads (Sigma-Aldrich) for 2 h. The beads were then washed three times with lysis buffer and boiled in 2× SDS sample buffer for 5 min; this was followed by Western blotting. For immunoprecipitation of endogenous IRS1, incubation with anti-IRS1 antibody was followed by incubation with protein A beads.
His-LanCL2 pull down
For His-LanCL2 pull down of endogenous Akt, cells were lysed in His pull-down buffer (20 mM Tris-Cl, pH 8.0, 150 mM NaCl, 25 mM NaF, 25 mM β-glycerophosphate, 0.1 mM NaVO3, 20 mM imidazole, 0.3% Triton X-100, and 1× protease inhibitor cocktail [Sigma-Aldrich]) and incubated with 10 μg His-LanCL2 protein for 2 h at 4°C; this was followed by incubation with cobalt beads for another 1 h. The beads were then washed three times with the lysis buffer and boiled in 2× SDS sample buffer for 5 min. For LanCL2-Akt in vitro binding, His-LanCL2 and GST-Akt were directly mixed in His pull-down buffer for 2 h; this was followed by incubation with cobalt beads. The beads were then washed and boiled as described above.
mTORC1 and mTORC2 kinase assay
mTORC1 and mTORC2 were immunoprecipitated from cell lysates with anti-raptor or anti-rictor antibody, respectively. The kinase assays were performed as previously described (Ikenoue et al., 2009). mTORC2 kinase assay was carried out at 37°C for 30 min in mTORC2 kinase buffer (25 mM HEPES, pH 7.4, 100 mM potassium acetate, 1 mM MgCl2, and 500 μM ATP) with 62 ng His-Akt as the substrate. mTORC1 kinase assay was carried out at 30°C for 30 min in mTORC1 kinase buffer (25 mM HEPES, pH 7.4, 50 mM KCl, 10 mM MgCl2, and 250 μM ATP) with 16 ng GST-S6K1 (aa 332–421) as the substrate. Reactions were stopped by adding 2× SDS buffer and boiling.
TUNEL assay and immunostaining
TUNEL assays were performed following the manufacturer's manual (Roche). For immunostaining, cells were fixed with 3.7% formaldehyde followed by permeabilization with 0.1% Triton X-100 and blocking with 3% bovine serum albumin in phosphate-buffered saline. Cells were then incubated with antibodies against cleaved PARP and HA for 2 h at room temperature, which was followed by incubation with Alexa Fluor–labeled secondary antibody and 4′,6-diamidino-2-phenylindole for 30 min. The stained cells were examined with a Leica DMI 4000B fluorescence microscope, and the fluorescent images were captured using a RETIGA EXi camera and analyzed with Q-capture Pro51 software (QImaging).
Supplementary Material
Acknowledgments
We thank MeeSup Yoon for providing GST-S6K1 protein and Nicholas Llewellyn for technical assistance. This work was supported by the Howard Hughes Medical Institute (W.A.v.d.D.) and the National Institutes of Health (grant no. GM089771 to J.C.).
Abbreviations used:
- CHX
cycloheximide
- co-IP
coimmunoprecipitation
- FBS
fetal bovine serum
- HA
hemagglutinin
- IRS
insulin receptor substrate
- LanC
lanthionine synthetase C
- LanC-like 2
lanthionine synthetase C–like 2
- mTORC2
mammalian target of rapamycin complex 2
- PARP
poly(ADP-ribose) polymerase
- PDK1
phosphoinositide-dependent kinase 1
- pen/strep
penicillin/streptomycin
- PI3K
phosphatidylinositide 3-kinase
- PP2
protein phosphatase 2
- shRNA
short hairpin RNA
- TNFα
tumor necrosis factor α
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick-end labeling
- WT
wild type
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-01-0004) on October 1, 2014.
REFERENCES
- Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- Anai M, Shojima N, Katagiri H, Ogihara T, Sakoda H, Onishi Y, Ono H, Fujishiro M, Fukushima Y, Horike N, et al. A novel protein kinase B (PKB)/AKT-binding protein enhances PKB kinase activity and regulates DNA synthesis. J Biol Chem. 2005;280:18525–18535. doi: 10.1074/jbc.M500586200. [DOI] [PubMed] [Google Scholar]
- Andjelkovic M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M, Cron P, Cohen P, Lucocq JM, Hemmings BA. Role of translocation in the activation and function of protein kinase B. J Biol Chem. 1997;272:31515–31524. doi: 10.1074/jbc.272.50.31515. [DOI] [PubMed] [Google Scholar]
- Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv Cancer Res. 2005;94:29–86. doi: 10.1016/S0065-230X(05)94002-5. [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Duronio V. The life of a cell: apoptosis regulation by the PI3K/PKB pathway. Biochem J. 2008;415:333–344. doi: 10.1042/BJ20081056. [DOI] [PubMed] [Google Scholar]
- Erbay E, Park IH, Nuzzi PD, Schoenherr CJ, Chen J. IGF-II transcription in skeletal myogenesis is controlled by mTOR and nutrients. J Cell Biol. 2003;163:931–936. doi: 10.1083/jcb.200307158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke TF. Intracellular signaling by Akt: bound to be specific. Sci Signal. 2008;1:pe29. doi: 10.1126/scisignal.124pe29. [DOI] [PubMed] [Google Scholar]
- Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005;18:13–24. doi: 10.1016/j.molcel.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Garcia-Martinez J M, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1) Biochem J. 2008;416:375–385. doi: 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- Gyorffy B, Serra V, Jurchott K, Abdul-Ghani R, Garber M, Stein U, Petersen I, Lage H, Dietel M, Schafer R. Prediction of doxorubicin sensitivity in breast tumors based on gene expression profiles of drug-resistant cell lines correlates with patient survival. Oncogene. 2005;24:7542–7551. doi: 10.1038/sj.onc.1208908. [DOI] [PubMed] [Google Scholar]
- Ikenoue T, Hong S, Inoki K. Monitoring mammalian target of rapamycin (mTOR) activity. Methods Enzymol. 2009;452:165–180. doi: 10.1016/S0076-6879(08)03611-2. [DOI] [PubMed] [Google Scholar]
- Knerr PJ, van der Donk WA. Discovery, biosynthesis, and engineering of lantipeptides. Annu Rev Biochem. 2012;81:479–505. doi: 10.1146/annurev-biochem-060110-113521. [DOI] [PubMed] [Google Scholar]
- Landlinger C, Salzer U, Prohaska R. Myristoylation of human LanC-like protein 2 (LANCL2) is essential for the interaction with the plasma membrane and the increase in cellular sensitivity to adriamycin. Biochim Biophys Acta. 2006;1758:1759–1767. doi: 10.1016/j.bbamem.2006.07.018. [DOI] [PubMed] [Google Scholar]
- Lawlor MA, Alessi DR. PKB/Akt: a key mediator of cell proliferation, survival and insulin responses. J Cell Sci. 2001;114:2903–2910. doi: 10.1242/jcs.114.16.2903. [DOI] [PubMed] [Google Scholar]
- Li B, Yu JP, Brunzelle JS, Moll GN, van der Donk WA, Nair SK. Structure and mechanism of the lantibiotic cyclase involved in nisin biosynthesis. Science. 2006;311:1464–1467. doi: 10.1126/science.1121422. [DOI] [PubMed] [Google Scholar]
- Magnone M, Sturla L, Jacchetti E, Scarfi S, Bruzzone S, Usai C, Guida L, Salis A, Damonte G, De Flora A, et al. Autocrine abscisic acid plays a key role in quartz-induced macrophage activation. FASEB J. 2012;26:1261–1271. doi: 10.1096/fj.11-187351. [DOI] [PubMed] [Google Scholar]
- Mitsuuchi Y, Johnson SW, Sonoda G, Tanno S, Golemis EA, Testa JR. Identification of a chromosome 3p14.3-21.1 gene, APPL, encoding an adaptor molecule that interacts with the oncoprotein-serine/threonine kinase AKT2. Oncogene. 1999;18:4891–4898. doi: 10.1038/sj.onc.1203080. [DOI] [PubMed] [Google Scholar]
- Murray JT, Campbell DG, Morrice N, Auld GC, Shpiro N, Marquez R, Peggie M, Bain J, Bloomberg GB, Grahammer F, et al. Exploitation of KESTREL to identify NDRG family members as physiological substrates for SGK1 and GSK3. Biochem J. 2004;384:477–488. doi: 10.1042/BJ20041057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers MG, Jr., Backer JM, Sun XJ, Shoelson S, Hu P, Schlessinger J, Yoakim M, Schaffhausen B, White MF. IRS-1 activates phosphatidylinositol 3′-kinase by associating with src homology 2 domains of p85. Proc Natl Acad Sci USA. 1992;89:10350–10354. doi: 10.1073/pnas.89.21.10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura A, Naito M, Tsuruo T, Fujita N. Freud-1/Aki1, a novel PDK1-interacting protein, functions as a scaffold to activate the PDK1/Akt pathway in epidermal growth factor signaling. Mol Cell Biol. 2008;28:5996–6009. doi: 10.1128/MCB.00114-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namikawa K, Honma M, Abe K, Takeda M, Mansur K, Obata T, Miwa A, Okado H, Kiyama H. Akt/protein kinase B prevents injury-induced motoneuron death and accelerates axonal regeneration. J Neurosci. 2000;20:2875–2886. doi: 10.1523/JNEUROSCI.20-08-02875.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, James CD. Lanthionine synthetase components C-like 2 increases cellular sensitivity to adriamycin by decreasing the expression of P-glycoprotein through a transcription-mediated mechanism. Cancer Res. 2003;63:723–727. [PubMed] [Google Scholar]
- Park SH, Zarrinpar A, Lim WA. Rewiring MAP kinase pathways using alternative scaffold assembly mechanisms. Science. 2003;299:1061–1064. doi: 10.1126/science.1076979. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S, Nakamura N, Vazquez F, Batt DB, Perera S, Roberts TM, Sellers WR. Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc Natl Acad Sci USA. 1999;96:2110–2115. doi: 10.1073/pnas.96.5.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries V, Henchcliffe C, Kareva T, Rzhetskaya M, Bland R, During MJ, Kholodilov N, Burke RE. Oncoprotein Akt/PKB induces trophic effects in murine models of Parkinson's disease. Proc Natl Acad Sci USA. 2006;103:18757–18762. doi: 10.1073/pnas.0606401103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruderman NB, Kapeller R, White MF, Cantley LC. Activation of phosphatidylinositol 3-kinase by insulin. Proc Natl Acad Sci USA. 1990;87:1411–1415. doi: 10.1073/pnas.87.4.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Scheid MP, Marignani PA, Woodgett JR. Multiple phosphoinositide 3-kinase-dependent steps in activation of protein kinase B. Mol Cell Biol. 2002;22:6247–6260. doi: 10.1128/MCB.22.17.6247-6260.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturla L, Fresia C, Guida L, Bruzzone S, Scarfi S, Usai C, Fruscione F, Magnone M, Millo E, Basile G, et al. LANCL2 is necessary for abscisic acid binding and signaling in human granulocytes and in rat insulinoma cells. J Biol Chem. 2009;284:28045–28057. doi: 10.1074/jbc.M109.035329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Cron P, Good VM, Thompson V, Hemmings BA, Barford D. Crystal structure of an activated Akt/protein kinase B ternary complex with GSK3-peptide and AMP-PNP. Nat Struct Biol. 2002;9:940–944. doi: 10.1038/nsb870. [DOI] [PubMed] [Google Scholar]
- Yang Q, Inoki K, Ikenoue T, Guan KL. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. 2006;20:2820–2832. doi: 10.1101/gad.1461206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Suraokar M, Darnay BG, Hollier BG, Shaiken TE, Asano T, Chen CH, Chang BH, Lu Y, Mills GB, et al. BSTA promotes mTORC2-mediated phosphorylation of Akt1 to suppress expression of FoxC2 and stimulate adipocyte differentiation. Sci Signal. 2013;6:ra2. doi: 10.1126/scisignal.2003295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wang L, Liu Y, Xu J, Zhu G, Cang H, Li X, Bartlam M, Hensley K, Li G, et al. Structure of human lanthionine synthetase C-like protein 1 and its interaction with Eps8 and glutathione. Genes Dev. 2009;23:1387–1392. doi: 10.1101/gad.1789209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.