

Genomic variations such as gene fusions are directly or indirectly associated with human diseases. A method is presented combining gene expression and interactome data analyses to identify specific targets in leukemia. The Myc network and the mRNA export machinery are perturbed in ETV6-RUNX1 and TCF3-PBX1 subtypes of leukemia.
Abstract
Genomic variations such as point mutations and gene fusions are directly or indirectly associated with human diseases. They are recognized as diagnostic, prognostic markers and therapeutic targets. However, predicting the functional effect of these genetic alterations beyond affected genes and their products is challenging because diseased phenotypes are likely dependent of complex molecular interaction networks. Using as models three different chromosomal translocations—ETV6-RUNX1 (TEL-AML1), BCR-ABL1, and TCF3-PBX1 (E2A-PBX1)—frequently found in precursor-B-cell acute lymphoblastic leukemia (preB-ALL), we develop an approach to extract perturbed molecular interactions from gene expression changes. We show that the MYC and JunD transcriptional circuits are specifically deregulated after ETV6-RUNX1 and TCF3-PBX1 gene fusions, respectively. We also identified the bulk mRNA NXF1-dependent machinery as a direct target for the TCF3-PBX1 fusion protein. Through a novel approach combining gene expression and interactome data analysis, we provide new insight into TCF3-PBX1 and ETV6-RUNX1 acute lymphoblastic leukemia.
INTRODUCTION
The development of every cancer is characterized by frequent genomic aberrations. Investigations focused on specific human neoplasms have identified numerous sequence variants in which mutations are implicated in oncogenesis. These human cancer genes are listed in the Cancer Genome Project database, with genes encoding protein kinase and transcriptional regulation domains highly represented (Futreal et al., 2004). Characterization of the biological properties of some mutated genes, such as the breakpoint cluster region-v-Abelson murine leukemia viral oncogene homologue 1 (BCR-ABL1), has led to the development of successful targeted therapies (Lynch et al., 2004; Gazdar, 2009; Quintas-Cardama and Cortes, 2009; Agrawal et al., 2010; Kaulfuss et al., 2013). The most prevalent category among the known cancer genes are chromosomal translocations, often involving immunoglobulin, T-cell receptor, and transcription factor genes (Futreal et al., 2004). Although these rearrangements represent important diagnostic markers that are used to define cancer subtypes (Mitelman et al., 2004; Maher et al., 2009), their molecular interactions and the pathways affected by the result of gene fusions are poorly characterized.
Genes and their products do not act in isolation but as part of complex molecular networks in which most genes play their roles through several molecular functions or interactions. The changes induced by gene fusions and other genetic alterations, as well as modifications of expression levels, do not lead to a complete loss of the gene products and are thus very likely to alter the different interactions of the same gene or protein in distinct fashions (Zhong et al., 2009). Classically, genome-wide transcriptomic studies have been used to identify genes or gene expression signatures in order to characterize and classify cancer types or subtypes (Golub et al., 1999; Andersson et al., 2005; Gandemer et al., 2007; Den Boer et al., 2009; Li et al., 2009; Fuka et al., 2011). Although very useful to identify oncogenes and for diagnostic purposes, these methods are limited in their ability to understand the underlying molecular biology, as they are focused on genes, transcripts, and proteins, neglecting the interactions between them.
In this study, we propose a strategy that uses gene expression profiles to identify genes, molecular interactions, and pathways that are important in a specific genetic alteration. We use as models two chromosomal translocations found in precursor-B-cell acute lymphoblastic leukemia (preB-ALL) and involving key specific transcription factors regulating hematopoietic development: 1) the Ets transcription factor variant 6 (ETV6)–runt-related transcription factor 1 (RUNX1) fusion (also known as TEL-AML1) and 2) the transcription factor 3 (TCF3)–pre-B-cell leukemia homeobox 1 (PBX1) fusion (also known as E2A-PBX1; Okuda et al., 1996; Zhou et al., 2012; Tijchon et al., 2013). These chromosomal rearrangements alone are insufficient for leukemogenesis but may support leukemia when additional molecular perturbations are present (Andreasson et al., 2001; Seto, 2010). We thus extracted perturbed molecular interactions and showed that MYC and JunD interactomes are specifically deregulated after ETV6-RUNX1 and TCF3-PBX1 gene fusions, respectively. Furthermore, we demonstrated that the TCF3-PBX1 fusion could impair the normal mRNA export machinery.
RESULTS
Predicting perturbed interactions linked to gene fusions
To predict perturbed molecular interactions specifically linked to ETV6-RUNX1, TCF3-PBX1, and BCR-ABL1 gene fusions, we used the human B-cell interactome (HBCI; Lefebvre, 2007, 2010) and expression data sets from two microarray series (Den Boer et al., 2009; Mullighan et al., 2009), including 24 samples with BCR-ABL1 fusion, 77 with ETV6-RUNX1 fusion, 16 with TCF3-PBX1 fusion, and 248 samples with other different genetic subtypes. Expression data were first normalized by frozen robust multiarray analysis (fRMA; McCall and Irizarry, 2011). For each interaction in HBCI, we computed the difference between the correlation of expression profiles in a group of samples exhibiting a genotype of interest and in the control samples (groups of samples with other genotypes). Because interacting genes/proteins are likely to be involved in similar biological processes and are likely coexpressed (Ge et al., 2001), we selected interactions with significant differences of correlation as deregulated (corrected p < 0.05; Figure 1A).
FIGURE 1:

Prediction of perturbed interactions. (A) Flowchart of the method. Arrows show the flow of data analysis: black for microarrays, and green and red for HBCI and Pathway commons interactome, respectively. For each interaction in the B-cell or pathways interactome, we computed the differential in correlation between genotypes. Significance of the difference in correlation is estimated from randomized data. Interactions with corrected p < 0.05 are predicted as perturbed. Dashed lines represent perturbed interactions. (B) Venn diagram representing the number of detected perturbed interactions (DPIs) in the B-cell interactome for ETV6-RUNX1, BCR-ABL1, and TCF3-PBX1.
We detected 2550 perturbed interactions (∼4.5% of interactions in the HBCI, involving 664 human genes) and 3334 (∼5.8% of the HBCI, involving 1022 human genes) in the ETV6-RUNX1 and TCF3-PBX1 ALL samples, respectively (Supplemental Tables S1 and S2). We found only 74 (0.13%) overlapping interactions between ETV6-RUNX1 and TCF3-PBX1 ALL samples, showing the specificity of the method (Figure 1B). For BCR-ABL1 genotype, which does not involve direct translocation of a transcription factor–coding gene, we detected only 10 (∼0.018%) potentially perturbed interactions (Supplemental Table S3). Our next analyses thus will compare perturbed networks for ETV6-RUNX1 and TCF3-PBX1 fusions.
We ranked proteins/genes according to the number of perturbed interactions, and identified MYC (∼46% of HBCI) as the most perturbed in the ETV6-RUNX1 subtype of preB-ALL. To confirm the direct link between MYC network alteration and the presence of ETV6-RUNX1 fusion protein, we used HEK293 cells stably expressing ETV6-RUNX1 and control cells expressing similar amounts of MYC (Figure 2A). We performed chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) in cells expressing the ETV6-RUNX1 fusion protein to detect the MYC-binding sites at a genome scale. We identified 557 MYC target genes in both cell lines (Figure 2B and Supplemental Table S4, HEK293 +ETV6-RUNX1 anti MYC and HEK293 anti MYC), representing 19% of MYC target genes reported in the human B-cell interactome (Lefebvre, 2007, 2010). As predicted, this experiment showed a high modification of MYC targets in the presence of ETV6-RUNX1 fusion, with ∼88% (489 of 557) of the targets being different between the two cell lines. Among these, 52% were also identified as MYC- perturbed interactions by our method (Figure 2C and Supplemental Table S1), further supporting the use of differences of correlation between expression profiles to predict perturbed interactions.
FIGURE 2:
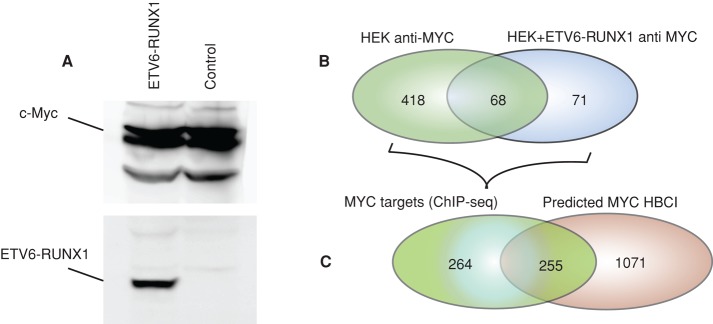
ETV6-RUNX1 expression perturbs MYC binding to its targets. (A) HEK 293T expressing V5-ETV6-RUNX1 and control cells were subjected to Western blot analysis using anti-MYC and anti-V5 antibodies. (B) Chromatin immunoprecipitation was performed using an anti-MYC antibody, followed by massively parallel sequencing of isolated DNA fragments. Venn diagrams indicate the comparison of MYC target genes identified in HEK vs. HEK+ETV6-RUNX1 cell lines. (C) Venn diagrams showing a comparison between the numbers of perturbed MYC targets identified by ChIP-seq and those predicted by computing the differences of correlation between expression profiles.
Topological analysis of the perturbed networks
To determine whether the structure of the network is modified after ETV6-RUNX1 or TCF3-PBX1 fusions, we analyzed network topology perturbations using three metrics: characteristic path length (cpl), edge betweenness centrality (ebc), and edge-clustering coefficient (ecc).
We sequentially removed edges corresponding to perturbed interactions by decreasing order of significance, calculated the cpl, average ebc, and average ecc of the resulting network at each step, and compared these metrics to those obtained by removing random edges (Figure 3, red lines). For ETV6-RUNX1 fusion, we observed a significant increase of cpl and ebc, whereas ecc decreased, indicating that edge perturbations in ETV6-RUNX1 fusion leads to a less compact network but with a globally higher, more evenly distributed communication potential and a lower local connectivity on high-degree nodes than expected at random (Figure 3, compare green to red lines). In the case of TCF3-PBX1 fusion, on the contrary, the perturbed network becomes more compact, with a slightly lower communication potential and local connectivity than expected at random (Figure 3, compare blue and red lines). We also compared ebc, ecc, and the edge shortest path length (espl) of the network composed of perturbed edges with the rest of the network (the network of not-perturbed edges). The espl was computed as the mean of all shortest path lengths between the vertices of an edge and all other vertices in the network. It appeared that perturbed edges after ETV6-RUNX1 fusion are characterized by significantly higher ebc, higher ecc, and lower espl than other edges in the network (Table 1A). Similar local metrics for the TCF3-PBX1 fusion showed minor changes (Table 1B).
FIGURE 3:

Topological analysis of the perturbed BCI network. We show the evolution of three network metrics while removing predicted perturbed interactions in their order of significance (highest difference of correlation first) for ETV6-RUNX1 and TCF3-PBX1 fusions. Abscissa, number of removed edges (perturbed interactions); ordinate, values of the metric. Green, blue, and red curves represents the distributions of the values of the metric removing edges sequentially in ETV6-RUNX1, TCF3-PBX1, and randomly (100 random iterations), respectively. Vertical bars, SEs of random iterations. (A) Characteristic path length (cpl). (B) Edge- betweenness centrality (ebc). (C) Edge-clustering coefficient (ecc).
TABLE 1:
Comparison of local metrics between perturbed edges and the rest of the network for (A) ETV6-RUNX1 and (B) TCF3-PBX1 fusion.
| Metrics | Perturbed edges | Other edges | p |
|---|---|---|---|
| A. ETV6-RUNX1 | |||
| Edge shortest path length (espl) | 2.084 | 2.288 | <2.22E-16 |
| Edge betweenness (ebc) | 1838 | 572 | <2.22E-16 |
| Edge clustering coefficient | 0.683 | 0.451 | <2.22E-16 |
| B. TCF3-PBX1 | |||
| Edge shortest path length (espl) | 2.259 | 2.280 | 3.5219E-13 |
| Edge betweenness (ebc) | 614 | 628 | 1.859E-12 |
| Edge clustering coefficient (ecc) | 0.457 | 0.462 | 0.4265 |
Difference of the means (p) is assessed through a Mann–Whitney U test.
Taken together, our network topology analysis suggests that ETV6-RUNX1 chromosomal translocation may lead to disruption of molecular interactions important for B-cell communication circuits, whereas TCF3-PBX1 fusion only slightly modifies the structure of the network.
Specific deregulation of transcription factor networks
We ranked nodes based on the proportion of their perturbed interactions in HBCI, and highlighted the 10 most deregulated nodes in ETV6-RUNX1 and TCF3-PBX1 fusion subtypes of ALL. It appeared that, for both ALL subtypes, top deregulated nodes correspond to diverse transcription regulators (Figure 4, A and B). This result suggests that ETV6-RUNX1 and TCF3-PBX1 fusions support oncogenesis mostly by specifically deregulating other transcriptional regulators. We therefore analyzed the interaction networks of transcription factors (TFs) that have at least one interaction predicted as deregulated and categorized the TFs according to the number of perturbed interactions and the published classification of human TFs (Wingender et al., 2013). We found that two classes of transcription factors, basic helix-loop-helix (bHLH) and leucine zipper (bZIP), account for the majority of perturbed interactions for ETV6-RUNX1 and TCF3-PBX1 fusions, respectively (Figure 5, A and B, red arrows). In particular, the majority of bZIP members of the activating protein-1 (AP-1) complexes, including JunD, JunB, c-Jun, c-Fos, Fra-1 (FOSL1), FosB, ATF2, ATF3, CREB1, and CREBL2, seems affected after TCF3-PBX1 fusion (Figures 4B and 5B).
FIGURE 4:
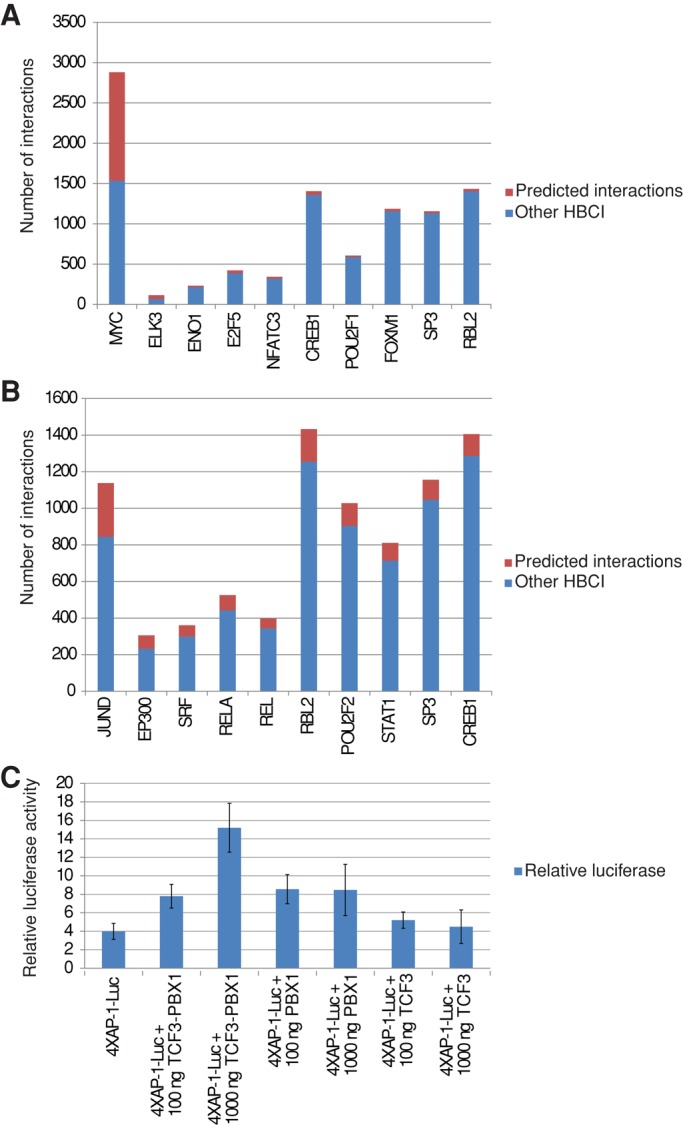
Deregulation of transcription factor interactions. (A) ETV6-RUNX1. (B) TCF3-PBX1. Ten most perturbed nodes, that is, genes/proteins showing the highest number of perturbed interactions. Colors: red bars (pred_inter) represent perturbed interactions, blue bars (other HBCI) represent other interactions in the HBCI database. (C) Transactivation of an AP-1 reporter by TCF3-PBX1. HEK293 cells were transfected with the reporter plasmid p4XAP1-luc and indicated amounts of effector plasmids (pFlag-TCF3-PBX1, pFlag-TCF3, pFlag-PBX1). Firefly luciferase data were normalized to Renilla luciferase counts, and data are reported as mean and SE of three independent experiments in triplicate.
FIGURE 5:

Network of TFs in (A) ETV6-RUNX1 and (B) TCF3-PBX1 fusion. Left, circles represent TFs that have at least one protein–DNA interaction (PDI) predicted as perturbed; colors correspond to the class of transcription factor. Red/gray edges represent the perturbed/not-perturbed interactions, respectively. Right, chart showing the number of perturbed interactions for each transcription factor class according to TF class (Wingender et al., 2013).
To validate our analysis experimentally, we investigated TCF3-PBX1–induced transcriptional regulation of the Jun/AP-1 pathway. HEK293 cells were cotransfected with a luciferase reporter construct harboring AP-1–binding sites (Samuel et al., 2008) and increasing amounts of expression vectors for TCF3-PBX1, TCF3, or PBX1 transcription factor. As shown on Figure 4C, AP-1 transactivation is significantly increased in the presence of the oncogenic fusion TCF3-PBX1, in accordance with previous reports that demonstrated functional interplay between TCF3-PBX1 and the CREB-binding protein (CBP) and p300 transcriptional coactivators (LeBrun, 2003; Bayly et al., 2004; Denis et al., 2012; Hyndman et al., 2012) and that CBP/p300 and AP-1 factors have many partners in common (Denis et al., 2012).
Among the bHLH TFs, the proto-oncogene MYC was the top-ranked gene, with ∼46% of HBCI perturbed interactions (Figure 5A). By comparison, only a small minority (1%) of MYC interactions is perturbed in TCF3-PBX1–associated ALL samples, suggesting a specific interplay between MYC and ETV6-RUNX1 transcriptional regulation. This is also illustrated by the network of MYC transcriptional regulators, showing that the majority of protein–DNA interactions (such as STAT5 and the MYC promoter; Denis et al., 2012; Ott et al., 2012; Mangolini et al., 2013) and protein–protein interactions (such as SP1 and MYC coregulation of some target genes; Zhang et al., 2012; Wang et al., 2013, 2014) are specifically deregulated in ETV6-RUNX1 compared with TCF3-PBX1 ALL subtypes (Figure 6). To further validate MYC deregulation in ETV6-RUNX1 compared with TCF3-PBX1 subtypes of ALL, we analyzed the relative expression of MYC and its targets involved in cell cycle regulation, including cyclin-dependent kinase inhibitors (CKIs), which are known targets repressed by the MYC/Miz1 complex (Figure 6C and Supplemental Table S5). Among these key cell cycle regulators, TP53, MYC, and CDKN3 were relatively underexpressed, whereas CDKN1A (p21WAF1/CIP1) expression was unchanged in ETV6-RUNX1 samples compared with other groups of samples. As recently reported for the MDM2 promoter (Kaindl et al., 2014), this result indicates a p53-independent mode of CDKN1A promoter regulation in ETV6-RUNX1 ALL samples. We thus hypothesized that the ETV6-RUNX1 fusion could compete with the formation of MYC/Miz1 complex or the binding to its down-regulated targets, including the CDKN1A promoter. To test the latter hypothesis, we analyzed the CDKN1A promoter sequence (−499 to +100 base pairs around the start site) using the eukaryotic promoter database (EPD; epd.vital-it.ch) and the TFsearch program (Heinemeyer et al., 1998) and identified two putative RUNX1-binding sites. The ETV6-RUNX1 chromosomal translocation conserves the RUNX1 DNA–binding domain (Supplemental Figure 1A), suggesting that any putative RUNX1-binding site would also be a potential target sequence for ETV6-RUNX1 fusion protein, as previously demonstrated (Wotton et al., 2008; Krapf et al., 2010; Kaindl et al., 2014). We therefore performed ChIP assays using leukemic REH cells (or REH cells silenced in ETV6-RUNX1 expression by short hairpin RNA [shRNA; REH-G1]; Fuka et al., 2012; Supplemental Figure 1B) and a specific antibody to ETV6 transcription factor. We specifically amplified a genomic CDKN1A promoter fragment encompassing both putative RUNX1-binding sites, indicating that ETV6-RUNX1 fusion protein directly binds the CDKN1A promoter (Supplemental Figure S1C). In a transcriptional reporter assay, we also demonstrated that the activation of the CDKN1A promoter by a phorbol ester (PMA; Zeng and el-Deiry, 1996) could be inhibited by overexpression of ETV6-RUNX1 fusion protein (Supplemental Figure 1D), similar to the previously reported repressor effect of MYC on CDKN1A promoter (Seoane et al., 2002; Wu et al., 2003). Finally, using HEK293 cells stably expressing ETV6-RUNX1, we compared MYC and ETV6-RUNX1 ChIP-seq data sets and showed that both transcription regulators could target similar genes (Supplemental Table S9). Taken together, these results support the idea that the ETV6-RUNX1 fusion protein might interplay with the MYC transcriptional network.
FIGURE 6:

MYC regulators and the cell cycle. Network showing HBCI interactions for transcription factors regulating Myc. Red edges represents interactions detected as perturbed; gray edges, interactions not detected as perturbed. (A) For ETV6-RUNX1. (B) For TCF3-PBX1. (C) Network showing HBCI interactions for cyclins (CCNs; light magenta), cyclin-dependent kinases (CDK; light blue), and cyclin-dependent kinase inhibitors (CDKNs; light green) relative to MYC, RUNX1, ETV6, and MIZ-1.
Perturbations in signaling pathways
To explore signaling pathways potentially disturbed after ETV6-RUNX1 and TCF3-PBX1 gene fusions, we adopted two strategies. First, we performed a gene enrichment pathway analysis using the 664 or 1024 human genes involved in ETV6-RUNX1 or TCF3-PBX1 perturbed interactions, respectively. As stated earlier, this data set is restricted to genes involved in B-cell interactions reported in the HBCI database. We performed a functional enrichment analysis using the Database for Annotation, Visualization and Integrated Discovery (DAVID; Huang da et al., 2009). We linked perturbed interactions in ETV6-RUNX1 or TCF3-PBX1 ALL samples to similar signaling pathways, including pathways in cancer, T- and B-cell receptor signaling, Toll-like and growth factor signaling, mitogen-activated protein kinase signaling, and cell cycle and cell adhesion (Figure 7, A and D). However, the proportions of perturbed interactions (Figure 7, compare B and E) and the pathway subnetwork profiles (Figure 7, compare C and F) are very different. Of interest, pathways involved in B-cell migration (chemokine receptor signaling) and cell adhesion were specific to the ETV6-RUNX1 fusion, consistent with previous studies showing that ALL cells, including REH, are highly motile and capable of rapid migration within lymphoid tissues (Makrynikola et al., 1994; Gandemer et al., 2007). Alternatively, we predicted NF-κB pathway deregulation for TCF3-PBX1, in accordance with the significant proportion of perturbed interactions involving NF-κB RELA (p65) and REL subunits (16 and 14% of HBCI reported interactions, respectively).
FIGURE 7:

Pathways enrichment of genes having at least one interaction predicted as perturbed in 1) ETV6-RUNX1 and 2) TCF3-PBX1 fusions. (A, D) The size of the circle represents the number of genes involved in the pathway, and the edges size represents the number of shared genes. The size of a green/red circle represents the total number of interactions in HBCI/perturbed interactions between genes involved in the pathway, respectively. The edge size represents the shared interactions (gray for interactions in BCI and red for the perturbed ones) between pathways. (B, E) Proportion of perturbed interactions in each pathway. (C, F) Networks of predicted perturbed interactions in each pathway. Circles represent genes; edges, interactions.
Next we used Pathway commons (www.pathwaycommons.org), which is a collection of pathways from multiple sources and organisms. Compared to the cell-context HBCI database, we reasoned that Pathway Commons might allow us to uncover unexpected novel functions for TCF3-PBX1 or ETV6-RUNX1 fusion proteins. We thus considered Pathway Commons as a single network and predicted disrupted interactions in the same way as for the HBCI. This analysis revealed 61 and 45 perturbed pathways for ETV6-RUNX1 and TCF3-PBX1 fusions, respectively (Supplemental Tables S6 and S7). Confirming our foregoing results, several pathways linked to MYC transcription factor were predicted as perturbed by the ETV6-RUNX1 fusion. Of interest, we highlighted a potential deregulation of pathways linked to RNA transport machinery after TCF3-PBX1 (Table 2). Perturbed interactions involve several proteins important for RNA processing, including mRNA export proteins such as the eukaryotic translation factor 4A3 (eIF4A3), the nuclear pore complex (NCP/NUP), and the nuclear export receptor NXF1/TAP (Siddiqui and Borden, 2012; Supplemental Table S8). Eukaryotic mRNA is exported from the nucleus either by the bulk export NXF1-dependent pathway or via more specialized factors such as the chromosome region maintenance 1 (CRM1, also called exportin-1 [XPO1]; Hutten and Kehlenbach, 2007; Siddiqui and Borden, 2012). Because most TCF3-PBX1 perturbed interactions involved the NXF1 rather than the CRM1 pathway, we analyzed potential interaction between TCF3-PBX1 fusion protein and NXF1 by examining the subcellular localization of both proteins. We showed that both NXF1 and TCF3-PBX1 colocalize in the nucleoplasm (Supplemental Figure S2A), indicating functional interplay. To test whether TCF3-PBX1 fusion protein could interfere with RNA localization, we visualized RNA molecules in cells transfected with the TCF3-PBX1 fusion and observed colocalization between TC3-PBX1 and RNA molecules, and, most important, RNA was delocalized from the nucleoli to the nucleoplasm (Supplemental Figure S2B). Together these results suggest a potential deregulating role of TCF3-PBX1 in the mRNA export machinery.
TABLE 2:
The 10 top perturbed pathways after (A) ETV6-RUNX1 and (B) TCF3-PBX1 gene fusion.
| Pathway name | Number of perturbed interactions | Number of interactions in the current pathway | Corrected p |
|---|---|---|---|
| A. ETV6-RUNX1 | |||
| Validated targets of c-Myc transcriptional activation | 48 | 489 | 2.92E-25 |
| Validated targets of c-Myc transcriptional repression | 27 | 272 | 7.26E-14 |
| Regulation of nuclear SMAD2 3 signaling | 41 | 716 | 2.56E-13 |
| RNA polymerase II transcription termination | 31 | 576 | 3.43E-9 |
| Cleavage of growing transcript in the termination region | 31 | 576 | 3.43E-9 |
| Postelongation processing of the transcript | 31 | 576 | 3.43E-9 |
| Postelongation processing of intron-containing pre-mRNA | 24 | 406 | 6.79E-8 |
| mRNA 3′-end processing | 24 | 406 | 6.79E-8 |
| c-Myc pathway | 13 | 106 | 1.15E-7 |
| Regulation of nuclear β-catenin signaling and target gene transcription | 22 | 362 | 1.56E-7 |
| B. TCF3-PBX1 | |||
| RNA transport | 209 | 1742 | 5.55E-17 |
| Transport of mature transcript to cytoplasm | 138 | 1004 | 9.85E-16 |
| Transport of mature mRNA derived from an intron-containing transcript | 126 | 934 | 8.65E-14 |
| Noncoding RNA metabolism | 101 | 703 | 1.09E-12 |
| Small nuclear ribonucleoprotein assembly | 101 | 703 | 1.09E-12 |
| mRNA surveillance pathway | 78 | 595 | 8.85E-8 |
| Transport of the stem-loop binding protein–dependent mature mRNA | 56 | 378 | 2.87E-7 |
| Regulation of nuclear SMAD2 3 signaling | 86 | 716 | 6.49E-7 |
| Transport of mature mRNAs derived from intronless transcripts | 57 | 404 | 9.90E-7 |
| TNFα | 244 | 2784 | 3.27E-6 |
Corrected p value is computed using Benjamini–Hochberg multitesting correction.
DISCUSSION
As genome-wide expression profiling and interactomic data accumulate and are stored in public databases, the integration to drive interpretation of genotype–phenotype relationships and identify genes and pathways associated with specific diseases remains challenging. Several approaches have been conducted on cancer samples to identify tumor markers and gene expression signatures and to classify cancer types or subtypes. However, functional perturbations arising from expression changes are rarely interpreted in the context of molecular network perturbations, which may be sensitive to subtle transcriptional changes.
In this study, we integrated data from gene expression in B-cell ALL subtypes, molecular interaction networks from the human B-cell interactome, and Pathway Commons databases to provide novel hypotheses about deregulated molecular interactions and pathways. We detected 0.018, 4.5, and 5.8% of perturbed interactions in the human B-cell interactome after chromosomal translocations of BCR-ABL1, ETV6-RUNX1, and TCF3-PBX1 fusions in ALL, respectively. Potential perturbed interactions were ranked according to the magnitude of change in gene expression for a pair of interacting partners (Supplemental Tables S1–S3).
The relatively low number of specific perturbed interactions for BCR-ABL subtype (Supplemental Table S3) is unexpected and implies that, for most pairs of interactions, 1) gene expression profiles are too different between BCR-ABL1 ALL samples, consistent with the fact that several breakpoints on chromosomes 9 (for the ABL gene) and 22 (for the BCR gene) may generate kinases with different outcome on the downstream signaling transduction molecules (Pendergast et al., 1993; Skorski et al., 1995; Ren, 2005); and 2) the common changes are not observable in BCR-ABL1 ALL samples and may have been missed. Those unidentified changes found in all subtypes could define the major networks implicated in BCR-ABL1, ETV6-RUNX1, and TCF3-PBX1 leukemia subtypes.
Although we showed that our method is useful in the identification of previously unknown mutant-specific deregulated biological processes, this strategy, like any other system biology model that predicts perturbations, presents some limitations: 1) To predict interactome network perturbations, we calculated the difference of correlation between expression profiles of two genes coding for proteins involved in a protein–protein or protein–DNA interaction. High-throughput data concerning other variables that may influence an interaction, such as mutations in coding sequences, proteins localization and translocation, protein modifications (phosphorylation, acetylation, glycosylation, etc.), and mRNA processing (transport, degradation, stability, etc.), were not included. 2) We applied our methodology to the analysis of ETV6-RUNX1 or TCF3-PBX1 chromosomal rearrangements restricted to precursor-B-cell leukemia, whereas we interrogated a mature-B-cell interactome data set (HBCI), which is an interaction network assembled from a collection of 254 B-cell gene expression profiles representing 24 distinct phenotypes of normal and diseased B cells (Lefebvre et al., 2010). To the best of our knowledge, similar cell-context interactomes for all stages of B-cell development, including precursor B cells, are not yet available. Some important interactions specific for precursor B cells may be missed and some irrelevant interactions may be included in our analysis. 3) The accuracy of our predictions depends on the technical quality of transcriptome and interactome data sets. In our study, transcriptome data sets were from published microarray hybridization data (Den Boer et al., 2009; Mullighan et al., 2009). High-throughput RNA sequencing should provide more precise measurement of gene expression levels and increase the accuracy of our predictions (Wang et al., 2009). In addition, the human B-cell interaction data were obtained by either reverse engineering of transcriptome data or literature curation of interactions. For technical reasons, both methods capture a number of false-positive and false-negative interactions and do not give a complete view of interactomes (Cusick et al., 2009; Dreze et al., 2010; Lefebvre et al., 2010; Yu et al., 2011; Tsang et al., 2014). To summarize, our strategy, like other systems biology predicting models, will improve over time as more accurate cell-specific interactome and transcriptome data are available. In all cases, biological validations are necessary to confirm perturbations of interactome networks in cancer subtypes of interest.
The identification of the MYC network as specifically deregulated after ETV6-RUNX1 fusion could not be anticipated. Of interest, we did not observe dramatic changes in MYC transcript expression levels in ETV6-RUNX1 compared with other subtype of B-ALL, and at the protein level, we show that ectopic expression of ETV6-RUNX1 does not affect MYC expression. We thus speculate that the deregulation of MYC network may be attributed to functional interplay between MYC and ETV6-RUNX1 transcriptional activities. MYC forms highly stable heterodimers with MYC interacting factor X (Max) through their respective basic helix-loop-helix leucine zipper (bHLHZ) domains, which specifically bind the E-box (5′-CACGTG-3′) DNA sequences (Nair and Burley, 2003) and recruit different cofactors for transcriptional activation or repression (Conacci-Sorrell et al., 2014; Diolaiti et al., 2014). Our analysis demonstrated that, in ETV6-RUNX1 samples, MYC/Max interaction was not affected, whereas some MYC interactions with cofactors, such as Miz-1, were perturbed (Figure 6, red vs. blue lines). Perturbation of MYC/Miz-1 interaction may suggest that the ETV6-RUNX1 fusion could preferentially target the repression function of MYC. It is possible that ETV6-RUNX1 fusion interferes with formation of MYC/Miz-1 complex and recruitment of MYC to target gene transcriptional initiators, as previously reported for the interplay between transforming growth facto-β/Smad signaling pathway and MYC/Miz-1 complex to control p15INK4b and p21WAF1 CDK inhibitors (Seoane et al., 2001, 2002, 2004). In addition, ETV6-RUNX1 may also exert its effect on the MYC network by binding to its target promoters (Supplemental Table S10), as shown here for the CDKN1A promoter (Supplemental Figure 1, C and D).
Another important result from this study is the possible involvement of the TCF3-PBX1 fusion protein in mRNA transport. RNA export is a central process in gene expression regulation and is an exciting new field in cancer biology. Although overexpression of some components of the mRNA export machinery, such as nucleoporins Nup88 and Nup214 (von Lindern et al., 1992; Xu and Powers, 2009; Kohler and Hurt, 2010), CRM1 (Noske et al., 2008), eIF4E (Borden and Culjkovic-Kraljacic, 2010), and GANP, the nuclear adapter for NXF1 (Fujimura et al., 2005), have been associated with other types of cancer, including B lymphomas, our data constitute the first report implicating TCF3-PBX1 in RNA localization and interaction with an export factor, NXF1 (Supplemental Figure S2). Similar to overexpression of eIF4E being efficiently inhibited by ribavirin in acute myelogenous leukemia (Kentsis et al., 2004), targeting TCF3-PBX1/mRNA export pathway interactions could lead to effective ALL therapies.
In conclusion, our study establishes the feasibility of predicting specific perturbations of molecular interactions based on gene expression profiles from multiple experiments and different biological conditions. Of importance, we show that integration of interactome data with differences of correlation between expression profiles could classify subtypes within the same lineage and provide specific potential targets.
MATERIALS AND METHODS
Experimental data
We downloaded from Gene Expression Omnibus (GEO) the Affymetrix HG-U133A expression data sets (GSE13425, GSE12995), comprising 190 and 175 ALL samples, respectively. These data sets contain 24 samples with BCR-ABL1 fusion, 77 with ETV6-RUNX1 fusion, 16 with TCF3-PBX1 fusion, and 248 with various other genetic subtypes (Den Boer et al., 2009; Mullighan et al., 2009)
The list of protein interactions was retrieved from the HBCI (Lefebvre, 2007, 2010), among which were 21,156 protein–protein interactions (PPIs), 41,568 protein–DNA interactions (PDIs), and 1925 transcription factor–modulator interactions (TFMIs). We also analyzed the network composed of known cellular pathways in Pathway Commons (Cerami et al., 2011) to predict affected cellular pathways. The Pathway Commons version of 27 October 2011 used in our study contains 2308 pathways collected from multiple sources (HumanCyc, Reactome, NCI-Pathways Interactions Database, Biocarta, and KEGG; Romero et al., 2005; Matthews et al., 2009; Schaefer et al., 2009; Kanehisa et al., 2012).
Prediction of disrupted interactions
Inspired by an oncogene prediction method (Mani et al., 2008), we detected changes in correlation of expression between gene pairs in different groups of patients: gene pairs whose expression correlation values show significantly different values between a test group (e.g., the ALL associated to a particular gene fusion) and a control group of samples (e.g., all other ALL samples).
First, microarray expression profiles are normalized using fRMA (McCall and Irizarry, 2011). Second, for each genotype (fusion) and each interaction in the HBCI, we computed the difference of correlations of expression profiles between a genotype of interest (exhibiting a genotype of interest) and other genotypes. We selected interactions showing significant differences of correlation for both Pearson and Spearman rank correlation coefficients. To detect interactions with significant differences of correlation, we generated 10,000 data sets permuting expression values across the whole table of the original data set. For each random set, we computed the difference of correlation in the same way as for the original data and computed the p value and corrected p value (using Benjamini–Hochberg multiple testing correction) from the distribution of all differences of correlation values. Interactions with corrected p < 0.05 for both Pearson and Spearman correlation measures are predicted as perturbed.
Difference of correlation between expression profiles
We computed Spearman's rank correlation (ρ; Best and Roberts, 1975) and Pearson's correlation (r; Pearson, 1895) using the following formulas:
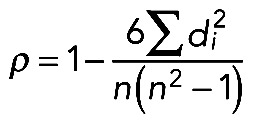 |
 |
where X = (x1,…,xn) and Y = (y1,…,yn) are vectors representing expression profiles of two genes/proteins in interaction; n is the number of samples; i = {1,…,n}; di = xi − yi represents the difference between ranks; and  and
and 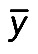 are the sample means of the X and Y vectors, respectively.
are the sample means of the X and Y vectors, respectively.
The difference of correlations (Δcor) of two genes between a genotype of interest (gi) and other genotypes (og) was computed as
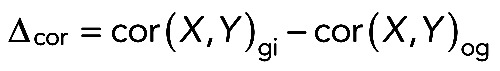 |
where cor(.,.) represents the correlation function (Spearman's rank or Pearson's correlation).
Topological analysis
The characteristic path length (cpl) of a graph G is the average length of the shortest paths between all distinct pairs of vertices in the graph (Watts and Strogatz, 1998). In a nondirected graph, the characteristic path length L(G) is computed as follows:
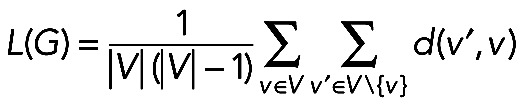 |
where V is the set of vertices and d(v, v′) is distance between vertices v and v′, that is, the length of the shortest path joining them.
The edge-betweenness centrality (ebc) or B(e) is defined as
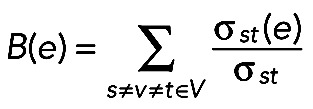 |
where σst(e) is the number of shortest paths between vertex s and t that pass through the given edge (Newman, 2010).
The edge clustering coefficient (EC) is computed as the number of triangles to which a given edge belongs, divided by the number of triangles in which the edge may possibly participate at most, given the degrees of the adjacent nodes (Wang et al., 2012):
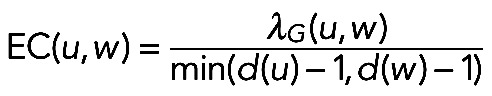 |
where λG denotes the number of triangles that include the edge (u, w) and d(u) and d(w) are degrees of u and w, respectively. The min(d(u) − 1, d(w) − 1) is the number of triangles in which the edge (u,w) may possibly participate at most.
Cell culture and transfection
HeLa and HEK293 cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, and penicillin/streptomycin. The same medium was used to culture ETV6-RUNX1 (E/R)–expressing HEK 293T clones generated as previously described (Fuka et al., 2012). The ETV6-RUNX1 (E/R)–positive leukemia cell line REH was cultured in RPMI 1640 supplemented with 10–20% FBS and antibiotics as recommended by the distributor (DSMZ, Braunschweig, Germany). Knockdown of E/R was performed as previously described (Fuka et al., 2012) using validated lentiviral vector encoding a U6 promoter-driven shRNA targeting the E/R fusion sequence. Knockdown of E/R was monitored by quantitative reverse transcription PCR using published primers and probe sets (Fuka et al., 2012), and cells with at least 50% reduction were used in ChIP and Western blot experiments.
Plasmids
Plasmids pFlag-TCF3, pFlag-PBX1, and pYFP-NXF1 were generated from the corresponding entry clones (human ORFeome 7.1) by LR recombination into pDEST1899 (Flag N-ter vector) or pDEST491 (YFP N-ter vector; gift of James L. Hartley and Dominic Esposito, Protein Expression Laboratory, Frederick National Laboratory for Cancer Research, Frederick, MD). TCF3-PBX1 (gift of David P. LeBrun, Queen's University, Kingston, Canada) and ETV6-RUNX1 (gift of Guy Leclerc, University of Miami, Miami, FL) cDNA clones were also subcloned by Gateway technology into pDEST1899 Flag N-ter vector.
Luciferase reporter assays
HEK293 cells were transfected with 1 μg of reporter plasmid (p4XAP1-luc, pkB-luc, CMV-luc, pCDKN1A-luc), different amounts of effector plasmids (pFlag-TCF3, pFlag-PBX1, pFlag-TCF3-PBX1 or pFlag-ETV6-RUNX1, pMX-MYC), and 100 ng of a control Renilla luciferase construct using polyethyleneimine (Polysciences Europe, Eppelheim, Germany) at 3 μg/μg of DNA. For CDKN1A promoter activation, cells were treated with 100 μg/ml of PMA. At 24 h posttransfection, cells were lysed and luciferase activities determined.
Chromatin immunoprecipitation
We collected 107 REH-G1, REH-C, HEK293-E/R-V5, or HEK295-V5 cells, performed DNA–protein cross-linking using 1% formaldehyde for 8 min at room temperature, and stopped the fixation by adding 125 mM glycine for 5 min at room temperature. Cells were collected and lysed using lysis buffers iL1 and iL2 according to the manufacturer's instructions (Diagenode, Liege, Belgium), and chromatin DNA was sheared by sonication for two or three runs of 10–30 cycles (depending on the cell line: two runs of 10 cycles for HEK293 and three runs of 30 cycles for REH cells), using the Bioruptor (Diagenode). DNA–protein complexes were immunoprecipitated overnight using validated specific ChIP antibodies for MYC or ETV6 proteins (Seitz et al., 2011; Torrano et al., 2011; N-262 and N-19, respectively; Santa Cruz Biotechnology, Santa Cruz, CA) and positive and negative control antibodies (histone H3 rabbit and normal rabbit immunoglobulin G). An aliquot (10%) was used for regular PCR amplification using specific primers, human RPL30 Exon 3 (7014; Cell Signaling, Danvers, MA) as positive control for histone H3 immunoprecipitation and CDKN1A-specific primers (forward, 5′-ACTGCCCCTATTTGGGAC-3′; and reverse, 5′-GATCACATACCCTGTTCA-3′). The remaining samples (10–20 ng of immunoprecipitated DNA) were used for ChIP-seq library sample preparation and subjected to HiSeq Illumina sequencing according to the manufacturer's instructions (Illumina, San Diego, CA).
The resulting reads were mapped to the human genome (GRCh 37/hg19) using BWA, version 0.6 (Li and Durbin, 2009). We used SWEMBL, version 3.3.1 (ebi.ac.uk/∼swilder/SWEMBL/), to identify regions of the genome where multiple reads align (peaks). We adjusted parameters for ChIP-seq and reference (Input) sequence relative to the number of reads in the samples, with a relative gradient of 0.002 (R parameter). The resulting peaks were submitted to GREAT, version 2.0.2 (McLean et al., 2010), to identify gene targets. We assigned each gene to a “regulatory domain” (Dostie et al., 2006; Lieberman-Aiden et al., 2009; Schoenfelder et al., 2010) of a minimum distance of 5.0 kb upstream and 1.0 kb downstream from its transcription start site. We set the extension of the regulatory domain up to 1000.0 kb in both directions. Then each DNA-binding region was associated with all genes whose regulatory domain it overlaps. Comparison between identified target gene lists and statistical analysis were performed using the R statistical package.
Immunofluorescence and confocal microscopy
HeLa cells were seeded onto coverslips in a 24-well plate and transfected with 1 μg of pFlag-TCF3-PBX1 and/or pYFP-NXF1 plasmids using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). At 24 h posttransfection, cells were washed with phosphate-buffered saline (PBS), fixed in 3.7% formaldehyde for 20 min at room temperature, permeabilized with 0.5% Triton X-100 for 15 min at room temperature, and incubated with anti-flag M2 antibody (Sigma-Aldrich, St. Louis, MO) diluted in immunofluorescence (IF) buffer (5% FBS, 0.1% Tween-20 in PBS) for 1 h at room temperature. After extensive washes, cells were incubated with Alexa 568 secondary antibody diluted in the IF buffer and for 1 h at room temperature). Where indicated, cells were also incubated with the SYTO RNASelect marker (Invitrogen) and mounted with Prolong gold Antifade reagent with 4′,6-diamidino-2-phenylindole (Invitrogen). Slides were analyzed by confocal microscopy using the Nikon A1R system (Melville, NY) and images processed with Imaris software (Bitplane, Zurich, Switzerland).
Supplementary Material
Acknowledgments
We thank Jacques van Helden (Technological Advances for Genomics and Clinics, Université de la Méditerranée, Marseille, France) for careful and critical reading of the manuscript. We thank J. L. Hartley, D. Esposito, D.P. LeBrun, G. Leclerc, and C. G. Mullighan and J. R. Downing (St. Jude Children's Research Hospital, Memphis, TN) for plasmid DNA constructs We also thank the GIGA-Interactomics, GIGA-Imaging and Flow Cytometry, and GIGA-Genotranscriptomics platforms for technical support. L.J.H., S.D., M.M., F.D., N.S., and T.J.C. are members of the Fonds National de la Recherche Scientifique. This project was supported by Fonds National de la Recherche Scientifique Televie Grant 7.4624.09 to T.J.C. and N.S. and a Fondation contre le Cancer Technical Research Platform and Heavy Equipment grant to F.D. and T.J.C.
Abbreviations used:
- ETV6
Ets transcription factor variant 6
- HBCI
human B-cell interactome
- PBX1
pre–B-cell leukemia homeobox 1
- RUNX1
runt-related transcription factor 1
- TCF3
transcription factor 3.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-06-1038) on October 1, 2014.
REFERENCES
- Agrawal M, Garg RJ, Cortes J, Quintas-Cardama A. Tyrosine kinase inhibitors: the first decade. Curr Hematol Malig Rep. 2010;5:70–80. doi: 10.1007/s11899-010-0045-y. [DOI] [PubMed] [Google Scholar]
- Andersson A, Olofsson T, Lindgren D, Nilsson B, Ritz C, Eden P, Lassen C, Rade J, Fontes M, Morse H, et al. Molecular signatures in childhood acute leukemia and their correlations to expression patterns in normal hematopoietic subpopulations. Proc Natl Acad Sci USA. 2005;102:19069–19074. doi: 10.1073/pnas.0506637102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasson P, Schwaller J, Anastasiadou E, Aster J, Gilliland DG. The expression of ETV6/CBFA2 (TEL/AML1) is not sufficient for the transformation of hematopoietic cell lines in vitro or the induction of hematologic disease in vivo. Cancer Genet Cytogenet. 2001;130:93–104. doi: 10.1016/s0165-4608(01)00518-0. [DOI] [PubMed] [Google Scholar]
- Bayly R, Chuen L, Currie RA, Hyndman BD, Casselman R, Blobel GA, LeBrun DP. E2A-PBX1 interacts directly with the KIX domain of CBP/p300 in the induction of proliferation in primary hematopoietic cells. J Biol Chem. 2004;279:55362–55371. doi: 10.1074/jbc.M408654200. [DOI] [PubMed] [Google Scholar]
- Best DJ, Roberts DE. Algorithm AS 89: the upper tail probabilities of Spearman's rho. J R Stat Soc C Appl Stat. 1975;24:377–379. [Google Scholar]
- Borden KL, Culjkovic-Kraljacic B. Ribavirin as an anti-cancer therapy: acute myeloid leukemia and beyond. Leuk Lymphoma. 2010;51:1805–1815. doi: 10.3109/10428194.2010.496506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami EG, Gross BE, Demir E, Rodchenkov I, Babur O, Anwar N, Schultz N, Bader GD, Sander C. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. 2011;39(Database issue):D685–D690. doi: 10.1093/nar/gkq1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conacci-Sorrell M, McFerrin L, Eisenman RN. An overview of MYC and its interactome. Cold Spring Harb Perspect Med. 2014;4:a014357. doi: 10.1101/cshperspect.a014357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusick ME, Yu H, Smolyar A, Venkatesan K, Carvunis AR, Simonis N, Rual JF, Borick H, Braun P, Dreze M, et al. Literature-curated protein interaction datasets. Nat Methods. 2009;6:39–46. doi: 10.1038/nmeth.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Den Boer ML, van Slegtenhorst M, De Menezes RX, Cheok MH, Buijs-Gladdines JG, Peters ST, Van Zutven LJ, Beverloo HB, Van der Spek PJ, Escherich G, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009;10:125–134. doi: 10.1016/S1470-2045(08)70339-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denis CM, Chitayat S, Plevin MJ, Wang F, Thompson P, Liu S, Spencer HL, Ikura M, LeBrun DP, Smith SP. Structural basis of CBP/p300 recruitment in leukemia induction by E2A-PBX1. Blood. 2012;120:3968–3977. doi: 10.1182/blood-2012-02-411397. [DOI] [PubMed] [Google Scholar]
- Diolaiti D, McFerrin L, Carroll PA, Eisenman RN. Functional interactions among members of the MAX and MLX transcriptional network during oncogenesis. Biochim Biophys Acta. 2014 doi: 10.1016/j.bbagrm.2014.05.016. S1874-9399(14)00122-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, Rubio ED, Krumm A, Lamb J, Nusbaum C, et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006;16:1299–1309. doi: 10.1101/gr.5571506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreze M, Monachello D, Lurin C, Cusick ME, Hill DE, Vidal M, Braun P. High-quality binary interactome mapping. Methods Enzymol. 2010;470:281–315. doi: 10.1016/S0076-6879(10)70012-4. [DOI] [PubMed] [Google Scholar]
- Fujimura S, Xing Y, Takeya M, Yamashita Y, Ohshima K, Kuwahara K, Sakaguchi N. Increased expression of germinal center-associated nuclear protein RNA-primase is associated with lymphomagenesis. Cancer Res. 2005;65:5925–5934. doi: 10.1158/0008-5472.CAN-04-3259. [DOI] [PubMed] [Google Scholar]
- Fuka G, Kantner HP, Grausenburger R, Inthal A, Bauer E, Krapf G, Kaindl U, Kauer M, Dworzak MN, Stoiber D, et al. Silencing of ETV6/RUNX1 abrogates PI3K/AKT/mTOR signaling and impairs reconstitution of leukemia in xenografts. Leukemia. 2012;26:927–933. doi: 10.1038/leu.2011.322. [DOI] [PubMed] [Google Scholar]
- Fuka G, Kauer M, Kofler R, Haas OA, Panzer-Grumayer R. The leukemia-specific fusion gene ETV6/RUNX1 perturbs distinct key biological functions primarily by gene repression. PLoS One. 2011;6:e26348. doi: 10.1371/journal.pone.0026348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nat Rev Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandemer V, Rio AG, de Tayrac M, Sibut V, Mottier S, Ly Sunnaram B, Henry C, Monnier A, Berthou C, Le Gall E, et al. Five distinct biological processes and 14 differentially expressed genes characterize TEL/AML1-positive leukemia. BMC Genomics. 2007;8:385. doi: 10.1186/1471-2164-8-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazdar AF. Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene. 2009;28(Suppl 1):S24–S31. doi: 10.1038/onc.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge H, Liu Z, Church GM, Vidal M. Correlation between transcriptome and interactome mapping data from Saccharomyces cerevisiae. Nat Genet. 2001;29:482–486. doi: 10.1038/ng776. [DOI] [PubMed] [Google Scholar]
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- Heinemeyer T, Wingender E, Reuter I, Hermjakob H, Kel AE, Kel OV, Ignatieva EV, Ananko EA, Podkolodnaya OA, Kolpakov FA, et al. Databases on transcriptional regulation: TRANSFAC, TRRD and COMPEL. Nucleic Acids Res. 1998;26:362–367. doi: 10.1093/nar/26.1.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutten S, Kehlenbach RH. CRM1-mediated nuclear export: to the pore and beyond. Trends Cell Biol. 2007;17:193–201. doi: 10.1016/j.tcb.2007.02.003. [DOI] [PubMed] [Google Scholar]
- Hyndman BD, Thompson P, Bayly R, Cote GP, LeBrun DP. E2A proteins enhance the histone acetyltransferase activity of the transcriptional co-activators CBP and p300. Biochim Biophys Acta. 2012;1819:446–453. doi: 10.1016/j.bbagrm.2012.02.009. [DOI] [PubMed] [Google Scholar]
- Kaindl U, Morak M, Portsmouth C, Mecklenbrauker A, Kauer M, Zeginigg M, Attarbaschi A, Haas OA, Panzer-Grumayer R. Blocking ETV6/RUNX1-induced MDM2 overexpression by Nutlin-3 reactivates p53 signaling in childhood leukemia. Leukemia. 2014;28:600–608. doi: 10.1038/leu.2013.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40(Database issue):D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaulfuss S, Seemann H, Kampe R, Meyer J, Dressel R, Konig B, Scharf JG, Burfeind P. Blockade of the PDGFR family together with SRC leads to diminished proliferation of colorectal cancer cells. Oncotarget. 2013;4:1037–1049. doi: 10.18632/oncotarget.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kentsis A, Topisirovic I, Culjkovic B, Shao L, Borden KL. Ribavirin suppresses eIF4E-mediated oncogenic transformation by physical mimicry of the 7-methyl guanosine mRNA cap. Proc Natl Acad Sci USA. 2004;101:18105–18110. doi: 10.1073/pnas.0406927102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler A, Hurt E. Gene regulation by nucleoporins and links to cancer. Mol Cell. 2010;38:6–15. doi: 10.1016/j.molcel.2010.01.040. [DOI] [PubMed] [Google Scholar]
- Krapf G, Kaindl U, Kilbey A, Fuka G, Inthal A, Joas R, Mann G, Neil JC, Haas OA, Panzer-Grumayer ER. ETV6/RUNX1 abrogates mitotic checkpoint function and targets its key player MAD2L1. Oncogene. 2010;29:3307–3312. doi: 10.1038/onc.2010.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBrun DP. E2A basic helix-loop-helix transcription factors in human leukemia. Front Biosci. 2003;8:s206–s222. doi: 10.2741/1030. [DOI] [PubMed] [Google Scholar]
- Lefebvre C, Lim WK, Basso K, Dalla Favera R, Califano A. A context-specific network of protein-DNA and protein-protein interactions reveals new regulatory motifs in human B cells. Lecture Notes Comput Sci. 2007;4532:42–56. [Google Scholar]
- Lefebvre C, Rajbhandari P, Alvarez MJ, Bandaru P, Lim WK, Sato M, Wang K, Sumazin P, Kustagi M, Bisikirska BC, et al. A human B-cell interactome identifies MYB and FOXM1 as master regulators of proliferation in germinal centers. Mol Syst Biol. 2010;6:377. doi: 10.1038/msb.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Zhang W, Wu M, Zhu S, Gao C, Sun L, Zhang R, Qiao N, Xue H, Hu Y, et al. Gene expression-based classification and regulatory networks of pediatric acute lymphoblastic leukemia. Blood. 2009;114:4486–4493. doi: 10.1182/blood-2009-04-218123. [DOI] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- Maher CA, Kumar-Sinha C, Cao X, Kalyana-Sundaram S, Han B, Jing X, Sam L, Barrette T, Palanisamy N, Chinnaiyan AM. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458:97–101. doi: 10.1038/nature07638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makrynikola V, Bianchi A, Bradstock K, Gottlieb D, Hewson J. Migration of acute lymphoblastic leukemia cells into human bone marrow stroma. Leukemia. 1994;8:1734–1743. [PubMed] [Google Scholar]
- Mangolini M, de Boer J, Walf-Vorderwulbecke V, Pieters R, den Boer ML, Williams O. STAT3 mediates oncogenic addiction to TEL-AML1 in t(12;21) acute lymphoblastic leukemia. Blood. 2013;122:542–549. doi: 10.1182/blood-2012-11-465252. [DOI] [PubMed] [Google Scholar]
- Mani KM, Lefebvre C, Wang K, Lim WK, Basso K, Dalla-Favera R, Califano A. A systems biology approach to prediction of oncogenes and molecular perturbation targets in B-cell lymphomas. Mol Syst Biol. 2008;4:169. doi: 10.1038/msb.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews L, Gopinath G, Gillespie M, Caudy M, Croft D, de Bono B, Garapati P, Hemish J, Hermjakob H, Jassal B, et al. Reactome knowledgebase of human biological pathways and processes. Nucleic Acids Res. 2009;37(Database issue):D619–D622. doi: 10.1093/nar/gkn863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall MN, Irizarry RA. Thawing frozen robust multi-array analysis (fRMA) BMC Bioinformatics. 2011;12:369. doi: 10.1186/1471-2105-12-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitelman F, Johansson B, Mertens F. Fusion genes and rearranged genes as a linear function of chromosome aberrations in cancer. Nat Genet. 2004;36:331–334. doi: 10.1038/ng1335. [DOI] [PubMed] [Google Scholar]
- Mullighan CG, Su X, Zhang J, Radtke I, Phillips LA, Miller CB, Ma J, Liu W, Cheng C, Schulman BA, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360:470–480. doi: 10.1056/NEJMoa0808253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair SK, Burley SK. X-ray structures of Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell. 2003;112:193–205. doi: 10.1016/s0092-8674(02)01284-9. [DOI] [PubMed] [Google Scholar]
- Newman M. Networks: An Introduction, Oxford: Oxford University Press. 2010 [Google Scholar]
- Noske A, Weichert W, Niesporek S, Roske A, Buckendahl AC, Koch I, Sehouli J, Dietel M, Denkert C. Expression of the nuclear export protein chromosomal region maintenance/exportin 1/Xpo1 is a prognostic factor in human ovarian cancer. Cancer. 2008;112:1733–1743. doi: 10.1002/cncr.23354. [DOI] [PubMed] [Google Scholar]
- Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84:321–330. doi: 10.1016/s0092-8674(00)80986-1. [DOI] [PubMed] [Google Scholar]
- Ott CJ, Kopp N, Bird L, Paranal RM, Qi J, Bowman T, Rodig SJ, Kung AL, Bradner JE, Weinstock DM. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood. 2012;120:2843–2852. doi: 10.1182/blood-2012-02-413021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson K. Mathematical contributions to the theory of evolution—III. Regression, heredity, panmixia. Philos Trans R Soc Lond. 1895;187:253–318. [Google Scholar]
- Pendergast AM, Quilliam LA, Cripe LD, Bassing CH, Dai Z, Li N, Batzer A, Rabun KM, Der CJ, Schlessinger J, et al. BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor protein. Cell. 1993;75:175–185. [PubMed] [Google Scholar]
- Quintas-Cardama A, Cortes J. Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood. 2009;113:1619–1630. doi: 10.1182/blood-2008-03-144790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer. 2005;5:172–183. doi: 10.1038/nrc1567. [DOI] [PubMed] [Google Scholar]
- Romero P, Wagg J, Green ML, Kaiser D, Krummenacker M, Karp PD. Computational prediction of human metabolic pathways from the complete human genome. Genome Biol. 2005;6:R2. doi: 10.1186/gb-2004-6-1-r2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel S, Twizere JC, Beifuss KK, Bernstein LR. Nucleolin binds specifically to an AP-1 DNA sequence and represses AP1-dependent transactivation of the matrix metalloproteinase-13 gene. Mol Carcinog. 2008;47:34–46. doi: 10.1002/mc.20358. [DOI] [PubMed] [Google Scholar]
- Schaefer CF, Anthony K, Krupa S, Buchoff J, Day M, Hannay T, Buetow KH. PID: the Pathway Interaction Database. Nucleic Acids Res. 2009;37(Database issue):D674–D679. doi: 10.1093/nar/gkn653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfelder S, Sexton T, Chakalova L, Cope NF, Horton A, Andrews S, Kurukuti S, Mitchell JA, Umlauf D, Dimitrova DS, et al. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat Genet. 2010;42:53–61. doi: 10.1038/ng.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz V, Butzhammer P, Hirsch B, Hecht J, Gutgemann I, Ehlers A, Lenze D, Oker E, Sommerfeld A, von der Wall E, et al. Deep sequencing of MYC DNA-binding sites in Burkitt lymphoma. PLoS One. 2011;6:e26837. doi: 10.1371/journal.pone.0026837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoane J. p21(WAF1/CIP1) at the switch between the anti-oncogenic and oncogenic faces of TGFbeta. Cancer Biol Ther. 2004;3:226–227. doi: 10.4161/cbt.3.2.717. [DOI] [PubMed] [Google Scholar]
- Seoane J, Le HV, Massague J. Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature. 2002;419:729–734. doi: 10.1038/nature01119. [DOI] [PubMed] [Google Scholar]
- Seoane J, Pouponnot C, Staller P, Schader M, Eilers M, Massague J. TGFbeta influences Myc, Miz-1 and Smad to control the CDK inhibitor p15INK4b. Nat Cell Biol. 2001;3:400–408. doi: 10.1038/35070086. [DOI] [PubMed] [Google Scholar]
- Seto M. Genomic profiles in B cell lymphoma. Int J Hematol. 2010;92:238–245. doi: 10.1007/s12185-010-0662-1. [DOI] [PubMed] [Google Scholar]
- Siddiqui N, Borden KL. mRNA export and cancer. Wiley Interdiscip Rev RNA. 2012;3:13–25. doi: 10.1002/wrna.101. [DOI] [PubMed] [Google Scholar]
- Skorski T, Kanakaraj P, Nieborowska-Skorska M, Ratajczak MZ, Wen SC, Zon G, Gewirtz AM, Perussia B, Calabretta B. Phosphatidylinositol-3 kinase activity is regulated by BCR/ABL and is required for the growth of Philadelphia chromosome-positive cells. Blood. 1995;86:726–736. [PubMed] [Google Scholar]
- Tijchon E, Havinga J, van Leeuwen FN, Scheijen B. B-lineage transcription factors and cooperating gene lesions required for leukemia development. Leukemia. 2013;27:541–552. doi: 10.1038/leu.2012.293. [DOI] [PubMed] [Google Scholar]
- Torrano V, Procter J, Cardus P, Greaves M, Ford AM. ETV6-RUNX1 promotes survival of early B lineage progenitor cells via a dysregulated erythropoietin receptor. Blood. 2011;118:4910–4918. doi: 10.1182/blood-2011-05-354266. [DOI] [PubMed] [Google Scholar]
- Tsang JS, Schwartzberg PL, Kotliarov Y, Biancotto A, Xie Z, Germain RN, Wang E, Olnes MJ, Narayanan M, Golding H, et al. Global analyses of human immune variation reveal baseline predictors of postvaccination responses. Cell. 2014;157:499–513. doi: 10.1016/j.cell.2014.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Lindern M, Fornerod M, van Baal S, Jaegle M, de Wit T, Buijs A, Grosveld G. The translocation (6;9), associated with a specific subtype of acute myeloid leukemia, results in the fusion of two genes, dek and can, and the expression of a chimeric, leukemia-specific dek-can mRNA. Mol Cell Biol. 1992;12:1687–1697. doi: 10.1128/mcb.12.4.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HB, Liu GH, Zhang H, Xing S, Hu LJ, Zhao WF, Xie B, Li MZ, Zeng BH, Li Y, et al. Sp1 and c-Myc regulate transcription of BMI1 in nasopharyngeal carcinoma. FEBS J. 2013;280:2929–2944. doi: 10.1111/febs.12299. [DOI] [PubMed] [Google Scholar]
- Wang J, Li M, Wang H, Pan Y. Identification of essential proteins based on edge clustering coefficient. IEEE/ACM Trans Comput Biol Bioinform. 2012;9:1070–1080. doi: 10.1109/TCBB.2011.147. [DOI] [PubMed] [Google Scholar]
- Wang X, Yan Z, Fulciniti M, Li Y, Gkotzamanidou M, Amin SB, Shah PK, Zhang Y, Munshi NC, Li C. Transcription factor-pathway coexpression analysis reveals cooperation between SP1 and ESR1 on dysregulating cell cycle arrest in non-hyperdiploid multiple myeloma. Leukemia. 2014;28:894–903. doi: 10.1038/leu.2013.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts DJ, Strogatz SH. Collective dynamics of “small-world” networks. Nature. 1998;393:440–442. doi: 10.1038/30918. [DOI] [PubMed] [Google Scholar]
- Wingender E, Schoeps T, Donitz J. TFClass: an expandable hierarchical classification of human transcription factors. Nucleic Acids Res. 2013;41(Database issue):D165–D170. doi: 10.1093/nar/gks1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wotton S, Terry A, Kilbey A, Jenkins A, Herzyk P, Cameron E, Neil JC. Gene array analysis reveals a common Runx transcriptional programme controlling cell adhesion and survival. Oncogene. 2008;27:5856–5866. doi: 10.1038/onc.2008.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Cetinkaya C, Munoz-Alonso MJ, von der Lehr N, Bahram F, Beuger V, Eilers M, Leon J, Larsson LG. Myc represses differentiation-induced p21CIP1 expression via Miz-1-dependent interaction with the p21 core promoter. Oncogene. 2003;22:351–360. doi: 10.1038/sj.onc.1206145. [DOI] [PubMed] [Google Scholar]
- Xu S, Powers MA. Nuclear pore proteins and cancer. Semin Cell Dev Biol. 2009;20:620–630. doi: 10.1016/j.semcdb.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Tardivo L, Tam S, Weiner E, Gebreab F, Fan C, Svrzikapa N, Hirozane-Kishikawa T, Rietman E, Yang X, et al. Next-generation sequencing to generate interactome datasets. Nat Methods. 2011;8:478–480. doi: 10.1038/nmeth.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng YX, el-Deiry WS. Regulation of p21WAF1/CIP1 expression by p53-independent pathways. Oncogene. 1996;12:1557–1564. [PubMed] [Google Scholar]
- Zhang J, Lou X, Yang S, He S, Yang L, Liu M, Zhu H, Shan Q, Su S, Zhan Q, et al. BAG2 is a target of the c-Myc gene and is involved in cellular senescence via the p21(CIP1) pathway. Cancer Lett. 2012;318:34–41. doi: 10.1016/j.canlet.2011.11.033. [DOI] [PubMed] [Google Scholar]
- Zhong Q, Simonis N, Li QR, Charloteaux B, Heuze F, Klitgord N, Tam S, Yu H, Venkatesan K, Mou D, et al. Edgetic perturbation models of human inherited disorders. Mol Syst Biol. 2009;5:321. doi: 10.1038/msb.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou MH, Gao L, Jing Y, Xu YY, Ding Y, Wang N, Wang W, Li MY, Han XP, Sun JZ, et al. Detection of ETV6 gene rearrangements in adult acute lymphoblastic leukemia. Ann Hematol. 2012;91:1235–1243. doi: 10.1007/s00277-012-1431-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.