

Abstract
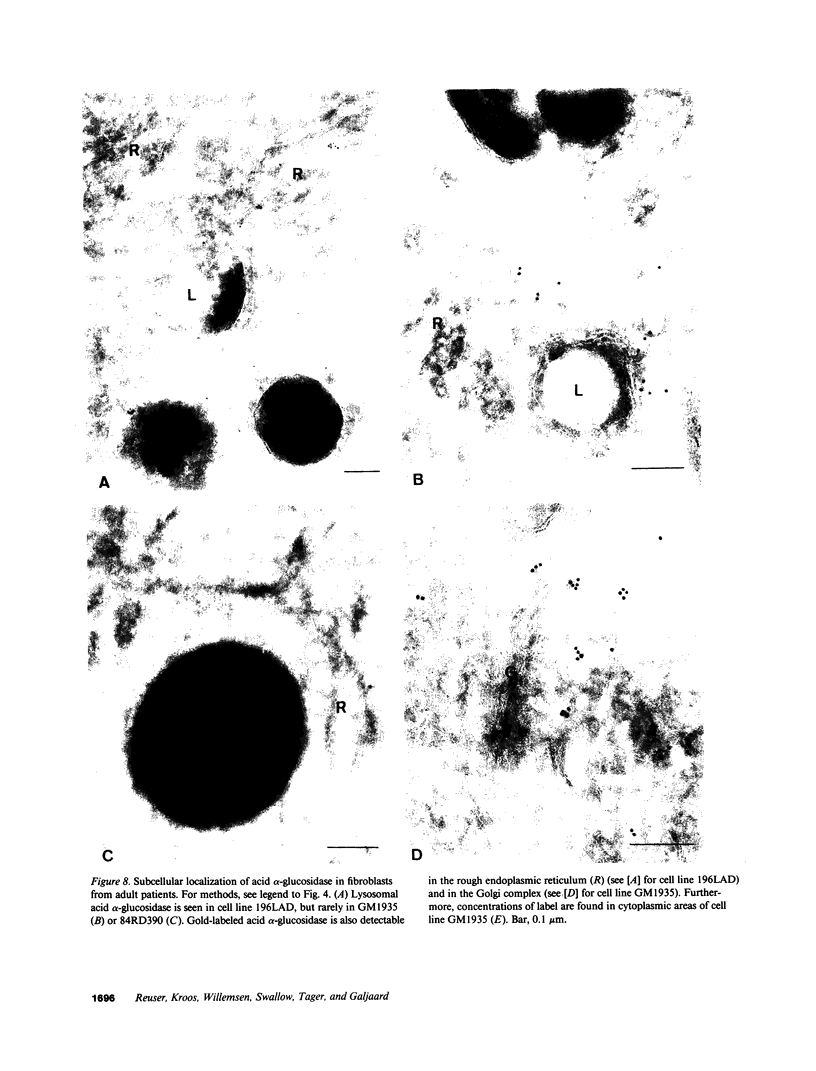
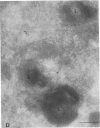
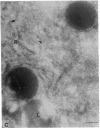
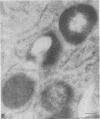
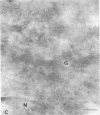
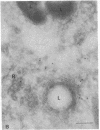
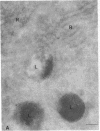
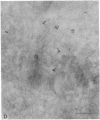
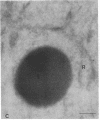
The molecular basis of clinical diversity in glycogenosis type II (Pompe's disease) was investigated by comparing the nature of acid alpha-glucosidase deficiency in cultured fibroblasts from 30 patients. Biosynthetic forms of acid alpha-glucosidase with different molecular mass were separated electrophoretically and identified by immunoblotting. Immuno-electron microscopy was employed to determine the intracellular localization of mutant enzyme. Our studies illustrate that maturation of acid alpha-glucosidase is associated with transport to the lysosomes. Deficiency of catalytically active mature enzyme in lysosomes is common to all clinical phenotypes but, in the majority of cases, is more profound in early onset than in late onset forms of the disease. Thus, the results suggest that the clinical course of glycogenosis type II is primarily determined by the amount of functional acid alpha-glucosidase. The role of secondary factors can, however, not be excluded because three adult patients were identified with very low activity and little enzyme in the lysosomes.
Full text
PDF


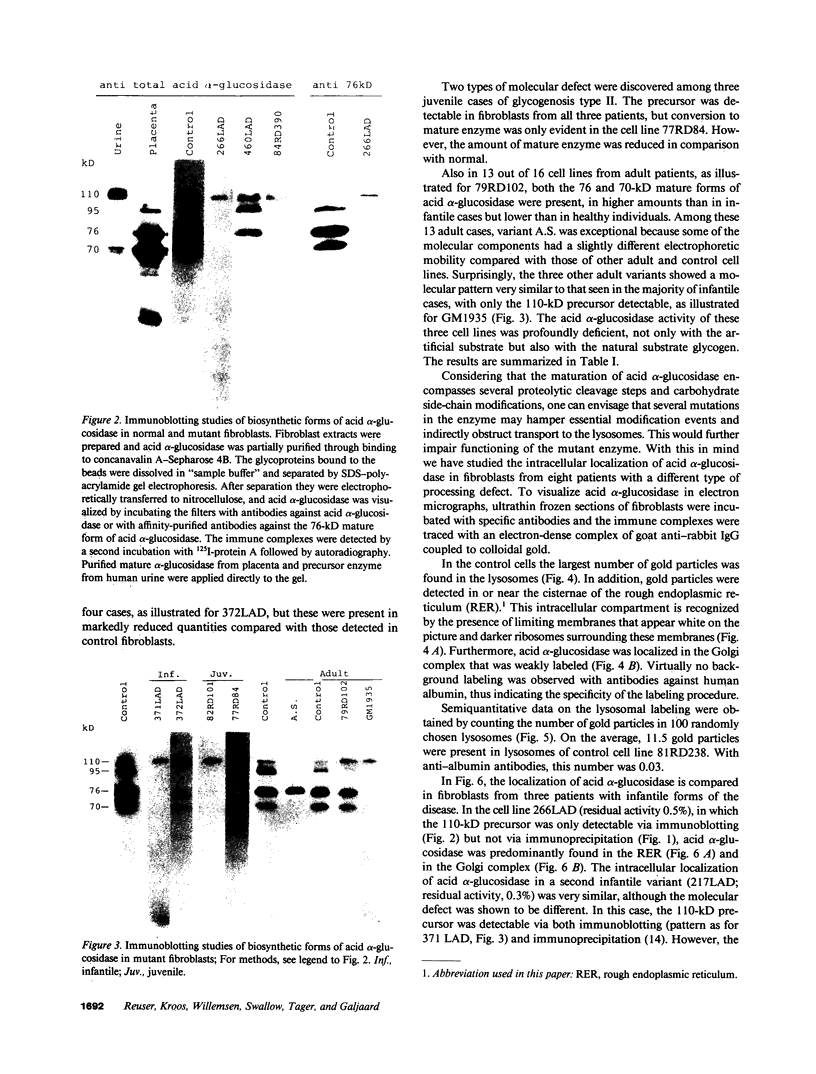
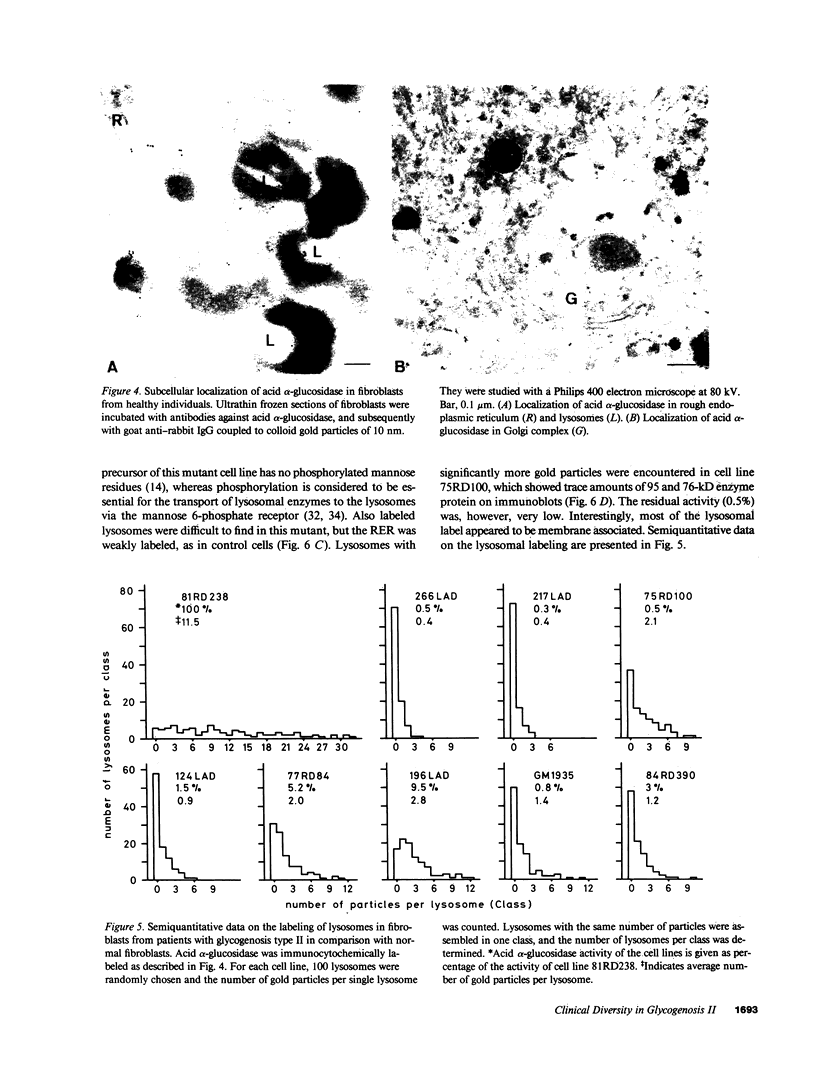
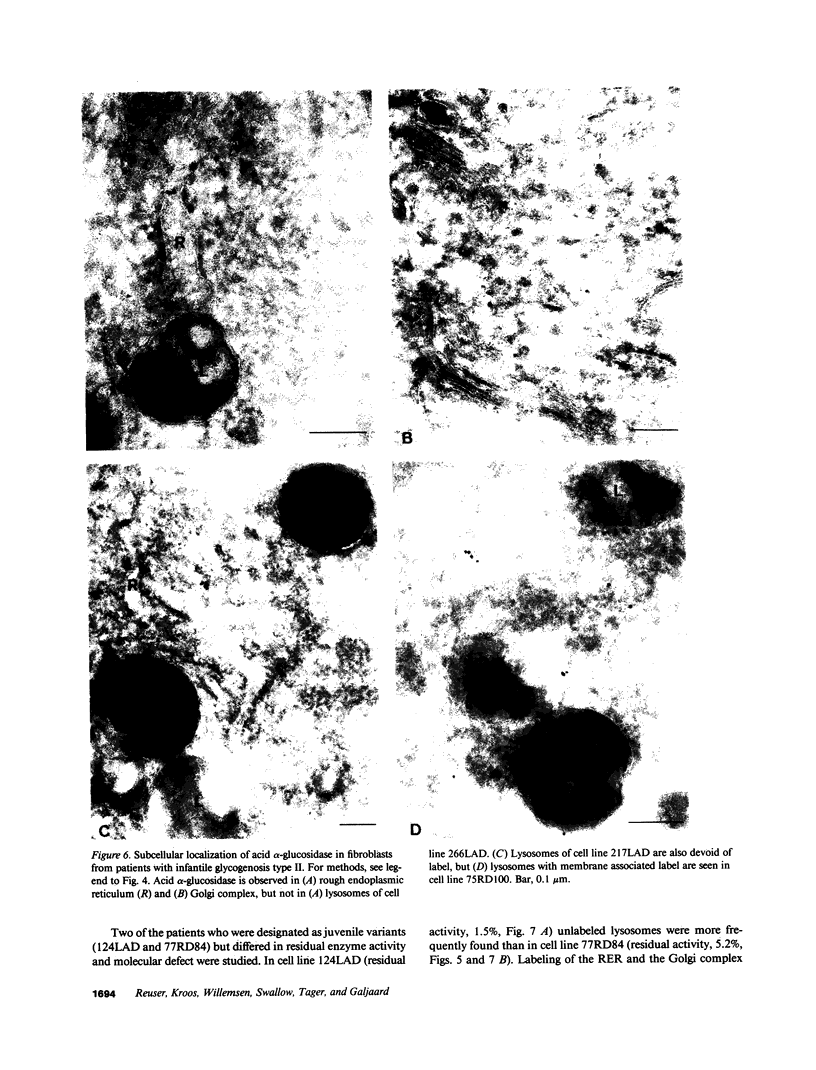
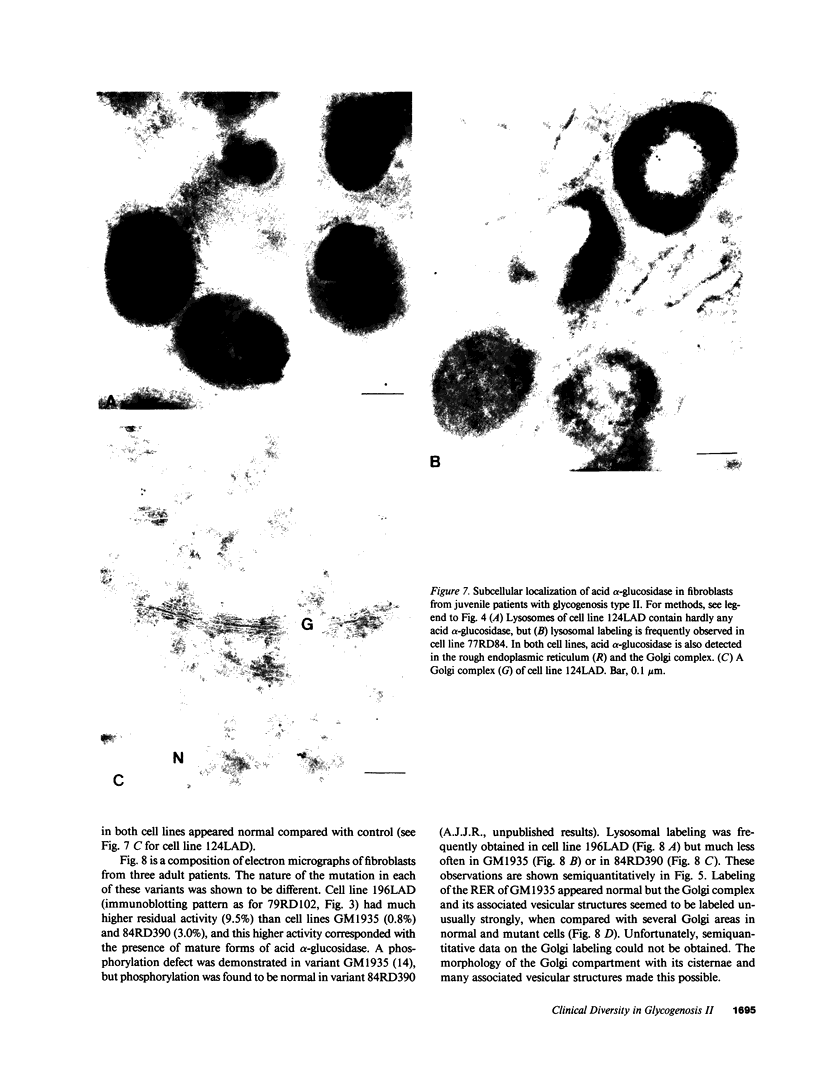



Images in this article

Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Angelini C., Engel A. G. Comparative study of acid maltase deficiency. Biochemical differences between infantile, childhood, and adult types. Arch Neurol. 1972 Apr;26(4):344–349. doi: 10.1001/archneur.1972.00490100074007. [DOI] [PubMed] [Google Scholar]
- Beratis N. G., LaBadie G. U., Hirschhorn K. Acid alpha-glucosidase: kinetic and immunologic properties of enzyme variants in health and disease. Isozymes Curr Top Biol Med Res. 1983;11:25–36. [PubMed] [Google Scholar]
- Beratis N. G., LaBadie G. U., Hirschhorn K. Characterization of the molecular defect in infantile and adult acid alpha-glucosidase deficiency fibroblasts. J Clin Invest. 1978 Dec;62(6):1264–1274. doi: 10.1172/JCI109247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beratis N. G., LaBadie G. U., Hirschhorn K. Genetic heterogeneity in acid alpha-glucosidase deficiency. Am J Hum Genet. 1983 Jan;35(1):21–33. [PMC free article] [PubMed] [Google Scholar]
- DI SANT'AGNESE P. A., ANDERSEN D. H., MASON H. H., BAUMAN W. A. Glycogen storage disease of the heart. I. Report of 2 cases in siblings with chemical and pathologic studies. Pediatrics. 1950 Sep;6(3):402–424. [PubMed] [Google Scholar]
- Engel A. G. Acid maltase deficiency in adults: studies in four cases of a syndrome which may mimic muscular dystrophy or other myopathies. Brain. 1970;93(3):599–616. doi: 10.1093/brain/93.3.599. [DOI] [PubMed] [Google Scholar]
- Hasilik A., Neufeld E. F. Biosynthesis of lysosomal enzymes in fibroblasts. Phosphorylation of mannose residues. J Biol Chem. 1980 May 25;255(10):4946–4950. [PubMed] [Google Scholar]
- Hasilik A., Neufeld E. F. Biosynthesis of lysosomal enzymes in fibroblasts. Synthesis as precursors of higher molecular weight. J Biol Chem. 1980 May 25;255(10):4937–4945. [PubMed] [Google Scholar]
- Hasilik A., Waheed A., von Figura K. Enzymatic phosphorylation of lysosomal enzymes in the presence of UDP-N-acetylglucosamine. Absence of the activity in I-cell fibroblasts. Biochem Biophys Res Commun. 1981 Feb 12;98(3):761–767. doi: 10.1016/0006-291x(81)91177-3. [DOI] [PubMed] [Google Scholar]
- LOWRY O. H., ROSEBROUGH N. J., FARR A. L., RANDALL R. J. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951 Nov;193(1):265–275. [PubMed] [Google Scholar]
- Laemmli U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970 Aug 15;227(5259):680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Matsuishi T., Yoshino M., Terasawa K., Nonaka I. Childhood acid maltase deficiency. A clinical, biochemical, and morphologic study of three patients. Arch Neurol. 1984 Jan;41(1):47–52. doi: 10.1001/archneur.1984.04050130053022. [DOI] [PubMed] [Google Scholar]
- Mehler M., DiMauro S. Residual acid maltase activity in late-onset acid maltase deficiency. Neurology. 1977 Feb;27(2):178–184. doi: 10.1212/wnl.27.2.178. [DOI] [PubMed] [Google Scholar]
- Ninomiya N., Matsuda I., Matsuoka T., Iwamasa T., Nonaka I. Demonstration of acid alpha-glucosidase in different types of Pompe disease by use of an immunochemical method. J Neurol Sci. 1984 Nov-Dec;66(2-3):129–139. doi: 10.1016/0022-510x(84)90001-7. [DOI] [PubMed] [Google Scholar]
- Oude Elferink R. P., Brouwer-Kelder E. M., Surya I., Strijland A., Kroos M., Reuser A. J., Tager J. M. Isolation and characterization of a precursor form of lysosomal alpha-glucosidase from human urine. Eur J Biochem. 1984 Mar 15;139(3):489–495. doi: 10.1111/j.1432-1033.1984.tb08032.x. [DOI] [PubMed] [Google Scholar]
- Oude Elferink R. P., Van Doorn-Van Wakeren J., Strijland A., Reuser A. J., Tager J. M. Biosynthesis and intracellular transport of alpha-glucosidase and cathepsin D in normal and mutant human fibroblasts. Eur J Biochem. 1985 Nov 15;153(1):55–63. doi: 10.1111/j.1432-1033.1985.tb09266.x. [DOI] [PubMed] [Google Scholar]
- Reitman M. L., Varki A., Kornfeld S. Fibroblasts from patients with I-cell disease and pseudo-Hurler polydystrophy are deficient in uridine 5'-diphosphate-N-acetylglucosamine: glycoprotein N-acetylglucosaminylphosphotransferase activity. J Clin Invest. 1981 May;67(5):1574–1579. doi: 10.1172/JCI110189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuser A. J., Koster J. F., Hoogeveen A., Galjaard H. Biochemical, immunological, and cell genetic studies in glycogenosis type II. Am J Hum Genet. 1978 Mar;30(2):132–143. [PMC free article] [PubMed] [Google Scholar]
- Reuser A. J., Kroos M. Adult forms of glycogenosis type II. A defect in an early stage of acid alpha-glucosidase realization. FEBS Lett. 1982 Sep 20;146(2):361–364. doi: 10.1016/0014-5793(82)80953-8. [DOI] [PubMed] [Google Scholar]
- Reuser A. J., Kroos M., Oude Elferink R. P., Tager J. M. Defects in synthesis, phosphorylation, and maturation of acid alpha-glucosidase in glycogenosis type II. J Biol Chem. 1985 Jul 15;260(14):8336–8341. [PubMed] [Google Scholar]
- Schram A. W., Brouwer-Kelder B., Donker-Koopman W. E., Loonen C., Hamers M. N., Tager J. M. Use of immobilized antibodies in investigating acid alpha-glucosidase in urine in relation to Pompe's disease. Biochim Biophys Acta. 1979 Apr 12;567(2):370–383. doi: 10.1016/0005-2744(79)90123-2. [DOI] [PubMed] [Google Scholar]
- Seiler D., Kelleter R., Kölmel H. W., Heene R. Alpha-1,4-glucosidase activity in leucocytes and lymphocytes of 2 adult patients with glycogen-storage disease type II, (Pompe's disease). Experientia. 1973 Aug 15;29(8):972–973. doi: 10.1007/BF01930409. [DOI] [PubMed] [Google Scholar]
- Shanske S., Bresolin N., DiMauro S. Multiple neutral maltase activities in normal and acid maltase-deficient human muscle. Exp Neurol. 1984 Jun;84(3):565–578. doi: 10.1016/0014-4886(84)90204-8. [DOI] [PubMed] [Google Scholar]
- Smith D. E., Fisher P. A. Identification, developmental regulation, and response to heat shock of two antigenically related forms of a major nuclear envelope protein in Drosophila embryos: application of an improved method for affinity purification of antibodies using polypeptides immobilized on nitrocellulose blots. J Cell Biol. 1984 Jul;99(1 Pt 1):20–28. doi: 10.1083/jcb.99.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J., Zellweger H., Afifi A. K. Muscular form of glycogenosis, type II (Pompe). Neurology. 1967 Jun;17(6):537–549. doi: 10.1212/wnl.17.6.537. [DOI] [PubMed] [Google Scholar]
- Steckel F., Gieselmann V., Waheed A., Hasilik A., von Figura K., Oude Elferink R., Kalsbeek R., Tager J. M. Biosynthesis of acid alpha-glucosidase in late-onset forms of glycogenosis type II (Pompe's disease). FEBS Lett. 1982 Dec 13;150(1):69–76. doi: 10.1016/0014-5793(82)81306-9. [DOI] [PubMed] [Google Scholar]
- Swaiman K. F., Kennedy W. R., Sauls H. S. Late infantile acid maltase deficiency. Arch Neurol. 1968 Jun;18(6):642–648. doi: 10.1001/archneur.1968.00470360064006. [DOI] [PubMed] [Google Scholar]
- Towbin H., Staehelin T., Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979 Sep;76(9):4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trend P. S., Wiles C. M., Spencer G. T., Morgan-Hughes J. A., Lake B. D., Patrick A. D. Acid maltase deficiency in adults. Diagnosis and management in five cases. Brain. 1985 Dec;108(Pt 4):845–860. doi: 10.1093/brain/108.4.845. [DOI] [PubMed] [Google Scholar]
- van Dongen J. M., Willemsen R., Ginns E. I., Sips H. J., Tager J. M., Barranger J. A., Reuser A. J. The subcellular localization of soluble and membrane-bound lysosomal enzymes in I-cell fibroblasts: a comparative immunocytochemical study. Eur J Cell Biol. 1985 Nov;39(1):179–189. [PubMed] [Google Scholar]