

Abstract
Asunaprevir (ASV), an isoquinoline-based competitive inhibitor targeting the hepatitis C virus (HCV) NS3/4A protease, is very potent in vivo. However, the potency is significantly compromised by the drug resistance mutations R155K and D168A. In this study three crystal structures of ASV and an analogue were determined to analyze the structural basis of drug resistance susceptibility. These structures revealed that ASV makes extensive contacts with Arg155 outside the substrate envelope. Arg155 in turn is stabilized by Asp168, and thus when either residue is mutated, the enzyme’s interaction with ASV’s P2* isoquinoline is disrupted. Adding a P1–P3 macrocycle to ASV enhances the inhibitor’s resistance barrier, likely due to poising the inhibitor to its bound conformation. Macrocyclic inhibitors with P2* extension moieties avoiding interaction with the protease S2 residues including Arg155 must be chosen for future design of more robust protease inhibitors.
The hepatitis C virus (HCV) is the causal agent of viral hepatitis, cirrhosis, and hepatocellular carcinoma. In the United States, HCV infects an estimated 3.2 million individuals with 17 thousand new cases annually. Globally, the HCV pandemic is a true public health problem and touches all corners of the world, infecting an estimated 170 million people.1,2
While most of the steps in the viral lifecycle are currently targeted in antiviral therapy, NS3/4A protease is a key target especially as combination therapy becomes the paradigm in patient therapy.3 The NS3/4A protease is a bifunctional enzyme consisting of an N-terminal serine protease domain and a C-terminal domain that is a member of the DExH/D-box helicase superfamily II with NTPase, nucleic acid binding, and helicase unwinding activities. The protease is responsible for processing the viral polyprotein and cleaving host factors involved in the immune response.4−6 Thus, inhibiting NS3/4A serves a dual purpose by preventing viral maturation and restoring the immune response.
There are currently three protease inhibitors (PIs) approved by the FDA (boceprevir, telaprevir, and most recently simeprevir) and several in advanced clinical trials, which are of a variety of chemical classes. Boceprevir and telaprevir are linear ketoamide compounds that form a reversible, covalent bond with the catalytic serine of NS3/4A protease.7,8 Noncovalent inhibitors include both linear (asunaprevir (ASV),9 BI 20133510) and macrocyclic compounds, containing either a P1–P3 (danoprevir,11 simeprevir12) or a P2–P4 (vaniprevir,13 MK-517214) macrocycle. However, despite often having nanomolar potency against the wildtype (WT) enzyme, drug-resistant mutants rapidly emerge.15 With the current therapy handicapped by drug resistance, suboptimal pharmacokinetics, and a lack of cross-genotype activity, the need for robust PIs with high barriers to resistance is paramount. Drug resistance in HCV NS3/4A is a complex interplay of molecular events whereby a change in the protease results in a decrease in inhibitor potency while retaining substrate processing and viral maturation. Most HCV PIs have a common peptidomimetic P1–P4 scaffold but differ in their P2* moiety. We have previously shown that the nature of this P2* extension moiety, which often protrudes from the substrate envelope,16 accounts for much of resistance conferred by single-site mutations at residues Arg155, Ala156, and Asp168. In addition, we have characterized how resistance susceptibility and potency of the NS3/4A inhibitors are dependent on both the P2* extension and the macrocyclization.17
ASV, a potent linear PI, is currently under development by Bristol-Myers Squibb and in phase III clinical trials. ASV is chemically characterized by a P1′ acylsulfonamide-linked cyclopropyl moiety, P2* isoquinoline, and a P1–P4 peptidomimetic backbone shared by most PIs.18 ASV has promising pharmacokinetics and as monotherapy lowers viremia by 2.8 to 3.5 log10 within 48 h of treatment.19 However, resistance challenges ASV due to the rapid evolution of the virus as relapse or viral breakthrough occurs mid-therapy. R155K, D168G, and I170T conferred low to moderate in genotype 1a and mutations at D168 conferred high resistance (16- to 280-fold) levels in genotype 1b.18,19 Thus, ASV needs to be supplemented with Peg-IFNα/RBV or another direct-acting antiviral (DAA).20 Consequently, understanding ASV’s binding mode in the WT and multi-drug resistant (MDR) variants of NS3/4A translates into unraveling the molecular basis for isoquinoline inhibitors’ drug resistance profile and sets the stage for improved structure-based drug design efforts.
In our previous study, we studied the impact of drug resistance mutations on the potency of ASV and ASV’s P1–P3 macrocyclic analogue (ASVmc)17 to WT, R155K, R155K/V36M, A156T, and D168A variants in both enzymatic studies and viral replicon assays. Both compounds had extremely good potency in replicon assays with 0.9 and 0.23 nM EC50’s but lost 70/35- (R155K) and 341/84- (D168A) fold in affinity for ASV and ASVmc, respectively. In this study, we report the crystal structures of WT-ASV, R155K-ASV, and WT-ASVmc complexes. ASV makes extensive intermolecular contacts with the catalytic residues His57 and Asp81, and the P2* isoquinoline forms an aromatic stacking on Arg155 with a salt bridge between Arg155–Asp168. Drug resistance occurs when this electrostatic network is disrupted, thus triggering a domino-like effect to alter the van der Waals (vdW) contacts, especially at Asp81, and compromises the stacking interaction. We also show that adding a P1–P3 macrocycle to ASV enhances the inhibitor’s resistance barrier by restraining flexibility to poise the inhibitor for binding. Through a detailed structural analysis, we describe the atomic basis for ASV’s resistance to R155K and show how macrocyclizing this inhibitor enhances its resistance barrier.
Results and Discussion
To structurally characterize the binding of ASV and assess the impact of mutations and macrocyclization on drug resistance, the crystal structures of three HCV-NS3/4A genotype 1a protease complexes were determined. These were WT-ASV, R155K-ASV, and WT-ASVmc, which diffracted to resolutions of 1.66–2.70 Å, respectively, in the P212121 space group (Supplementary Table S1).
Detailed Structural Analysis of ASV Binding
The HCV NS3/4A inhibitor ASV binds in the active site extensively packing around the catalytic His57 and Asp81 (Figure 1b) with the P2* isoquinoline moiety stacking both with the catalytic D81 and R155. ASV makes contacts with 27 protease residues (3 of which are minor) contributing to a total vdW energy of −44.5 kcal/mol (Figure 2 and Supplementary Figure S2). While contact with His 57 is the most extensive, Asp81, Ser139, Arg155, Ala156, and Ala157 also form extensive contacts, followed by residues 135–137. In contrast Asp168 makes relatively little direct contact with the inhibitor, suggesting that the large loss of affinity to the D168A variant results from indirect effects.
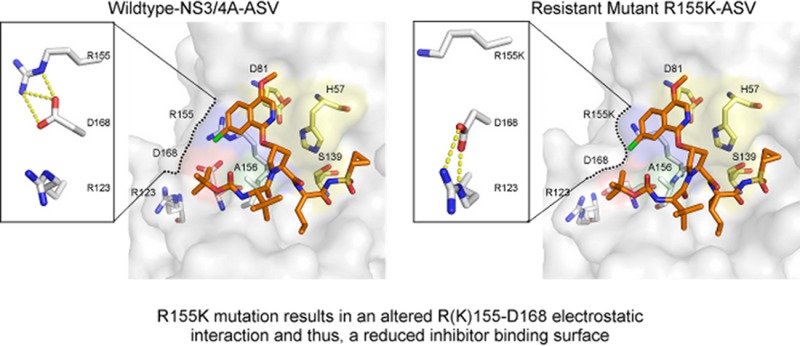
Figure 1.

Asunaprevir’s binding mode is reliant on protease S2 residues: structure of HCV NS3/4A protease in complex with ASV. (a) ASV P2* isoquinoline protrudes from the substrate envelope (blue volume), interacting with protease S2 residues Arg155 and Asp168 (orange). (b) In the WT complex (PDB id: 4WF8), ASV (orange sticks) engages in stacking interactions with the catalytic D81 and S2 residue R155. (c) In the R155K complex (PDB id: 4WH6), Lys155 is no longer stabilized by Asp168, which rotates toward R123 for electrostatic interaction. Consequently, ASV’s P2* isoquinoline loses important interaction binding surface (black dashed lines) and is destabilized. (d) ASVmc (PDB id: 4WH8, pink sticks) adopts a similar binding mode as ASV.
Figure 2.
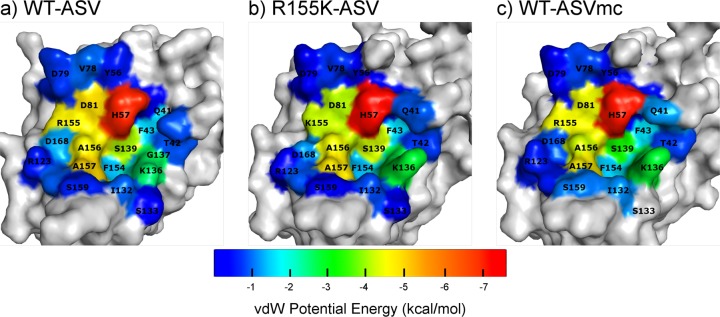
vdW contacts are reduced in the drug-resistant mutant structure but maintained in ASVmc. Surface representation of protease-inhibitor vdW contact energy, where cool and warm colors indicate low and high vdW energy, respectively. (a) In the WT-ASV structure, high contact energy is exhibited with the catalytic His57, Asp81 and S2 R155, while low to moderate contact energy is observed in the P1′–P3 pocket. (b) In addition to altering this energy landscape by reducing inhibitor-protease vdW contacts at Asp81 and Lys155, the R155K mutation also changes the amino acid packing around Gln41, Val78, Asp168 and Arg123. (c) WT-ASVmc exhibits similar amino acid packing as WT-ASV around the S2 and P1′–P3 pockets.
Eleven direct intermolecular and three water-mediated hydrogen bonds (Hbonds) are formed between the protease and ASV (Supplementary Table S2), in particular at P1′, P1, and P3 moieties. As in other HCV NS3/4A PIs, the P1′ acylsulfonamide oxygens accept Hbonds from Ser139 and the oxyanion hole residue Gly137, while the nitrogen (N45) provides a hydrogen for His57 Nε2. The ASV P1 carbonyl oxygen accepts three Hbonds from Ser139, Gly137, and Ser138, while ASV’s P1 amide nitrogen is a donor for Arg155’s backbone oxygen. Finally, the P3 carbonyl oxygen and amide hydrogen atoms both participate in Hbonds with the protease Ala157 backbone NH and CO, respectively. Additionally, three hydrogen bonds are formed between neighboring water molecules and ASV’s P2 carbonyl oxygens, acylsulfonamide oxygens, and P4 carbamate oxygen.
Upon binding, ligands often induce conformational changes in the protein. Compared to the unliganded apo crystal structure (PDB id: 3RC6),21 such changes are observed throughout the WT-ASV complex, specifically within the backbone Cα, illustrated by the presence of major peaks in the Cα distance difference plot (Figure 3). In the WT-ASV complex, Asp168 has two ionic interactions with Arg155, in contrast to the apo structure of HCV NS3/4A where Asp168 is rotated away from Arg155 to interact with Arg123 instead (Supplementary Figure S1a). Thus, in the WT-ASV complex, Asp168 stabilizes Arg155, thereby providing a binding surface for the isoquinoline and facilitating the aromatic stacking on the catalytic Asp81.
Figure 3.

P1–P3 macrocyclization of Asunaprevir restrains protease conformation: Cα displacement analysis of inhibitor complexes relative to the unliganded WT protease (PDB id: 3RC6). The backbone in WT-ASVmc (b) is less disturbed than that in the WT-ASV complex (a). Mapping the displacement onto the apo protease structure shows the various secondary structural elements whose conformations are allosterically affected by inhibitor binding (c and d).
Structural Changes Leading to Resistance
ASV is most susceptible to resistance mutations at amino acids 155, 156, and 168. These amino acids are all located at the NS3/4A S2 binding pocket, where the inhibitor’s isoquinoline moiety protrudes from the substrate envelope22 (Figure 1a) and interacts extensively with the protease. In the R155K-ASV structure (Figure 1b), the absence of a Nε on amino acid 155 prevents Lys155 from interacting with Asp168’s Oδ, thus disrupting the 168–155 electrostatics, which provides an additional binding surface for the isoquinoline. As a result, Asp168 shifts toward Arg123 for electrostatic interactions (Supplementary Figure S1b). This alteration affects the entire binding interface with ASV as reflected in the altered van der Waals interactions (Figure 2b and Supplementary Figure S3), while the inhibitor–protease hydrogen bonding network is maintained (Supplementary Table S2). The extensive disruption of the ASV binding interface in the R155K-ASV complex is in agreement with inhibitor potency drop from 2.7 to 142.7 nM. A similar mechanism may also explain the loss of inhibitor potency in the D168A variant. While attempts to crystallize the ASV in complex with the D168A variant were unsuccessful, the interactions of the inhibitor are expected to be significantly disrupted as the Asp168A–Arg155 ionic interaction will not exist. Consequently, ASV’s affinity for the D168A variant drops by 3 orders of magnitude.17 This structural resistance mechanism helps to explain the observed low to moderate resistance patterns in genotype 1a. Resistance to mutations at residues Arg155 and Asp168 is not unique to ASV, but rather these are signatures of resistance for many NS3/4A inhibitors such as danoprevir, vaniprevir, sovaprevir, and faldaprevir.23−25 However, in other genotypes, such as genotype 1b, the patterns of resistance are altered; for instance, to acquire the R155K mutation two nucleotide substitutions would be required,26 thus genetically altering the resistance barrier.
Improving Asunaprevir’s Resistance Barrier
Although the crystal structure of WT protease with the P1–P3 ASV macrocyclic analogue revealed a similar binding mode as the WT-ASV complex (Figure 1d), several differences were observed. Compared with the apo structure, the WT-ASVmc complex does not show as extensive changes in the protease backbone relative to the WT-ASV structure, perhaps accounting for some of the enhanced binding affinity (Figure 3). Compared to ASV, the macrocyclic ASVmc has altered vdW interactions at the binding site (Figure 2c). Specifically, subtle alterations between the structures were observed at P1′, where the cyclopropyl group rotates toward the catalytic histidine, the P2 proline bears a less puckered pyrrolidine ring, and the P3–P4 peptidomimetic bond is displaced out of the active site by 1 Å. Within the active site, the major difference lies in the S2 pocket electrostatic network. While both carbonyl oxygens of Asp168 in WT-ASV are oriented toward R155’s Nε and Nη (Supplementary Figure S1a), Asp168 in WT-ASVmc rotates by 8.7° about the Cα–Cβ–O angle, leading to the formation of an extensive electrostatic network connecting Arg155–Asp168–Arg123 (Supplementary Figure S1c).
In agreement with our structural data, ASV and ASVmc displayed similar enzyme inhibition constants with the WT protease (2.6 and 1.2 nM, respectively).17 While not significantly improving potency to the WT protease, the addition of this macrocycle considerably improved the inhibitor’s resistance barrier. Indeed, all three MDR variants R155K, A156T, and D168A experienced reduced drug resistance susceptibility with lower fold changes in EC50’s.17 In the free state, small molecules typically adopt a dynamic ensemble of conformations; this is particularly true for those containing proline amide groups, which have the ability to form cis and trans conformations. The P1–P3 macrocycle restricts ASVmc in a trans conformation, thereby potentially decreasing the entropic penalty associated with conformational reconfiguration required for binding compared to the linear ASV.
Conclusion
In summary, asunaprevir is a potent competitive inhibitor targeting the HCV NS3/4A protease, with clinical and in vitro resistance susceptibility to protease mutations at Arg155, Asp168, and Ala156. Similar to other inhibitors that we and others have analyzed,15,16,22 this resistance profile is the result of the inhibitor’s reliance on packing to the S2 site residues including Arg155 beyond the substrate envelope. Avoiding such extensive S2 interactions and restraining the trans conformation via macrocyclization may be viable strategies to design inhibitors that are more robust against resistance.
Methods
Protein Handling and Data Collection
Protein expression, crystallization, and data collection and processing were performed as previously reported,22 with the WT-ASV data set collected at the Advanced Photon source LS-CAT 21-ID-F and the WT-ASVmc collected in-house with a Rigaku Saturn 944 HG CCD detector.
Structural Analysis
Superpositions were performed in PyMOL using the Cα atoms of the active site protease residues 137–139 and 154–160. The A chain of the WT-ASV complex was used as the reference structure for each alignment. During the preparation of this manuscript, a WT-ASV structure (PDB id: 4NWL) similar to our WT-ASV was released.27 We chose to use our structure for analysis because of its improved statistics and resolution.
van der Waals contact energy potential and distance difference plots were determined as previously described.28,29
Acknowledgments
We thank Dr. D. Smith of the Argonne National Library, Advanced Photon Source LS-CAT beamline for his help with data collection. We also thank Dr. N. Kurt Yilmaz for critical editing and K. Prachanronarong for technical assistance. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). This work was supported by the National Institute of Allergy and Infectious Disease (R01-AI085051), the National Institute of General Medical Sciences of the NIH (F31-GM103259), as well as the Biomedical Science Career Program (BSCP) HOPE Scholarship.
Supporting Information Available
This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The PDB codes for the structures used in this study are 4WF8 for WT-ASV, 4WH6 for R155K-ASV, and 4WH8 for WT-ASVmc.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Modi A. A.; Liang T. J. (2008) Hepatitis C: a clinical review. Oral Diseases 14, 10–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alter M. J. (2007) Epidemiology of hepatitis C virus infection. World J. Gastroenterol. 13, 2436–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong A. D. (2014) The HCV revolution did not happen overnight. ACS Med. Chem. Lett. 5, 214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z.; Benureau Y.; Rijnbrand R.; Yi J.; Wang T.; Warter L.; Lanford R. E.; Weinman S. A.; Lemon S. M.; Martin A.; Li K. (2007) GB virus B disrupts RIG-I signaling by NS3/4A-mediated cleavage of the adaptor protein MAVS. J. Virol. 81, 964–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K.; Foy E.; Ferreon J. C.; Nakamura M.; Ferreon A. C.; Ikeda M.; Ray S. C.; Gale M. Jr.; Lemon S. M. (2005) Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. U.S.A. 102, 2992–2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. D.; Sun L.; Seth R. B.; Pineda G.; Chen Z. J. (2005) Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. U.S.A. 102, 17717–17722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong A. D.; Kauffman R. S.; Hurter P.; Mueller P. (2011) Discovery and development of telaprevir: an NS3–4A protease inhibitor for treating genotype 1 chronic hepatitis C virus. Nat. Biotechnol. 29, 993–1003. [DOI] [PubMed] [Google Scholar]
- Rotella D. P. (2013) The discovery and development of boceprevir. Expert Opin. Drug Discovery 8, 1439–1447. [DOI] [PubMed] [Google Scholar]
- McPhee F. (2010) Identification and preclinical profile of the novel HCV NS3 protease inhibitor BMS-650032. J. Hepatol. 52, S296. [Google Scholar]
- White P. W.; Llinàs-Brunet M.; Amad M. a.; Bethell R. C.; Bolger G.; Cordingley M. G.; Duan J.; Garneau M.; Lagacé L.; Thibeault D.; Kukolj G. (2010) Preclinical characterization of BI 201335, a C-terminal carboxylic acid inhibitor of the hepatitis C virus NS3-NS4A protease. Antimicrob. Agents Chemother. 54, 4611–4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiwert S. D.; Andrews S. W.; Jiang Y.; Serebryany V.; Tan H.; Kossen K.; Rajagopalan P. T.; Misialek S.; Stevens S. K.; Stoycheva A.; Hong J.; Lim S. R.; Qin X.; Rieger R.; Condroski K. R.; Zhang H.; Do M. G.; Lemieux C.; Hingorani G. P.; Hartley D. P.; Josey J. A.; Pan L.; Beigelman L.; Blatt L. M. (2008) Preclinical characteristics of the hepatitis C virus NS3/4A protease inhibitor ITMN-191 (R7227). Antimicrob. Agents Chemother. 52, 4432–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T.-I.; Lenz O.; Fanning G.; Verbinnen T.; Delouvroy F.; Scholliers A.; Vermeiren K.; Rosenquist A.; Edlund M.; Samuelsson B.; Vrang L.; de Kock H.; Wigerinck P.; Raboisson P.; Simmen K. (2009) In vitro activity and preclinical profile of TMC435350, a potent hepatitis C virus protease inhibitor. Antimicrob. Agents Chemother. 53, 1377–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liverton N. J.; Carroll S. S.; DiMuzio J.; Fandozzi C.; Graham D. J.; Hazuda D.; Holloway M. K.; Ludmerer S. W.; McCauley J. A.; McIntyre C. J.; Olsen D. B.; Rudd M. T.; Stahlhut M.; Vacca J. P. (2010) MK-7009, a potent and selective inhibitor of hepatitis C virus NS3/4A protease. Antimicrob. Agents Chemother. 54, 305–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper S.; McCauley J. A.; Rudd M. T.; Ferrara M.; DiFilippo M.; Crescenzi B.; Koch U.; Petrocchi A.; Holloway M. K.; Butcher J. W.; Romano J. J.; Bush K. J.; Gilbert K. F.; McIntyre C. J.; Nguyen K. T.; Nizi E.; Carroll S. S.; Ludmerer S. W.; Burlein C.; DiMuzio J. M.; Graham D. J.; McHale C. M.; Stahlhut M. W.; Olsen D. B.; Monteagudo E.; Cianetti S.; Giuliano C.; Pucci V.; Trainor N.; Fandozzi C. M.; Rowley M.; Coleman P. J.; Vacca J. P.; Summa V.; Liverton N. J. (2012) Discovery of MK-5172, a macrocyclic hepatitis C virus NS3/4a protease inhibitor. ACS Med. Chem. Lett. 3, 332–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrazin C.; Zeuzem S. (2010) Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology 138, 447–462. [DOI] [PubMed] [Google Scholar]
- Romano K. P.; Ali A.; Royer W. E.; Schiffer C. A. (2010) Drug resistance against HCV NS3/4A inhibitors is defined by the balance of substrate recognition versus inhibitor binding. Proc. Natl. Acad. Sci. U.S.A. 107, 20986–20991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali A.; Aydin C.; Gildemeister R.; Romano K. P.; Cao H.; Ozen A.; Soumana D.; Newton A.; Petropoulos C. J.; Huang W.; Schiffer C. A. (2013) Evaluating the role of macrocycles in the susceptibility of hepatitis C virus NS3/4A protease inhibitors to drug resistance. ACS Chem. Biol. 8, 1469–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhee F.; Sheaffer A. K.; Friborg J.; Hernandez D.; Falk P.; Zhai G.; Levine S.; Chaniewski S.; Yu F.; Barry D.; Chen C.; Lee M. S.; Mosure K.; Sun L. Q.; Sinz M.; Meanwell N. A.; Colonno R. J.; Knipe J.; Scola P. (2012) Preclinical profile and characterization of the hepatitis C virus NS3 protease inhibitor asunaprevir (BMS-650032). Antimicrob. Agents Chemother. 56, 5387–5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhee F.; Friborg J.; Levine S.; Chen C.; Falk P.; Yu F.; Hernandez D.; Lee M. S.; Chaniewski S.; Sheaffer A. K.; Pasquinelli C. (2012) Resistance analysis of the hepatitis C virus NS3 protease inhibitor asunaprevir. Antimicrob. Agents Chemother. 56, 3670–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everson G. T.; Sims K. D.; Rodriguez-Torres M.; Hézode C.; Lawitz E.; Bourlière M.; Loustaud-Ratti V.; Rustgi V.; Schwartz H.; Tatum H.; Marcellin P.; Pol S.; Thuluvath P. J.; Eley T.; Wang X.; Huang S.-P.; McPhee F.; Wind-Rotolo M.; Chung E.; Pasquinelli C.; Grasela D. M.; Gardiner D. F. (2014) Efficacy of an interferon- and ribavirin-free regimen of daclatasvir, asunaprevir, and BMS-791325 in treatment-naive patients with HCV genotype 1 Infection. Gastroenterology 146, 420–429. [DOI] [PubMed] [Google Scholar]
- Romano K. P.; Laine J. M.; Deveau L. M.; Cao H.; Massi F.; Schiffer C. A. (2011) Molecular mechanisms of viral and host cell substrate recognition by hepatitis C virus NS3/4A protease. J. Virol. 85, 6106–6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano K. P.; Ali A.; Aydin C.; Soumana D.; Ozen A.; Deveau L. M.; Silver C.; Cao H.; Newton A.; Petropoulos C. J.; Huang W.; Schiffer C. A. (2012) The molecular basis of drug resistance against hepatitis C virus NS3/4A protease inhibitors. PLoS Pathog. 8, e1002832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manns M. P.; Gane E.; Rodriguez-Torres M.; Stoehr A.; Yeh C.-T.; Marcellin P.; Wiedmann R. T.; Hwang P. M.; Caro L.; Barnard R. J. O.; Lee A. W.; (2012) Vaniprevir with pegylated interferon alpha-2a and ribavirin in treatment-naïve patients with chronic hepatitis C: A randomized phase II study. Hepatology 56, 884–893. [DOI] [PubMed] [Google Scholar]
- Manns M. P.; Bourlière M.; Benhamou Y.; Pol S.; Bonacini M.; Trepo C.; Wright D.; Berg T.; Calleja J. L.; White P. W.; Stern J. O.; Steinmann G.; Yong C.-L.; Kukolj G.; Scherer J.; Boecher W. O. (2011) Potency, safety, and pharmacokinetics of the NS3/4A protease inhibitor BI201335 in patients with chronic HCV genotype-1 infection. J. Hepatol. 54, 1114–1122. [DOI] [PubMed] [Google Scholar]
- Svarovskaia E. S.; Martin R.; McHutchison J. G.; Miller M. D.; Mo H. (2012) Abundant drug-resistant NS3 mutants detected by deep sequencing in hepatitis C virus-infected patients undergoing NS3 protease inhibitor monotherapy. J. Clin. Microbiol. 50, 3267–3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cento V.; Mirabelli C.; Salpini R.; Dimonte S.; Artese A.; Costa G.; Mercurio F.; Svicher V.; Parrotta L.; Bertoli A.; Ciotti M.; Di Paolo D.; Sarrecchia C.; Andreoni M.; Alcaro S.; Angelico M.; Perno C. F.; Ceccherini-Silberstein F. (2012) HCV genotypes are differently prone to the development of resistance to linear and macrocyclic protease inhibitors. PLoS One 7, e39652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scola P. M.; Sun L. Q.; Wang A. X.; Chen J.; Sin N.; Venables B. L.; Sit S. Y.; Chen Y.; Cocuzza A.; Bilder D. M.; D’Andrea S. V.; Zheng B.; Hewawasam P.; Tu Y.; Friborg J.; Falk P.; Hernandez D.; Levine S.; Chen C.; Yu F.; Sheaffer A. K.; Zhai G.; Barry D.; Knipe J. O.; Han Y. H.; Schartman R.; Donoso M.; Mosure K.; Sinz M. W.; Zvyaga T.; Good A. C.; Rajamani R.; Kish K.; Tredup J.; Klei H. E.; Gao Q.; Mueller L.; Colonno R. J.; Grasela D. M.; Adams S. P.; Loy J.; Levesque P. C.; Sun H.; Shi H.; Sun L.; Warner W.; Li D.; Zhu J.; Meanwell N. A.; McPhee F. (2014) The discovery of asunaprevir (BMS-650032), an orally efficacious NS3 protease inhibitor for the treatment of hepatitis C virus infection. J. Med. Chem. 57, 1730–1752. [DOI] [PubMed] [Google Scholar]
- Özen A.; Sherman W.; Schiffer C. A. (2013) Improving the resistance profile of hepatitis C NS3/4A inhibitors: Dynamic substrate envelope guided design. J. Chem. Theory Comput. 9, 5693–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabu-Jeyabalan M.; Nalivaika E. A.; Romano K.; Schiffer C. A. (2006) Mechanism of substrate recognition by drug-resistant human immunodeficiency virus type 1 protease variants revealed by a novel structural intermediate. J. Virol. 80, 3607–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.