

Abstract
In this article we outline the molecular findings of select odontogenic tumors. In each section, we briefly review selected the clinicoradiographic, histologic, immunologic features, focusing on the molecular findings and their applications in practice. The understanding of molecular pathobiology at various other organ sites has developed quite rapidly in recent years, however much remains unknown about the genetic profile of odontogenic tumors. Improved understanding of mutations in odontogenic tumors may clarify classification schema and elucidate targets for novel therapies. Molecular testing will no doubt improve our understanding of odontogenic tumor pathogenesis and will likely be, someday, an important component of routine clinical practice and its role will only increase in the coming years.
Keywords: Odontogenic tumors, Molecular pathology, Translocation, Mutation, EWSR1, BRAF, PTCH
In this article we outline the molecular findings of select odontogenic tumors. Arising within the jaws or tooth-bearing areas of the oral mucosa, odontogenic tumors comprise a heterogeneous group of lesions encountered with variable frequency and typically in a subspecialist setting. These lesions are derived from epithelial, ectomesenchymal, and/or mesenchymal elements of the tooth forming apparatus [1]. While the understanding of molecular pathobiology at various other organ sites has developed quite rapidly in recent years, much remains unknown about the genetic profile of odontogenic tumors. Improved understanding of mutations in odontogenic tumors may clarify classification schema and elucidate targets for novel therapies. In each section, we briefly review selected the clinicoradiographic, histologic, immunologic features, focusing on the molecular findings and their applications in practice.
Ameloblastic Fibroma, Ameloblastic Fibro-Odontoma, and Ameloblastic Fibrosarcoma
Clinicoradiographic, Histologic, and Immunophenotypic Features
Ameloblastic fibroma (AF), ameloblastic fibro-odontoma (AFO), and ameloblastic fibrosarcoma (AFS) are uncommon, closely related, odontogenic neoplasms of mixed epithelial and ectomesenchymal origin [2]. AF and AFO are benign lesions, while AFS exhibits a malignant mesenchymal component which may arise de novo, or within a pre-existing AF or AFO [3]. Most tumors in all three categories occur in the mandible [3, 4]. AF and AFO tend to develop more frequently in the first or second decade of life while the mean age for AFS is 27.5 years [4]. AF and AFO are typically asymptomatic, or may cause swelling, whereas pain and swelling are characteristic of AFS. Radiographically, AF and AFO frequently surround the crown of an unerupted tooth, presenting as a well-defined radiolucent (AF) or mixed radiolucent-radiopaque (AFO) lesion [4]. AFS, on the other hand, often shows a destructive appearance. The histologic hallmark of these mixed lesions is the combination of mesenchymal and epithelial elements. AF and AFO usually show thin, elongated islands of odontogenic epithelium set in a myxoid mesenchymal stroma reminiscent of the primitive dental papilla. AFO also show dental hard tissue formation. In contrast, AFS contains benign epithelial islands scattered within a cellular background of hyperchromatic and pleomorphic spindle cells. Numerous mitotic figures are typically present. Lesions that arise from a pre-existing benign neoplasm will demonstrate features recognizable as AF or AFO [4].
Molecular Findings
The molecular features of AF, AFS, and AFO are not well studied. A very limited study examining loss of heterozygosity (LOH) at tumor suppressor gene loci on 3p, 9p, 11p, 11q, and 17p of AF, AFO, and AFS revealed a higher mean fraction of allelic loss (FAL) in AFS (74.6 %) compared to AF and AFO combined [5]. Furthermore, the FAL average was higher in AFO (36.6 %) than AF (13.2 %). Overall, for both benign and malignant lesions, LOH of 17p13.1 loci involving the p53 and CHRNB1 genes were most frequently encountered (62 % for p53 and 55 % for CHRNB1).
Diffuse positivity for p53 and c-kit also has been reported in the sarcomatous component of an AFS ex AF, although molecular analysis failed to show genetic mutations in the KIT gene in exons 9, 11, 13, and 17 [6]. These observations could indicate a possible role of p53 and c-kit in malignant progression of AF to AFS.
While only 2 AF and 1 AFO cases have been tested, early data has shown the BRAF V600E mutation present in these tumors [7].
Applications in Practice
Current molecular findings are of little utility in routine diagnosis. Overexpression of p53, Bcl-2, Ki-67, PCNA and c-kit may be observed in AFS and might be useful in distinguishing between benign and malignant neoplasms. AFS that overexpress c-kit might respond favorably to the tyrosine kinase inhibitor imatinib (Gleevec), although the usefulness of this pharmaceutical agent in the treatment of AFS has yet to be demonstrated [6].
Ameloblastoma
Clinicoradiographic, Histologic, and Immunophenotypic Features
The central solid or multicystic ameloblastoma is a slow growing but aggressive benign neoplasm with a high recurrence rate [1]. The mean age of presentation is 36 with an even gender distribution, and a predilection for the posterior mandible. Small tumors may be asymptomatic, while larger lesions may show significant expansion of the jaw and tooth resorption. Radiographically, ameloblastoma may present as a unilocular or multilocular radiolucency, which may surround the crown of an unerupted tooth [1, 4]. While different histologic patterns have been described, tumors typically contain epithelial structures reminiscent of the enamel organ, with tall prominent peripheral cells exhibiting reverse nuclear polarity and central stellate reticulum-like tissue [4]. SOX2, a transcription factor involved in ectodermal development, is involved in the development of odontogenic epithelium [8]. Immunohistochemistry has shown a stronger staining pattern for SOX2 in ameloblastic carcinomas than ameloblastomas [9]. EGFR reactivity is common in ameloblastomas [10]. EGFR overexpression is correlated with SOX2 expression in ameloblastic carcinomas.
Molecular Findings
EGFR-targeted therapy has been shown to be effective in diminishing cellular proliferation in ameloblastoma cell cultures. In the presence of BRAF V600E mutations, a statistically significant difference was seen in diminished sensitivity to the EGFR tyrosine kinase inhibitors [11]. In one study, 63 % (15 of 24) of mandibular ameloblastomas were found to be positive for a BRAF V600E mutation using immunohistochemistry [11]. Another series of 28 cases, including both maxillary and mandibular ameloblastomas, detected somatic mutations in the Hedgehog and mitogen-activated protein kinase (MAPK) pathways. 39 % (11/28) had SMO mutations and 46 % (13/28) had BRAF mutations (Fig. 1a, b). Interestingly, there was a significant correlation between the mutation and location as well as histologic subtype. SMO mutation was present in 9/11 maxillary tumors while only present in 1/13 mandibular cases, as well as 8/10 ameloblastomas with a plexiform pattern [12]. A series of 84 ameloblastomas and 40 various other odontogenic tumors were tested for MAPK pathway mutations. Via polymerase chain reaction (PCR) 62 % (31/50) of ameloblastomas were found to have BRAF V600E mutations. A 100 % concordance was found between molecular detection and immunohistochemical analysis of BRAF V600E. 88 % of ameloblastomas were found to have a BRAF, RAS, or FGFR2 mutation and these were found to be mutually exclusive [7].
Fig. 1.
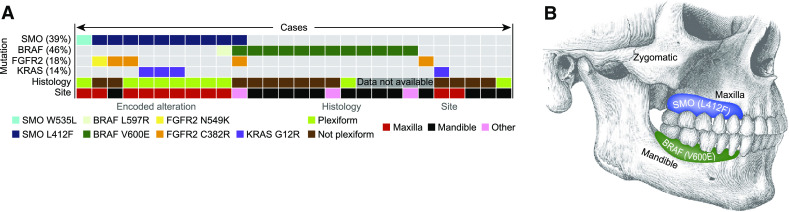
Reprinted by permission from Macmillan Publishers Ltd: Nature Genetics, Sweeney et al. [12]. a Ameloblastoma mutation status for four genes correlated with histology and site. b Graphical representation of ameloblastoma distribution and tumor mutation
Applications in Practice
The presence of BRAF V600E mutation in a high proportion of mandibular ameloblastomas as well as the identification of other mutations has the potential to change our perspectives, classification, and even treatment of ameloblastoma. These mutations may correlate with clinical behavior and provide targets for drugs and allow for more personalized therapy to reduce surgical morbidity. Much of the research in the molecular pathology of ameloblastomas is recent, and there is a need for additional studies in this area.
Clear Cell Odontogenic Carcinoma
Clinicoradiographic, Histologic, and Immunophenotypic Features
Clear cell odontogenic carcinoma (CCOC) is a locally aggressive low grade malignancy found in the gnathic region, presenting as an ill-defined lucency of the jaw, most commonly the mandible with a slight female predilection (Fig. 2a, b) [13]. Clinically, about one-third of tumors recur locally and metastases to distant sites, most commonly the lungs, have been reported. The tumor morphologically resembles salivary (hyalinizing) clear cell carcinoma (CCC) and is comprised of sheets, cords, or nests of polygonal cells separated by a hyalinized to fibrous stroma. Clear cells, which may predominate, are PAS positive and diastase sensitive, demonstrating the presence of intracytoplasmic glycogen [14]. A vague histologic feature generally seen in CCOCs but not CCC of salivary gland origin is peripheral palisading, though only 58.8 % CCOCs exhibit this feature (Fig. 2c) [13].
Fig. 2.

Clear cell odontogenic carcinoma. a Aggressive tumor presenting as an ill-defined radiolucency of the mandible, b cross section of fresh resection specimen, c H&E, ×20; cords and nests of cell in a hyalinized stroma. Focal peripheral palisading is seen (arrow). d Fluorescence in situ hybridization dual-color break apart probe indicated the presence of EWSR1 (22q12) gene rearrangement in clear cell odontogenic carcinoma by the split red and green color signal
CCOCs are classically considered to be of odontogenic origin. As with many odontogenic lesions, CCOC are phenotypically squamous and like CCC, express high molecular weight keratins and p63. They may also show focal keratin pearl formation, and ultrastructurally demonstrate tonofilaments [15].
Molecular Findings
In addition to sharing morphologic and immunophenotypic features with salivary CCC, CCOC also appear to share EWSR1-ATF1 translocation, which may imply that CCOC is better considered a ‘central’ CCC. In one study, 83.3 % of CCOCs harbor a EWSR1 rearrangement (Fig. 2d) and one case was tested and positive for the ATF1 rearrangement [16]. EWSR1-ATF1 has also been reported subsequently in a CCOC case report [17]. To date, specific clear cell tumors found to be negative for this translocation include other primary clear cell salivary, odontogenic, and squamous neoplasms [16].
Applications in Practice
EWSR1 testing is readily available using breakapart fluorescence in situ hybridization (FISH) methodology used for most types of EWSR1 rearrangements. If nucleic acid of sufficient quality for RT-PCR testing can be obtained from a specimen, assessment for EWSR1-ATF1 fusion transcript is plausible as well. As noted above, the presence of this translocation can distinguish CCOC from other clear cell lesions. Specific clear cell tumors tested to date and negative for this translocation include: clear cell calcifying epithelial odontogenic tumor, clear cell squamous carcinoma, epithelial–myoepithelial carcinoma, sinonasal renal cell-like adenocarcinoma, and clear cell mucoepidermoid carcinoma.
Glandular Odontogenic Cyst Versus Central Mucoepidermoid Carcinoma
Clinicoradiographic, Histologic, and Immunophenotypic Features
The term glandular odontogenic cyst (GOC) was coined by Gardner in 1988 [18], although the lesion was described initially by Padayachee and Van Wyk as a sialo-odontogenic cyst [19]. Radiologically they present as either unilocular or multilocular well defined lesions, 80 % of which arise in the mandible, usually anteriorly [20]. They are somewhat aggressive lesions, often causing cortical perforation and less commonly root displacement or resorption. They occur across a wide age range with the mean age at presentation in the 5th decade [20]. Recurrence rates range from 30 to 50 % [20, 21] with larger, multicystic lesions treated by curettage having higher risk for recurrence [22]. Five major histologic criteria that have been proposed for GOCs are: (1) a squamous lining with a flat interface to the adjacent tissue, (2) plaque-like thickenings with whorled epithelium (3) cuboidal eosinophilic hobnail cells (4) mucous or goblet cells and (5) duct-like structures. Other features include papillae, ciliated cells, multicystic architecture, and clear cells in the basal or spinous layer [20, 23]. The histologic differential diagnosis of GOC includes a dentigerous cyst with mucous prosoplasia, and central mucoepidermoid carcinoma. In contrast to GOC and cMEC, the lining of dentigerous cysts is usually overall thinner without whorled plaques. While mucous prosoplasia and cilia may be present, they are not prominent.
Central mucoepidermoid carcinomas (cMECs) share a number of histologic features with GOCs. They can also have a squamous (epidermoid) component, cystic areas, and intracellular mucin making distinction from GOC challenging on small biopsies. These tend to be more architecturally complex with pericystic sclerosis in comparison to cMEC. Although cMECs represent only 2–3 % of mucoepidermoid carcinomas they are the most common intra-osseous salivary type malignancy. It is unclear whether they arise from heterotopic salivary rests or represent salivary analogues of odontogenic origin [24, 25]. Interestingly, earlier literature cited an association with odontogenic cysts [26], complicating the diagnosis.
Molecular Findings
For the diagnosis of more challenging MEC cases, FISH may be performed to identify the t(11;19)(q21;p13) translocation, which results in a CRTC1-MAML2 fusion. CRTC1 activates CREB mediated transcription, whereas MAML2 is involved in Notch signaling pathways. Two recent series have shown the absence of the CRTC1-MAML2 translocation in all GOCs analyzed (n = 9 and n = 21, respectively) [27, 28]. Interestingly, there has been a claim that half of dentigerous cysts with mucous cell prosoplasia showed a subpopulation of MAML2 rearranged cells [29]. However, this fraction is so low that most laboratories would consider it negative, and this study did not validate this finding by another methodology.
Applications in Practice
In clinical practice, FISH for CRTC1-MAML2 can be useful in assisting in the discrimination of a cMEC and an architecturally complex GOC or a small biopsy sample of GOC. Future studies are needed to substantiate the findings of MAML2 rearrangement in odontogenic cysts with mucous prosoplasia.
Keratocystic Odontogenic Tumor
Clinicoradiographic, Histologic, and Immunophenotypic Features
Keratocystic odontogenic tumor (KCOT) is a relatively common cystic neoplasm of the jaws which accounts for 3–11 % of all gnathic cystic lesions derived from remnants of the tooth-forming apparatus [4]. Also known as odontogenic keratocyst, it was renamed KCOT by the World Health Organization in 2005 to better account for its clonal nature and high recurrence rate [1]. KCOT favors the posterior jaws, with the mandibular posterior body and ascending ramus accounting for almost half of the cases [4, 30]. Any age group can be affected, although most sporadic KCOTs occur in young to middle-aged adults, and there is a slight male predilection [30]. Clinically, a large KCOT may cause bony expansion and result in obvious facial asymmetry, but many lesions are clinically imperceptible, owing either to the size of the lesion or to a tendency of KCOT to grow in an antero-posterior direction without altering the lateral dimension of the bone [30]. On panoramic radiograph, KCOT presents as a well-defined, frequently corticated, uni- or multilocular radiolucency. Up to 40 % of lesions develop around the crown of an impacted tooth [4]. Histologically, KCOT consists of a cystic structure lined by parakeratinized stratified squamous epithelium. The epithelial lining is of uniform thickness, typically 6-8 cell layers thick, with prominent, hyperchromatic and palisaded basal cells [4]. The keratin layer is frequently corrugated or ‘wavy,’ and keratin scales may be found within the cyst lumen. Satellite (or daughter) cysts are commonly encountered in the adjacent connective tissue wall. This, in addition to easy fragmentation of the cyst lining during surgical manipulation, may account for KCOT’s high recurrence rate [31, 32].
KCOT most commonly occurs as a sporadic lesion of the jaws, although there is a well-known association of KCOT with the nevoid basal cell carcinoma syndrome (NBCCS) [33]. NBCCS, also known as Gorlin syndrome, Gorlin–Goltz syndrome, and basal cell nevus syndrome, is an autosomal dominant disorder characterized by multiple basal cell carcinomas (BCCs), skeletal anomalies of the ribs and vertebrae, calcification of the falx cerebri, palmar and plantar pits, and an increased incidence of tumors, particularly childhood medulloblastoma (primitive neuroectodermal tumor) and ovarian and cardiac fibromas [33]. The presence of multiple jaw keratocysts are a major diagnostic criterion for the syndrome and are found in approximately 90 % of affected individuals. In this patient population, KCOTs typically develop at a younger age than in non-syndromic cases [4]. NBCCS-associated KCOTs exhibit the same characteristic microscopic features as those observed in sporadic cases, although satellite cysts and intramural remnants are more frequently seen in syndrome-associated lesions [4].
Molecular Findings
Mutations in the PTCH1 gene, mapped to 9q22.3-q31, are known to cause NBCCS and related tumors, including KCOTs [34]. PTCH1 is the human homologue of the Drosophila fly patched gene [34]. PTCH1 encodes for a transmembrane glycoprotein that acts as a receptor in the hedgehog signaling pathway. In vertebrates, three hedgehog genes have been identified, Sonic (Shh), Desert, and Indian hedgehog [35]. Of these, Shh has been the most widely studied. The Shh pathway is most important during embryologic development, responsible for growth and patterning of the limbs, neural tube, axial skeleton, lungs, skin, hair, and teeth [35, 36]. During odontogenesis, it affects epithelial cell proliferation and might also play a role in epithelial–mesenchymal interactions [37]. PTCH1 appears to be inactivated in various odontogenic cysts and tumors, such as dentigerous cysts, calcifying epithelial odontogenic tumors, and ameloblastomas, including one malignant ameloblastoma [38–40]. In the absence of Shh, the PTCH1 transmembrane receptor acts as a gatekeeper of the hedgehog signaling pathway by inhibiting a neighboring transmembrane protein, SMO (Fig. 3a) [41, 42]. Binding of Shh to PTCH1 activates the pathway and results in upregulation of downstream target genes, such as Wnt and genes related to bone morphogenetic proteins (Fig. 3b) [42]. The Shh signaling pathway might also interact with and aid in activation of bcl-2 [43]. Transcriptional activation of downstream genes is linked to tumorigenesis.
Fig. 3.

a PTCH1 is a transmembrane receptor which inhibits SMO in the absence of Shh. b Binding of Shh to PTCH1 releases the inhibitory effect on SMO, activating the Shh pathway and associated downstream targets. During embryogenesis, this leads to growth and patterning of the limbs and of various tissues. c Mutation of PTCH1 results in loss of SMO inhibition. Tumorigenesis appears to result from constitutive activation of Shh-related proteins such as Gli1, Gli2, Gli3, and Wnt
PTCH1 therefore appears to act as a tumor suppressor gene by inhibiting the hedgehog pathway (Fig. 3c) [34, 42]. While haploinsufficiency alone is adequate for the developmental anomalies observed in NBCCS patients, tumors are likely to occur with inactivation of the remaining allele [42]. Both familial and sporadic BCCs show LOH of PTCH1 [34]. Similarly, LOH on chromosome 9q has been noted in both NBCCS-associated and sporadic KCOTs, although allelic loss appears to be less common in sporadic versus hereditary lesions [44]. This suggests that other mechanisms might be involved, such as point mutations in both copies of the gene or epigenetic changes, such as hypermethylation [44]. Inactivating PTCH1 mutations have also been linked to development of primitive neuroectodermal tumors (including medulloblastoma), trichoepithelioma, esophageal squamous cell carcinoma and transitional cell carcinoma [42].
LOH of common tumor suppressor genes has been observed in KCOT. In one study, LOH of at least one tumor suppressor gene was observed in 7 of 10 sporadic KCOTs [45]. Allelic loss was observed most frequently in PTCH, p16, p53, and MCC. In another study, expression of bcl-2 oncoprotein was significantly higher in both sporadic and hereditary KCOTs compared to other odontogenic cysts [43]. These findings support the argument that KCOTs are neoplastic. The authors also noted a significant correlation between LOH and the presence of satellite cysts. Epigenetic events have also been reported [46]. Methylation of the promoter region of the cyclin-dependent kinase inhibitor p21 was found in 3 of 10 KCOTs, but not in dental follicles or normal mucosa. Methylation of p16 was noted in 50 % of KCOTs, but was similarly present in normal and follicular tissues. P27 showed fewer epigenetic alterations in KCOTs than in non-tumor tissues. P53 and RB1 methylation was not seen in any of the KCOTs.
Applications in Practice
Various PTCH1 mutations have been described in both sporadic and hereditary KCOTs, although no hot spots have yet been identified (Table 1). Additionally, overexpression of PTCH1 protein product has been demonstrated with the use of immunohistochemistry in all hereditary and sporadic KCOTs studied thus far, although differences in the pattern of expression were noted [47, 48]. Syndromic KCOTs showed heavy cytoplasmic staining in the basal cell layers, whereas non-syndromic cysts showed staining exclusively in the superficial epithelial cells. Increased expression of downstream Gli-1 was also noted in most cysts and was noted to parallel the staining pattern of PTCH1 [47]. They explain these differing patterns of expression as indicative of a more differentiated phenotype in non-syndromic KCOT.
Table 1.
Specific mutations identified in KOT
| Study | NBCCS versus sporadic | Mutation |
|---|---|---|
| Barreto et al. [48] | NBCCS | Nonsense mutation (C2760A) |
| NBCCS | Missense mutation (G3499A) | |
| Sporadic | 5-base pair deletion exon 3 (518delAAGCG) | |
| Zedan et al. [47] | NBCCS | Tandem CC → TT substitution intron exon 2 (proximal to the immunogenic sequence) |
| NBCCS | AAA deletion intron exon 11, insertion exon 14 | |
| NBCCS | 8-base pair deletion exon 3 | |
| NBCCS | G deletion intron 17, tandem GG → TT substitution exon 17 | |
| Sporadic | G → A substitution exon 8 | |
| Sporadic | C insertion exon 13 | |
| Sporadic | C insertion exon 8 |
Current experimental strategies for the treatment of BCCs SMO inhibitors (cyclopamine, CUR61414, GDC-0449, vitamin D3, LDE225, and LEQ506) and other Shh pathway antagonists (vismodegib, hedgehog interacting protein, MYCN inhibitors, rapamycin, statins, and itraconazole) [49, 50]. Such targeted therapies might someday be of use in the treatment of KCOT, particularly in hereditary cases, or in patients with recurrent lesions.
Odontogenic Tumors with Ghost Cells
Clinicoradiographic, Histologic, and Immunophenotypic Features
Odontogenic tumors with ghost cells include calcifying cystic odontogenic tumor (CCOT) (formerly termed calcifying odontogenic cyst or Gorlin cyst) and dentinogenic ghost cell tumor, as well as some odontomas. Ghost cell odontogenic carcinoma is the malignant counterpart of CCOT. CCOTs were first described by Gorlin who described the presence eosinophilic ghost-cells, comparing them to the calcifying epithelioma of Malherbe (pilomatrixoma) [51]. These ghost cells have a tendency to calcify. The simple cystic CCOT is comprised of an epithelial lining 4–10 cell layers thick with a palisaded layer of cuboidal to columnar basal cells that may exhibit reverse polarity, with areas of epithelial proliferation seen [52].
Odontomas are considered the most common odontogenic tumors, more common than all others combined. However, many pathologists consider odontomas to be hamartomatous, as the tissue from which they are comprised is morphologically relatively normally, although aberrantly arranged. Clinically, odontomas present in the jaws of children or adolescents often after radiographic evaluation for an unerupted tooth [53]. They are slow growing asymptomatic lesions with the potential for tooth displacement, occasionally causing expansion of the jaw. The radiographic features are usually distinctive, presenting as a radiopacity surrounded by a thin radiolucent line. The compound odontoma is comprised of malformed tooth-like structures, whereas the complex odontoma is an amorphous radiopaque mass. Microscopically, enamel, dentin, cementum, and pulp tissue can be identified with tooth-like structures seen in the compound odontoma and a haphazard arrangement of tissue found in the complex odontoma. Ghost cells are present in 16 % of complex odontomas and 3 % of compound odontomas [54, 55]. When seen in association with another tumor, the most common combination is an odontoma-associated CCOT, seen in approximately 24 % of CCOT [56].
Molecular Findings
The Wnt signaling pathway regulates multipotent stem cell development and is important for tooth development [57]. CTNNB1 is a regulatory gene in this pathway that encodes β-catenin, a downstream transcriptional activator of Wnt. β-catenin accumulates in the nucleus and forms complexes with DNA binding proteins such as TCF and LEF-1 [58]. β-catenin can thus be considered a transcriptional co-activator that interacts with LEF-1. β-catenin positivity has been reported in CCOTs and odontomas [59, 60].
Patients with familial adenomatous polyposis (FAP) have mutations in the APC gene which may be inherited in an autosomal dominant pattern and may present with extraintestinal manifestations including odontomas (as well as jaw osteomas and supernumerary teeth) [61].
Applications in Practice
β-catenin and LEF-1 are not utilized in the diagnosis of CCOT or odontomas with ghost cells. However, there is an association of β-catenin mutations in tumors with ghost cells and the Wnt signaling pathway may be involved in their pathogenesis. In patients with multiple odontomas, genetic testing for FAP should be considered [62].
Odontogenic Myxoma
Clinicoradiographic, Histologic, and Immunophenotypic Features
Odontogenic myxoma (OM) is an uncommon benign neoplasm of the jaws derived from the ectomesenchyme of the tooth forming apparatus. It accounts for approximately 3 % of all odontogenic tumors [63]. Any gnathic site can be affected, although OM shows a predilection for the posterior mandible [64]. The lesion most commonly develops in young adults, but can affect a broad age group. A slight female predilection has been observed [64]. Unlike myxomas of other sites, OMs are invariably sporadic and are not a feature of Carney complex or Mazabraud syndrome. On panoramic radiograph, OM characteristically presents as a multilocular radiolucency, with wispy septations imparting a “soap-bubble”-like appearance [65]. Histologically, OMs are composed of abundant, pale-staining, myxoid ground substance with a paucicellular infiltrate of bland-appearing, thin spindle-shaped cells. Small rests of odontogenic epithelium may be observed within the stroma. The microscopic borders of the tumor are typically poorly delineated, with focal infiltration of soft tissue and bone. This finding likely accounts for the approximately 25 % recurrence rate associated with these tumors [4].
Molecular Findings
One study noted increased expression of anti-apoptotic proteins Bcl-2 and Bcl-XL in OM cells compared to control tissue using standard IHC procedures [66]. In this study, myxomatous dental follicular and pulpal tissues were used as controls. This would suggest that dysregulated apoptosis might play a role in neoplastic growth of OM. The authors also found that 90 % of tumor cells stained positively for a gelatinase-type matrix metalloproteinase, MMP-2, compared to 10 % of cells staining in the control tissue. Production of MMP-2 by tumor cells would facilitate spread of the lesion within the surrounding tissue. The authors noted a Ki-67 proliferative index of less than 1 %.
Mazabraud syndrome (MS) is a condition characterized by the presence of intramuscular myxomas along with fibrous dysplasia of bone [67]. Guanine nucleotide-binding protein, α-stimulating activity polypeptide 1 (GNAS1) mutations have been implicated in the pathogenesis of McCune–Albright syndrome and have been identified in some patients with MS [68]. GNAS1 mutations have also been reported in sporadic and syndromic intramuscular myxomas [69]. However, based on one study of 7 cases, OM does not appear to be associated with GNAS1 mutations [70].
Carney complex (CNC) is a familial multiple neoplasia syndrome characterized by skin and mucosal pigmentation, endocrine abnormalities, as well as cutaneous and cardiac myxomas [71]. Osteochondromyxoma of bone and myxomas at other sites have also been described in association with this condition [72]. Tumorigenesis in patients with CNC has been linked in part to dysregulation of the cAMP signaling pathways via alterations of protein kinase A (PKA) regulatory subunit type 1A (PRKAR1A, on chromosome 17q22-24) which encodes the RIα subunit of PKA type I [72]. PRKAR1A inactivating mutations are encountered in up to one-half of patients with familial CNC [72]. Interestingly, sporadic (non-CNC) cardiac myxomas have not shown an association with PRKAR1A gene mutations. Investigation into the role of PRKAR1A in the pathogenesis of OM indicates a possible commonality of at least a subset of OM with CNC-related tumors. Sequencing of PRKAR1A confirmed heterozygous mutations in 2 cases of OM [73]. Furthermore, LOH in the PRKAR1A gene is found within tumor cells in patients with CNC and indicate that RIα might act as a tumor suppressor gene in certain tissues [72, 74]. However, the tumorigenesis is likely more complex and the exact mechanism by which mutations in PRKAR1A lead to tumorigenesis in CNC and OM has not been fully elucidated. Dysregulation of PKA activity appears to play a role. Despite decreased RIα levels, CNC tumors show increased responsiveness to cAMP compared to non-CNC control tumors, and total PKA activity is preserved. This is likely due to a compensatory overexpression of RIβ and an increase in PKA II activity [72, 74].
Applications in Practice
Perdigão et al. [73] found significantly decreased immunohistochemical expression of RIα in 9 of 17 OM (with compensatory increased expression of RIIα and/or RIIβ), when compared to expression of the protein in non-tumor cells within the surrounding normal tissue. Sequencing of PRKAR1A confirmed heterozygous mutations in 2 cases. One tumor showed a missense mutation in exon 6 (c.725C > A) and the other harbored a single-pair deletion (del774C), also in exon 6. Interestingly, these mutations identified in OMs have not been detected in patients with CNC. At the present time, molecular studies are of limited value in the diagnosis of OM.
Conclusion
In summary, while recent advances have been made in molecular pathology of odontogenic tumors, including the identification of BRAF V600E and SMO mutations in ameloblastomas and the EWSR1-ATF1 translocation in clear cell odontogenic carcinomas, much remains unknown. Clinically, molecular testing has the ability to aid in diagnosis of only a limited subset of tumors and is currently of limited applicability in practice. However, improved understanding of molecular biology may help refine tumor classification and perhaps even allow for targeted therapeutic approaches. Molecular testing will no doubt improve our understanding of odontogenic tumor pathogenesis and someday will likely be an important component of routine clinical practice and its role will only increase in the coming years. With time, we anticipate more widespread integration of molecular pathology into clinical practice as costs decrease, technology advances, and more is known about the clinical significance of these findings.
References
- 1.Barnes L, Eveson J, Reichart P, Sidransky D. Pathology and genetics of head and neck tumours. Lyon: IARC Press; 2005. [Google Scholar]
- 2.Takeda Y. Ameloblastic fibroma and related lesions: current pathologic concept. Oral Oncol. 1999;35(6):535–540. doi: 10.1016/S1368-8375(99)00039-1. [DOI] [PubMed] [Google Scholar]
- 3.Muller S, Parker DC, Kapadia SB, Budnick SD, Barnes EL. Ameloblastic fibrosarcoma of the jaws. A clinicopathologic and DNA analysis of five cases and review of the literature with discussion of its relationship to ameloblastic fibroma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1995;79(4):469–477. doi: 10.1016/S1079-2104(05)80130-1. [DOI] [PubMed] [Google Scholar]
- 4.Neville BW, Allen CM, Bouquot JE. Odontogenic cysts and tumors. Oral and maxillofacial pathology. 3rd ed. St. Louis: Saunders Elsevier; 2009. p. 678–740.
- 5.Galvao CF, Gomes CC, Diniz MG, Vargas PA, de Paula AM, Mosqueda-Taylor A, et al. Loss of heterozygosity (LOH) in tumour suppressor genes in benign and malignant mixed odontogenic tumours. J Oral Pathol Med. 2012;41(5):389–393. doi: 10.1111/j.1600-0714.2011.01115.x. [DOI] [PubMed] [Google Scholar]
- 6.Williams MD, Hanna EY, El-Naggar AK. Anaplastic ameloblastic fibrosarcoma arising from recurrent ameloblastic fibroma: restricted molecular abnormalities of certain genes to the malignant transformation. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007;104(1):72–75. doi: 10.1016/j.tripleo.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 7.Brown NA, Rolland D, McHugh JB, Weigelin HC, Zhao L, Lim MS, et al. Activating FGFR2-RAS-BRAF mutations in ameloblastoma. Clin Cancer Res. 2014 doi: 10.1158/1078-0432.CCR-14-1069. [DOI] [PubMed] [Google Scholar]
- 8.Sarkar A, Hochedlinger K. The sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell Stem Cell. 2013;12(1):15–30. doi: 10.1016/j.stem.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lei Y, Jaradat JM, Owosho A, Adebiyi KE, Lybrand KS, Neville BW et al. Evaluation of SOX2 as a potential marker for ameloblastic carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol. 2014;117(5):608–16e1. doi:10.1016/j.oooo.2014.01.017. [DOI] [PubMed]
- 10.Heikinheimo K, Voutilainen R, Happonen RP, Miettinen PJ. EGF receptor and its ligands, EGF and TGF-alpha, in developing and neoplastic human odontogenic tissues. Int J Dev Biol. 1993;37(3):387–396. [PubMed] [Google Scholar]
- 11.Kurppa KJ, Caton J, Morgan PR, Ristimaki A, Ruhin B, Kellokoski J, et al. High frequency of BRAF V600E mutations in ameloblastoma. J Pathol. 2014;232(5):492–498. doi: 10.1002/path.4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sweeney RT, McClary AC, Myers B, Biscocho J, Neahring L, Kwei KA, et al. Identification of recurrent SMO and BRAF mutations in ameloblastomas. Nat Genet. 2014;46(7):722–725. doi: 10.1038/ng.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bilodeau EA, Hoschar AP, Barnes EL, Hunt JL, Seethala RR. Clear cell carcinoma and clear cell odontogenic carcinoma: a comparative clinicopathologic and immunohistochemical study. Head Neck Pathol. 2011;5(2):101–107. doi: 10.1007/s12105-011-0244-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li TJ, Yu SF, Gao Y, Wang EB. Clear cell odontogenic carcinoma: a clinicopathologic and immunocytochemical study of 5 cases. Arch Pathol Lab Med. 2001;125(12):1566–1571. doi: 10.5858/2001-125-1566-CCOC. [DOI] [PubMed] [Google Scholar]
- 15.Weinreb I. Hyalinizing clear cell carcinoma of salivary gland: a review and update. Head Neck Pathol. 2013;7(Suppl 1):S20–S29. doi: 10.1007/s12105-013-0466-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bilodeau EA, Weinreb I, Antonescu CR, Zhang L, Dacic S, Muller S, et al. Clear cell odontogenic carcinomas show EWSR1 rearrangements: a novel finding and a biological link to salivary clear cell carcinomas. Am J Surg Pathol. 2013;37(7):1001–1005. doi: 10.1097/PAS.0b013e31828a6727. [DOI] [PubMed] [Google Scholar]
- 17.Yancoskie AE, Sreekantaiah C, Jacob J, Rosenberg A, Edelman M, Antonescu CR, et al. EWSR1 and ATF1 rearrangements in clear cell odontogenic carcinoma: presentation of a case. Oral Surg Oral Med Oral Pathol Oral Radiol. 2014 doi: 10.1016/j.oooo.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 18.Gardner D, Kessler H, Morency R, Schaffner D. The glandular odontogenic cyst: an apparent entity. J Oral Pathol. 1988;17:359–366. doi: 10.1111/j.1600-0714.1988.tb01298.x. [DOI] [PubMed] [Google Scholar]
- 19.Padayachee A, Van Wyk CW. Two cystic lesions with features of both the botryoid odontogenic cyst and the central mucoepidermoid tumour: sialo-odontogenic cyst? J Oral Pathol. 1987;16(10):499–504. doi: 10.1111/j.1600-0714.1987.tb00680.x. [DOI] [PubMed] [Google Scholar]
- 20.Fowler CB, Brannon RB, Kessler HP, Castle JT, Kahn MA. Glandular odontogenic cyst: analysis of 46 cases with special emphasis on microscopic criteria for diagnosis. Head Neck Pathol. 2011;5(4):364–375. doi: 10.1007/s12105-011-0298-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaplan I, Anavi Y, Hirshberg A. Glandular odontogenic cyst: a challenge in diagnosis and treatment. Oral Dis. 2008;14(7):575–581. doi: 10.1111/j.1601-0825.2007.01428.x. [DOI] [PubMed] [Google Scholar]
- 22.Kaplan I, Gal G, Anavi Y, Manor R, Calderon S. Glandular odontogenic cyst: treatment and recurrence. J Oral Maxillofac Surg. 2005;63(4):435–441. doi: 10.1016/j.joms.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 23.Kaplan I, Anavi Y, Manor R, Sulkes J, Calderon S. The use of molecular markers as an aid in the diagnosis of glandular odontogenic cyst. Oral Oncol. 2005;41(9):895–902. doi: 10.1016/j.oraloncology.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 24.Bouquot JE, Gnepp DR, Dardick I, Hietanen JH. Intraosseous salivary tissue: jawbone examples of choristomas, hamartomas, embryonic rests, and inflammatory entrapment: another histogenetic source for intraosseous adenocarcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90(2):205–217. doi: 10.1067/moe.2000.107058. [DOI] [PubMed] [Google Scholar]
- 25.Brookstone MS, Huvos AG. Central salivary gland tumors of the maxilla and mandible: a clinicopathologic study of 11 cases with an analysis of the literature. J Oral Maxillofac Surg. 1992;50(3):229–236. doi: 10.1016/0278-2391(92)90317-S. [DOI] [PubMed] [Google Scholar]
- 26.Eversole L, Sabes W, Rovin S. Aggressive growth and neoplastic potential of odontogenic cysts. Cancer. 1975;35:270–282. doi: 10.1002/1097-0142(197501)35:1<270::AID-CNCR2820350134>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 27.Garcia J, Barnes E, Cieply K, Dacic S, Jordan R, Seethala R. Utility of MAML2 rearrangement detection using fluorescent in situ hybridization to distinguish glandular odontogenic cyst from central mucoepidermoid carcinoma. Mod Pathol. 2001;24:277A–278A. [Google Scholar]
- 28.Bishop J, Yonescu R, Batista D, Westra W. Glandular odontogenic cysts consistently lack the MAML2 Rearrangements that are frequently found in central mucoepidermoid carcinomas. Modern Pathol. 2014;27:315A–316A. doi: 10.1007/s12105-014-0534-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Argyris PP, Wehrs RN, Garcia JJ, Koutlas IG. Fluorescence in situ hybridization identifies mastermind-like 2 (MAML2) rearrangement in odontogenic cysts with mucous prosoplasia. A Pilot Study. Histopathology. 2014 doi: 10.1111/his.12526. [DOI] [PubMed] [Google Scholar]
- 30.Brannon RB. The odontogenic keratocyst. A clinicopathologic study of 312 cases. Part I. Clinical features. Oral Surg Oral Med Oral Pathol. 1976;42(1):54–72. doi: 10.1016/0030-4220(76)90031-1. [DOI] [PubMed] [Google Scholar]
- 31.Morgan TA, Burton CC, Qian F. A retrospective review of treatment of the odontogenic keratocyst. J Oral Maxillofac Surg. 2005;63(5):635–639. doi: 10.1016/j.joms.2004.07.026. [DOI] [PubMed] [Google Scholar]
- 32.Evans D, Farndon, PA. Nevoid basal cell carcinoma syndrome. Internet: University of Washington, Seattle 1993–2014. [PubMed]
- 33.Gorlin RJ. Nevoid basal cell carcinoma syndrome. Dermatol Clin. 1995;13(1):113–125. [PubMed] [Google Scholar]
- 34.Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85(6):841–851. doi: 10.1016/S0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- 35.Riddle RD, Johnson RL, Laufer E, Tabin C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell. 1993;75(7):1401–1416. doi: 10.1016/0092-8674(93)90626-2. [DOI] [PubMed] [Google Scholar]
- 36.Bellusci S, Furuta Y, Rush MG, Henderson R, Winnier G, Hogan BL. Involvement of Sonic hedgehog (Shh) in mouse embryonic lung growth and morphogenesis. Development. 1997;124(1):53–63. doi: 10.1242/dev.124.1.53. [DOI] [PubMed] [Google Scholar]
- 37.Cobourne MT, Hardcastle Z, Sharpe PT. Sonic hedgehog regulates epithelial proliferation and cell survival in the developing tooth germ. J Dent Res. 2001;80(11):1974–1979. doi: 10.1177/00220345010800110501. [DOI] [PubMed] [Google Scholar]
- 38.Levanat S, Pavelic B, Crnic I, Oreskovic S, Manojlovic S. Involvement of PTCH gene in various noninflammatory cysts. J Mol Med (Berl). 2000;78(3):140–146. doi: 10.1007/s001090000090. [DOI] [PubMed] [Google Scholar]
- 39.Peacock ZS, Cox D, Schmidt BL. Involvement of PTCH1 mutations in the calcifying epithelial odontogenic tumor. Oral Oncol. 2010;46(5):387–392. doi: 10.1016/j.oraloncology.2010.02.023. [DOI] [PubMed] [Google Scholar]
- 40.Farias LC, Gomes CC, Brito JA, Galvao CF, Diniz MG, de Castro WH, et al. Loss of heterozygosity of the PTCH gene in ameloblastoma. Hum Pathol. 2012;43(8):1229–1233. doi: 10.1016/j.humpath.2011.08.026. [DOI] [PubMed] [Google Scholar]
- 41.Stone DM, Hynes M, Armanini M, Swanson TA, Gu Q, Johnson RL, et al. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature. 1996;384(6605):129–134. doi: 10.1038/384129a0. [DOI] [PubMed] [Google Scholar]
- 42.Wicking C, Smyth I, Bale A. The hedgehog signalling pathway in tumorigenesis and development. Oncogene. 1999;18(55):7844–7851. doi: 10.1038/sj.onc.1203282. [DOI] [PubMed] [Google Scholar]
- 43.Vered M, Peleg O, Taicher S, Buchner A. The immunoprofile of odontogenic keratocyst (keratocystic odontogenic tumor) that includes expression of PTCH, SMO, GLI-1 and bcl-2 is similar to ameloblastoma but different from odontogenic cysts. J Oral Pathol Med. 2009;38(7):597–604. doi: 10.1111/j.1600-0714.2009.00778.x. [DOI] [PubMed] [Google Scholar]
- 44.Levanat S, Gorlin RJ, Fallet S, Johnson DR, Fantasia JE, Bale AE. A two-hit model for developmental defects in Gorlin syndrome. Nat Genet. 1996;12(1):85–87. doi: 10.1038/ng0196-85. [DOI] [PubMed] [Google Scholar]
- 45.Agaram NP, Collins BM, Barnes L, Lomago D, Aldeeb D, Swalsky P, et al. Molecular analysis to demonstrate that odontogenic keratocysts are neoplastic. Arch Pathol Lab Med. 2004;128(3):313–317. doi: 10.5858/2004-128-313-MATDTO. [DOI] [PubMed] [Google Scholar]
- 46.Moreira PR, Guimaraes MM, Guimaraes AL, Diniz MG, Gomes CC, Brito JA, et al. Methylation of P16, P21, P27, RB1 and P53 genes in odontogenic keratocysts. J Oral Pathol Med. 2009;38(1):99–103. doi: 10.1111/j.1600-0714.2008.00718.x. [DOI] [PubMed] [Google Scholar]
- 47.Zedan W, Robinson PA, Markham AF, High AS. Expression of the Sonic Hedgehog receptor “PATCHED” in basal cell carcinomas and odontogenic keratocysts. J Pathol. 2001;194(4):473–477. doi: 10.1002/path.940. [DOI] [PubMed] [Google Scholar]
- 48.Barreto DC, Bale AE, De Marco L, Gomez RS. Immunolocalization of PTCH protein in odontogenic cysts and tumors. J Dent Res. 2002;81(11):757–760. doi: 10.1177/154405910208101107. [DOI] [PubMed] [Google Scholar]
- 49.Iwasaki JK, Srivastava D, Moy RL, Lin HJ, Kouba DJ. The molecular genetics underlying basal cell carcinoma pathogenesis and links to targeted therapeutics. J Am Acad Dermatol. 2012;66(5):e167–e178. doi: 10.1016/j.jaad.2010.06.054. [DOI] [PubMed] [Google Scholar]
- 50.Dreier J, Felderer L, Barysch M, Rozati S, Dummer R. Basal cell carcinoma: a paradigm for targeted therapies. Expert Opin Pharmacother. 2013;14(10):1307–1318. doi: 10.1517/14656566.2013.798644. [DOI] [PubMed] [Google Scholar]
- 51.Gorlin RJ, Pindborg JJ, Odont, Clausen FP, Vickers RA. The calcifying odontogenic cyst—a possible analogue of the cutaneous calcifying epithelioma of Malherbe. An analysis of fifteen cases. Oral Surg Oral Med Oral Pathol. 1962;15:1235–1243. doi: 10.1016/0030-4220(62)90159-7. [DOI] [PubMed] [Google Scholar]
- 52.Reichart PA, Philipsen H. Calcifying ghost cell odontogenic cysts/tumors. Odontogenic Tumors and Allied Lesions. London: Quintessence; 2004. p. 155–78.
- 53.Philipsen HP, Reichart PA, Praetorius F. Mixed odontogenic tumours and odontomas. Considerations on interrelationship. Review of the literature and presentation of 134 new cases of odontomas. Oral Oncol. 1997;33(2):86–99. doi: 10.1016/S0964-1955(96)00067-X. [DOI] [PubMed] [Google Scholar]
- 54.Cho YA, Yoon HJ, Hong SP, Lee JI, Hong SD. Multiple calcifying hyperplastic dental follicles: comparison with hyperplastic dental follicles. J Oral Pathol Med. 2011;40(3):243–249. doi: 10.1111/j.1600-0714.2010.00968.x. [DOI] [PubMed] [Google Scholar]
- 55.Sedano HO, Pindborg JJ. Ghost cell epithelium in odontomas. J Oral Pathol. 1975;4(1):27–30. doi: 10.1111/j.1600-0714.1975.tb01737.x. [DOI] [PubMed] [Google Scholar]
- 56.Hirshberg A, Kaplan I, Buchner A. Calcifying odontogenic cyst associated with odontoma: a possible separate entity (odontocalcifying odontogenic cyst) J Oral Maxillofac Surg. 1994;52(6):555–558. doi: 10.1016/0278-2391(94)90087-6. [DOI] [PubMed] [Google Scholar]
- 57.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14(15):1837–1851. [PubMed] [Google Scholar]
- 58.Miyake T, Tanaka Y, Kato K, Tanaka M, Sato Y, Ijiri R, et al. Gene mutation analysis and immunohistochemical study of beta-catenin in odontogenic tumors. Pathol Int. 2006;56(12):732–737. doi: 10.1111/j.1440-1827.2006.02039.x. [DOI] [PubMed] [Google Scholar]
- 59.Sekine S, Sato S, Takata T, Fukuda Y, Ishida T, Kishino M, et al. Beta-catenin mutations are frequent in calcifying odontogenic cysts, but rare in ameloblastomas. Am J Pathol. 2003;163(5):1707–1712. doi: 10.1016/S0002-9440(10)63528-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tanaka A, Okamoto M, Yoshizawa D, Ito S, Alva PG, Ide F, et al. Presence of ghost cells and the Wnt signaling pathway in odontomas. J Oral Pathol Med. 2007;36(7):400–404. doi: 10.1111/j.1600-0714.2007.00550.x. [DOI] [PubMed] [Google Scholar]
- 61.Wijn MA, Keller JJ, Giardiello FM, Brand HS. Oral and maxillofacial manifestations of familial adenomatous polyposis. Oral Dis. 2007;13(4):360–365. doi: 10.1111/j.1601-0825.2006.01293.x. [DOI] [PubMed] [Google Scholar]
- 62.Hegde M, Ferber M, Mao R, Samowitz W, Ganguly A. Working Group of the American College of Medical G et al. ACMG technical standards and guidelines for genetic testing for inherited colorectal cancer (Lynch syndrome, familial adenomatous polyposis, and MYH-associated polyposis). Genet Med. 2014;16(1):101–16. doi:10.1038/gim.2013.166. [DOI] [PubMed]
- 63.Regezi JA, Kerr DA, Courtney RM. Odontogenic tumors: analysis of 706 cases. J Oral Surg. 1978;36(10):771–778. [PubMed] [Google Scholar]
- 64.Simon EN, Merkx MA, Vuhahula E, Ngassapa D, Stoelinga PJ. Odontogenic myxoma: a clinicopathological study of 33 cases. Int J Oral Maxillofac Surg. 2004;33(4):333–337. doi: 10.1016/j.ijom.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 65.Peltola J, Magnusson B, Happonen RP, Borrman H. Odontogenic myxoma—a radiographic study of 21 tumours. Br J Oral Maxillofac Surg. 1994;32(5):298–302. doi: 10.1016/0266-4356(94)90050-7. [DOI] [PubMed] [Google Scholar]
- 66.Bast BT, Pogrel MA, Regezi JA. The expression of apoptotic proteins and matrix metalloproteinases in odontogenic myxomas. J Oral Maxillofac Surg. 2003;61(12):1463–1466. doi: 10.1016/j.joms.2003.06.002. [DOI] [PubMed] [Google Scholar]
- 67.Zoccali C, Teori G, Prencipe U, Erba F. Mazabraud’s syndrome: a new case and review of the literature. Int Orthop. 2009;33(3):605–610. doi: 10.1007/s00264-007-0483-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. Activating mutations of the stimulatory G protein in the McCune–Albright syndrome. N Engl J Med. 1991;325(24):1688–1695. doi: 10.1056/NEJM199112123252403. [DOI] [PubMed] [Google Scholar]
- 69.Delaney D, Diss TC, Presneau N, Hing S, Berisha F, Idowu BD, et al. GNAS1 mutations occur more commonly than previously thought in intramuscular myxoma. Mod Pathol. 2009;22(5):718–724. doi: 10.1038/modpathol.2009.32. [DOI] [PubMed] [Google Scholar]
- 70.Friedrich RE, Scheuer HA, Assaf AT, Grob T, Zustin J. Odontogenic myxomas are not associated with GNAS1 mutations. Anticancer Res. 2012;32(5):2169–2172. [PubMed] [Google Scholar]
- 71.Carney JA, Gordon H, Carpenter PC, Shenoy BV, Go VL. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine (Baltimore). 1985;64(4):270–283. doi: 10.1097/00005792-198507000-00007. [DOI] [PubMed] [Google Scholar]
- 72.Stratakis CA. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit (PRKAR1A) in patients with the “complex of spotty skin pigmentation, myxomas, endocrine overactivity, and schwannomas” (Carney complex) Ann N Y Acad Sci. 2002;968:3–21. doi: 10.1111/j.1749-6632.2002.tb04323.x. [DOI] [PubMed] [Google Scholar]
- 73.Perdigao PF, Stergiopoulos SG, De Marco L, Matyakhina L, Boikos SA, Gomez RS, et al. Molecular and immunohistochemical investigation of protein kinase a regulatory subunit type 1A (PRKAR1A) in odontogenic myxomas. Genes Chromosomes Cancer. 2005;44(2):204–211. doi: 10.1002/gcc.20232. [DOI] [PubMed] [Google Scholar]
- 74.Bossis I, Voutetakis A, Bei T, Sandrini F, Griffin KJ, Stratakis CA. Protein kinase A and its role in human neoplasia: the Carney complex paradigm. Endocr Relat Cancer. 2004;11(2):265–280. doi: 10.1677/erc.0.0110265. [DOI] [PubMed] [Google Scholar]