


Abstract
Thousands of unique mutations in transcription factors (TFs) arise in cancers, and the functional and biological roles of relatively few of these have been characterized. Here, we used structure-based methods developed specifically for DNA-binding proteins to systematically predict the consequences of mutations in several TFs that are frequently mutated in cancers. The explicit consideration of protein–DNA interactions was crucial to explain the roles and prevalence of mutations in TP53 and RUNX1 in cancers, and resulted in a higher specificity of detection for known p53-regulated genes among genetic associations between TP53 genotypes and genome-wide expression in The Cancer Genome Atlas, compared to existing methods of mutation assessment. Biophysical predictions also indicated that the relative prevalence of TP53 missense mutations in cancer is proportional to their thermodynamic impacts on protein stability and DNA binding, which is consistent with the selection for the loss of p53 transcriptional function in cancers. Structure and thermodynamics-based predictions of the impacts of missense mutations that focus on specific molecular functions may be increasingly useful for the precise and large-scale inference of aberrant molecular phenotypes in cancer and other complex diseases.
INTRODUCTION
The loss or aberration of protein function through mutation occurs in many diseases, including cancers. However, it is difficult to determine which genes and mutations are responsible for disease-linked phenotypes when many mutations are present (1,2). Mutations in signaling and gene regulatory proteins are prevalent and strongly linked to cancers due to their disruption of the cell cycle, apoptosis and the control of proliferation (1,3–4). The tumor suppressor p53 (encoded by the gene TP53), a sequence-specific transcriptional regulator of the cell cycle, apoptosis and genome integrity, is the most broadly and significantly mutated protein across all cancer types (1,5). Mutations in TP53 allow cancer cells to evade apoptosis and to avoid DNA repair (3,6–7). Mechanisms believed or demonstrated to be responsible for the deactivation of p53-mediated processes include losses in protein stability, losses in DNA binding and the dominant negative interference of wild-type p53 and other proteins by p53 mutants (8,9). Additional transcription factors appear significantly mutated in one or more cancer types, albeit at lower frequencies than TP53. These include RUNX1, CBFB, CTCF, GATA3, MYB, SMAD2, SMAD4, FOXA1, FOXQ1 and several zinc-finger (ZNF) transcription factors (10,11). RUNX1, for example, was found to be significantly mutated in tumor samples from breast cancer and acute myeloid leukemia (10). Additional frequently mutated transcription factors in the Catalog of Somatic Mutations in Cancer (COSMIC (12)) and The Cancer Genome Atlas (13) include TP63, STAT1 and RBPJ (Table 1).
Table 1. Several frequently mutated transcription factors in The Cancer Genome Atlas and the changes in gene expression that were correlated with their mutation status.
| TF | Number of mutations Identified in TCGA | # of GEXP∼ΔTF links in TCGA | Pathways and genes correlated with TF loss of function mutations (DAVID/BIOCARTA) | ||||
|---|---|---|---|---|---|---|---|
| Total | MS | S | NS | FS/D | |||
| TP53 | 1409 | 871 | 97 | 174 | 181 | 100s to 1000s**, depending on cancer type, number of samples | Down (pathways): p53, WNT, BTG, CTCF, PML, ceramide, telomerase, BAD/BCL-2; Up (pathways): cell cycle, Ran; Down (genes)**: EDA2R, RPS27L, XPC, KIAA0141, GHDC, CCNG1, PAR-SN, PGPEP1, CYP4V2, KIAA1456…; Up (genes)**: FAM83D, CEP55, CDC20, TRIP13, MYBL2, CENPA, CDCA8, UBE2C, CKS1B, TTK… |
| RUNX1 | 121 | 63 | 24 | 7 | 23 | 29**; 71*; 5001 | Down (genes)**: NCF4, DPP3, HYAL3, OSBPL5, QSOX1, CCDC24, VAT1, TMEM216, MGC2752, KCTD1; Up (genes)**: SETBP1, OPALIN, TLL2, DNTT, PPP3CC, C1ORF21, AKT3, MPP6, LRCH1 |
| TP63 | 116 | 67 | 40 | 5 | 2 | 19* to 501 | Down (genes)*: NEBL, SHANK2, PBX1, RPS6KA6, THRB, MYO6, NPNT, MKL2, CPE, AR, OCLN; Up (genes)*: SRM, PLOD3, PPAN, MED15, MFSD12, BIN3, TMEM201, MAD1L1 |
| STAT1 | 54 | 33 | 14 | 4 | 2 | 2* to 1861 | Down (genes)1: ZFP3, EMCN, MPV17L, ABCA3, TMEM136, MYCT1, CTSO, GJC3, ZNF302, RGS5…; Up (genes)1: TUBA1C, RPSAP58, SPC24, PPAN, CDK11A, BOP1, CDT1, SLC7A5P1, HIST1H2AJ, HIST1H2AH… |
| RBPJ | 35 | 21 | 6 | 2 | 5 | 1 to 1001 | Down (genes)1: PRDM5, LOC728819, ST6GALNAC6, LDOC1, TSPAN31, IQCA1, C2ORF40, TSLP, SMARCA4, SNAPIN…; Up (genes)1: ANAPC1, SPAG7, TYMS, ABCB10, SNORD114–3, C1ORF96, TSGA13, ZNF479, TWF1, C10ORF91… |
Mutation count statistics were compiled from The Cancer Genome Atlas (TCGA; Oct. 2012) and are annotated as follows: MS: missense; S: silent, NS: nonsense; FS/D: frameshifts and deletions. ‘GEXP∼ΔTF’ refers to statistically significant correlations between the expression of individual genes and TF mutation status, according to the predicted impact of the mutations present in paired genotype and expression data collected from thousands of human tissue samples. **: P-value cut off (corrected): 0.01/21 927 genes; *: P-value cut off (corrected): 0.05/21 927 genes; 1: P-value cut off: 0.001 (uncorrected).
The occurrence of significantly mutated transcription factors in cancers is believed to be due to the transcriptional dysregulation of downstream gene regulatory targets that are involved in critical cellular regulatory pathways (3). However, hypermutation also frequently occurs in cancer, and not all mutations are equally likely to contribute to disease phenotypes or the hallmarks of cancer (14). This complicates efforts to understand the molecular and systems genetics of various cancer types, and has prompted statistical methods to rapidly assess the disease-related impacts of large numbers of mutations (1,11,15–16). Such methods can be applied in various ways to better understand the relationship between mutations and disease.
Systematically predicting the roles of diverse transcription factor mutations in cancers is important for assessing the potential causes of changes in gene expression and cellular dysregulation. While decades of research have characterized the occurrence, roles and significance of transcription factor mutations and loss of function in cancer (3,5,17), mechanistic models that broadly explain gene expression changes and phenotypes in real cancers on the basis of mutation status remain incomplete. Existing methods to comprehensively predict the functional consequences of protein mutations include sequence-based and statistical approaches (15,18–23) as well as explicit thermodynamic modeling of mutations using high-resolution protein structures (24–33).
In this paper, we employed structure-based modeling of protein–DNA interactions (34,35), are able to a sequence-based estimator of mutation severity (15), and a multi-parameter classification-based method (20) to investigate links between mutations in several transcription factors (TP53, TP63, RBPJ, STAT1, RUNX1) and genome-wide expression changes in The Cancer Genome Atlas (TCGA) (13). We applied these methods to identify statistical links between transcription factor mutation status and gene expression changes across thousands of clinical samples collected for several different cancer types, and assessed the usefulness of these methods to mechanistically explain gene expression changes in hundreds of known and probable direct transcriptional targets of p53 (6,36–38).
MATERIALS AND METHODS
Data sources and statistics
Mutation statistics were obtained from the IARC (version R16) and COSMIC (v59) databases (5,17). In vitro measurements of the effects of mutations in p53 on protein folding stability were obtained from the IARC TP53 Database (17). Yeast one-hybrid transactivation data for a library of all SNV-accessible missense mutations in p53 were taken from Kato et al. (39). Somatic mutation and gene expression data for thousands of samples collected across several different cancer types were obtained from TCGA (13,40–47). All predictions of the protein-level impacts of missense mutations that were produced or employed in this study, including structure-based modeling of protein–DNA interactions (34,35), a sequence-based estimator of mutation severity (MutationAsessor) (15), and a multi-parameter classification-based method (PolyPhen2) (20), are available through the following url: http://shiny.systemsbiology.net/TFPredictions/.
Structure-based modeling of protein folding stability and protein–DNA interactions
All structure-based modeling was conducted using the macromolecular modeling program Rosetta (48). Methods developed specifically for the prediction of changes in free energies upon mutation in proteins and protein–DNA interfaces (34,35) were applied to predict the thermodynamic consequences of all possible single amino acid changes in the DNA-binding core domains of the transcription factors p53 (49), p63 (50), RBPJ (51), STAT1 (52) and RUNX1 (53) in complexes with their DNA recognition sequences. Missense mutations were modeled explicitly as amino acid substitutions in order to obtain the lowest energy conformation of the mutated amino acid and all neighboring wild-type amino acids and DNA in the structural complex. Changes in the energy of protein folding and formation of the complex, ΔΔGcomplex, were calculated as the difference in the energy between models containing the mutant and wild-type amino acids. Inter-subunit and protein–DNA interactions were implicit in these calculations due to the presence of interfaces in the structural complexes. For example, the use of a high-resolution crystal structure (pdb: 3KMD (49)) that includes a tetramer of p53 subunits bound in tandem to DNA allowed the implicit prediction of mutational impacts on some (though not all) of the protein–protein interactions between p53 monomers. The change in the free energy of binding to DNA, ΔΔGbinding, for each transcription factor mutation was calculated by subtracting the free energies of the unbound protein and DNA molecules from the free energy of the complex for both the wild-type and mutant proteins. The change in the energy of protein folding alone, ΔΔGprotein, was calculated by subtracting ΔΔGbinding from ΔΔGcomplex. Amino acid positions were considered to be in the protein core if the solvent-exposed surface area of the native amino acid was less than 40% of the solvent-exposed surface area of an unfolded equivalent of the same type of amino acid (27). All other amino acid positions were considered to be surface positions.
Comparison of loss of function predictions to mutation prevalence, and probabilistic conditioning of predicted impacts
Structure-based predictions of the thermodynamic impact of individual missense mutations were compared to the background-corrected prevalence of TP53 mutations in the IARC Database (17). The frequency of a protein mutation in cancer was assumed to be the result of disease-associated selection bias (or prevalence) of that mutation as well as the background rate of codon-specific nucleotide variation. The disease-associated prevalence of TP53 mutations was thus estimated by dividing the raw mutation counts for each unique amino acid substitution by the average background rates of mutation for all corresponding nine-mer nucleotide SNVs throughout the human genome. This did not take into account heterogeneous rates of mutation across the genome (1), but was applicable for background-corrected comparisons between different mutations occurring within a single gene. The expected number of observed counts for unique missense mutations in TP53 was computed by multiplying the total number of observed missense mutations in TP53 by the codon-specific background likelihood for each mutation.
To assess the predictive value of structure-based energy scores for various classes of protein mutations, a Bayesian predictor was used to condition structure-based ΔΔG predictions upon their ability to explain mutation prevalence for p53 mutations:
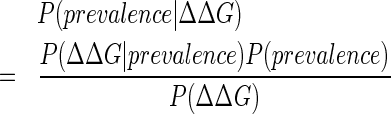 |
This class-based posterior likelihood model for TP53 was extended to condition ΔΔG predictions for other transcription factors whose own mutation frequencies were insufficient for direct conditioning.
Association of transcription factor mutations with gene expression changes in cancer
In a pan-cancer data set that included comprehensive genome or exome sequencing and gene expression measurements for several hundreds of samples collected for each of 19 different cancer types (13), pairwise Spearman rank-correlation tests were performed between the predicted impact of TF genotypes and matched genome-wide expression changes. Multiple methods to predict the functional impact of TF genotypes were compared, including: the presence or absence of missense or nonsense mutations (‘nonsilent’); the presence or absence of missense, nonsense or frameshift mutations (‘MNF’); the presence or absence of mutations in the DNA-binding domain (‘DBD’); MutationAssessor (15); PolyPhen2 (20); structure-based predictions (ΔΔGcomplex) (35); posterior probabilities of mutation impact and prevalence given ΔΔG (P(prev|ΔΔG)); and hybrid combinations comprising structure-based scores for structured positions and statistics-based scores for unstructured positions. For structure-based predictions, nonsense mutations were assumed to be equivalent to the worst possible energy of mutation that could occur throughout the structurally resolved regions of the protein. The significance of individual correlations between gene expression and genotype scores was corrected for multiple hypotheses with 21 970 possible genes and 14 different mutation scoring metrics.
Definition and enrichment of known and probable direct p53 transcriptional targets
The set of known and multiple evidence-based transcriptional targets for p53 was estimated using a logical Bayesian approximation: the posterior probability P (known p53 target | evidence) for all human genes was estimated by conditioning (i) experimental measurements of genome-wide binding by p53 (36,37), (ii) the presence of high-scoring p53 DNA recognition sequences in 5 kbp upstream noncoding regulatory sequences FIMO (54) (P ≤ 1 × 10−5) and (iii) the co-occurrence of p53 DNA recognition sequences with high-resolution genome-wide DNAse hypersensitivity measurements (55) based upon their ability P (evidence | known p53 target) to recapitulate previously curated direct p53 transcriptional targets (6,36). The enrichment of known transcriptional targets for p53 among genes whose expression changes were correlated with TF mutation impacts in TCGA was calculated using a hypergeometric test for known and probable direct transcriptional target genes of p53 among all genes whose expression was correlated with TP53 mutation status in TCGA data (Spearman P-value < = 0.001).
RESULTS
Structurally predicted impacts of TP53 mutations recapitulate experimental results and critically involve protein–DNA interactions
The breadth, pervasiveness and nonuniformity of TP53 mutations in cancers provide a deep set of observations to be addressed by de novo mutation assessment and classification methods (Figure 1). 20 945 out of the 28 581 (73%) somatic mutations observed in TP53 are missense mutations, representing 1458 unique individual amino acid substitutions distributed over 344 protein positions (IARC TP53 database R16, 17) (Figure 1). 1409 TP53 mutations were observed in TCGA by the time of this study, 871 of which were missense mutations (Table 1). The molecular and functional impacts of a small portion (n = 59) of TP53 mutations have been characterized to determine their impact on protein stability and function, with results suggesting that the p53 core domain is relatively unstable (56–62). This instability may partly explain why widespread mutations appear to compromise its function. However, the impacts of thousands of additional unique missense mutations have been observed in cancers, and these remain unexplained. Comprehensive new predictions may help to understand the diverse spectrum of mutations that occur throughout this and other proteins.
Figure 1.

The mutation prevalence and predicted thermodynamic impacts of all known missense mutations in the core DNA-binding domain of the p53 protein. The raw counts (A) and background-corrected relative prevalence (B) of all unique mutations are indicated by the darkness of each amino acid position in the high-resolution crystal structure of the p53 tetramer bound to a pair of p53 DNA recognition sequences (PDB accession: 3KMD).
Using an explicit structure-based method developed to estimate the impact of amino acid variations in DNA-binding proteins (35), the thermodynamic consequences of all possible single amino acid substitutions in the p53 core domain on protein stability (Figure 1C) and DNA binding (Figure 1D) were predicted based on a crystal structure of the p53 protein tetramer bound to a full consensus DNA site (49). This comprised a set of 3762 hypothetical single amino acid substitutions distributed over the core DNA-binding domain (positions 93–290), 1359 of which are accessible by single nucleic acid variations (SNVs) based on the human TP53 mRNA sequence (NM_000546.5). These structure-based predictions were significantly correlated with in vitro protein folding measurements of losses in protein folding stability (57–62) (Pearson R2 = 0.49, P = 2.3 × 10−7, Supplementary Figure S1), and were able to explain the basis of loss-of-function for highly prevalent p53 mutations (Supplementary Table S1, Supplementary Figure S2). Loss of DNA binding was successfully predicted for highly prevalent mutations: for example, R273H is the third most frequent mutation in the IARC database (Supplementary Table S1) and the predicted loss in DNA-binding energy for this mutation was +3.1 kcal/mol (96th percentile over all residues at the protein–DNA interface). A similar mutation, R273L, is the most prevalent p53 mutation after taking into account background SNP mutation rates (Supplementary Table S1). The predicted loss in DNA-binding energy for this mutation was +2.8 kcal/mol (95th percentile). In both cases, structure-based modeling provided a quantitative and accurate prediction of the impact of these mutations on the function of the protein, in which protein–DNA interactions were an essential feature. Structure-based predictions were also able to explain highly prevalent mutations on the basis of protein stability, V157F (Supplementary Figure S2C) and E286K (Supplementary Figure S2D). The predicted loss of function for p53 mutations also generally agreed with a previously conducted comprehensive assay in which all possible p53 missense mutations were tested for their ability to drive expression in a yeast-based assay (Supplementary Figure S3) (39). Scores obtained by additional mutation assessment methods also appeared predictive of function in this assay, albeit with distinctly different distributions (Supplementary Figure S3C-E).
Individual TP53 mutation prevalence is quantitatively related to loss-of-protein stability and DNA binding in the core domain of p53
The nonuniform distribution of prevalence for individual TP53 mutations in cancers (Figure 2A) suggests that certain mutations in this gene are under selection for the loss of p53-mediated functions (e.g. apoptosis) that limit oncogenesis in evolving populations of cancer cells. We found that the most prevalent mutations in cancer were predicted on average to be more destabilizing of the p53–DNA complex (Figure 2B), which is consistent with this theory. Expectedly, the explicit consideration of protein–DNA interactions was crucial for structure-based predictions to explain the basis of loss of function for almost a third (8/30) of the most prevalent missense mutations in TP53 (Supplementary Table S2 and Supplementary Figure S4).
Figure 2.

TP53 mutation prevalence in cancers is quantitatively related to protein instability and loss of DNA binding. (A) The frequency distribution of the prevalence of TP53 missense mutations in cancers after normalizing for genome-wide background codon mutation rates. Numbers indicate the numbers of unique mutations in each prevalence bin. (B) The predicted thermodynamic impact (ΔΔG) of missense mutations in the core DNA-binding domain of TP53 on protein folding stability and DNA binding steadily increases on average for mutations that are more prevalent in cancers. The ΔΔG values shown here are the sum of the estimated effects of mutations on protein stability and DNA binding. Many nondisruptive missense mutations are also predicted, particularly for less prevalent mutations.
In addition to highly prevalent loss-of-function mutations, hundreds of less prevalent missense mutations in TP53 were predicted to be functionally silent, implicating them to be passenger mutations that may have accumulated through hypermutation rather than by selection. The estimated thermodynamic impacts of TP53 mutations were predictive of existing mutation prevalence at various thresholds (Supplementary Figure S5). In order to investigate the advantage of this relationship for the purposes of mutation assessment in cancer, the posterior probability P(prevalence|ΔΔG) for all TP53 missense mutations in cancer was estimated using a Bayesian predictor that conditioned structure-based ΔΔG predictions upon their ability to explain mutation prevalence. This likelihood increased with greater ΔΔG, particularly for amino acids (i) in the core of the protein, (ii) at the protein–DNA interface, (iii) mutations of charged wild-type amino acids and (iv) mutations to nonpolar amino acids (Figure 3, Supplementary Table S3). Thus, structural and thermodynamic predictions of the impacts of missense mutations were predictive of their prevalence and putative impact) in cancers.
Figure 3.

Structure-based estimates of the impact of mutations on protein stability and DNA binding predict the prevalence of missense mutations in TP53. Colored lines indicate the posterior probability (y-axis) that a mutation in the core DNA-binding domain of p53 exceeds prevalence thresholds (colored line labels), given the predicted change in energy upon mutation (ΔΔG, x-axis). Mutations that are not predicted to be destabilizing (ΔΔG < 0) are much less likely to be prevalent than those that are predicted to be destabilizing (ΔΔG > +2) for mutations of charged amino acids (A), mutations to nonpolar amino acids (B), and mutations in the DNA interface (C).
The impacts of some prevalent TP53 missense mutations are not well predicted or explained by protein-based predictions
Incongruence between protein-based predictions and the prevalence of TP53 mutations in cancer may be indicative either of modeling errors, or of functions and properties of the protein that are not yet fully captured. Not all relevant functional information is contained solely within the three-dimensional structures of individual proteins; for example, the impacts of mutating amino acids on the protein surface that are responsible for unseen protein–protein interactions may be difficult to estimate. In the case of the p53 core domain (protein positions 93–290), 92% of prevalent mutations are buried in the protein core or are involved in protein–DNA interactions (165 out of 180 total mutations which were at least 2-fold more prevalent in IARC mutation statistics than expected, based on codon-specific background rates of mutation). Out of the 15 most prevalent mutations on the surface of p53 core domain (Supplementary Table S4A), only four (P152L, R181L, R202L and R202S) were predicted to cause a small or negligible effect on protein stability (ΔΔG ≤ +2 kcal/mol) according to structure-based modeling. In the case of R181L (4.5-fold more prevalent than expected; +1.3 kcal/mol predicted), it appears that the impacts of this mutation on the interaction between p53 subunits may be underestimated. In the case of P152L (2.5-fold more prevalent than expected; +1.4 kcal/mol predicted), the mutation occurs in a loop region, where it is possible that either a proline residue is necessary for proper loop structure, or that these loops may interact with other unseen proteins. Some, but not all mutations at its neighboring residue, P151, are also both more prevalent (six mutations, 0.6- to 6.4-fold higher than expected, mean 2.8-fold), and also predicted to be highly destabilizing of the protein (+1.4 to +14.2 kcal/mol predicted, mean 6.9 kcal/mol) (Supplementary Table S4B). In the case of the surface mutations R202L and R202S (2.2- and 3-fold more prevalent than expected; +1.4 and +0.9 kcal/mol predicted), the loss of an arginine residue may unbalance an electrostatic salt bridge made with E221 more strongly than predicted. No other unexplained prevalent mutations could be identified that would implicate them in unseen conserved protein–protein interaction domains. The impacts predicted for these 15 prevalent surface mutations according to other mutation assessment scores were also mixed (Supplementary Table S4).
An additional source of inaccuracy in structure-based predictions is the energetics of water-mediated contacts between proteins and DNA. For example, five distinct missense mutations at R248, which makes a water-mediated contact with the minor groove of the p53-DNA recognition site (Supplementary Figure S6), are prevalent in TP53 mutation statistics (18- to 96-fold more prevalent than expected; Supplementary Table S4C). Structure-based predictions indicated unambiguously negative impacts for only three out of five mutations at R248 that were observed in IARC or TCGA (Supplementary Table S4C). In the case of R248Q (23-fold more prevalent than expected), only a minor impact on DNA binding was predicted (+1.0 kcal/mol), and in the case of R248G (19-fold more prevalent than expected), a considerable impact on DNA binding (+2.1 kcal/mol) was predicted to be balanced out by a reduction in the entropy of the protein complex (-3.6 kcal/mol). These may be inaccurate predictions. While both implicit and explicit solvation (63,64) and the implicit entropy of amino acid side chains (65) were considered in the structure-based modeling approach, the energetic balances and changes in entropy upon mutation for these interactions may require further refinement and data-driven training in order to maximize prediction accuracy.
Prevalent RUNX1 mutations involve the loss of protein–DNA interactions
In cancers, there are fewer occurrences of missense mutations in transcription factor genes other than TP53 (Table 1). Nevertheless, several additional TFs are significantly mutated in one or more cancers (1), and many of these mutations are presumed to be impactful. In the core DNA-binding domain of RUNX1, 144 occurrences of 58 unique missense mutations have been observed at 39 different amino acid positions (COSMIC Database) (5). We found that the frequency of observed mutations in RUNX1 was not correlated with predictions of protein folding stability (Supplementary Figure S7A), but was correlated with the predicted losses in DNA-binding energy (Supplementary Figure S7B). This is because the top five most frequently observed RUNX1 missense mutations (Arg174Gln, Asp171Asn, Arg80Cys, Asp171Gly, Arg177Gln) are involved in direct interactions with DNA. Thus the explicit consideration of protein–DNA interactions was also essential to capture the impacts of these mutations on the function of RUNX1.
Predictions of mutational impacts broadly link genome-wide expression to transcription factor mutation status across thousands of tumor samples
Determining statistically significant and mechanistic links between genotypes and phenotypes in cancer is an important goal in cancer systems biology. An expected effect of oncogenic mutations in transcription factors is a change in the expression levels of their transcriptional regulatory targets. We tested for correlations between the expression of all genes versus the predicted degree of loss of function for each of five transcription factors (TP53, RUNX1, TP63, STAT1, RBPJ), using data collected from thousands of samples collected across several different cancer types in The Cancer Genome Atlas. Specifically, pairwise correlations were calculated between whole-genome mRNA transcript levels and mutation scores for sample-specific TF genotypes across hundreds of tumor samples in TCGA. This identified genes whose expression changes in tumors were correlated with the predicted severity of mutations in particular transcription factors. The number of genes whose expression levels (‘GEXP’) were correlated to TF mutation status (ΔTF) according to at least one mutation assessment method are summarized in Table 1. The expression levels of hundreds to thousands of genes in cancers were significantly correlated with the mutation status of TP53, depending on the type of cancer and the amount of data available for each cancer type. Genes involved in the p53, WNT, BTG, CTCF, PML, telomerase and BAD/BCL-2 signaling pathways were decreased in expression in correlation with the severity of missense mutations in TP53, while genes involved in the regulation of proliferation (CEP55, FAM83D, TRIP13, CENPA) and cell cycle (CDC20, MYBL2, CDCA8, CKS1B) increased in expression. Among some of the most significantly correlated genes were well-known cancer biomarkers and targets of p53 regulation, including MDM2, DDB2, FDXR, CDKN1A, PHLDA3 and EDA2R. The expression levels of these genes are highly indicative of cancer and directly related to the pathologies of the disease (66–69).TP53 transcript levels themselves were consistently and significantly lower in abundance in samples specifically containing nonsense or frameshift mutations in the tetramerization domain of p53 (protein positions 318–358) in each of several different cancer types (breast invasive carcinomas (brca, P = 1.4 × 10−19), lung adenocarcinomas (luad, P = 1.1 × 10−17), head and neck squamous carcinomas (hnsc, P = 2.2 × 10−15), colon and rectal adenocarcinomas (coad/read, P = 1.4 × 10−9), lung squamous cell carcinomas (lusc, P = 1.7 × 10−7), uterine endometrioid carcinomas (ucec, P = 8.7 × 10−3), bladder urothelial carcinomas (blca, P = 3.5 × 10−3) and ovarian carcinomas (ov, P = 1.4 × 10−3)). Among several scoring metrics assessed for TP53 mutations, the presence of nonsense and frameshift mutations in the tetramerization domain of p53 was the most significant correlate for TP53 transcript levels. This effect could possibly be due to the loss of p53 tetramerization or to dominant-negative phenotypes that truncated and frameshifted domains may have on wild-type p53 in heterozygotes. While structure-based predictions of the effects of individual mutations in the tetramerization domain of the p53 protein could not be produced due to the lack of high-resolution structural data for this region of the protein, these mutations broadly impact the formation of p53 oligomers according to comprehensive experimental characterization (70).
Because TP53 is the most significantly mutated gene in most cancers (1), gene expression changes that co-occur with TP53 mutations may do so for indirect and noncausal reasons. To estimate the extent to which the GEXP∼ΔTP53 associations discovered in TCGA data are due directly to losses of function in the p53 protein, we specifically examined genes for which evidence of direct transcriptional regulation by p53 exists (Supplementary Table S5). Hundreds of known transcriptional targets of p53 were included and significantly enriched among genes whose transcript levels in cancers were correlated with predicted losses of function in p53 (Table 2). This enrichment of known p53 transcriptional targets varied between different mutation assessment methods (Figure 4). GEXP∼ΔTP53 associations based on structure-based predictions identified known p53 transcriptional targets with higher specificity than those based on simply the presence or absence of TP53 mutations (Figure 4: nonsilent, NMF, DBD). In colon and rectal adenocarcinomas, for example, structure-based predictions alone recovered over 90% of the p53-regulated genes identified using a logical mutation classifier (the presence of nonsilent mutations), with a 30% reduction in false positives (Figure 5). The reduced false positive rate observed when using a quantitative protein function based impact assessment in place of a simple logical classifier likely is due to the ability of the former to accurately distinguish functionally silent or conservative missense mutations from those that are highly destabilizing. Thus, the use of explicit structure based methods to predict changes in protein stability and protein–DNA interactions improved the detection of direct and mechanistic effects of transcription factor mutations on gene expression in cancers.
Table 2. Transcriptional targets of p53 were enriched among genes whose expression levels in cancers were correlated with the mutational loss of function of TP53.
| Cancer type | Total numbers of GEXP∼ΔTP53 | Number of GEXP∼ΔTP53 correlations which are possible p53 transcriptional targets | Enrichment in all GEXP∼ΔTP53's (hypergeom. P) | Number of negative correlations (GEXP lower | ΔTP53) | Number of positive correlations (GEXP higher | ΔTP53) |
|---|---|---|---|---|---|
| [all19] | 15 648 | 362 | 2.42 × 10-14 | 147 | 232 |
| brca | 7449 | 195 | 2.44 × 10-08 | 93 | 103 |
| lgg | 3059 | 70 | 0.044 | 41 | 29 |
| luad | 2193 | 67 | 3.96 × 10-05 | 32 | 36 |
| ucec | 2058 | 57 | 0.0017 | 29 | 28 |
| gbm | 365 | 27 | 5.04 × 10-10 | 20 | 7 |
| hnsc | 519 | 27 | 9.01 × 10-07 | 20 | 8 |
| coad/ read | 224 | 24 | 1.41 × 10-12 | 22 | 2 |
| skcm | 63 | 19 | 0 | 19 | 0 |
| stad | 270 | 10 | 0.015 | 9 | 1 |
| blca | 12 | 6 | 6.35 × 10-10 | 6 | 0 |
Shown are statistics for the identification of probable p53 transcriptional targets whose expression levels were correlated with TP53 mutation status (ΔTP53) in several distinct cancer types. Four hundred and eighteen genes were estimated to be probable p53 transcriptional targets based on multiple ChIP experiments, the presence of accessible p53 transcription factor DNA-binding site sequences in gene upstream regions, and previously curated evidence (Supplementary Table S4).
Figure 4.

Enrichment of p53 transcriptional targets in GEXP∼ΔTP53 correlations. The enrichment of known and probable transcriptional targets for p53 is shown for different mutation scoring metrics across several cancer types in TCGA. Four hundred and eighteen genes were assumed to be transcriptional targets for p53 based on multiple sources of evidence (Supplementary Table S1). The higher enrichment of known p53 targets in GEXP∼ΔTP53 associations when using continuous scoring metrics (MutationAssessor [‘MA’], PolyPhen [‘PP’], Rosetta) compared to simple logical classifiers (nonsilent, DBD) was due primarily to a reduction in the number of non-p53-regulated genes whose expression levels appear to correlate with TP53 mutation.
Figure 5.

Structure-based predictions more accurately identify correlations between TP53 genotype and the transcript levels of known p53 transcriptional targets. Results from colon and rectal adenocarcinomas (coad/read) are shown as an example. Each point indicates a gene whose transcript level in cancer (‘GEXP’) was significantly correlated with the mutation impact of TP53 genotype (ΔTP53), according to at least one of the following two mutation scoring metrics: (i) the presence of nonsilent mutations in the TP53 gene (P-values for GEXP∼ΔTP53 correlation on vertical axis), or (ii) a structure-based mutation scoring metric (P-values for GEXP∼ΔTP53 horizontal axis). Both axes express negative log10 Spearman P-values for the correlation of changes in gene expression to the predicted impact of co-occurring mutations in TP53 across hundreds of tissue samples. A diagonal dashed line represents a linear fit of the data. Circular points to the right of the dashed line indicate genes whose expression in cancer was more highly correlated with structure-based predictions of the impact of TP53 mutations, and points to the left of the dashed line indicate genes whose expression in cancer was more highly correlated with the presence of nonsilent mutations in TP53. Genes whose transcription is or may be directly regulated by p53 are shown as triangles. Inset: structure-based predictions (**; P = 2 × 10−11) resulted in a higher enrichment of known p53-regulated genes in these associations, compared to the results obtained when using the simple logical classifier (*; P = 7 × 10−8), due primarily to an increase in statistical specificity.
Structure-based mutation prediction methods are limited by the availability of functionally relevant macromolecular structure data, both in terms of coverage and functional relevance. In cases where mutations arise in regions of proteins for which high-resolution structural information does not exist, orthogonal methods must be used to predict their consequences. In our analysis, a hybrid metric comprising structure-based ΔΔGs for the structurally well-resolved region of p53 (positions 93–290) and MutationAssessor (15) scores for the rest of the protein (positions 1–92, 291–393) yielded superior accuracy in terms of the recovery of known p53 transcriptional targets in cancer data than either metric alone (Figure 4, ‘A’ U ‘MA’).
Mutations in additional transcription factors are linked to expression changes in TCGA data
For four additional transcription factors besides TP53 (TP63, STAT1, RUNX1, RBPJ), fewer gene expression changes were significantly correlated with the impact of mutations in these TFs (Table 1). The ability to detect the transcriptional effects of mutations in these TFs is currently limited by the fewer numbers of mutations that have been observed thus far in the genes that encode them. Nevertheless, the expression levels of at least 29 genes in acute myeloid leukemia (AML or laml) were significantly correlated with the presence and severity of mutations in the DNA-binding domain of the RUNX1 protein. Similar associations for RUNX1 were not seen in other cancer types, indicating a specific role for the mutation of RUNX1 in AML, as has been previously reported (71). Hundreds of genes were detected at lower statistical thresholds in for TP63, STAT1 and RBPJ mutations, some of which may represent new regulatory roles for these TFs in cancer. Future increases in the total number of mutations observed in these and other TFs should improve the power with which their mechanistic effects can be detected in the future.
DISCUSSION
In order to better understand and utilize high-throughput genomics, transcriptomics and other kinds of system-wide data collected from human cancers, it is essential to accurately assess the functional and mechanistic impacts of large numbers of mutations in proteins. While many prediction methods are applicable to the assessment of disease-linked mutations, few of them predict and demonstrate how these mutations result in mechanistic and measurable links between molecular features. In this study, multiple protein function based prediction methods outperformed the ability of a simple logical classifier (the presence of nonsilent mutations) to identify known targets of p53 among gene expression changes that occur across thousands of tumor samples in the TCGA, and we found that protein structure based predictions that explicitly consider protein–DNA interactions were well suited for this task. Our results suggest that while statistical solutions to the problem of classifying known disease-linked mutations exist (20), physics-based and thermodynamic predictions of specific protein-level functions may be particularly useful for predicting quantitative phenotypes in disease.
Existing methods to comprehensively predict the functional consequences of protein mutations include sequence-based and statistical approaches (15,18–23) as well explicit thermodynamic modeling of mutations using high-resolution protein structures (24–33). While none of these methods perfectly predict the true consequences of mutations on functions such as transcriptional activity (Supplementary Figure S3), each of them presents distinct advantages. Sequence-based methods require only sequence-based information as input, whereas structure-based methods take advantage of high-resolution structural information, and do not necessarily require deep existing examples of variation. Machine learning-based classification methods (e.g. PolyPhen2 (20)) take advantage of both sequence- and structure-based information as predictors; however, not all types of protein functions are necessarily captured in this way, and training of multi-parameter models as classifiers on binomial data (known deleterious versus nondeleterious mutations) results in bimodal and non-thermodynamic prediction scores that may be less than optimal for the purpose of making quantitative or mechanistic predictions. The accuracy and applicability of explicit structure-based modeling methods to predict specific kinds of protein functions are increasing (25,27,31,35,72), and high-resolution structural and biophysical models have been particularly effective for studying proteins whose specific modes of function depend on precise interactions, such as protein–protein (72) or protein–DNA interfaces (34,35). Thus, new hybrid approaches wherein thermodynamic predictions are given higher weight in specific contexts may be appropriate for improving mutation assessment.
While this analysis focused solely on the prediction of the impacts of mutations on protein stability and DNA binding, the inference of mutational impacts beyond losses of function in proteins will be required to fully capture the effects of all disease-linked mutations. ‘Gain of function’ mutations in the p53 protein, for example, are believed to be prevalent in cancers, and to cause mutant p53 proteins to engage in new or aberrant molecular interactions with other proteins, DNA or regulatory pathways in the cell (8,9). This includes the ability of destabilized or nonfunctional mutant p53 proteins to directly interfere with the function of other tumor suppressors (including wild-type p53) in multimeric protein complexes (73,74). These dominant-negative effects may contribute to the high occurrence of TP53 missense mutations in cancers, though the extent to which they statistically explain disease remains uncertain (75). Additional functional impacts of mutations that must be systematically predicted for transcription factors include altered DNA binding specificities and transactivation patterns, intra- and intermolecular kinetics, signal sensitivity and subcellular localization. Similarly, additional gain-of-function mechanisms in other protein classes (such as auto-activation in the RAS family of GTPases) will be essential to understand and predict the consequences of rapidly expanding mutational spectra in cancers and other diseases.
CONCLUSION
In this analysis, we employed multiple protein mutation assessment methods to predict the functional impacts of mutations in transcription factors that are frequently mutated in cancers, and identified links between the impacts of large numbers of TF mutations and gene expression measurements in The Cancer Genome Atlas (TCGA). A protein structure based prediction method developed for DNA-binding proteins was particularly useful to explain the roles and prevalence of mutations in TP53 and RUNX1, and resulted in a greater specificity to detect known p53-regulated genes among genetic associations between TP53 genotypes and genome-wide expression changes in pan-cancer data. While several mutation assessment methods resulted in correlations between TF genotypes and the aberrant expression of oncogenes in cancer data, the highest enrichment of direct transcriptional targets of p53 among these associations was obtained using a hybrid statistical and structure-based approach. Additionally, biophysical predictions indicated that the relative prevalence of individual TP53 mutations in cancer is proportional to their thermodynamic impacts of protein stability and DNA binding, which is consistent with the theory of direct negative selection against the transcriptional activity of p53 during the progression of cancers. Structure-based predictions of the impacts of protein mutations that focus on specific molecular functions may become increasingly useful for the precise and large-scale inference of aberrant molecular phenotypes in cancer, as well as in other genetically complex diseases. The integration of function-specific structure-based predictions into omic analyses may also help to elucidate the mechanistic underpinnings of disease-perturbed networks and cellular phenotypes in complex disease.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We would like to thank Gustavo Glusman for helpful discussions and feedback on this manuscript.
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
FUNDING
National Institutes of Health [5U24CA143835 to I.S., 5P50GM076547 to N.S.B.]. Gordon and Betty Moore Foundation Fellow of the Life Sciences Research Foundation [to J.A.]. American Cancer Society fellowship [to C.L.P.]. Funding for open access charge: The Life Sciences Research Foundation on behalf of the Gordon and Betty Moore Foundation.
Conflict of interest statement. None declared.
REFERENCES
- 1.Lawrence M.S., Stojanov P., Polak P., Kryukov G.V., Cibulskis K., Sivachenko A., Carter S.L., Stewart C., Mermel C.H., Roberts S.A., et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCarthy M.I., Abecasis G.R., Cardon L.R., Goldstein D.B., Little J., Ioannidis J.P.A., Hirschhorn J.N. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat. Rev. Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Reimand J., Bader G.D. Systematic analysis of somatic mutations in phosphorylation signaling predicts novel cancer drivers. Mol. Syst. Biol. 2013;9:637. doi: 10.1038/msb.2012.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Forbes S.A., Bindal N., Bamford S., Cole C., Kok C.Y., Beare D., Jia M., Shepherd R., Leung K., Menzies A., et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2010;39:D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riley T., Sontag E., Chen P., Levine A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008;9:402. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- 7.Weinberg R.L., Veprintsev D.B., Bycroft M., Fersht A.R. Comparative binding of p53 to its promoter and DNA recognition elements. J. Mol. Biol. 2005;348:589–596. doi: 10.1016/j.jmb.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 8.Freed-Pastor W.A., Prives C. Mutant p53: one name, many proteins. Genes Dev. 2012;26:1268–1286. doi: 10.1101/gad.190678.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muller P.A.J., Vousden K.H. p53 mutations in cancer. Nat. Cell Biol. 2013;15:2–8. doi: 10.1038/ncb2641. [DOI] [PubMed] [Google Scholar]
- 10.Lawrence M.S., Stojanov P., Mermel C.H., Robinson J.T., Garraway L.A., Golub T.R., Meyerson M., Gabriel S.B., Lander E.S., Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kandoth C., McLellan M.D., Vandin F., Ye K., Niu B., Lu C., Xie M., Zhang Q., McMichael J.F., Wyczalkowski M.A., et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bamford S., Dawson E., Forbes S., Clements J., Pettett R., Dogan A., Flanagan A., Teague J., Futreal P.A., Stratton M.R., et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer. 2004;91:355–358. doi: 10.1038/sj.bjc.6601894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cancer Genome Atlas Research Network. Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R.M., Ozenberger B.A., Ellrott K., Shmulevich I., Sander C., Stuart J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bozic I., Antal T., Ohtsuki H., Carter H., Kim D., Chen S., Karchin R., Kinzler K.W., Bogelstein B., Nowak M.A. Accumulation of driver and passenger mutations during tumor progression. Proc. Natl. Acad. Sci. U.S.A. 2010;107:18545–18550. doi: 10.1073/pnas.1010978107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reva B., Antipin Y., Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39:e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Samocha K.E., Robinson E.B., Sanders S.J., Stevens C., Sabo A., McGrath L.M., Kosmicki J.A., Rehnström K., Mallick S., Kirby A., et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 2014;46:944–950. doi: 10.1038/ng.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petitjean A., Mathe E., Kato S., Ishioka C., Tavtigian S.V., Hainaut P., Olivier M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007;28:622–629. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- 18.Gorlov I.P., Gorlova O.Y., Amos C.I. Predicting the oncogenicity of missense mutations reported in the International Agency for Cancer Research (IARC) mutation database on p53. Hum. Mutat. 2005;26:446–454. doi: 10.1002/humu.20242. [DOI] [PubMed] [Google Scholar]
- 19.Tavtigian S.V., Greenblatt M.S., Lesueur F., Byrnes G.B. In silico analysis of missense substitutions using sequence-alignment based methods. Hum. Mutat. 2008;29:1327–1336. doi: 10.1002/humu.20892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chan P.A., Duraisamy S., Miller P.J., Newell J.A., McBride C., Bond J.P., Raevaara T., Ollila S., Nyström M., Grimm A.J., et al. Interpreting missense variants: comparing computational methods in human disease genes CDKN2A, MLH1, MSH2, MECP2, and tyrosinase (TYR) Hum. Mutat. 2007;28:683–693. doi: 10.1002/humu.20492. [DOI] [PubMed] [Google Scholar]
- 22.Goldgar D.E., Easton D.F., Byrnes G.B., Spurdle A.B., Iversen E.S., Greenblatt M.S. Genetic evidence and integration of various data sources for classifying uncertain variants into a single model. Hum. Mutat. 2008;29:1265–1272. doi: 10.1002/humu.20897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greenblatt M.S., Beaudet J.G., Gump J.R., Godin K.S., Trombley L., Koh J., Bond J.P. Detailed computational study of p53 and p16: using evolutionary sequence analysis and disease-associated mutations to predict the functional consequences of allelic variants. Oncogene. 2003;22:1150–1163. doi: 10.1038/sj.onc.1206101. [DOI] [PubMed] [Google Scholar]
- 24.Danziger S.A., Swamidass S.J., Zeng J., Dearth L.R., Lu Q., Chen J.H., Cheng J., Hoang V.P., Saigo H., Luo R., et al. Functional census of mutation sequence spaces: the example of p53 cancer rescue mutants. IEEEACM Trans. Comput. Biol. Bioinforma. IEEE ACM. 2006;3:114–125. doi: 10.1109/TCBB.2006.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fowler D.M., Araya C.L., Fleishman S.J., Kellogg E.H., Stephany J.J., Baker D., Fields S. High-resolution mapping of protein sequence-function relationships. Nat. Methods. 2010;7:741–746. doi: 10.1038/nmeth.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guerois R., Nielsen J.E., Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J. Mol. Biol. 2002;320:369–387. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 27.Kellogg E.H., Leaver-Fay A., Baker D. Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins. 2011;79:830–838. doi: 10.1002/prot.22921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin A.C.R., Facchiano A.M., Cuff A.L., Hernandez-Boussard T., Olivier M., Hainaut P., Thornton J.M. Integrating mutation data and structural analysis of the TP53 tumor-suppressor protein. Hum. Mutat. 2002;19:149–164. doi: 10.1002/humu.10032. [DOI] [PubMed] [Google Scholar]
- 29.Shi Z., Moult J. Structural and functional impact of cancer-related missense somatic mutations. J. Mol. Biol. 2011;413:495–512. doi: 10.1016/j.jmb.2011.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yip Y.L., Zoete V., Scheib H., Michielin O. Structural assessment of single amino acid mutations: application to TP53 function. Hum. Mutat. 2006;27:926–937. doi: 10.1002/humu.20379. [DOI] [PubMed] [Google Scholar]
- 31.Alibes A., Nadra A.D., De Masi F., Bulyk M.L., Serrano L., Stricher F. Using protein design algorithms to understand the molecular basis of disease caused by protein-DNA interactions: the Pax6 example. Nucleic Acids Res. 2010;38:7422–7431. doi: 10.1093/nar/gkq683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hurst J.M., McMillan L.E.M., Porter C.T., Allen J., Fakorede A., Martin A.C.R. The SAAPdb web resource: a large-scale structural analysis of mutant proteins. Hum. Mutat. 2009;30:616–624. doi: 10.1002/humu.20898. [DOI] [PubMed] [Google Scholar]
- 33.Kiel C., Serrano L. Structure-energy-based predictions and network modelling of RASopathy and cancer missense mutations. Mol. Syst. Biol. 2014;10:727. doi: 10.1002/msb.20145092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Havranek J.J., Duarte C.M., Baker D. A simple physical model for the prediction and design of protein-DNA interactions. J. Mol. Biol. 2004;344:59–70. doi: 10.1016/j.jmb.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 35.Ashworth J., Baker D. Assessment of the optimization of affinity and specificity at protein-DNA interfaces. Nucleic Acids Res. 2009;37:e73. doi: 10.1093/nar/gkp242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Botcheva K., McCorkle S.R., McCombie W.R., Dunn J.J., Anderson C.W. Distinct p53 genomic binding patterns in normal and cancer-derived human cells. Cell Cycle Georget. Tex. 2011;10:4237–4249. doi: 10.4161/cc.10.24.18383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smeenk L., van Heeringen S.J., Koeppel M., van Driel M.A., Bartels S.J.J., Akkers R.C., Denissov S., Stunnenberg H.G., Lohrum M. Characterization of genome-wide p53-binding sites upon stress response. Nucleic Acids Res. 2008;36:3639–3654. doi: 10.1093/nar/gkn232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Veprintsev D.B., Fersht A.R. Algorithm for prediction of tumour suppressor p53 affinity for binding sites in DNA. Nucleic Acids Res. 2008;36:1589–1598. doi: 10.1093/nar/gkm1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kato S., Han S.-Y., Liu W., Otsuka K., Shibata H., Kanamaru R., Ishioka C. Understanding the function–structure and function–mutation relationships of P53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci. U.S.A. 2003;100:8424–8429. doi: 10.1073/pnas.1431692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–322. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cancer Genome Atlas Research Network. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leaver-Fay A., Tyka M., Lewis S.M., Lange O.F., Thompson J., Jacak R., Kaufman K., Renfrew P.D., Smith C.A., Sheffler W., et al. Rosetta3 an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen Y., Dey R., Chen L. Crystal structure of the p53 core domain bound to a full consensus site as a self-assembled tetramer. Struct. Lond. Engl. 1993. 2010;18:246–256. doi: 10.1016/j.str.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen C., Gorlatova N., Herzberg O. Pliable DNA conformation of response elements bound to transcription factor p63. J. Biol. Chem. 2012;287:7477–7486. doi: 10.1074/jbc.M111.315820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Choi S.H., Wales T.E., Nam Y., O'Donovan D.J., Sliz P., Engen J.R., Blacklow S.C. Conformational locking upon cooperative assembly of notch transcription complexes. Struct. Lond. Engl. 1993. 2012;20:340–349. doi: 10.1016/j.str.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen X., Vinkemeier U., Zhao Y., Jeruzalmi D., Darnell J.E., Kuriyan J. Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell. 1998;93:827–839. doi: 10.1016/s0092-8674(00)81443-9. [DOI] [PubMed] [Google Scholar]
- 53.Bravo J., Li Z., Speck N.A., Warren A.J. The leukemia-associated AML1 (Runx1)–CBFβ complex functions as a DNA-induced molecular clamp. Nat. Struct. Mol. Biol. 2001;8:371–378. doi: 10.1038/86264. [DOI] [PubMed] [Google Scholar]
- 54.Grant C.E., Bailey T.L., Noble W.S. FIMO: scanning for occurrences of a given motif. Bioinforma. Oxf. Engl. 2011;27:1017–1018. doi: 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thurman R.E., Rynes E., Humbert R., Vierstra J., Maurano M.T., Haugen E., Sheffield N.C., Stergachis A.B., Wang H., Vernot B., et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bullock A.N., Henckel J., DeDecker B.S., Johnson C.M., Nikolova P.V., Proctor M.R., Lane D.P., Fersht A.R. Thermodynamic stability of wild-type and mutant p53 core domain. Proc. Natl. Acad. Sci. U.S.A. 1997;94:14338–14342. doi: 10.1073/pnas.94.26.14338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ang H.C., Joerger A.C., Mayer S., Fersht A.R. Effects of common cancer mutations on stability and DNA binding of full-length p53 compared with isolated core domains. J. Biol. Chem. 2006;281:21934–21941. doi: 10.1074/jbc.M604209200. [DOI] [PubMed] [Google Scholar]
- 58.Bullock A.N., Henckel J., Fersht A.R. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy. Oncogene. 2000;19:1245–1256. doi: 10.1038/sj.onc.1203434. [DOI] [PubMed] [Google Scholar]
- 59.Joerger A.C., Ang H.C., Veprintsev D.B., Blair C.M., Fersht A.R. Structures of p53 cancer mutants and mechanism of rescue by second-site suppressor mutations. J. Biol. Chem. 2005;280:16030–16037. doi: 10.1074/jbc.M500179200. [DOI] [PubMed] [Google Scholar]
- 60.Joerger A.C., Ang H.C., Fersht A.R. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc. Natl. Acad. Sci. U.S.A. 2006;103:15056–15061. doi: 10.1073/pnas.0607286103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nikolova P.V., Henckel J., Lane D.P., Fersht A.R. Semirational design of active tumor suppressor p53 DNA binding domain with enhanced stability. Proc. Natl. Acad. Sci. U.S.A. 1998;95:14675–14680. doi: 10.1073/pnas.95.25.14675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nikolova P.V., Wong K.B., DeDecker B., Henckel J., Fersht A.R. Mechanism of rescue of common p53 cancer mutations by second-site suppressor mutations. EMBO J. 2000;19:370–378. doi: 10.1093/emboj/19.3.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lazaridis T., Karplus M. Effective energy function for proteins in solution. Proteins Struct. Funct. Bioinforma. 1999;35:133–152. doi: 10.1002/(sici)1097-0134(19990501)35:2<133::aid-prot1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 64.Jiang L., Kuhlman B., Kortemme T., Baker D. A ‘solvated rotamer’ approach to modeling water-mediated hydrogen bonds at protein-protein interfaces. Proteins. 2005;58:893–904. doi: 10.1002/prot.20347. [DOI] [PubMed] [Google Scholar]
- 65.Dunbrack R.L., Cohen F.E. Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci. Publ. Protein Soc. 1997;6:1661–81. doi: 10.1002/pro.5560060807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gartel A.L., Tyner A.L. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol. Cancer Ther. 2002;1:639–649. [PubMed] [Google Scholar]
- 67.Kattan Z., Marchal S., Brunner E., Ramacci C., Leroux A., Merlin J.L., Domenjoud L., Dauça M., Becuwe P. Damaged DNA binding protein 2 plays a role in breast cancer cell growth. PloS One. 2008;3:e2002. doi: 10.1371/journal.pone.0002002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Momand J., Jung D., Wilczynski S., Niland J. The MDM2 gene amplification database. Nucleic Acids Res. 1998;26:3453–3459. doi: 10.1093/nar/26.15.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu J., Marsh S., Ahluwalia R., McLeod H.L. Ferredoxin reductase: pharmacogenomic assessment in colorectal cancer. Cancer Res. 2003;63:6170–6173. [PubMed] [Google Scholar]
- 70.Kamada R., Nomura T., Anderson C.W., Sakaguchi K. Cancer-associated p53 tetramerization domain mutants: quantitative analysis reveals a low threshold for tumor suppressor inactivation. J. Biol. Chem. 2011;286:252–258. doi: 10.1074/jbc.M110.174698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ito Y. Oncogenic potential of the RUNX gene family: ‘overview’. Oncogene. 2004;23:4198–4208. doi: 10.1038/sj.onc.1207755. [DOI] [PubMed] [Google Scholar]
- 72.Kortemme T., Morozov A.V., Baker D. An orientation-dependent hydrogen bonding potential improves prediction of specificity and structure for proteins and protein-protein complexes. J. Mol. Biol. 2003;326:1239–1259. doi: 10.1016/s0022-2836(03)00021-4. [DOI] [PubMed] [Google Scholar]
- 73.Chan W.M., Siu W.Y., Lau A., Poon R.Y.C. How many mutant p53 molecules are needed to inactivate a tetramer. Mol. Cell. Biol. 2004;24:3536–3551. doi: 10.1128/MCB.24.8.3536-3551.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu J., Reumers J., Couceiro J.R., Smet F., Gallardo R., Rudyak S., Cornelis A., Rozenski J., Zwolinska A., Marine J.-C., et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011;7:285–295. doi: 10.1038/nchembio.546. [DOI] [PubMed] [Google Scholar]
- 75.Monti P., Perfumo C., Bisio A., Ciribilli Y., Menichini P., Russo D., Umbach D.M., Resnick M.A., Inga A., Fronza G. Dominant-negative features of mutant TP53 in germline carriers have limited impact on cancer outcomes. Mol. Cancer Res. 2011;9:271–279. doi: 10.1158/1541-7786.MCR-10-0496. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.