

Abstract
• Background DNA flow cytometry describes the use of flow cytometry for estimation of DNA quantity in cell nuclei. The method involves preparation of aqueous suspensions of intact nuclei whose DNA is stained using a DNA fluorochrome. The nuclei are classified according to their relative fluorescence intensity or DNA content. Because the sample preparation and analysis is convenient and rapid, DNA flow cytometry has become a popular method for ploidy screening, detection of mixoploidy and aneuploidy, cell cycle analysis, assessment of the degree of polysomaty, determination of reproductive pathway, and estimation of absolute DNA amount or genome size. While the former applications are relatively straightforward, estimation of absolute DNA amount requires special attention to possible errors in sample preparation and analysis.
• Scope The article reviews current procedures for estimation of absolute DNA amounts in plants using flow cytometry, with special emphasis on preparation of nuclei suspensions, stoichiometric DNA staining and the use of DNA reference standards. In addition, methodological pitfalls encountered in estimation of intraspecific variation in genome size are discussed as well as problems linked to the use of DNA flow cytometry for fieldwork.
• Conclusions Reliable estimation of absolute DNA amounts in plants using flow cytometry is not a trivial task. Although several well-proven protocols are available and some factors controlling the precision and reproducibility have been identified, several problems persist: (1) the need for fresh tissues complicates the transfer of samples from field to the laboratory and/or their storage; (2) the role of cytosolic compounds interfering with quantitative DNA staining is not well understood; and (3) the use of a set of internationally agreed DNA reference standards still remains an unrealized goal.
Keywords: Flow cytometry, nuclear genome size, DNA C-value, nuclear DNA amount, DNA staining, intraspecific variation
INTRODUCTION
Early attempts to estimate DNA amounts in cell nuclei (Caspersson and Schultz, 1938) preceded the discovery of its central role in heredity. Shortly after, a constancy of DNA amount per organism was established and the term ‘C-value’ was coined by Swift (1950), referring to the DNA content of an unreplicated haploid chromosome complement (n). Hence, a nucleus in G1 phase of the cell cycle, with two copies of unreplicated genome has a 2C DNA amount. Subsequently, it was found that there was no relationship between DNA C-value and organismic complexity (Mirsky and Ris, 1951). The lack of correlation was later termed ‘C-value paradox’ by Thomas (1971). The discovery of non-coding DNA provided a clue to the paradox, but the origin, function and significance of variation in DNA content remain enigmatic. Several theories have been proposed to explain the ‘C-value enigma’ (Gregory, 2001). Scientific disciplines, which profit from the knowledge of C-values, are numerous and include molecular biology, systematics and ecology (Bennett et al., 2000a). Despite its importance, C-values are known for only a fraction of all plant species, e.g. only 1·4 % of angiosperms (Hanson et al., 2003), but nonetheless are easily accessible online at http://www.rbgkew.org.uk/cval/homepage.html. As the usefulness of published data depends on their reliability, appropriate methods for C-value measurement and their careful use are of prime importance.
There are two approaches towards the determination of 2C DNA content of a given organism: analysis of DNA extracted from a large number of cells, and measurement of individual nuclei. Chemical analysis (Schmidt and Thannhauser, 1945) and reassociation kinetics (Britten and Kohne, 1968) represent examples of the first approach. As the source tissue may contain cells at different phases of the cycle and with different DNA amounts, estimates provided by chemical analysis do not represent the 2C DNA amount. The so-called Cot curves obtained after reassociation of DNA fragments are hard to interpret in terms of C-values due to the presence of different types of repetitive DNA sequences. The second (‘single nuclei’) approach offers much higher precision, but is technically more demanding. Early measurements of individual nuclei relied on the absorption of UV light by the DNA molecule. Later, the nuclei were stained by the Feulgen method (Feulgen and Rossenbeck, 1924), which is considered specific for DNA, and the absorption of visible monochromatic light was quantified (Swift, 1950). Efforts to eliminate errors due to irregularly shaped nuclei and chromosomes with non-homogeneously stained chromatin lead to the development of scanning microspectrophotometry (Deeley, 1955). The absorption was then measured in many small areas across the object and the values integrated. DNA image cytometry may be seen as an electronic alternative to microspectrophotometry, where the absorption is determined by estimating grey level values of pixels of an image grabbed by a video camera (Vilhar et al., 2001).
Unlike microspectrophotometry and image cytometry, flow cytometry analyses microscopic particles in suspension, which are constrained to flow in single file within a fluid stream through the focus of intense light. Pulses of scattered light and fluorescence are collected and converted to electric current pulses by optical sensors and classified. Because the particles are analysed individually and at high speed, large populations can be measured in a short time and the presence of subpopulations may be detected (Shapiro, 2003). Since there is no need to employ tissues with dividing cells, the ease of sample preparation, and the ability to measure DNA quickly in large populations of cells, made flow cytometry an attractive alternative to microspectrophotometry. Indeed, there has been a shift towards flow cytometry during the last decade (Bennett and Leitch, 1995; Bennett et al., 2000a). This review focuses on the use of flow cytometry for estimation of nuclear DNA content (DNA flow cytometry) in plants with a special emphasis on the estimation of DNA in absolute units (genome size).
FLOW CYTOMETRY OF NUCLEAR DNA
The first flow cytometers quantified DNA in human cells by measuring absorbance of UV light (Kamentsky et al., 1965). This approach was soon abandoned for fluorescence (Dittrich and Göhde, 1969; Van Dilla et al., 1969) and until the present day DNA content has been determined indirectly by measuring fluorescence emission. To estimate nuclear DNA content, suspensions of nuclei and/or permeabilized cells are stained with a DNA-specific fluorochrome and the amount of light emitted by each nucleus is quantified. The result of the analysis is usually displayed in the form of a histogram of relative fluorescence intensity, representing relative DNA content. Because large populations of cells may be measured in a short time, DNA flow cytometry has been used extensively in biomedical research to detect aneuploidy (Kawara et al., 1999), apoptotis (Vermes et al., 2000) and monitor cell cycle kinetics and its perturbations (Rabinovitch, 1994).
Attempts to apply the method in plants were hampered by difficulties in preparation of suspensions of intact cells and nuclei suitable for flow cytometry. In the first successful experiment, Heller (1973) prepared suspensions of field bean nuclei from alcohol acetic acid-fixed root tips after enzymatic treatments with pectinase and pepsin. Nuclear DNA was stained with ethidium bromide and the analysis of relative fluorescence intensity indicated a potential for analysis of cell cycle kinetics. For almost a decade, others did not follow this work, perhaps because in those days flow cytometers were expensive machines with applications largely limited to biomedical research. The sample preparation was laborious, and the fact that the paper was written in German probably did not help it reach a wide audience.
Subsequent reports appeared only in the early 1980s. One, less popular strategy explored the possibility of estimating the DNA content of nuclei within intact cells. The presence of a rigid cell wall, which is autofluorescent and confers an irregular cell shape that disturbs the fluid stream, makes isolated plant cells unsuitable for estimation of DNA content using flow cytometry. Removal of the cell wall using hydrolytic enzymes (cellulases, pectinases) in the presence of an inert osmoticum converts cells to protoplasts, which are spherical and behave regularly within the flow stream. Puite and Ten Broeke (1983) showed that nuclear DNA could be stained in plant protoplasts. However, the histograms of fluorescence intensity could not be interpreted in terms of cell cycle distribution. This was probably the effect of cytoplasmic autofluorescence and low permeability of plasma membrane. Fixation with ethanol–acetic acid permeabilizes cell membrane and decreases the autofluorescence. Nevertheless, the quality of resulting histograms is rather poor (Galbraith and Shields, 1982; Puite and Ten Broeke, 1983), probably due to the ‘off-centre’ position of the nucleus (Galbraith, 1990). A more successful approach relies on the analysis of intact nuclei, which may be released from protoplasts by lysis either in the presence of a detergent or in a hypotonic medium, and leads to very good histograms of DNA content (Puite and Ten Broeke, 1983; Galbraith, 1984; Ulrich et al., 1988).
The early experiments demonstrated that DNA content in plants could be estimated with sufficient precision by only using nuclei isolated from protoplasts. This approach is time consuming, laborious, and cannot be easily applied to a broad range of species. This stimulated Galbraith et al. (1983) to come up with a radically practical solution, in which the suspensions of intact nuclei are prepared by chopping a small amount of fresh tissue in a suitable isolation buffer. Histograms of high quality could be obtained within a short time. Unlike the previous methods, this was incredibly simple, convenient and rapid. The ability to estimate DNA content stimulated a vast array of applications, which ranged from basic research to breeding and seed production, and included estimation of nuclear genome size (Hülgenhof et al., 1988), ploidy screening (De Laat et al., 1987), detection mixoploidy (Roux et al., 2001) and aneuploidy (Roux et al., 2003), assessment of the degree of polysomaty (Barow and Meister, 2003), reproductive pathways (Matzk et al., 2000), and cell cycle kinetics (Sandoval et al., 2003). These studies involved the analysis of intact plants and plant populations as well as cells and tissues cultured in vitro and plants regenerated from them. It is therefore not an exaggeration to say that the work of Galbraith et al. (1983) marked the real beginning of DNA flow cytometry in plants.
ESTIMATION OF NUCLEAR GENOME SIZE
Most of the above-mentioned applications of DNA flow cytometry are quite straightforward and, at least conceptually, their application does not represent a problem. Unfortunately, the opposite is true for the estimation of absolute DNA amount, or nuclear genome size, which is the subject of this review. As flow cytometry analyses relative fluorescence intensity, and hence relative DNA content, the genome size of an unknown sample may be determined only after a comparison with nuclei of a reference standard, whose genome size is known.
This can be achieved by several ways. External standardization involves separate analyses of nuclei of unknown sample and the standard. Even if the instrument settings are kept unchanged, the analysis may be compromised by random instrument drift and by variation in the sample preparation and staining. This is avoided by internal standardization, in which the nuclei of the standard and the sample are isolated, stained and analysed simultaneously (Doležel, 1991; Fig. 1). Some authors employed a compromise between the two, sometimes referred to as ‘pseudo-internal standardization’, in which the nuclei of the target and the reference standard are isolated and stained separately before being mixed and analysed (Price and Johnston, 1996; Price et al., 1998; Johnston et al., 1999). Clearly, this approach does not eliminate the errors due to variation in nuclei isolation and staining.
Fig. 1.

Estimation of absolute nuclear DNA amount (genome size) in Ensete gilletii. The histogram of relative DNA content was obtained after flow cytometric analysis of propidium iodide-stained nuclei of Ensete and soybean, which were isolated, stained and analysed simultaneously. Soybean (Glycine max ‘Polanka’, 2C = 2·50 pg DNA) served as internal reference standard. The gain of the cytometer was adjusted so that the G1 peak of soybean was positioned on channel 200. The ratio of G1 peak means (Ensete : soybean) was equal to 0·484 and hence the 2C DNA amount of E. gilletii was estimated as 1·210 pg. Note that a reliable estimation of genome size of a species requires that several randomly selected plants are analysed, each of them several times and on different days. The replicate measurements of the same plant facilitate the detection of variation in the procedure and estimation of variation between plants.
The measurements of relative fluorescence intensity of stained nuclei are performed on a linear scale and typically, 5000–20 000 nuclei are analysed for each sample (Galbraith et al., 1998). The absolute DNA amount of a sample is calculated based on the values of the G1 peak means:
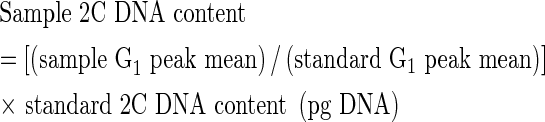 |
Absolute DNA amounts are traditionally reported in pg DNA. However, with the advent of molecular biology and progress in genome sequencing projects, there has been a trend to express DNA amounts in terms of the number of base pairs (bp), and to use the term genome size. Unfortunately, this term lacks a precise definition, having been used to describe the DNA amount in G1 phase nucleus as well as in unreplicated haploid chromosome set (n). The issue becomes more complicated in polyploids, where genome size has been used to describe the haploid (n) and monoploid chromosome set(s) (x). Clearly there is need for the agreement on the terminology (Greilhuber et al., 2005). Surprisingly, also a conversion from pg DNA to bp and vice versa shows some, albeit minor inconsistencies. The calculations assume a 1 : 1 ratio of AT : GC pairs and ignore the presence of modified nucleotides in the DNA molecule. Nevertheless, the errors should be <1 % (Doležel et al., 2003). Strangely, authors also differed in the estimation of the mean relative weight of a nucleotide pair, and thus conversion factors ranging from 0·965 × 109 bp to 0·980 × 109 bp for 1 pg DNA have been used (Straus, 1971; Cavalier-Smith, 1985). Considering the 1:1 ratio of AT:GC pairs and ignoring the presence of modified nucleotides, Doležel et al. (2003) showed that 1 pg DNA = 0·978 × 109 bp.
To estimate genome size of a species, several randomly selected plants are analysed, and each is analysed several times. The replicate measurements of the same plant facilitate the detection of the variation in the procedure, while the analysis of several plants permits monitoring of intraspecific variation. The number of plants and replicate measurements vary among different studies. While the number of replicates is lower in large-scale screening experiments (Suda et al., 2003), it is generally assumed that a minimum of three plants should be analysed, each of them three times (Greilhuber and Obermayer, 1997; Lysák et al., 1999) when intraspecific genome size variation is studied. Several other conditions must be fulfilled to estimate genome size reliably: (a) the nuclei must be isolated in sufficient quantity, they must be intact, and their DNA must not be degraded or modified; (b) DNA staining must be specific and stoichiometric for both the target and standard nuclei; (c) the genome size of the reference standard must be known. Unfortunately, none of the three conditions is easy to satisfy, which may lead to erroneous results.
Preparation of nuclei suspensions
Preparation of suspensions of intact nuclei for estimation of absolute DNA amounts has been almost universally performed following the method of Galbraith et al. (1983). In this procedure, the nuclei are released into a nuclei isolation buffer by mechanical homogenization of a small amount of fresh plant tissue. The composition of the isolation buffer is critical to facilitate the release of nuclei free of cytoplasm and in sufficient quantities, maintain the integrity of isolated nuclei, protect their DNA against endonucleases, and facilitate DNA staining.
The chemical composition of the six most popular nuclear isolation buffers is given in Table 1. Nuclear chromatin may be stabilized by magnesium ions in magnesium chloride buffers (Galbraith et al., 1983; Pfosser et al., 1995) and magnesium sulfate buffers (Arumuganathan and Earle, 1991) and by spermine in the polyamine buffer (Doležel et al., 1989). In Marie's isolation buffer, the presence of glucose helps maintain nuclear integrity and prevents their clumping (Marie and Brown, 1993). EDTA, a metal chelator is used to bind divalent cations, which serve as nuclease cofactors (Doležel et al., 1989; Marie and Brown, 1993). Sodium citrate, a mild chelating agent, is also included in some buffers (Galbraith et al., 1993; Marie and Brown, 1993). Inorganic salts (KCl, NaCl) have been included by some to achieve adequate ionic strength (Doležel et al., 1989; Marie and Brown, 1993). The pH of the solutions varies within a limited range (7·0–8·0), which is compatible with common DNA fluorochromes, and is stabilized by organic buffers (TRIS, MOPS, HEPES). Two non-ionic detergents, Triton X-100 and Tween 20, are included to facilitate nuclear release from the cytoplasm, remove cytoplasmic remnants from the surface of isolated nuclei, disperse chloroplasts and decrease a tendency of nuclei and cytoplasmic debris to aggregate. Reducing agents (β-mercaptoethanol, dithiothreitol) preserve chromatin proteins and counteract the interference of phenolic compounds with DNA staining. As the mercaptoethanol may be harmful to human health, other compounds have been used such as potassium metabisulfite and polyvinyl pyrrolidone (Bogunic et al., 2003).
Table 1.
The most popular buffers used for preparation of nuclei suspensions
| Buffer |
Composition* |
References |
|---|---|---|
| Galbraith's buffer | 45 mm MgCl2; 30 mm sodium citrate; 20 mm MOPS; 0·1 % (w/v) Triton X-100; pH 7·0 | Galbraith et al. (1983) |
| LB01 | 15 mm TRIS; 2 mm Na2EDTA; 0·5 mm spermine.4HCl; 80 mm KCl; 20 mm NaCl; 15 mm β-mercaptoethanol; 0·1 % (v/v) Triton X-100; pH 7·5 | Doležel et al. (1989) |
| Arumuganathan and Earle | 9·53 mm MgSO4.7H2O; 47·67 mm KCl; 4·77 mm HEPES; 6·48 mm DTT; 0·25 % (w/v) Triton X-100; pH 8·0 | Arumuganathan and Earle (1991) |
| Marie's nuclear isolation buffer | 50 mm glucose; 15 mm KCl; 15 mm NaCl; 5 mm Na2EDTA; 50 mm sodium citrate; 0·5 % (v/v) Tween 20; 50 mm HEPES; 0·5 % (v/v) β-mercaptoethanol; pH 7·2 | Marie and Brown (1993) |
| Otto buffers† | Otto I buffer: 100 mm citric acid; 0·5 % (v/v) Tween 20 (pH approx. 2·3) | Otto (1990) |
| Otto II buffer: 400 mm Na2HPO4.12H2O (pH approx. 8·9) | Doležel and Göhde (1995) | |
| Tris-MgCl2 | 200 mm TRIS; 4 mm MgCl2.6H2O; 0·5 % (v/v) Triton X-100; pH 7·5 | Pfosser et al. (1995) |
Final concentrations are given (MOPS = 4-morpholinepropane sulfonate; DTT = dithiothreitol; TRIS = tris-(hydroxymethyl)-aminomethane; EDTA = ethylenediaminetetraacetic acid; HEPES = 4-(hydroxymethyl)piperazine-1-ethanesulfonic acid). For details of the buffer preparation and use see the original articles.
pH of the buffers is not adjusted. The nuclei are isolated in Otto I buffer; DNA staining is done in a mixture of Otto I and Otto II buffers (1 : 4) with the final pH approx. 7·3 (modifications of the protocol can be found at: http://www.ueb.cas.cz/olomouc1). Baranyi and Greilhuber (1996) modify the Otto II buffer by adding 10 mm sodium citrate and 25 mm sodium sulfate.
The procedure of Otto (1990), which was introduced to plant flow cytometry by Ulrich and Ulrich (1991) and modified for use with non-fixed nuclei by Doležel and Göhde (1995), consists of separate nuclear isolation and staining steps. The nuclei are released into the Otto I buffer, within which they are fixed by citric acid. Staining is performed in a mixture of Otto I and Otto II buffers (1 : 4), which together comprise a phosphate/citric acid buffer of pH 7·3. In most plant species, this procedure results in DNA content histograms with unsurpassed resolution. This is most probably due to the citric acid step, which improves chromatin accessibility and ‘homogenizes’ chromatin structure, thus greatly eliminating differences in staining intensity amongst populations of nuclei with the same DNA content but different chromatin states (F. J. Otto, pers. comm.). It has been found that it is possible to keep isolated nuclei in the Otto I buffer at room temperature for prolonged periods of time without a negative influence on DNA staining. On the other hand, the nuclei should be measured shortly after adding the Otto II buffer to nuclei in the Otto I, since nuclei of some species deteriorate rapidly after this step (J. Doležel, unpubl. res.).
Considering the diversity in tissue anatomy and chemistry among plant species, it is not surprising that there is no single isolation buffer which works well with all species. This may be documented by two extreme examples. In some species, the Otto I buffer precipitates mucous substances, and nuclei released from cells adhere to the precipitate (J. Doležel, unpubl. res.). On the other hand, nuclear isolation from Oxalis, which is characterized by highly acidic cytoplasm, was only successful in the acidic Otto I buffer, and hence the characteristic low pH of the leaves of this species was not a problem. Other buffers failed most probably because the buffering capacity of the solutions was exceeded (Emshwiller, 2002). Although other cases may not be so dramatic, it is worth testing various buffers to identify the best one. Further improvements may be obtained by subtle changes in buffer composition (Zoldoš et al., 1998) and its pH (Rival et al., 1997; Noirot et al., 2000). In some difficult species, it may be necessary to increase the concentration of a detergent (Morgan et al., 1995; Rival et al., 1997). When used at higher concentrations (0·5–1 %), the detergents lyse chloroplasts, which are no longer detected as significant objects by the flow cytometer. Isolation of nuclei from tissues rich in phenols may require addition of a reducing agent (Blondon et al., 1994; Zoldoš et al., 1998) or polyvinylpyrrolidone (Morgan et al., 1995; Yokoya et al., 2000; Thiem and Sliwinska, 2003).
The quality of nuclear suspension is best judged by analysing a histogram of relative nuclear DNA content. The histogram should contain minimal amounts of background debris, G1 (G2) peaks should be symmetrical and the variation should be low. The variation is usually expressed as the coefficient of variation (CV) = standard deviation/peak mean × 100 %. Unlike the standard deviation, CV does not depend on peak mean and hence the precision of measurements with peaks at different positions may be directly compared. As shown by Doležel and Göhde (1995), histograms with peak CVs lower than 1 % may be obtained under specific conditions. In most cases, CVs below 3 % are considered fully acceptable (Marie and Brown, 1993; Galbraith et al., 1998). Such precision may not be attainable in ‘difficult’ species, where CVs below 5 % are considered acceptable. The CV of DNA peaks quantifies a precision of individual measurements but says nothing about the reproducibility of DNA content estimation. As discussed above, this may be assessed only based on replicate measurements. Since DNA flow cytometry involves measurements of fluorescence intensity, the quality of DNA content histograms depends not only on the quality of nuclei suspension but also on DNA staining.
Fluorescent staining of nuclear DNA
The early experiments with DNA content estimation using flow cytometry used a whole range of fluorescent dyes to stain nuclear DNA, which included ethidium bromide (Heller, 1973), mithramycin (Galbraith et al., 1983) and Hoechst dyes (Puite and Ten Broeke, 1983). Little attention was paid to the mode by which the fluorochromes bound to DNA. Ethidium bromide (EB) quantitatively intercalates into double-stranded DNA, and its binding does not seem to be affected by DNA base composition (Le Pecq and Paoletti, 1967). However, EB binds also to double-stranded RNA and the samples must be treated with ribonuclease to provide meaningful DNA content measurements. While this may not be critical for leaf tissues, it is of prime importance for tissues undergoing high levels of protein synthesis such as root tips. Mithramycin, together with other fluorescent antibiotics (chromomycin, olivomycin) is highly specific for double-stranded DNA, binding preferentially to GC-rich regions (Van Dyke and Dervan, 1983). Hoechst dyes (33342, 33258) are specific to double-stranded DNA, and bind to its AT-rich segments (Portugal and Waring, 1988).
Since in most instances the AT : GC ratio of the standard and the sample DNA is not known (and hence may differ), this generated many incorrect estimates of absolute DNA amounts. Beyond the work of Galbraith et al. (1983), mithramycin has not been used to a considerable extent. On the other hand, the Hoechst dyes and, particularly, DAPI, which also binds preferentially to AT-rich regions (Portugal and Waring, 1988), became popular, presumably due to two reasons: (1) like the Hoechst dyes, DAPI is specific for double-stranded DNA and its binding to DNA is not influenced by chromatin structure, which results in low peak CVs (Cowden and Curtis, 1981); (2) many plant scientists preferred using arc-lamp-based flow cytometers, with which DAPI fluorescence was particularly easy to excite and measure.
In 1991, Michaelson et al. (1991a) described a strong correlation between DNA amounts determined using Feulgen microspectrophotometry and by flow cytometry using a DNA intercalator. However, as the study did not involve a base-specific fluorochrome, many assumed that DAPI was equally suitable. This was disproved by Doležel et al. (1992), who showed that the use of fluorochromes binding preferentially to AT- or GC-rich DNA may cause errors approaching 100 %, and recommended DNA intercalators for absolute DNA measurements. Both Michaelson et al. (1991a) and Doležel et al. (1992) used propidium iodide (PI), a DNA intercalator introduced by Crissman and Steinkamp (1973) as a homologue of ethidium bromide. The DNA binding properties of PI are similar to EB (Waring, 1970), and many authors believe that PI produces histograms with lower variation compared with EB. More recent results obtained independently in four different laboratories showed a perfect agreement between flow cytometry of PI-stained samples and Feulgen microspectrophotometry; the opposite was found for DAPI-stained samples (Doležel et al., 1998). Thus, a current consensus is that only DNA intercalators (EB, PI) should be used for absolute DNA measurements.
Despite the fact that EB and PI have been recommended for absolute DNA measurements, one should be aware of their limitations. The sensitivity of EB and PI to chromatin structure (Prosperi et al., 1991; Rayburn et al., 1992) implies that alterations in chromatin condensation as a function of growth state or tissue type might affect DNA content estimation. Galbraith et al. (1998) recommended that the target and standard nuclei should be isolated from tissues of similar metabolic and developmental state. Nevertheless, Blondon et al. (1994) and Kamaté et al. (2001) did not find significant differences between DNA amounts estimated for various organs. It remains to be seen whether this is true for all plant organs at all developmental stages. On the other hand, fixed samples with changed chromatin structure and hence DNA accessibility should be considered with caution (Becker and Mikel, 1990; Holtfreter and Cohen, 1990). To achieve maximal fluorescence and highest resolution, EB and PI are used at saturating concentrations (Taylor and Milthorpe, 1980, Giangare et al., 1989). As dye accessibility of the standard and target nuclei may differ, it has been recommended that optimal dye concentrations should be determined for each given pair of species (Arumuganathan and Earle, 1991; Barre et al., 1996). By doing this it should be remembered that it is the dye : DNA ratio which is critical. It has been noted that the extent of incubation with PI does not improve staining intensity and may lead to increased levels of background debris. Therefore, shorter staining times (2–20 min) have been preferred (Michaelson et al., 1991a; Barre et al., 1996).
While AT- or GC-binding dyes are not suitable for estimating absolute DNA content, some authors have taken advantage of their binding mode to determine genomic base composition (Marie and Brown, 1993; Ricroch and Brown, 1997; Zoldoš et al., 1998). These studies assumed a curvilinear relationship between the fluorescence of base-specific fluorochromes and base content, and used a simple formula to determine AT : GT content (Godelle et al., 1993). However, the recent results of Barow and Meister (2002) indicate that fluorescence of base-specific dyes is influenced by a non-random distribution of bases in the DNA molecule. The issue would be best clarified by comparing flow cytometric data with those obtained biochemically.
Interference with DNA staining
Chromatin is exposed to cytosolic compounds during nuclear isolation. As the nuclei are usually stained within a crude homogenate, the staining is influenced not only by the composition of the nuclear isolation buffer, but also by the compounds present in the cytosol. Problems with the presence of phenolic compounds have been noted in various species and, in most cases, alleviated by incorporating into the nuclear isolation buffer antioxidants such as β-mercaptoethanol (Doležel et al., 1994) and potassium metabisulfite (Ricroch and Brown, 1997; Zoldoš et al., 1998), or polyvinyl pyrrolidone, which binds phenolic compounds (Yokoya et al., 2000; Thiem and Sliwinska, 2003). Nevertheless, it seems that the effect of cytosolic compounds on absolute DNA measurements has been underestimated, leading potentially to erroneous measurements, and that the interference may be far more complex.
Michaelson et al. (1991b) reported that DNA content varied up to 48 % among leaves of individual sunflower plants. In an attempt to explain the nature of this variation, Price and Johnston (1996) found that the mean 2C DNA content of the first leaf was influenced by the quality and the quantity of light under which the plants were grown. However, the ‘fluidity’ of the sunflower genome was not confirmed in a subsequent study, which indicated a role for environmentally induced inhibitor(s) interfering with the fluorescence emission of PI and/or its binding to DNA (Price et al., 2000). In an elegant series of papers, Noirot et al. (2000, 2002, 2003) clearly demonstrated the effect of cytosol on PI fluorescence in several species. When compared with four other species, unidentified compounds in yam leaves had the most pronounced effect and decreased the fluorescence of petunia nuclei by 20 %. The cytosol of Coffea exhibited a less dramatic effect on petunia nuclei (Noirot et al., 2000). Nevertheless, it was sufficient to cause an apparent intraspecific variation in genome size in Coffea, when petunia was used as DNA reference standard (Noirot et al., 2002). An interesting observation was made by Noirot et al. (2003), who found that a negative effect of chlorogenic acid (a precursor of polyphenols) on PI staining of petunia nuclei was counteracted by caffeine, which complexes with polyphenols and inhibits their action. While this observation raised hopes that antagonists of DNA staining inhibitors might be identified, it shows that the fluorescence of PI is influenced by the interaction of DNA staining inhibitors and their antagonists; the presence and amounts of both being determined environmentally and genetically.
Future work should concentrate on the identification of interfering compounds biochemically and by genetic mapping (Noirot et al., 2002). For the time being, testing of various nuclei isolation buffers is recommended. Some of them differ dramatically in pH (Table 1), which may influence the activity of secondary metabolites. The buffers should be supplemented with known inhibitors of secondary metabolites, and internal standardization, in which the target and sample nuclei are exposed to the same environment, should be employed to minimize the risk of errors (Price et al., 2000; Noirot et al., 2003). Dilution of nuclear suspensions or cytosol removal by centrifugation helps to reduce the effect of cytosolic compounds (Noirot et al., 2000; Price et al., 2000). However, this is time consuming and may not avoid modification of chromatin at the moment of nuclear isolation. The approach of checking several standard species for sensitivity to the effect of cytosol of the target species should also be considered. When working with a new species, the best practice is to test the presence of an interfering substance by comparing the DNA peak positions of the standard nuclei measured alone and after simultaneous isolation and staining with the target nuclei. Any difference indicates a presence of interfering compounds and the results should be interpreted with caution.
DNA REFERENCE STANDARDS AND THEIR USE
An ideal DNA reference standard should have a genome size close to the target species. This avoids the risk of nonlinearity and offset errors (Vindeløv et al., 1983; Bagwell et al., 1989). The experiments with DNA staining mentioned above imply that the chromatin structure of the standard and sample nuclei should be similar, and that the nuclei should react in a similar way to compounds interfering with DNA staining. The standard should be genetically stable with constant genome size, easy to use, and available in sufficient quantities. Last, but not least, the genome size of the standard should be known with sufficient precision. These requirements are hard to satisfy and, as a result, different authors have used different standards, including human (Lysák et al., 2000), domestic chicken (Galbraith et al., 1983) and rainbow trout (Turpeinen et al., 1999), as well as various plant species such as petunia (Marie and Brown, 1993), alfalfa (Martel et al., 1997) and oats (Morgan et al., 1995). The standards were not calibrated against each other, and this, in addition to methodological errors in sample preparation and analysis, makes it difficult to compare data obtained in different laboratories.
Clearly, there is a need for agreement on reference standards for DNA flow cytometry. As the genome size in plants ranges over 1000-fold (http://www.rbgkew.org.uk/cval/homepage.html), a set of reference standards is needed with genome size distributed at appropriate intervals. Doležel et al. (1992) calibrated a set of six plant species with 2C amounts ranging from 1·11 to 34·76 pg DNA, suitable as reference standards. In their study, male human leucocytes served as the primary reference standard with 2C = 7·0 pg DNA (Tiersch et al., 1989). The list was extended to nine species in the following study (Doležel et al., 1998). Soon after, Johnston et al. (1999) presented another set of standards consisting of 12 species calibrated against barley (2C = 11·12 pg DNA) and with 2C-values ranging from 1·08 to 32·97 pg DNA. A comparison between the two publications shows a surprisingly good agreement between the estimates for species, which were included in both studies (Table 2). The differences could be, at least in part, due to the use of different primary reference standards.
Table 2.
Plant reference standards that have been used in DNA flow cytometry
|
Doležel et al. (1998) |
|||||||
|---|---|---|---|---|---|---|---|
| Species |
Doležel et al. (1992) |
Lamp* |
Laser† |
Mean |
Johnston et al. (1999) |
||
| Arabidopsis thaliana | 0·34 | 0·43 | 0·39 | ||||
| Oryza sativa | 1·08 | ||||||
| Raphanus sativus | 1·11 | 1·11 | 1·41 | 1·26 | |||
| Vigna radiata | 1·40 | ||||||
| Sorghum bicolor | 1·74 | ||||||
| Lycopersicon esculentum | 1·96 | ||||||
| Glycine max‡ | 2·39 | 2·91 | 2·65 | ||||
| Zea mays | 5·72 | 5·44 | 5·82 | 5·67 | 5·73 | ||
| Lactuca sativa | 5·95 | ||||||
| Pisum sativum§ | 9·07 | 9·09 | 9·09 | 9·09 | 9·39 | ||
| Nicotiana tabacum | 10·04 | ||||||
| Hordeum vulgare | 10·44 | 10·43 | 10·43 | 11·12 | |||
| Secale cereale | 16·44 | 15·95 | 16·19 | 16·65 | |||
| Triticum durum | 21·50 | ||||||
| Vicia faba | 26·90 | 27·84 | 26·17 | 27·00 | 26·66 | ||
| Triticum aestivum | 31·90 | ||||||
| Allium cepa | 34·76 | 36·45 | 33·34 | 34·89 | 32·97 | ||
Arc lamp-based flow cytometers.
Laser-based flow cytometers.
Doležel et al. (1994) estimated 2C DNA amount of Glycine max ‘Polanka’ as 2·5 pg.
Used as a reference standard by Doležel et al. (1998).
Doležel et al. (1992, 1998) used male human leucocytes as a primary reference standard, anticipating the completion of sequencing of the human genome. This goal of the Human Genome Project has not yet been achieved, and it is improbable that repetitive regions will be fully sequenced in the near future. The study of Johnston et al. (1999) demonstrated difficulties when using chicken as reference standard. The authors estimated 2C-values ranging from 2·49 to 3·01 pg DNA for different blood isolates and different strains of chicken. The absence of agreement on genome size on chicken with published 2C-values ranging from 2·33 to 2·5 pg DNA (Galbraith et al., 1983; Tiersch et al., 1989) puts its use as a primary reference in question. The only animal species whose genome size is known with sufficient precision is the nematode worm, Caenorhabditis elegans, for which 1C = 100·25 Mbp (http://wormbase.org/). Bennett et al. (2003) took the advantage of this fact and compared the genome of C. elegans with that of Arabidopsis thaliana, the first plant species to be sequenced. Surprisingly, the results indicated a larger genome size (1C = 157 Mbp) as compared with the value of 125 Mbp given by the Arabidopsis Genome Initiative (2000).
Due to their small genome size, neither C. elegans nor A. thaliana seems a good primary reference standard. As it is not realistic to expect that a larger animal or plant genome will be fully sequenced in the near future, an option is to agree on a single plant species, which would serve as a universal primary reference standard. Garden pea (Pisum sativum) appears to be the most suitable candidate. The current estimates of its 2C-value range from 8·84 to 9·39 pg DNA (Baranyi and Greilhuber, 1996; Doležel et al., 1998; Johnston et al., 1999), which is in the middle of the known range of genome sizes in plants. This would facilitate calibration of reference standards with higher or lower genome sizes. The nuclear genome of pea seems to be stable (Baranyi and Greilhuber, 1995; Baranyi et al., 1996), the plants are easy to grow and multiply, and high quality nuclei suspensions may be prepared easily from leaves, which appear to be free of compounds interfering with PI staining. A fixed 2C-value should be agreed for a pea cultivar, and the standard should be used to recalibrate the existing plant reference standards. If other standards are added to already existing lists (Doležel et al., 1992, 1998; Johnston et al., 1999), care should be taken to select seed-propagated species that are easy to multiply so that sufficient quantities of seed may be produced. As many laboratories do not have facilities to produce seeds on their own, an ideal solution is that one or few centres distribute them for others, thereby ensuring that different laboratories use the same genotype. The authors' laboratory has been providing this service on a cost-free basis over the last 10 years (http://www.ueb.cas.cz/olomouc1).
For quite some time, there has been a discussion on the use of animal reference standards. Although opinions are varied (Arumuganathan and Earle, 1991; Michaelson et al., 1991a), the discussion above indicates a rather limited advantage of animal over plant standards. With the exception of C. elegans, their exact genome sizes are not known, and thus they do not offer any advantage over plants as primary reference standards. Animal reference standards are easy to use when in a tube in a form of a nuclear suspension. However, the nuclei are added only after preparing plant nuclei suspension, thus violating the concept of internal standardization. Their preparation is more difficult and international distribution may pose serious problems. Last but not least, intraspecific variation and sex-related differences in genome size have been described in human and chicken (De Vita et al., 1994; Mefford et al., 1997), the two most frequently used standards.
A need to agree on a unified set of cross-calibrated reference standards is obvious. Nevertheless, the results of Doležel et al. (1998) indicate that even this may not lead to perfect agreement between estimates obtained in different laboratories. In their study, DNA peak ratios obtained in four laboratories for otherwise identical species pairs differed from between 1·8 and 15·6 %. When the ratios were used to calculate genome size, the differences were higher (1·8–33·3 %) due to propagation of error when using intermediate standards. The data were more homogenous (maximum differences being 9·6 % for peak ratio and 14·0 % for genome size) when laboratories using the same type of instrument were compared; indicating a systematic difference between the estimates obtained using arc lamp-based flow cytometers equipped with enclosed flow chamber and laser-based flow cytometers with a ‘jet-in-air’ design (Table 2). For instance, higher 2C-values were obtained using laser-based instruments for maize, soybean, raphanus and arabidopsis when pea was used as a primary reference standard. Barrow and Meister (2002) used a laser-based instrument and, with the same standard, obtained similarly higher 2C-values for the four species, independently confirming the consistence of this difference. Instruments with both laser and lamp excitation are available and it would of highest importance to compare data obtained with lamp and laser excitation by one instrument.
Although the reason for the discrepancies between laboratories and different instruments are not known, they show that absolute DNA amounts determined in different laboratories should be compared with caution, and in no way should the differences be interpreted in terms of intraspecific variation in genome size. If more than one reference standard has to be used in a study, the standards should be recalibrated against each other for the purpose of that study, facilitating reliable comparisons.
INTRASPECIFIC VARIATION IN GENOME SIZE
Plant species where intraspecifc variation in genome size has been reported include soybean (Graham et al., 1994; Rayburn et al., 1997), sunflower (Michaelson et al., 1991b), pea (Arumuganathan and Earle, 1991) and maize (Rayburn et al., 1989), and the variation as high as 32 % was described (Michaelson et al., 1991b); in some cases correlated with environmental gradients or growth conditions. On the other hand, great stability of the nuclear genome has been reported for geographically isolated populations of Sesleria albicans (Lysák et al., 2000), in various species of Settaria (Le Thierrry d'Ennequin et al., 1998), Cistus (Ellul et al., 2002), Capsicum (Moscone et al., 2003), and in cultivars of pea and onion (Baranyi and Greilhuber, 1995; Bennett et al., 2000b).
The advanced understanding of nuclear genome and its components provides clues as to the mechanisms that could be responsible for genome increase, including the activation of Class I retrotransposons (Bennetzen and Kellogg, 1997), and for genome decrease by deletions (Petrov, 1997; Gregory, 2003; Bennetzen et al., 2005). Recently, variation in BARE-1 retrotransposon copy number was observed in populations of wild barley in response to differing microclimates. However, differences between C-values of different populations estimated by flow cytometry were not statistically significant and genome size was only weakly associated with microclimatic gradient (Kalendar et al., 2000). Despite this, the study was perceived as supporting previous reports on large intraspecific variations in genome size (Wendel and Wessler, 2000). One cannot overlook the growing number of reports, which do not confirm the original observations. Thus intraspecific variation in soybean has not been confirmed by Greilhuber and Obermayer (1997) and Obermayer and Greilhuber (1999), the pea genome was found to be stable (Baranyi and Greilhuber, 1995, 1996), and the variation in sunflower was found to be due to the effect of inhibitors of DNA staining (Price et al., 2000).
Intraspecific variation in genome size has been critically reviewed by Greilhuber (1998, 2005) who distinguishes ‘orthodox’ variation due to non-recognized taxonomic heterogeneity of the material (taxonomical artefacts) and ‘unorthodox’ variation due to increase or decrease in the copy number of DNA sequences. This review focuses specifically on the issues related to the use of DNA flow cytometry. A growing number of cases, in which the original reports were not confirmed, suggests that most were artefacts. After having considered various methodological aspects of DNA flow cytometry, it is evident that reliable detection of intraspecific variation in genome size is not a trivial task.
Doležel et al. (1998) demonstrated that differences in DNA content estimates observed between different laboratories cannot be interpreted in terms of intraspecific variation, and that small differences may only be identified when using a single instrument. Furthermore, minor instrument drifts (e.g. due to slight differences in instrument alignment) may result in very small but statistically significant differences between estimates produced on different days (J. Doležel, unpubl. res.). A good practice is therefore to perform replicate measurements on different days. The sample preparation and the standardization procedure deserve the highest attention. Internal standardization should be used exclusively with the target and standard processed simultaneously and under the same experimental conditions for all samples. The effect of chromatin condensation on DNA staining is best avoided by isolating target and standard nuclei from tissues of similar metabolic and developmental state. As cytosol composition may change in response to changes in the environment, the plants should be grown under identical conditions and a test for the presence of DNA staining inhibitors should be performed.
DNA FLOW CYTOMETRY AND FIELD WORK
The success of flow cytometry and its ever-increasing use in plant taxonomy, systematics and ecology may pose unexpected problems. As for most of the other analytical methods, the materials to be analysed by flow cytometry are sampled and then dispatched to the laboratory. This causes no difficulties if the plants are grown within a reasonable distance of the laboratory. The problems start with expeditions to more distant areas, when the transport and/or the maintenance of the material become an issue. Leaf samples (the most popular tissue for DNA flow cytometry) may be transported when bagged in humid paper tissue and kept at low temperature. The workers in the authors' laboratory have valuable experience in analysing leaf samples of Musa spp. which have been delivered rapidly via a courier from tropical countries of America, Africa and Asia. However, it may not be feasible to dispatch samples rapidly from remote areas; for some species, the leaves may deteriorate quickly. If a capacity of the laboratory is saturated with leaf samples, they may deteriorate after arrival and before being analysed.
Brown (1993) was probably the first to suggest the opposite, namely to bring the flow cytometer to the plants. This proposal, termed ‘wheel-barrow cytometry’ or ‘bush flow cytometry’, which included various other applications, appears particularly applicable to the current discussion. Ten years ago, this would be hard to do. Today, compact and portable flow cytometers are available that can be operated using a single 12 V car battery. It is thus not a problem to establish a field laboratory for on-site sample preparation and flow analysis. In fact, marine biologists analysing phytoplankton often carry flow cytometry laboratories on research vessels (Sosik and Olson, 2002) or use cytometers free floating in the ocean (Dubelaar and Gerritzen, 2000). Yet, this change of concept will not solve all problems of field DNA flow cytometry. The cost of transporting or establishing the laboratory may be prohibitive in certain areas. However, the most important issues would concern the actual estimation of genome size in accessions of species not analysed before. This would require transportation (or growth) of a series of plant standards and, in addition, running preliminary experiments to identify an optimal sample preparation protocol, including a test for interference of cytosolic compounds with DNA staining. To conclude, bush flow cytometry is a very attractive tool for ploidy screening on-site, where the same protocol is used for hundreds or thousands of individuals representing a limited number of species. Its value for systematic on-site estimation of absolute DNA amounts in large numbers of species remains to be established.
There has been continuing debate on the use of fixed materials for absolute DNA measurements. There are two types of fixation used in modern cytogenetics: non-additive, represented by methanol–acetic acid and additive, represented by formaldehyde. Unfortunately, the latter interferes with quantitative staining using DNA intercalators (Overton and McCoy, 1994). Few authors have isolated nuclei from tissues or cells fixed by methanol–acetic acid by enzymatic digestion (Pfosser, 1989); the protocol involves enzymatic digestion, it is time consuming and labour intensive. It needs to be shown that fixation does not alter the accessibility of the target and the sample nuclei to EB or PI in a different way. It is also known that the fixation may release tannins from vacuoles. Released tannins bind strongly to chromatin and interfere with quantitative DNA staining using the Feulgen reaction (Greilhuber, 1988). As it is highly probable that EB or PI staining is disturbed too, the use of non-additive fixatives should be considered with caution. Doležel (1991) complained that the effect of fixatives on (fluorescent) DNA staining was largely neglected. Unfortunately, the same is true more than 10 years later.
What then is the way forward for field DNA flow cytometry? Probably the most elegant approach is to collect and transport seeds as shown recently by Suda et al. (2003), who analysed 104 Macronesian angiosperms in their Prague laboratory. An important advantage of this strategy is that roots may be collected for chromosome counting and further observation of plants is possible, which may help classification. Vegetatively propagated seedless species are a problem and the only solution—transportation of offshoots and/or small plantlets—may be in conflict with phytosanitary rules and the Convention on Biological Diversity (http://www.biodiv.org/).
THE FUTURE OF DNA FLOW CYTOMETRY
Twenty years after the breakthrough paper of Galbraith et al. (1983), plant DNA flow cytometry is a very popular method with applications ranging from basic and applied research to industry. New applications continue to emerge, and there are several areas where the potential of the method has not been fully explored. Taxonomy, population biology and ecology require the analysis of large populations of plants, for which flow cytometry is ideally suited. It seems highly probable that a growing number of applications will be seen in these areas. While the estimation of relative DNA amounts for ploidy screening and some other applications usually does not represent serious problems, the use of flow cytometry for estimation of genome size is a greater challenge.
The problems that need to be solved fall in two categories: (1) general problems that concern any method used to estimate genome size; and (2) problems related to flow cytometry, which is considered complementary to Feulgen microspectrophotometry and DNA image densitometry. General problems concern mainly the use of DNA standards. Agreement is needed both on a set of DNA reference standards and their genome sizes. Hand in hand with this, the terminology employed for genome size and DNA C-values needs to be clarified. Problems specific to DNA flow cytometry primarily concern the sample preparation. The role of cytosolic compounds interfering with quantitative staining of DNA is a hot topic. Future work should lead to the improvement in buffer composition and in sample preparation that will control the effects of these compounds. Another difficulty with flow cytometry is the requirement for fresh plant samples. Compact and portable flow cytometers suitable for the analysis of EB- or PI-stained nuclei became available and may be used in the field. It is also possible to dispatch fresh samples to a geographically distant laboratory. As both options may not be ideal under certain circumstances, currently the most convenient approach is to dispatch seeds and grow plantlets close to the cytometry facility. Nevertheless, a development of a procedure for sample storage is highly desirable.
Acknowledgments
We thank David W. Galbraith (Tucson) and Spencer C. Brown (Gif sur Yvette) for critical reading and helpful suggestions. This work was supported by research grants no. 522/03/0354 and 204/04/1207 from the Grant Agency of the Czech Republic.
LITERATURE CITED
- Arabidopsis Genome Initiative. 2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana Nature 408: 796–815. [DOI] [PubMed] [Google Scholar]
- Arumuganathan K, Earle ED. 1991. Estimation of nuclear DNA content of plants by flow cytometry. Plant Molecular Biology Reporter 9: 229–241. [Google Scholar]
- Bagwell CB, Baker D, Whetstoe S, Munson M, Hitchcox S, Ault K, Lovett EJ. 1989. A simple and rapid method for determining the linearity of a flow cytometer amplification system. Cytometry 10: 689–694. [DOI] [PubMed] [Google Scholar]
- Baranyi M, Greilhuber J. 1995. Flow cytometric analysis of genome size variation in cultivated and wild Pisum sativum (Fabaceae). Plant Systematics and Evolution 194: 231–239. [Google Scholar]
- Baranyi M, Greilhuber J. 1996. Flow cytometric and Feulgen densitometric analysis of genome size variation in Pisum Theoretical and Applied Genetics 92: 297–307. [DOI] [PubMed] [Google Scholar]
- Baranyi M, Greilhuber J, Swiecicki WK. 1996. Genome size in wild Pisum species. Theoretical and Applied Genetics 93: 717–721. [DOI] [PubMed] [Google Scholar]
- Barow M, Meister A. 2002. Lack of correlation between AT frequency and genome size in higher plants and the effect of nonrandomness of base sequences on dye binding. Cytometry 47: 1–7. [DOI] [PubMed] [Google Scholar]
- Barow M, Meister A. 2003. Endopolyploidy in seed plants is differently correlated to systematics, organ, life strategy and genome size. Plant, Cell and Environment 26: 571–584. [Google Scholar]
- Barre P, Noirot M, Louarn J, Duperray C, Hamon S. 1996. Reliable flow cytometric estimation of Nuclear DNA content in coffee trees. Cytometry 24: 32–38. [DOI] [PubMed] [Google Scholar]
- Becker RL, Mikel UV. 1990. Interrelation of formalin fixation, chromatin compactness and DNA values as measured by flow cytometry and image cytometry. Analytical and Quantitative Cytology and Histology 12: 333–341. [PubMed] [Google Scholar]
- Bennett MD, Leitch I. 1995. Nuclear DNA amounts in angiosperms. Annals of Botany 76: 113–176. [Google Scholar]
- Bennett MD, Bhandol P, Leitch I. 2000. Nuclear DNA amounts in angiosperms and their modern uses: 807 new estimates. Annals of Botany 86: 859–909. [Google Scholar]
- Bennett MD, Johnston S, Hodnett GL, Price HJ. 2000b.Allium cepa L. cultivars from four continents compared by flow cytometry show nuclear DNA constancy. Annals of Botany 85: 351–357. [Google Scholar]
- Bennett MD, Leitch IJ, Price HJ, Johnston JS. 2003. Comparison with Caenorhabditis (∼100 Mb) and Drososphila (∼175 Mb) using flow cytometry show genome size in Arabidopsis to be ∼157 Mb and thus ∼25 % larger than the Arabidopsis Genome Initiative Estimate of ∼125 Mb. Annals of Botany 91: 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen JL, Kellogg EA. 1997. Do plants have a one-way ticket to genomic obesity? The Plant Cell 9: 1509–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen J, Ma J, Devos KM. 2005. Mechanisms of recent genome size variation in flowering plants. Annals of Botany 95: 127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondon F, Marie D, Brown S, Kondorosi A. 1994. Genome size and base composition in Medicago sativa and M. truncatula species. Genome 37: 264–270. [DOI] [PubMed] [Google Scholar]
- Britten RJ, Kohne DE. 1968. Repeated sequences in DNA. Science 161: 529–540. [DOI] [PubMed] [Google Scholar]
- Bogunic F, Muratovic E., Brown SC, Siljak-Yakovkev S. 2003. Genome size and base composition of five Pinus species from Balkan region. Plant Cell Reports 22: 59–63. [DOI] [PubMed] [Google Scholar]
- Brown SC. 1993. Cytometrie tout terrain or bush DNA cytometry. In: Sablon A, ed. Flow cytometry, Jacquemin-NATO ASI Series, Vol. H67. Heidelberg: Springer-Verlag, 227–241. [Google Scholar]
- Caspersson T, Schultz J. 1938. Nucleic acid metabolism of the chromosomes in relation to gene reproduction. Nature 142: 294. [Google Scholar]
- Cavalier-Smith T. 1985.The evolution of genome size. Chichester: John Wiley & Sons. [Google Scholar]
- Cowden RR, Curtis SK. 1981. Microfluorometeric investigations of chromatin structure. I. Evaluation of nine DNA-specific fluorochromes as probes of chromation organization. Histochemistry 72: 11–23. [DOI] [PubMed] [Google Scholar]
- Crissman HA, Steinkamp JA. 1973. Rapid simultaneous measurement of DNA, protein and cell volume in single cells from large mammalian cell populations. Journal of Cell Biology 59: 766–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeley EM. 1955. An integrating micodensitometer for biological cells. Journal of Cell Instrumentation 32: 263–267. [Google Scholar]
- De Latt AMM, Göhde W, Vogelzang MJD. 1987. Determination of ploidy of single plants and plant populations by flow cytometry. Plant Breeding 99: 303–307. [Google Scholar]
- De Vita R, Cavallo D, Eleuteri P, Dellomo G. 1994. Evaluation of interspecific DNA content variation and sex identification in falconiformes and strigiformes by flow cytometric analysis. Cytometry 16: 346–350. [DOI] [PubMed] [Google Scholar]
- Dittrich W, Göhde W. 1969. Impulsfluorometrie bei Einzellen in Suspensionen. Zeitschrift für Naturforschung 24b: 360–361. [PubMed] [Google Scholar]
- Doležel J. 1991. Flow cytometric analysis of nuclear DNA content in higher plants. Phytochemical Analysis 2: 143–154. [Google Scholar]
- Doležel J, Bartoš J, Voglmayr H, Greilhuber J. 2003. Nuclear DNA content and genome size of trout and human. Cytometry 51: 127–128. [DOI] [PubMed] [Google Scholar]
- Doležel J, Binarová P, Lucretti S. 1989. Analysis of nuclear DNA content in plant cells by flow cytometry. Biologia Plantarum 31: 113–120. [Google Scholar]
- Doležel J, Doleželová M, Novák FJ. 1994. Flow cytometric estimation of nuclear DNA amount in diploid bananas (Musa acuminata and M. balbisiana). Biologia Plantarum 36: 351–357. [Google Scholar]
- Doležel J, Göhde W. 1995. Sex determination in dioecious plants Melandrium album and M. rubrum using high-resolution flow cytometry. Cytometry 19: 103–106. [DOI] [PubMed] [Google Scholar]
- Doležel J, Greilhuber J, Lucretti S, Meister A, Lysák MA, Nardi L, Obermayer R. 1998. Plant genome size estimation by flow cytometry: inter-laboratory comparison. Annals of Botany 82 (Suppl. A): 17–26. [Google Scholar]
- Doležel J, Sgorbati S, Lucretti S. 1992. Comparison of three DNA fluorochromes for flow cytometric estimation of nuclear DNA content in plants. Physiologia Plantarum 85: 625–631. [Google Scholar]
- Dubelaar GBJ, Gerritzen PL. 2000. CytoBuoy: a step forward towards using flow cytometry in operational oceanography. Scientia Marina 64: 255–265. [Google Scholar]
- Ellul P, Boscaiu M, Vicente O, Moreno V, Roselló JA. 2002. Intra- and interspecific variation in DNA content in Cistus (Cistaceae). Annals of Botany 90: 345–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emshwiller E. 2002. Ploidy levels among species in the ‘Oxalis tuberosa Alliance’ as inferred by flow cytometry. Annals of Botany 89:741–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feulgen R, Rossenbeck H. 1924. Mikroskopisch-chemischer Nachweis einer Nukleinsäure von Typus der Thymonucleinsäure und die darauf beruhende selektive Färbung von Zellkernen in mikroskopischen Präparaten. Zeitschrift für physikalische Chemie, Stöchiometrie und Verwandtschaftslehre 135: 203–248. [Google Scholar]
- Galbraith DW. 1984. Flow cytometric analysis of the cell cycle in higher plants. In: Vasil, IK, ed. Cell culture and somatic cell genetics of plant. New York; Academic Press, 765–777. [Google Scholar]
- Galbraith DW. 1990. Flow cytometric analysis of plant genomes. Methods in Cell Biology 33: 549–562. [DOI] [PubMed] [Google Scholar]
- Galbraith DW, Harkins KR, Maddox JM, Ayres NM, Sharma DP, Firoozabady E. 1983. Rapid flow cytometric analysis of the cell cycle in intact plant tissues. Science 220: 1049–1051. [DOI] [PubMed] [Google Scholar]
- Galbraith DW, Lambert GM, Macas J, Doležel J. 1998. Analysis of nuclear DNA content and ploidy in higher plants. In: Robinson, JP, Darzynkiewicz, Z, Dean, PN, Dressler, LG, Orfao, A, Rabinovitch, PS, Stewart, CC, Tanke, HJ, Wheeless, LL, eds. Current protocols in cytometry. New York: John Wiley & Sons, 7.6.1–7.6.22. [Google Scholar]
- Galbraith DW, Shields BA. 1982. The effect of inhibitors of cell wall synthesis on tobacco protoplast development. Physiologia Plantarum 55: 25–30. [Google Scholar]
- Giangare MC, Prosperi E, Pedralinov G, Bottiroli G. 1989. Flow cytometric evaluation of DNA stainability with propidium iodide after H1 extraction. Cytometry 10: 726–730. [DOI] [PubMed] [Google Scholar]
- Godelle B, Cartier D, Marie D, Brown SC, Siljak-Yakovlev S. 1993. Heterochromatin study demonstrating the non-linearity of fluorometry useful for calculating genomic base composition. Cytometry 14: 618–626. [DOI] [PubMed] [Google Scholar]
- Graham MJ, Nickell CD, Rayburn AL. 1994. Relationship between genome size and maturity group in soybean. Theoretical and Applied Genetics 88: 429–432. [DOI] [PubMed] [Google Scholar]
- Gregory TR. 2001. Coincidence, coevolution, or causation? DNA content, cell size, and the C-value enigma. Biological Reviews 76: 65–101. [DOI] [PubMed] [Google Scholar]
- Gregory TR. 2003. Is small indel bias a determinant of genome size? Trends in Genetics 19: 485–488. [DOI] [PubMed] [Google Scholar]
- Greilhuber J. 1988. ‘Self-tanning’—a new and important source of stoichiometric error in cytophotometric determination of nuclear DNA content in plants. Plant Systematics and Evolution 158: 87–96. [Google Scholar]
- Greilhuber J. 1998. Intraspecific variation in genome size: a critical reassessment. Annals of Botany 82 (Suppl. A): 27–35. [Google Scholar]
- Greilhuber J. 2005. Intraspecific variation in genome size in angiosperms: identifying its existence. Annals of Botany 95: 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greilhuber J, Doležel J, Lysák M, Bennett M.D. 2005. The origin, evolution and proposed stabilization of the terms ‘genome size’ and ‘C-value’ to describe nuclear DNA contents. Annals of Botany 95: 255–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greilhuber J, Obermayer R. 1997. Genome size and maturity group in Glycine max (soybean). Heredity 78: 547–551. [Google Scholar]
- Hanson L, Brown RL, Boyd A, Johnson MAT, Bennett MD. 2003. First nuclear DNA C-values for 28 Angiosperm genera. Annals of Botany 91: 31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller FO. 1973. DNS-Bestimmung an Keimwurzeln von Vicia faba L. mit Hilfe der Impulscytophotometrie. Bericht der Deutschen Botanischen Gesellschaft 86: 437–441. [Google Scholar]
- Holtfreter HB, Cohen N. 1990. Fixation-associated quantitative variation of DNA fluorescence observed in flow cytometric analysis of homopoietic cells from adult diploid frogs. Cytometry 11: 676–685. [DOI] [PubMed] [Google Scholar]
- Hülgenhof E, Weidhase RA, Schlegel R, Tewes A. 1988. Flow cytometric determination of DNA content in isolated nuclei of cereals. Genome 30: 565–569. [Google Scholar]
- Kalendar R, Tanskanen J, Irnmonen S, Nevo E, Schulman AH. 2000. Genome evolution of wild barley (Hordeum spontaneum) by BARE-1 retrotransposon dynamics in response to sharp microclimatic divergence. Proceedings of the National Academy of Sciences of the USA 97: 6603–6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamaté K, Brown S, Durand P, Bureau JM, De Nay D, Trinh TH. 2001. Nuclear DNA content and base composition in 28 taxa of Musa Genome 44: 622–627. [PubMed] [Google Scholar]
- Kamentsky LA, Melamed MR, Derman H. 1965. Spectrophotometer: new instrument for ultrarapid cell analysis. Science 150: 630–631. [DOI] [PubMed] [Google Scholar]
- Kawara S, Takata M, Takehara K. 1999. High frequency of DNA aneuploidy detected by DNA flow cytometry in Bowen's disease. Journal of Dermatological Science 21: 23–26. [DOI] [PubMed] [Google Scholar]
- Johnston JS, Bennett MD, Rayburn AL, Galbraith DW, Price HJ. 1999. Reference standards for determination of DNA content of plant nuclei. American Journal of Botany 86: 609–613. [PubMed] [Google Scholar]
- Le Pecq JB, Paoletti G. 1967. A fluorescent complex between ethidium bromide and nucleic acids. Physical-chemical characterization. Journal of Molecular Biology 27: 87–106. [DOI] [PubMed] [Google Scholar]
- Le Thierrry d'Ennequin M, Panaud O, Brown S, Siljak-Yakovlev A, Sarr A. 1998. First evaluation of DNA content in Setaria genus by flow cytometry. Journal of Heredity 89: 556–559. [Google Scholar]
- Lysák MA, Doleželová M, Horry JP, Swennen R, Doležel J. 1999. Flow cytometric analysis of nuclear DNA content in Musa Theoretical and Applied Genetics 98: 1344–1350. [Google Scholar]
- Lysák MA, Rostková A, Dixon JM, Rossi G, Doležel J. 2000. Limited genome size variation in Sesleria albicans Annals of Botany 86: 399–403. [Google Scholar]
- Marie D, Brown SC. 1993. A cytometric exercise in plant DNA histograms, with 2C values for 70 species. Biology of the Cell 78: 41–51. [DOI] [PubMed] [Google Scholar]
- Martel E, De Nay D, Siljak-Yakovlev S, Brown S, Sarr A. 1997. Genome size variation and basic chromosome number in pearl millet and fourteen related Pennisetum species. Journal of Heredity 88: 139–143. [Google Scholar]
- Matzk F, Meister A, Schubert I. 2000. An efficient screen for reproductive pathways using mature seeds of monocot and dicots. The Plant Journal 21: 97–108. [DOI] [PubMed] [Google Scholar]
- Mefford H, vandenEngh G, Friedman C, Trask BJ. 1997. Analysis of variation in chromosome size among diverse human populations by bivariate flow karyotyping. Human Genetics 100: 138–144. [DOI] [PubMed] [Google Scholar]
- Michaelson MJ, Price HJ, Ellison JR, Johnston JS. 1991. Comparison of plant DNA contents determined by Feulgen microspectrophotometry and laser flow cytometry. American Journal of Botany 78: 183–188. [Google Scholar]
- Michaelson MJ, Price HJ, Johnston JS, Ellison JR. 1991. Variation of nuclear DNA content in Helianthus annuus (Asteraceae). American Journal of Botany 78: 1238–1243. [Google Scholar]
- Mirsky AE, Ris H. 1951. The desoxyribonucleic acid content of animal cells and its evolutionary significance. Journal of General Physiology 34: 451–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan ER, Burge GK, Seelye JF, Grant JE, Hopping ME. 1995. Interspecific hybridization between Limonium perigrinum Bergius and Limonium purpuratum L. Euphytica 83: 215–224. [Google Scholar]
- Moscone EA, Baranyi M, Ebert I, Greilhuber J, Ehrendorfer F, Hunziker AT. 2003. Analysis of nuclear DNA content in Capsicum (Solanaceae) by flow cytometry and Feulgen densitometry. Annals of Botany 92: 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noirot M, Barre P, Duperray C, Louarn J, Hamon S. 2003. Effects of caffeine and chlorogenic acid on propidium iodide accessibility to DNA: consequences on genome size evaluation in coffee tree. Annals of Botany 92: 259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noirot M, Barre P, Louarn J, Duperray C, Hamon S. 2000. Nucleus–cytosol interactions: a source of stoichiometric error in flow cytometric estimation of nuclear DNA content in plants. Annals of Botany 86: 309–316. [Google Scholar]
- Noirot M, Barre P, Louarn J, Duperray C, Hamon S. 2002. Consequences of stoichiometric error on nuclear DNA content evaluation in Coffea liberica var. dewevrei using DAPI and propidium iodide. Annals of Botany 89: 385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermayer R, Greilhuber J. 1999. Genome size in Chinese soybean accessions: stable or variable? Annals of Botany 84: 259–262. [Google Scholar]
- Otto FJ. 1990. DAPI staining of fixed cells for high-resolution flow cytometry of nuclear DNA. In: Darzynkiewickz Z, Crissman HA, eds. Methods in cell biology, Vol. 33. San Diego: Academic Press, 105–110. [DOI] [PubMed] [Google Scholar]
- Overton WR, McCoy JP. 1994. Reversing the effect of formalin on the binding of propidium iodide to DNA. Cytometry 16: 351–356. [DOI] [PubMed] [Google Scholar]
- Petrov D. 1997. Slow but steady: reduction of genome size through biased mutation. The Plant Cell 9: 1900–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfosser M. 1989. Improved method for critical comparison of cell cycle data on asynchronously dividing and synchronized cell cultures of Nicotiana tabacum Journal of Plant Physiology 134: 741–745. [Google Scholar]
- Pfosser M, Amon A, Lelley T, Heberle-Bors E. 1995. Evaluation of sensitivity of flow cytometry in detecting aneuploidy in wheat using disomic and ditelosomic wheat-rye addition lines. Cytometry 21: 387–393. [DOI] [PubMed] [Google Scholar]
- Portugal J, Waring MJ. 1988. Assignment of DNA binding sites for4′,6-diamidino-2-phenylindole and bisbenzimide (Hoechst 33258). A comparative footprinting study. Biochim. Biophys. Acta 949: 158–168. [DOI] [PubMed] [Google Scholar]
- Price HJ, Hodnet G, Johnston JS. 2000. Sunflower (Helianthus annuus) leaves contain compounds that reduce nuclear propidium iodide fluorescence. Annals of Botany 86: 929–934. [Google Scholar]
- Price HJ, Johnston JS. 1996. Influence of light on DNA content of Helinathus annuus Linnaeus. Proceedings of the National Academy of Sciences of the USA 93: 11264–11267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price HJ, Morgan PW, Johnston JS. 1998. Environmentally correlated variation in 2C nuclear DNA content measurements in Helianthus annuus L. Annals of Botany 82 (Suppl. A): 95–98. [Google Scholar]
- Prosperi E, Giangare MC, Bottiroli G. 1991. Nuclease induced DNA structural changes assessed by flow cytometry with the intercalating dye propidium iodide. Cytometry 12: 323–329. [DOI] [PubMed] [Google Scholar]
- Puite KJ, Ten Broeke WRR. 1983. DNA staining of fixed and non-fixed plant protoplasts for flow cytometry with Hoechst 33342. Plant Science Letters 32: 79–88. [Google Scholar]
- Rabinovitch PS. 1994. DNA content histogram and cell-cycle analysis. In: Darzynkiewicz Z, Robinson JP, Crissman HA, eds. Methods in cell biology: flow cytometry, Vol. 41. San Diego: Academic Press, 263–296. [DOI] [PubMed] [Google Scholar]
- Rayburn AL, Auger JA, Benzinger ES, Hepburn AG. 1989. Detection of intraspecific DNA content variation in Zea mays L. by flow cytometry. Journal of Experimental Botany 40: 1179–1183. [Google Scholar]
- Rayburn AL, Auger JA, McMurphy LM. 1992. Estimating percentage constitutive heterochromatin by flow cytometry. Experimental Cell Research 198: 175–178. [DOI] [PubMed] [Google Scholar]
- Rayburn AL, Biradar DP, Bullock DG, Nelson RL, Gourmet C, Wetzel JB. 1997. Nuclear DNA content diversity in Chinese soybean introductions. Annals of Botany 80: 321–325. [Google Scholar]
- Ricroch A, Brown SC. 1997. DNA base composition of Allium genomes with different chromosome numbers. Gene 205: 255–260. [DOI] [PubMed] [Google Scholar]
- Rival A, Beule T, Barre P, Hamon S, Duval Y, Noirot M. 1997. Comparative flow cytometric estimation of nuclear DNA content in oil palm (Elaeis guineensis Jacq) tissue cultures and seed-derived plants. Plant Cell Reports 16: 884–887. [DOI] [PubMed] [Google Scholar]
- Roux N, Doležel J, Swennen R, Zapata-Arias FJ. 2001. Effectiveness of three micropropagation techniques to dissociate cytochimeras in Musa spp. Plant Cell, Tissue and Organ Culture 66: 189–197. [Google Scholar]
- Roux N, Toloza A, Radecki Z, Zapata-Arias FJ, Doležel J. 2003. Rapid detection of aneuploidy in Musa using flow cytometry. Plant Cell Reports 21: 483–490. [DOI] [PubMed] [Google Scholar]
- Sandoval A, Hocker V, Verdeil JL. 2003. Flow cytometric analysis of the cell cycle in different coconut palm (Cocos nucifera L.) tissues cultured in vitro Plant Cell Reports 22: 25–31. [DOI] [PubMed] [Google Scholar]
- Schmidt G, Thannhauser SJ. 1945. A method for the determination of desoxyribonucleic acid, ribonucleic acid, and phosphoproteins in animal tissues. Journal of Biological Chemistry 161: 83–89. [PubMed] [Google Scholar]
- Shapiro HM. 2003.Practical flow cytometry, 4th edn. New York: Wiley-Liss. [Google Scholar]
- Sosik HM, Olson RJ. 2002. Phytoplankton and iron limitation of photosynthetic efficiency in the Southern Ocean during late summer. Deep Sea Research. Part I. Oceanographic Research Papers 49: 1195–1216. [Google Scholar]
- Straus NA. 1971. Comparative DNA renaturation kinetics in amphibians. Proceedings of the National Academy of Sciences of the USA 68: 799–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda J, Kyncl T, Freiová R. 2003. Nuclear DNA amounts in Macronesian angiosperms. Annals of Botany 92: 153–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift H. 1950. The constancy of desoxyribose nucleic acid in plant nuclei. Proceedings of the National Academy of Sciences of the USA 36: 643–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor IW, Milthorpe BK. 1980. An evaluation of DNA fluorochromes, staining techniques, and analysis for flow cytometry. I. Unperturbed cell populations. Journal of Histochemistry and Cytochemistry 28: 1224–1232. [DOI] [PubMed] [Google Scholar]
- Thiem B, Sliwinska E. 2003. Flow cytometric analysis of nuclear DNA content in cloudberry (Rubus chamaemorus L.) in vitro cultures. Plant Science 164: 129–134. [Google Scholar]
- Thomas CA. 1971. The genetic organisation of chromosomes. Annual Review of Genetics 5: 237–256. [DOI] [PubMed] [Google Scholar]
- Tiersch TR, Chandler RW, Wachtel SS, Elias S. 1989. Reference standards for flow cytometry and application in comparative studies of nuclear DNA content. Cytometry 10: 706–710. [DOI] [PubMed] [Google Scholar]
- Turpeinen T, Kulmala J, Nevo E. 1999. Genome size variation in Hordeum spontaneum populations. Genome 42: 1094–1099. [DOI] [PubMed] [Google Scholar]
- Ulrich I, Fritz B, Ulrich W. 1988. Application of DNA fluorochromes for flow cytometric DNA analysis of plant protoplast. Plant Science 55: 151–158. [Google Scholar]
- Ulrich I, Ulrich W. 1991. High-resolution flow cytometry of nuclear DNA in higher plants. Protoplasma 165: 212–215. [Google Scholar]
- Van Dilla MA, Trujillo TT, Mullaney PF, Coulter JR. 1969. Cell microfluorometry: a method for rapid fluorescence measurement. Science 163: 1213–1214. [DOI] [PubMed] [Google Scholar]
- Van Dyke MW, Dervan PB. 1983. Chromomycin, mithramycin, and olivomycin binding sites on heterogenous deoxyribonucleic acid. Footprinting with (methidiumpropyl-EDTA) iron(II). Biochemistry 22: 2373–2377. [DOI] [PubMed] [Google Scholar]
- Vermes I, Haanen C, Reutelingsperger C. 2000. Flow cytometry of apoptotic cell death. Journal of Immunological Methods 243:167–190. [DOI] [PubMed] [Google Scholar]
- Vilhar B, Greilhuber J, Dolenc Koce J, Temsch EM, Dermastia M. 2001. Plant genome size measurement with DNA image cytometry. Annals of Botany 87: 719–728. [Google Scholar]
- Vindeløv LL, Christensen IJ, Jensen GJ, Nissen NI. 1983. Limits of detection of nuclear DNA abnormalities by flow cytometric DNA analysis. Results obtained by a set of methods for sample-storage, staining and interval standardization. Cytometry 3: 332–339. [DOI] [PubMed] [Google Scholar]
- Waring M. 1970. Variation of the supercoils in closed circular DNA by binding of antibiotics and drugs: evidence for molecular models involving intercalation. Journal of Molecular Biology 54: 247–279. [DOI] [PubMed] [Google Scholar]
- Wendel JF, Wessler S. 2000. Retrotransposon-mediated genome evolution on a local ecological scale. Proceedings of the National Academy of Sciences of the USA 97: 6250–6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoya K, Roberts AV, Mottley J, Lewis R, Brandham PE. 2000. Nuclear DNA amounts in roses. Annals of Botany 85: 557–561. [Google Scholar]
- Zoldoš V, Papeš D, Brown SC, Panaud O, Šiljak-Yakovlev S. 1998. Genome size and base composition of seven Quercus species: inter- and intra-population variation. Genome 41: 162–168. [Google Scholar]