

Abstract
• Background and Aims Plant nuclear genomes vary tremendously in DNA content, mostly due to differences in ancestral ploidy and variation in the degree of transposon amplification. These processes can increase genome size, but little is known about mechanisms of genome shrinkage and the degree to which these can attenuate or reverse genome expansion. This research focuses on characterizing DNA removal from the rice and Arabidopsis genomes, and discusses whether loss of DNA has effectively competed with amplification in these species.
• Methods Retrotransposons were analyzed for sequence variation within several element families in rice and Arabidopsis. Nucleotide sequence changes in the two termini of individual retrotransposons were used to date their time of insertion.
• Key Results An accumulation of small deletions was found in both species, caused by unequal homologous recombination and illegitimate recombination. The relative contribution of unequal homologous recombination compared to illegitimate recombination was higher in rice than in Arabidopsis. However, retrotransposons are rapidly removed in both species, as evidenced by the similar apparent ages of intact elements (most less than 3 million years old) in these two plants and all other investigated plant species.
• Conclusions Differences in the activity of mechanisms for retrotransposon regulation or deletion generation between species could explain current genome size variation without any requirement for natural selection to act on this trait, although the results do not preclude selection as a contributing factor. The simplest model suggests that significant genome size variation is generated by lineage-specific differences in the molecular mechanisms of DNA amplification and removal, creating major variation in nuclear DNA content that can then serve as the substrate for fitness-based selection.
Keywords: Deletion, illegitimate recombination, insertion, retrotransposon, unequal recombination
INTRODUCTION
Although flowering plants (the Angiosperms) vary tremendously in nuclear DNA content, most of this variation is not associated with differences in gene number or gene size. Polyploidy is a rapid and dramatic mechanism that can double gene content and genome size in a single generation, and most or all flowering plants are either current polyploids or have a polyploidy origin (Wendel, 2000). However, molecular investigations of plant nuclear DNA content have shown that most genome size variability is associated with differences in repetitive DNA content (Flavell et al., 1974). In all plants investigated, the most significant contributions to genome size have been by a class of mobile DNA called retroelements, primarily the LTR- (long terminal repeat-) retrotransposons (SanMiguel et al., 1996; Vicient et al., 1999). In maize, LTR-retrotransposons make up over 70 % of the nuclear genome (SanMiguel and Bennetzen, 1998) and they are similarly major contributors to all other large plant genomes. The arrangements of these elements have been comprehensively investigated in only a few plant species. As in animal genomes, plant LTR-retrotransposons are preferentially associated with the large heterochromatic regions that flank functional centromeres (reviewed in Kumar and Bennetzen, 1999). In large genome species like maize (2400 Mb) and barley (4900 Mb), many of these elements are intermixed with genes, often as nested structures of LTR-retrotransposons inserted into LTR-retrotransposons (SanMiguel et al., 1996; Rostoks et al., 2002). The genic regions of smaller genomes like those of Arabidopsis (130 Mb), rice (430 Mb) and sorghum (750 Mb) usually have only single LTR-retrotransposons inserted in or near genes, although some clusters of elements can be found in gene-poor regions of their chromosomes (The Arabidopsis Genome Initiative, 2000).
Many studies have investigated the potential selective advantages or disadvantages of large genome size. These studies have an inherent flaw in that it is not possible to create isogenic comparisons. That is, we have no two plant lines that are identical for all alleles of genes yet differ appreciably for genome size. Hence, correlations of genome size with radiation resistance, cell size, generation time or endangered species status (for instance) provide compelling correlations that are not experimentally testable (Sparrow and Mischke, 1961; Bennett, 1972; Vinogradov, 2003). Moreover, the common cohabitation of repetitive DNAs with genes, centromeres, telomeres and possibly other chromosome features will usually make it difficult or impossible to isolate large deletions that significantly affect genome size but do not affect other important processes. This difficulty in removing large blocks of repetitive DNA in single steps without also removing important genes (for example) is as true for natural mutations as it is for induced mutations. Although this problem should not exist for small deletions of 10 kb or less, it is difficult to see how natural selection might act at the level of genome size to distinguish between an individual that has 1000 Mb of DNA and one that has 999·99 Mb of DNA. If fitness were directly proportional to genome size, then a difference in fitness of 0·001 % may be far below the level of effective selection (Petrov and Hartl, 2000).
In the absence of incremental selection for nuclear DNA content, variations in genome size must originate with lineage-specific differences in the molecular mechanisms that increase or decrease genome size. The first phenomena found to affect genome size, polyploidy and transposable element amplification, both lead to overall growth in nuclear DNA content. Bennetzen and Kellogg (1997) proposed that, in the absence of any vigorous mechanism to counteract these processes, plant genomes would be on an inevitable road to ‘genomic obesity’. This disconcerting proposition was meant as a call to search for mechanisms that might decrease genome size. This call has been answered with recent studies that have shown the ability of unequal homologous recombination and illegitimate recombination (Shirasu et al., 2000; Devos et al., 2002; Wicker et al., 2003) to generate abundant small deletions that can attenuate or reverse plant genome growth, in agreement with similar evidence for insect genomes (Petrov et al., 1996).
It still remains unclear whether major variations in genome size are primarily the products of differences in rates/mechanisms of genome growth or in the efficiency of DNA removal by illegitimate or homologous recombination processes. Moreover, the contributions of natural selection to the current spectrum of genome size variation are not understood. This article describes recent results relating to mechanisms of genome size variation, and presents a model suggesting how intrinsic differences in the efficiencies of DNA amplification and removal can lead to major changes in nuclear DNA content that can serve as the substrates for natural selection.
RESULTS AND DISCUSSION
The LTR-retrotransposons are particularly sensitive indicators of basic nucleic acid biochemistry in the nucleus for a number of reasons. First, they are numerous and broadly distributed. Hence, they can always be found, they can indicate molecular events in all parts of the genome, and comparisons of related elements can be used to reconstruct the parental element(s) that gave rise to current LTR-retrotransposon populations. Second, LTR-retrotransposons have consistent structural features that facilitate their discovery and often permit conclusions regarding the nature of any mutational events they have experienced. For instance, the absence of a conserved element feature (like the primer binding site [PBS] or polypurine tract [PPT], both needed for element replication and subsequent insertion) can be conclusively attributed to deletions that occurred after insertion of the element. Third, most individual LTR-retrotransposons appear to be selectively neutral. Hence, most or all mutational events that occur within an element should be observable, without the filter applied by natural selection against a subset of ‘deleterious’ events. Of course, some LTR-retrotransposons can affect the expression of adjacent genes (White et al., 1994; Kashkush et al., 2003), but these are relatively rare. Most LTR-retrotransposons are not inserted near genes, and alleles of the same gene that have very different transposable element populations nearby (Fu and Dooner, 2002) will usually have similar or identical expression profiles. Similarly, evolutionary geneticists have used synonymous changes in protein-encoding exons as relatively neutral phenomena, despite the fact that synonymous nucleotide changes that do not alter protein-coding capacity can cause changes in mRNA three-dimensional structure and codon bias that may affect transcript stability and/or translational efficiency. We feel that LTR-retrotransposons provide a superior, although not perfect, standard for monitoring change in neutral DNA molecules.
We have searched for LTR-retrotransposons in the nearly complete sequences for both the Arabidopsis and rice genomes. In order to simplify the analysis, only a small number of element families were selected in each species (12 in Arabidopsis, 11 in rice), but their selection was completely random. This random selection was based on the initial random choice of a small number of completely sequenced bacterial artificial chromosomes (BACs) containing inserts of Arabidopsis or rice DNA. Very careful annotation by a combination of automated and manual procedures (Devos et al., 2002) was used to find all of the intact and fragmented LTR-retrotransposons that were present on these BACs. These annotation procedures included homology searches to databases that contained previously identified LTR-retrotransposons, but also for any repeated sequence. A second, and key, criterion was the presence of various structural features (e.g. two LTRs in the correct locations and orientations, PPTs, PBSs, etc.) that are definitive for an LTR-retrotransposon. Our experience indicates that these approaches definitively identify many more LTR-retrotransposons than are usually annotated on these same BACs by other researchers, even those that use relatively rigorous and comprehensive procedures (Ma et al., 2004).
Our studies on Arabidopsis determined that the majority of the elements in this nuclear genome are highly truncated, most with deletions that remove half or more of the complete element (Devos et al., 2002). Because many of these deleted elements were highly fragmented, it seemed likely that they are often the products of multiple independent deletions or other rearrangement events. Precise definition of individual changes from inspection of current molecules would be impossible if these rearrangements often overlapped. Thus, we decided to analyse only those insertions or deletions (indels) that could be precisely defined by comparison to the known or reconstructed structure of the parental element. For this same reason, we limited ourselves to the study of indels that were 10 bp or larger. Although smaller deletions were highly numerous and commonly appeared to be an outcome of slipped strand replication (J. Ma and J. L. Bennetzen, unpub. obs.), we were often unable to precisely define their 5′ and 3′ ends, so they were omitted from our analysis.
The Arabidopsis data indicated that about 90 % of the indels were deletions. One class of deletion was exceptionally common, and these are represented by the presence of solo LTRs. Solo LTRs occur frequently in any genome that contains LTR-retrotransposons. In yeast, they have been shown to be the outcome of unequal intrastrand homologous recombination (Roeder et al., 1980), and the same is expected to be true in all other organisms. Other deletions that were derived from unequal homologous recombination were indicated by LTR-retrotransposon structures that lack the flanking direct target repeats. These events require unequal homologous recombination between two different (but homologous) LTR-retrotransposons. Although rare (less than 2 % of elements analyzed), these unequal recombinations between different LTR-retrotransposons are significant because they delete most internal components of each of the participating elements plus the DNA between the two LTR-retrotransposons (Devos et al., 2002).
Among the deletions that were not associated with unequal homologous recombination, sizes varied from 10 bp (the lower size limit that we set) up to 3766 bp in the 172 events that we characterized. The smaller deletions far outnumbered the larger ones, providing median and mean deletion sizes of 50 bp and 304 bp in Arabidopsis. However, if the deletions smaller than 10 bp had been included, these two averages would have been shifted to a much smaller size. Moreover, our analysis is limited to deletions that are internal to an LTR-retrotransposon, meaning that we will miss any events that are larger than the size of an intact element (i.e. usually less than 10 kb). Because deletions larger than 1 kb are very rare (Fig. 1), we expect that big deletions of 10 kb or more are exceedingly uncommon. Thus, it appears that accumulation of a very large number of small deletions is responsible for the highly fragmented status of most of the LTR-retrotransposons in Arabidopsis (Devos et al., 2002).
Fig. 1.
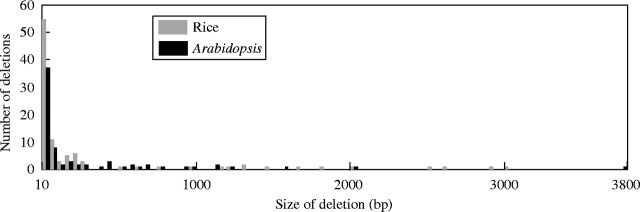
Spectrum of deletion sizes in LTR-retrotransposons of Arabidopsis and rice. Dark bars indicate Arabidopsis deletions and light bars indicate rice deletions. Deletions of specific size were pooled for this figure in ranges of 50 bp, except the first pool which records deletions of 10–50 bp. Subsequent pooled deletions sizes were 51–100 bp, 101–150 bp, 151–200 bp, etc. The data show that the respective mean and median deletion sizes were 304 bp and 50 bp for Arabidopsis, with 318 bp and 41 bp for rice.
A comparable study in rice found very similar results to those in Arabidopsis. In rice, the median and mean deletion sizes were 41 bp and 318 bp (Fig. 1). Hence, it does not appear that the more than three-fold difference in the sizes of these two genomes is caused by a comparable difference in the size or distribution of deletion events. However, the rice investigations also uncovered a high relative ratio of solo LTRs to intacts elements (Ma et al., 2004). This suggests that unequal homologous recombination may be relatively more active (compared to other deletion processes) in rice than in Arabidopsis. In both species, most of the deletions were found to be associated with short flanking repeats of anywhere from 2 bp to 15 bp. Such repeats, too small to allow homologous recombination, are commonly associated with illegitimate recombination. Illegitimate recombination, a term that describes a recombinational outcome that does not require the participation of a recA protein or large (>50 bp) stretches of sequence homology, can be the product of many different mechanisms including slipped strand repair and double-strand break repair.
The rice data were also analyzed to provide lower limits to the amount of LTR-retrotransposon DNA that had been removed by these small deletions over the last few million years. As a very minimum estimate, the data guaranteed that at least 190 Mb had been removed from the rice genome in the last 5 million years, leaving a genome that is now about 430 Mb (Ma et al., 2004). These analyses also indicate why LTR-retrotransposons in both Arabidopsis and rice appear to be so young. Our dating procedures require a largely intact element, with two LTRs, for a date to be ascertained. As deletions accumulate, we predict that the older elements will rarely have two surviving LTRs and thus can not be dated. In this regard, Ma and co-workers found that the oldest LTR-retrotranspons were the most severely deleted (as expected) and rarely had more than a few LTR segments conserved that could be used to establish an insertion date (Ma et al., 2004). Putting these facts into a coherent picture, the data indicated that LTR-retrotransposon sequences have a half-life of less than 6 million years in rice (Ma et al., 2004).
It is at least arguable that these deletion processes might be somehow limited to LTR-retrotransposons and possibly other repeated or heterochromatic DNA. However, analysis of sequence change in ‘homoeologous’ regions of the maize genome since its tetraploid origin ∼15 mya (million years ago, Gaut and Doebley, 1997) indicates that many genes have been removed by an accumulation of small deletions (Ilic et al., 2003). Moreover, analysis of indels that differentiate two races of Oryza sativa (japonica and indica), using Oryza glaberrima as an outgroup, has shown that these same types of deletions also affect genes and gene-flanking regions that do not contain any known transposons (J. Ma and J. L. Bennetzen, unpub. obs.).
The identification of processes that can efficiently increase or decrease genome size provides the mechanistic foundation for how genome size variation can be generated. However, it does not explain why genomes vary in size. If all plants have these mechanisms, then why aren't all plant genomes the same size? There are several possible, and non-exclusive, answers to this question. First, it is possible that some species may have greater activity of one of these mechanisms. For instance, LTR-retrotransposons might amplify more easily in some species than in others, perhaps because of a less-efficient silencing mechanism (Vance and Vaucheret, 2001). Alternatively, some species might have more frequent or larger deletions. Larger and more frequent deletions were observed in the small-genome insect Drosophila melanogaster than in the larger genome of the Hawaiian cricket (Petrov et al., 2000), while Puchta and co-workers have observed more frequent and larger deletions in Arabidopis than in tobacco, which has an approximately forty-fold larger genome (Kirik et al., 2000; Orel and Puchta, 2003). A second possibility is that the unique environment of a species might alter its exposure to genome-altering agents. For instance, some species might be more exposed to horizontal gene transfer by wide mating or insect herbivory (Bennetzen, 1996). Or an organism could have been exposed to a high level of radiation or other chromosome-breaking agent that can induce transposable element action (increasing genome size) or activate error-prone repair (that might decrease genome size). The third possible reason for major differences in genome size could be selection for or against large genomes. There is ample evidence to suggest that this must occur, at least where genome sizes vary by large amounts (Sparrow and Mischke, 1961; Bennett, 1972; Vinogradov, 2003; Knight et al. 2005).
In only one scenario, polyploidy, can we prove that a particular process is responsible for a dramatic difference in genome size between two closely related species. Much more research is needed before we can determine the foundations of other differences in nuclear DNA content. At the simplest level, it would be useful to determine if particular genomes have recently grown or shrunk. Phylogenetic analyses are particularly useful for this determination (Bennetzen and Kellogg, 1997; Leitch et al., 1998; Wendel et al., 2002; Leitch et al. 2005; Price et al. 2005).
One possible model for difference in genome size can be partially tested with current data sets. This model suggests that larger genomes have been more recently exposed to high levels of transposon amplification, particularly of the LTR-retrotransposons that account for so much of nuclear DNA content. Extensive evidence indicates that transposons can undergo transition from an inactive state to an active state in a single generation, leading to rapid amplification of element number and genome size. If this phenomenon accounted for most variation in genome size, then we might expect to see younger LTR-retrotransposons in larger genome species. However, our analyses of the ages of LTR-retrotransposons that can be dated by LTR divergence (SanMiguel et al., 1998) in multiple plant species have indicated that they all appear to be equally young in all plants investigated so far (Table 1). Because most LTR-retrotransposons older than 5 million years post-insertion are too severely fragmented (e.g. deleted) to be accurately dated (Ma et al., 2004), we cannot gain much information about ancient LTR-retrotransposon activity, but elements active in different periods within the last few million years can be distinguished. For instance, if one plant species had all of its elements active in the last one hundred thousand years, then this would be discerned as a great majority of elements with identical LTRs, while a species without any elements active for the last 2 million years or more would have very few retrotransposons with identical LTRs. There does seem to be variation for this trait (Table 1), so it will be useful to further investigate this question once a larger number of species have sufficiently deep databases. It may be that there are some instances where recentness of LTR-retrotransposon activity is a significant contributor to genome size variation between closely related plant species.
Table 1.
Recent insertion of LTR-retrotransposons in several plant species
| Ages of LTR-retrotransposons (mys)a |
|||||
|---|---|---|---|---|---|
| Species |
Genome size (Mb) |
No. of LTR-retrotransposons dated |
Range |
Average |
|
| Arabidopsis | 130 | 87b | 0·4–8·7 | 3·8 | |
| Rice | 430 | 260c | 0–11·7 | 2·5 | |
| Lotus | 470 | 12d | 0–1·7 | 0·3 | |
| Sorghum | 750 | 10e | 0·2–3·5 | 1·5 | |
| Maize | 2500 | 26f | 0·1–5·2 | 1·5 | |
| Barley | 4800 | 8g | 1·2–5·2 | 2·8 | |
| Diploid wheat | 5700 | 11h | 0·8–4·5 | 2·5 | |
The insertion times (million years) of LTR-retrotransposons were estimated in a manner similar to SanMiguel et al., 1998, using a mutation rate of 6·5 × 10−9 substitutions per synonymous site per year.
Sequence data are from: bDevos et al., 2002; cMa et al., 2004; eRamakrishna et al., 2002; eK. Ilic and J. L. Bennetzen, unpub. obs.; fSanMiguel et al., 1997; fIlic et al., 2003; gRostoks et al., 2002; and hSanMiguel et al., 2002. dLTR-retrotransposons were identified from randomly chosen BAC sequences in the GenBank non-redundant database.
Studies over the last several years have shown that polyploidy and retroelement amplification are the two processes responsible for most genome size increases within specific plant lineages. The number of species examined has been few, however, and significantly biased. Crop species and cereals have been the most frequently investigated. Obviously, more research is needed on species that cover a broader and more phylogenetically-informative set of lineages (Bennetzen, 2002). In addition, the reasons why some species have experienced more genome growth are not well understood. For instance, why would one lineage of plants show a consistently greater degree of LTR-retrotransposon amplification than another? One reasonable possibility is that some lineages are less effective in transposon inactivation by epigenetic silencing (Vance and Vaucheret, 2001). Comparative studies to investigate these questions would be appropriate, but they would not be simple given the inherent episodic natures of transposon activation, transgene silencing and other epigenetic phenomena.
Little is known at this time about the processes that remove DNA from the nuclear genome. Although unequal homologous recombination has been known as a generator of deletions for almost 80 years (Sturtevant, 1925), unequal homologous recombination between chromatids usually yields reciprocal duplication/deletions that do not provide any net change in DNA content. Unequal homologous recombination within a single chromatid (intrastrand recombination) appears to preferentially lead to deletions, but both of these processes can only remove the DNA between two large (>50 bp) direct repeats. Although such structures are found in LTR-retrotransposons, they are not that frequent in other genome components except tandem gene families. Illegitimate recombination, initially perceived as an exceedingly rare phenomenon in E. coli (Ehrlich, 1989), can act anywhere because it requires as little as 1 bp of sequence homology. These short repeated sequences are associated with deletions in many genomes, suggesting that illegitimate recombination is the major factor responsible for most DNA removal in those plant and animal species that have been investigated to date (Petrov and Hartl, 1997; Devos et al., 2002; Wicker et al., 2003; Ma et al., 2004). Hence, it seems likely that illegitimate recombination is a much more active process in higher eukaryotes than it is in E. coli. The actual molecular mechanism(s) of illegitimate recombination is not known in any investigated plant species, although DNA break repair is a likely candidate (Gorbunova and Levy, 1999). Future studies are needed to investigate these processes and their relative efficiencies in different plant lineages.
Investigations into the possible selective advantages or disadvantages of various nuclear DNA contents have dominated research into genome size variation. Because isogenic comparisons are not feasible, these studies have been unable to generate more than several very interesting correlations. Hence, we decided to take a step back and look at the molecular mechanisms underlying genome size variation, to see if these could provide insight into the level and type of variation that could be acted on by selection (Bennetzen and Kellogg, 1997). Our data have consistently shown that de novo genome size variation caused by transposable element amplification and accumulated small deletions can be massive, but will require generation over millions of years. Selection is not likely to act on the initial tiny changes, suggesting that variation in genome size within a population could accumulate exclusively by small and chance differences in transposon activity or by similar variability in mechanisms for DNA removal. These differences could lead to large degrees of genome size variation in an environment where genome size is not of selective significance. However, if the environment changes, then already-established major differences in genome size could now provide a selective advantage or disadvantage. For instance, tropical maize germplasm can vary greatly in genome size, and many lines carry more repetitive DNA than the average maize accession. When maize was adopted as a crop in North America, genome size was apparently of selective significance and led to the exclusive use of the smallest genome corns in the most northern parts of the continent (e.g. the Canadian flints; Cooper and Brink, 1937; Brown, 1949; Laurie and Bennett, 1985; Rayburn et al., 1985).
Plants provide a wonderful opportunity for the study of genome variation. This is partly true because so many important crop plants, and a few models, will have their genomes thoroughly investigated. Compared to similarly complex animals (e.g. the mammals), plants are growing, shrinking and otherwise rearranging their genomes at a tremendous pace. Hence, there is a wealth of opportunity for the discovery and characterization of both specific rearrangements and the mechanisms that drive these events. Future research will better define the roiling seas of repetitive DNA that surround plant gene islands, and perhaps indicate how we can utilize these complex genome geographies for basic study and crop improvement.
Acknowledgments
This research was supported by a grant from the Plant Genome Program at the National Science Foundation of the USA (#9975793).
LITERATURE CITED
- Bennett MD. 1972. Nuclear DNA content and minimum generation time in herbaceous plants. Proceedings of the Royal Society of London, Series B. Biological Sciences 181: 109–135. [DOI] [PubMed] [Google Scholar]
- Bennetzen JL. 1996. The contributions of retroelements to plant genome organization, function and evolution. Trends in Microbiology 4: 347–353. [DOI] [PubMed] [Google Scholar]
- Bennetzen JL. 2002. Opening the door to comparative plant biology. Science 296: 60–63. [DOI] [PubMed] [Google Scholar]
- Bennetzen JL, Kellogg EA. 1997. Do plants have a one way ticket to genomic obesity? The Plant Cell 9: 1509–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown WL. 1949. Numbers and distribution of chromosome knobs in United States maize. Genetics 34: 524–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DC, Brink RA. 1937. Chromosome homology in races of maize from different geographical regions. American Naturalist 71: 582–587. [Google Scholar]
- Devos KM, Brown JKM, Bennetzen JL. 2002. Genome size reduction through illegitimate recombination counteracts genome expansion in Arabidopsis Genome Research 12: 1075–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich SD. 1989. Illegitimate recombination in bacteria. In: Berg DE, Howe MM, eds. Mobile DNA. Washington, DC: American Society of Microbiology, 799–832. [Google Scholar]
- Flavell RB, Bennett MD, Smith JB, Smith DB. 1974. Genome size and proportion of repeated nucleotide DNA sequence in plants. Biochemical Genetics 12: 257–269. [DOI] [PubMed] [Google Scholar]
- Fu H, Dooner HK. 2002. Intraspecific violation of genetic colinearity and its implications in maize. Proceedings of the National Academy of Sciences, USA 99: 9573–9578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaut BS, Doebley JF. 1997. DNA sequence evidence for the segmental allotetraploid origin of maize. Proceedings of the National Academy of Sciences, USA 94: 6809–6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbunova V, Levy AA. 1997. How plants make ends meet: DNA double-strand break repair. Trends in Plant Science 4: 263–269. [DOI] [PubMed] [Google Scholar]
- Ilic K, SanMiguel PJ, Bennetzen JL. 2003. A complex history of rearrangement in an orthologous region of the maize, sorghum and rice genomes. Proceedings of the National Academy of Sciences, USA 100: 12265–12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashkush K, Feldman M, Levy AA. 2003. Transcriptional activation of retrotransposons alters the expression of adjacent genes in wheat. Nature Genetics 33: 102–106. [DOI] [PubMed] [Google Scholar]
- Kirik A, Salomon S, Puchta H. 2000. Species-specific double-strand break repair and genome evolution in plants. EMBO Journal 19: 5562–5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight CA, Molinari N, Petrov DA. 2005. The large genome constraint hypothesis: evolution, ecology and phenotype. Annals of Botany 95: 177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Bennetzen JL. 1999. Plant retrotransposons. Annual Review of Genetics 33: 479–532. [DOI] [PubMed] [Google Scholar]
- Laurie DA, Bennett MD. 1985. Nuclear DNA content in the genera Zea and Sorghum Intergeneric, interspecific and intraspecific variation. Heredity 55: 307–313. [Google Scholar]
- Leitch IJ, Chase MW, Bennett MD. 1998. Phylogenetic analysis of DNA C-value provides evidence for a small ancestral genome size in flowering plants. Annals of Botany 82: 85–94. [Google Scholar]
- Leitch IJ, Soltis DE, Soltis PS, Bennett MD. 2005. Evolution of DNA amounts across land plants (Embryophyta). Annals of Botany 95: 207–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Devos KM, Bennetzen JL. 2004. Analyses of LTR-retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Research 14: 860–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orel N, Puchta H. 2003. Differences in the processing of DNA ends in Arabidopsis thaliana and tobacco: possible implications for genome evolution. Plant Molecular Biology 51: 523–531. [DOI] [PubMed] [Google Scholar]
- Petrov DA, Hartl DL. 2000. Pseudogene evolution and natural selection for a compact genome. Journal of Heredity 91: 221–227. [DOI] [PubMed] [Google Scholar]
- Petrov DA, Lovovskaya ER, Hartl DL. 1996. High intrinsic rate of DNA loss in Drosophila Nature 384: 346–349. [DOI] [PubMed] [Google Scholar]
- Petrov DA, Sangster TA, Johnston JS, Hartl DL, Shaw KL. 2000. Evidence for DNA loss as a determinant of genome size. Science 287: 1060–1062. [DOI] [PubMed] [Google Scholar]
- Price HJ, Dillon SL, Hodnett G, Rooney WL, Ross L, Johnston JS. 2005. Genome evolution in the genus Sorghum (Poaceae). Annals of Botany 95: 207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayburn AL, Price HJ, Smith JD, Gold JR. 1985. C-band heterochromatin and DNA content in Zea mays American Journal of Botany 72: 1610–1617. [Google Scholar]
- Roeder GS, Farabaugh PJ, Chaleff DT, Fink GR. 1980. The origins of gene instability in yeast. Science 209: 1375–1380. [DOI] [PubMed] [Google Scholar]
- Rostoks N, Park Y-J, Ramakrishna W, Ma J, Druka A, Shiloff BA, SanMiguel PJ, Jiang Z, Brueggeman R, Sandhu D, Gill K, Bennetzen JL, Kleinhofs A. 2002. Genomic sequencing reveals gene content, genomic organization, and recombination relationships in barley. Functional and Integrative Genomics 2: 70–80. [DOI] [PubMed] [Google Scholar]
- SanMiguel P, Bennetzen JL. 1998. Evidence that a recent increase in maize genome size was caused by the massive amplification of intergene retrotransposons. Annals of Botany 82: 37–44. [Google Scholar]
- SanMiguel P, Gaut BS, Tikhonov A, Nakajima Y, Bennetzen JL. 1998. The paleontology of intergene retrotransposons of maize. Nature Genetics 20: 43–45. [DOI] [PubMed] [Google Scholar]
- SanMiguel P, Tikhonov A, Jin Y-K, Motchoulskaia N, Zakharov D, Melake-Berhan A, Springer PS, Edwards KJ, Lee M, Avramova Z, Bennetzen JL. 1996. Nested retrotransposons in the intergenic regions of the maize genome. Science 274: 765–768. [DOI] [PubMed] [Google Scholar]
- Shirasu K, Schulman AH, Lahaye T, Schulze-Lefert P. 2000. A contiguous 66-kb barley DNA sequence provides evidence for reversible genome expansion. Genome Research 10: 908–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow AH, Mischke JP. 1961. Correlation of nuclear volume and DNA content with higher plant tolerance to chronic radiation. Science 134: 282–283. [DOI] [PubMed] [Google Scholar]
- Sturtevant AH. 1925. The effects of unequal crossing over at the bar locus in Drosophila Genetics 10: 117–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Arabidopsis Genome Initiative. 2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana Nature 408: 796–815. [DOI] [PubMed] [Google Scholar]
- Vance V, Vaucheret H. 2001. RNA silencing in plants: defense and counterdefense. Science 292: 2277–2280. [DOI] [PubMed] [Google Scholar]
- Vicient CM, Suoniemi A, Anamthawat-Jonsson K, Tanskanen J, Beharav A, Nevo E, Schulman AH. 1999. Retrotransposon BARE-1 and its role in genome evolution in the genus Hordeum The Plant Cell 11: 1769–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradov AE. 2003. Selfish DNA is maladaptive: evidence from the plant Red List. Trends in Genetics 19: 609–614. [DOI] [PubMed] [Google Scholar]
- Wendel JF. 2000. Genome evolution in polyploids. Plant Molecular Biology 42: 225–249. [PubMed] [Google Scholar]
- Wendel JF, Cronn RC, Johnston JS, Price HJ. 2002. Feast and famine in plant genomes. Genetica 115: 37–47. [DOI] [PubMed] [Google Scholar]
- White SE, Habera LF, Wessler SR. 1994. Retrotransposons in the flanking regions of normal plant genes: a role for copia-like elements in the evolution of gene structure and expression. Proceedings of the National Academy of Sciences, USA 91: 11792–11796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T, Yahiaoui N, Guyot R, Schlagenhauf E, Liu Z, Dubcovsky J, Keller B. 2003. Rapid genome divergence at orthologous low molecular weight glutenin loci of the A and Am genomes of wheat. The Plant Cell 15: 1186–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]