

The male-specific lethal dosage compensation complex (MSL-DCC) selectively assembles on the X chromosome in Drosophila and activates gene transcription by twofold through histone acetylation. An MSL recognition element (MRE) sequence motif nucleates the initial MSL association. Here, Zheng et al. identified the CXC domain of MSL2 specifically recognizing the MRE motif and determined its crystal structure bound to specific and nonspecific DNAs. Specific DNA-binding mutants of MSL2 are impaired in MRE binding and X chromosome localization in vivo.
Keywords: dosage compensation, crystal structure, DNA–protein complex, nonspecific DNA binding
Abstract
The male-specific lethal dosage compensation complex (MSL-DCC) selectively assembles on the X chromosome in Drosophila males and activates gene transcription by twofold through histone acetylation. An MSL recognition element (MRE) sequence motif nucleates the initial MSL association, but how it is recognized remains unknown. Here, we identified the CXC domain of MSL2 specifically recognizing the MRE motif and determined its crystal structure bound to specific and nonspecific DNAs. The CXC domain primarily contacts one strand of DNA duplex and employs a single arginine to directly read out dinucleotide sequences from the minor groove. The arginine is flexible when bound to nonspecific sequences. The core region of the MRE motif harbors two binding sites on opposite strands that can cooperatively recruit a CXC dimer. Specific DNA-binding mutants of MSL2 are impaired in MRE binding and X chromosome localization in vivo. Our results reveal multiple dynamic DNA-binding modes of the CXC domain that target the MSL-DCC to X chromosomes.
The evolution of species with sexual dimorphism commonly involved converting a pair of autosomes into heteromorphic sex chromosomes. In humans and fruit flies, two X chromosomes define the female sex, whereas males have only one X in addition to the Y chromosome. Avoiding recombination between the sex chromosomes, the proto-Y chromosome lost most of its resident genes, leaving the proto-X monosomic in the males. This unbalanced situation diminishes the vitality of the organism and therefore generated an evolutionary pressure to compensate for the reduced dosage of X chromosomal genes. In mammals and fruit flies, this is achieved by selective transcriptional activation of X chromosomal genes through histone acetylation (Straub and Becker 2011; Deng et al. 2013). Whereas in Drosophila melanogaster, the X chromosome is only boosted in males, in mammals, all X chromosomes in both sexes are activated followed by the selective inactivation of one X in females (Disteche 2012).
One of the fundamental questions of outstanding interest is how an entire sex chromosome is molecularly distinguished from the autosomes. This question can be addressed conveniently in the Drosophila model, where a basic set of dosage compensation factors has been found following the male-specific lethal (MSL) loss-of-function phenotype. These so-called MSL proteins and noncoding roX (or RNA on the X) RNAs form a regulatory complex (the MSL dosage compensation complex [MSL-DCC]) that selectively associates with the X chromosome. The MSL-DCC consists of MSL1, MSL2, MSL3, the RNA helicase maleless (MLE), the histone acetyltransferase MOF (males absent on the first), and roX RNAs. MSL1 is a dimeric scaffolding protein that interacts with MSL2, MSL3, and MOF. The structural basis for these interactions has been determined for the homologous mammalian MSL complex (Kadlec et al. 2011; Hallacli et al. 2012).
According to the prevailing model, the X chromosome is marked by the presence of a relatively small number (150–250) of chromosomal entry sites (CESs) or high-affinity sites (HASs). These sites are able to autonomously recruit the MSL-DCC even if inserted into an ectopic, autosomal location (Kageyama et al. 2001; Park et al. 2002, 2010; Alekseyenko et al. 2008). Once bound, the complex is thought to distribute the activating histone H4 acetylation to active genes in the nuclear neighborhood (Gelbart and Kuroda 2009; Gorchakov et al. 2009). Within the HASs, a GA-rich sequence motif, the MSL recognition element (MRE), is important for MSL-DCC targeting (Alekseyenko et al. 2008; Straub et al. 2008). Interestingly, in the related species Drosophila miranda, large autosomal fragments have been fused to a proto-X chromosome relatively recently and are apparently on their way to “catch up” with dosage compensation by newly acquiring high-affinity MRE sequences from transposon-derived precursor sequences (Alekseyenko et al. 2013; Ellison and Bachtrog 2013).
The hallmark of an MRE is a degenerate, 21-base-pair (bp) GAGA-rich sequence motif, which is enriched ∼1.5-fold to 1.8-fold on the X chromosome versus the autosomes (Alekseyenko et al. 2008; Straub et al. 2008). The Drosophila genome harbors ∼12,000 MRE sequence motifs, yet only a small fraction of these (1%–3%) is actually bound by the MSL-DCC (Alekseyenko et al. 2012). Recently, a zinc (Zn) finger protein, CLAMP (chromatin-linked adaptor for MSL proteins), was found to regulate the assembly of the MSL complex on the X chromosome and be enriched in HASs (Larschan et al. 2012; Soruco et al. 2013). CLAMP shows synergism with the MSL-DCC in chromosomal interaction, and its in vivo and in vitro binding consensus closely matches MRE (Soruco et al. 2013). However, while it is true that CLAMP can be found at most HASs, it also binds thousands of other GAGA sequences on X and autosomes that do not qualify as MREs and do not attract the MSL-DCC. Therefore, the search for a specific determinant for HAS discrimination is still ongoing.
The critical determinant for HAS recognition must reside within the MSL-DCC itself. Early genetic studies had shown that among all MSL-DCC components, only MSL1 and MSL2 are required for initial association of the MSL complex to HASs (Lyman et al. 1997; Gu et al. 1998; Meller et al. 2000; Oh et al. 2003). More recently, we noted in studies aimed at determining the contact surface of the MSL-DCC with HASs in vivo that MSL2 and the RNA helicase MLE are the only subunits that can be cross-linked to HASs under the most stringent conditions (Straub et al. 2013). Our attention is thus drawn to MSL2, the only male-specific component of the MSL-DCC that initiates the assembly of the complex from its components. We found earlier in reporter gene assays that MSL2 can be recruited to HAS sequences in cells, but it remained unclear whether this binding was direct (Fauth et al. 2010). MSL2 contains an N-terminal RING finger domain and a small cysteine-rich CXC domain. The RING domain binds the coiled-coil domain of MSL1 and functions as a ubiquitin E3 ligase (Wu et al. 2011; Hallacli et al. 2012; Villa et al. 2012). The nuclear magnetic resonance (NMR) structure of the CXC domain shows that it binds three Zn ions and shares an unexpected structural resemblance with the pre-SET domain of histone methyltransferases (Zheng et al. 2012). The CXC domain enables MSL2 to bind DNA, but so far the specificity of this interaction could not be demonstrated (Fauth et al. 2010).
Here, we demonstrated that the isolated CXC domain specifically recognizes a model MRE motif. Through structural studies, we found that the CXC domain is able to interact with DNA in three distinct ways. Isolated CXC domains can engage with DNA in specific or nonspecific low-affinity interactions, which we define as the “search mode.” In addition, two CXC domains bind synergistically with the MRE in a high-affinity conformation that involves sequence-specific contacts through single arginines. Mutation of this contact arginine strongly impairs the ability of MSL2 to bind HAS sequences in vivo. Expression of the MSL2 mutant leads to disruption of the coherent dosage-compensated X chromosomal territory. Our data suggest an active participation of the MSL2 CXC domain in HAS recognition and point to conformational changes of the protein upon transition from the DNA sequence search mode to the high-affinity interaction at bona fide MSL response elements.
Results
The CXC domain of MSL2 specifically recognizes an MRE motif
To study DNA-binding properties of the CXC domain in detail, we titrated the isolated, 15N-labeled domain with a 12-bp DNA fragment, called S12, derived from the MRE motif CES11D1 (Fig. 1A), and monitored their binding by NMR (Fig. 1B). The CES11D1 sequence has been shown to recruit the MSL-DCC complex when inserted into autosomes, and S12 includes the core GA-rich region of CES11D1, which, when mutated, abolished the MSL recruitment (Alekseyenko et al. 2008).
Figure 1.

The MSL2 CXC domain specifically recognizes the MRE motif. (A) The CES11D1 MRE motif and derived DNA fragments used in this study. Nucleotides on each strand are numbered, with prime denoting the bottom strand. The dinucleotide sequences recognized by R543 of CXC domains 1–4 in the S15 complex structure are shaded green. Mutations are red. (B) Superimposition of 1H–15N HSQC spectra of the 15N-labeled CXC domain titrated with S12 DNA of the indicated molar ratios. Peaks of R526, G528, and G531 used for fitting are boxed. (C) The combined chemical shift changes Δδ of three residues upon titration of S12 and S12 mutant (S12mut) DNA are globally fit to a binding model with the indicated Kd values.
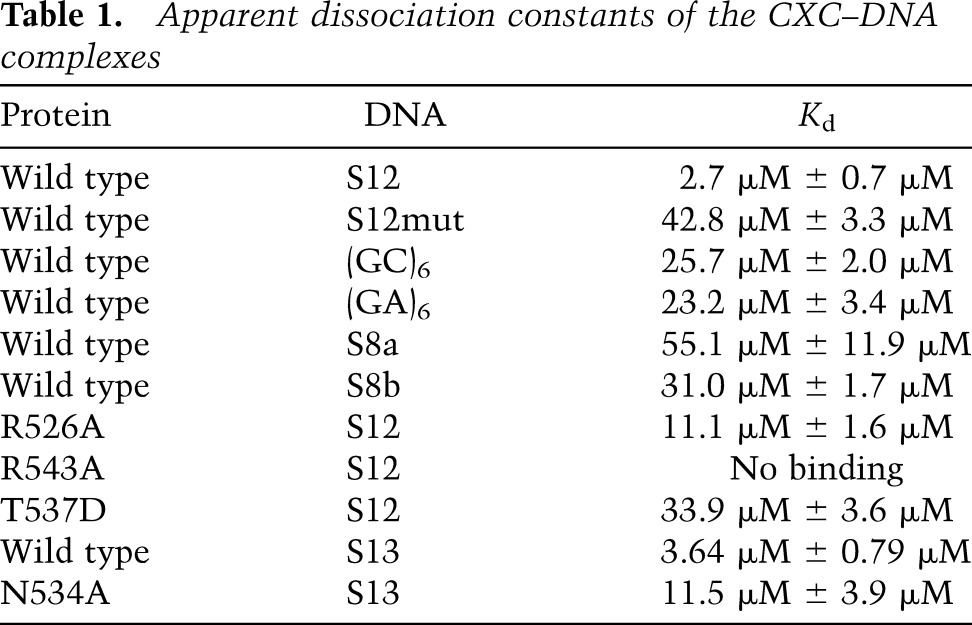
Many cross-peaks in the HSQC spectra continuously shifted upon addition of DNA (Fig. 1B), indicating that the CXC domain interacts with DNA and undergoes fast exchange between free and bound states on the NMR timescale. The S12 DNA appears to contain at least two binding sites, as the titration was nearly complete with 0.5 molar equivalents of DNA. We estimated the apparent dissociation constant (Kd) of the DNA complex to be 2.7 µM with quantitative analysis of chemical shift changes (Fig. 1C; Table 1). Mutation of the central GA repeat of CES11D1 to the A tract has been shown to block the MSL association in flies (Alekseyenko et al. 2008). Incorporation of the same mutation into S12 significantly reduced the binding affinity by 15-fold to 42.8 µM (Fig. 1C). These results indicate that the CXC domain specifically recognizes the MRE motif.
Table 1.
Apparent dissociation constants of the CXC–DNA complexes
Structure of the CXC domain bound to a specific MRE DNA
To understand the detailed mechanism of DNA recognition, we determined a crystal structure of the CXC domain bound to a 15-bp fragment of CES11D1 (S15) at 2.0 Å resolution and with an Rwork/Rfree of 0.197/0.254 (Fig. 2A; Table 2). In the complex structure, four CXC domains, numbered 1–4, bind the DNA in the asymmetric unit, providing four independent views of DNA recognition.
Figure 2.

Structure of a specific DNA complex of the CXC domain. (A) Overall structure of four CXC domains bound to S15 DNA. (B) Schematics of protein–DNA hydrogen bond interactions. (C–F) Detailed DNA interactions of CXC molecules 1 (C), 2 (D), 3 (E), and 4 (F). The 2fo − fc electron density map is shown at 1.0 σ level for R543 and R526. (G) The dimer interface between CXC molecules 1 and 2. Dashed lines denote hydrogen bonds. Four CXC molecules (1–4) are colored slate, green, cyan, and magenta; the top and bottom strand of DNA are colored yellow and wheat; and oxygen and nitrogen are colored red and blue.
Table 2.
Statistics on data collection and structure refinement
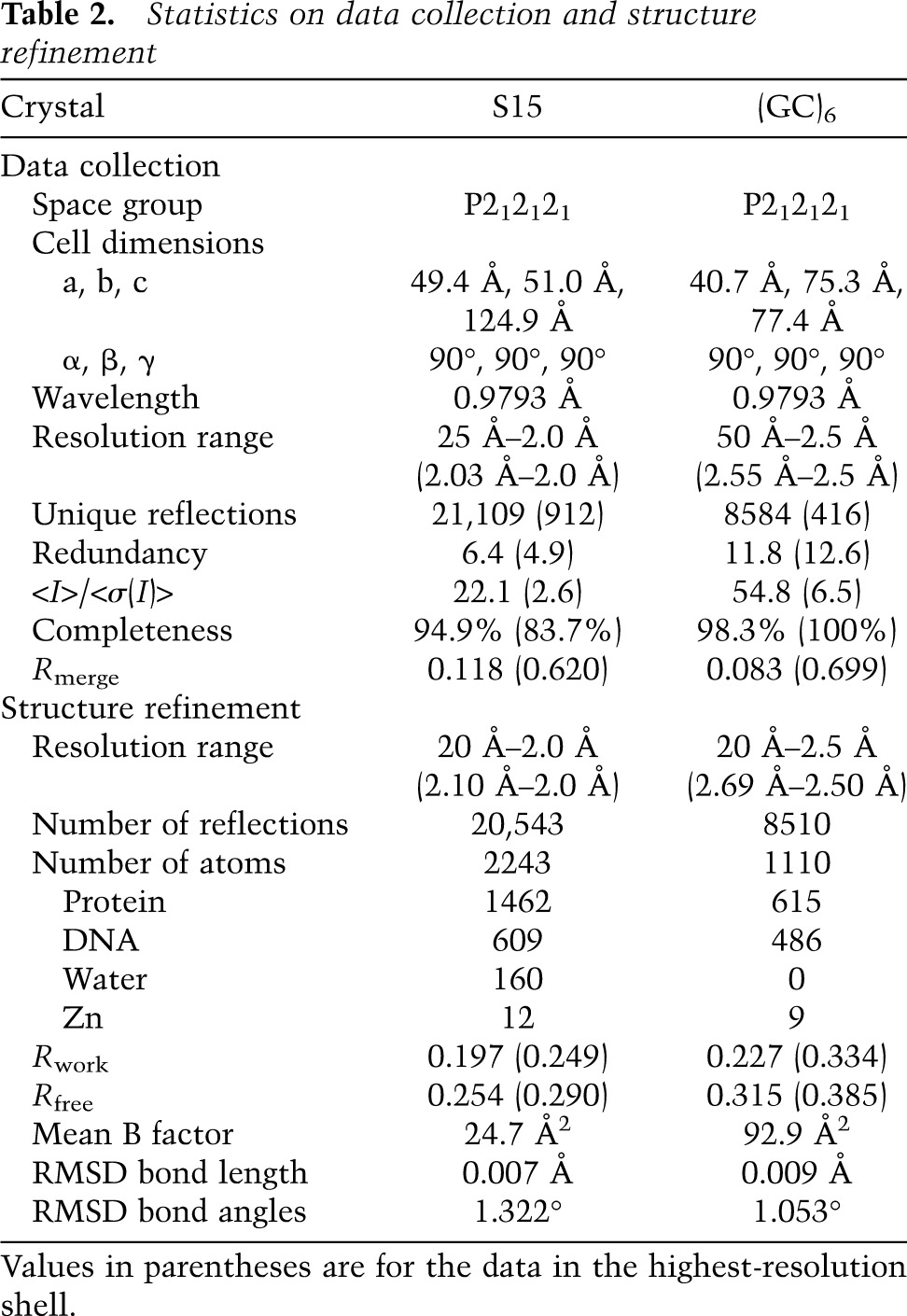
The crystal structure of the CXC domain closely resembles its previously determined NMR structure (Supplemental Fig. S1A; Zheng et al. 2012). Each CXC domain is composed of several loops and a short α helix that encircle three Zn ions by two rounds in a right-handed manner. Three Zn ions are coordinated by six terminal and three bridging cysteines (Supplemental Fig. S1B). The crystal structure validates the Zn coordination pattern previously determined by NMR and also provides more accurate measurements of the geometry of the unusual Zn3Cys9 cluster.
The DNA duplex adopts a standard B-form structure and stacks with symmetry-related DNA in a staggered manner (Fig. 2A). Each CXC domain spans nearly 6 bp of DNA and yet primarily binds with one strand. Molecules 1 and 3 bind the bottom strand with a 4-bp shift, whereas molecules 2 and 4 bind the top strand with a 5-bp shift. All molecules bind DNA in a generally similar way (Fig. 2B–F), but molecules 1 and 2 additionally contact each other, resulting in enhanced DNA binding. Each CXC domain strides on one strand of DNA duplex and inserts two prominent arginine “legs,” R526 and R543, into the major and minor groove, respectively.
The DNA-binding surface is composed of the N-terminal half of the polypeptide (residues 525–545) that can be divided into two parts placed at the major and minor groove sides, respectively. Residues 525–532 are placed over the major groove and contact intimately with the phosphate sugar backbone of DNA. Specifically, the backbone amide nitrogen atoms of R526 and G528 form hydrogen bonds with two consecutive phosphate groups. The side chain of R526 is well ordered at the major groove except in molecule 4 but does not directly form hydrogen bonds with sequence-encoding base edges. R526 of molecule 1 interacts with T19′ from a symmetry-related S15 DNA, but this interaction may be artificial, since the DNA helix is staggered at the junction (Fig. 2C).
Residues 534–545 are situated at the minor groove side. The side chain of R543 shows well-ordered electron density in all CXC molecules. It inserts vertically deep into the minor groove and forms two hydrogen bonds with the edges of two adjacent bases in the primary bound strand (Fig. 2C–F). R543 is the only residue in the CXC domain that directly reads out DNA sequence. Specifically, R543 of CXC domains 1–4 contacts with the dinucleotide sequences of TC, CG, TC, and TG (in 5′–3′ order), respectively (Fig. 1A). R543 of molecule 2 additionally makes a water-bridged interaction with C10′. The terminal nitrogen atoms of the guanidinium group of R543 hydrogen-bond with O2 of thymine and cytosine and N3 of guanine. The A-tract mutation would alter the binding site of molecules 2 and 3.
The MRE motif is enriched with GA dinucleotides (Alekseyenko et al. 2008; Straub et al. 2008). In the structure, TC, which is equivalent to GA in a complementary strand, is recognized twice, supporting that GA is a favorable target. However, a 12-bp DNA composed of six consecutive GA repeats does not constitute a strong binding target (Kd = 23.2 µM) (Table 1), suggesting that the sequence context around GA is also important.
In addition, residue S542 reaches out to contact a phosphate group of the other strand across the minor groove. This interaction is the only one directed to the other strand and could gauge the width of the minor groove and hence indirectly recognize the DNA sequence.
DNA titration in solution showed that residues that experienced significant chemical shift change are all located at the DNA-binding surface, validating the crystal structure (Fig. 1B; Supplemental Fig. S2A,B). The DNA-binding surface is positively charged, which would facilitate interaction with negatively charged DNA (Supplemental Fig. S2C).
The core region of the MRE motif cooperatively recruits a CXC dimer
Importantly, molecules 1 and 2, which bind to opposite DNA strands across the minor groove, additionally contact each other to form a dimer. Such an arrangement suggests that they bind cooperatively to the central GAGCG region of the MRE motif. They are roughly related by twofold symmetry and dimerize with an equivalent region (residues 532–537), but the dimer interface is asymmetric (Fig. 2G).
The side chain amide group of N534 in molecule 2 forms two hydrogen bonds with the side chain oxygen and backbone nitrogen of S542′ in molecule 1 (residues in molecule 1 are denoted by prime hereafter). This interaction pulls S542′ away from binding the phosphate of C11 but stabilizes N534 and its nearby S532 to bind the phosphate of G12. N534 and S532 are intrinsically flexible in the free protein (Zheng et al. 2012) and are disordered or do not contact DNA in the other three CXC molecules in the crystal. Protein dimerization appears to stabilize the flexible loop of molecule 2 to bind DNA. In addition, the dimer interface is stabilized by van der Waals and hydrogen bond interactions among residues L536 and T537 of molecule 2 and L536′, T537′, and N534′ of molecule 1. The protein interaction between two CXC domains and the enlarged DNA-binding region of molecule 2 would increase the affinity and specificity of DNA binding.
Our crystal structure suggests that the central GAGCG region constitutes the strongest binding site in the CES11D1 MRE motif and can simultaneously recruit two CXC domains. To test whether such binding also occurs in solution, we measured the chemical shift change of sugar H1′ protons of S12 and CES11D1 DNA upon binding with one molar equivalent of the CXC domain. The protein was of limiting amount compared with available binding sites of DNA to ensure that it selectively bound to high-affinity sites of DNA. The chemical shifts of H1′ protons were assigned through sequential NOE connections of ribose H1′ protons with their own and 5′-flanking base H6/8 protons in two-dimensional (2D) 1H–1H NOESY spectra (Supplemental Fig. S3A–D). We could assign H1′ protons for both strands in the 12-bp S12 DNA but only for the top strand in the 21-bp CES11D1 DNA due to a spectral overlap problem. Nevertheless, the results of S12 show that the chemical shift changes are highly correlated for two H1′ protons from the same base pairs (Fig. 3A).
Figure 3.

Chemical shift perturbation of MRE DNA upon binding of the CXC domain. (A,B) Chemical shift changes of H1′ protons in S12 (A) and CES11D1 (B) DNA induced by binding of one molar equivalent of the CXC domain. Chemical shift changes were calculated as δfree − δbound and are shown in red for the top strand and black for the bottom strand. The CES11D1 sequence is aligned at the bottom, with four R543-binding sites seen in the S15 complex structure shaded.
The two most significantly perturbed H1′ protons are located at positions 8 and 12 for both DNAs, which correspond to the 3′ nucleotides of dinucleotide sequences bound by molecules 1 and 2 in the S15 complex (Fig. 3A,B). These results strongly support that two CXC domains also preferentially bind at the GAGCG region in solution.
To provide biochemical evidence of cooperative binding at sites 1 and 2, we measured the Kd for two 8-bp DNAs (S8a and S8b) that contain separated binding sites for molecule 1 and molecules 2/3 (the overlapping binding sites for molecules 2 and 3 are not separable), respectively (Figs. 1A, 2B). Both DNAs displayed significantly reduced affinities (Kd = 55.1 and 31.0 µM) compared with S12 (Table 1). Hence, the strong binding of S12 does not stem from any of its subsites yet requires the juxtaposition and cooperative binding of sites 1 and 2/3.
The CXC–DNA interaction is required for X chromosome localization
To experimentally assess the importance of residues mediating DNA binding or protein dimerization, we carried out mutagenesis in vitro and in vivo. NMR titration showed that alanine mutation of R543 completely abolished the DNA binding, whereas alanine mutation of R526 reduced the binding affinity by fourfold (Fig. 4A; Table 1). Mutations at the protein dimer interface, N534A and T537D, also reduced DNA binding by 3.1-fold and 12.5-fold, indicating that the CXC domain dimerization is important for MRE recognition. These mutations did not grossly perturb the structure, as all mutant proteins still displayed a single folded conformation in HSQC spectra (Fig. 4A). The MSL2-GFP mutants showed similar expression level and stability (Supplemental Fig. S4B) and retained similar interaction with MSL1 in coimmunoprecipitation (data not shown).
Figure 4.

Mutational analysis of the CXC domain. (A) Titration of CXC mutants with S12 DNA. Selected regions of HSQC spectra are shown. (B) Immunofluorescence analysis of the X chromosomal territories in S2 cell lines stably expressing MSL2-GFP or its R543A mutant using antibodies against GFP and MSL3. Arrowheads indicate compact normal X territories. Unmarked GFP-expressing cells lack a distinct chromosomal territory and show delocalized MSL2-GFP and endogenous MSL3. The percentage of cells that show such defects is displayed at the right. (C) ChIP assays on S2 cells (CTRL) or stable cell lines expressing MSL2-GFP or its mutants (R543A, R526A, and N534A). The binding of MSL2 or one of the mutants to three high-affinity sites (CES11D1, roX1, and Set2-H) or to a control locus that has no MRE sequences (Set2-1) from two independent biological replicates is shown side by side and expressed as percentage of input.
The MSL-DCC is localized to distinct X chromosomal territories in male S2 cells (Straub et al. 2005). Expression of MSL2-GFP proteins bearing CXC point mutations in S2 cells allows monitoring of their functionality in vivo. MSL2-GFP wild type faithfully colocalizes with MSL3 to the X chromosome, which appears as a distinct, compact territory in 95% of the cases. In contrast, expressing the R543A mutated protein led to disruption of the dosage-compensated chromosomal territory and delocalization of both MSL2-R543A-GFP and endogenous MSL3 in 75% of the cases (Fig. 4B). The expression of R526A and N534A mutants showed similar, albeit less pronounced, effects (Supplemental Fig. S4A).
The localization of each mutant to the X chromosomal territory correlates with its DNA-binding capability. To study this in vivo, we subjected the MSL2-GFP proteins to chromatin immunoprecipitation (ChIP) and monitored their interactions with three selected HASs: CES11D1, roX1, and Set2-H (Alekseyenko et al. 2008; Straub et al. 2013). The robust binding of MSL2-GFP to all three sites in vivo was strongly compromised if R543 was mutated to alanine (Fig. 4C). The R526 and R534 mutants showed intermediate steady-state binding, as before. We conclude that the DNA-binding ability of the CXC domain is required for X targeting and the organization of the dosage-compensated X chromosomal territory.
Structure of the CXC domain bound to a nonspecific DNA
Our data show that the CXC domain is capable of binding nonspecific sequences with weak affinity. To understand the difference between the specific and nonspecific binding mode, we determined a crystal structure of the CXC domain bound to a 12-bp DNA composed of six GC repeats at 2.5 Å (Table 2). This DNA represents a nonspecific target, as it binds the CXC domain with a Kd value of 25.7 µM, 10-fold weaker than the same length DNA S12 (Table 1). In the complex structure, two CXC molecules bind separately to the same strand of DNA (Fig. 5A). A third CXC domain was associated with very poor electron density, likely because of structural flexibility or low occupancy. This molecule was not modeled except for three Zn ions, which could contribute to the relatively high Rfree value (0.312) of the current model. The interaction with the DNA phosphate backbone is generally similar between the nonspecific (GC)6 and specific S15 complex, but the conformation of R526 and R543 is notably different (Fig. 5B–D). R526 does not specifically contact base edges in the S15 complex structure but makes hydrogen bonds with the Hoogsteen edge of a guanine at the major groove in the (GC)6 complex structure. The side chain of R543 is associated with very weak electron density in the (GC)6 complex structure, suggesting that it is rather flexible. R543 would contact a GC dinucleotide if taking a specific binding mode; GC does not seem to be a favorable sequence. Nevertheless, R543 is essential for binding even nonspecific sequences (Fig. 4A; data not shown). R543 could interact dynamically with the minor groove and make electrostatic interaction with DNA phosphates in the presence of unfavorable sequences. The nonspecific complex structure further demonstrates the important role of R543 in sensing DNA sequences.
Figure 5.

Structure of a nonspecific DNA complex of the CXC domain. (A) Overall structure of three CXC domains bound to (GC)6 DNA. (B) Schematics of protein–DNA hydrogen bond interactions. (C,D) Detailed DNA interactions of CXC molecules 1 (C) and 2 (D). The 2fo − fc electron density map is shown at the 1.0 σ level for R543 and R526. Dashed lines denote hydrogen bonds. Two CXC molecules (1 and 2) are colored slate and green, the top and bottom strand of DNA are colored yellow and wheat, and oxygen and nitrogen are red and blue.
Discussion
We showed here that the CXC domain specifically recognizes a central element of the MRE motif, and this interaction is required for localization of the MSL complex to X chromosomes. Remarkably, the CXC domain uses a single arginine to directly read out dinucleotide sequences from the minor groove of DNA, distinct from other DNA-binding domains that commonly recognize DNA sequence from the major groove with large secondary structure elements (Freemont et al. 1991). Arginine has been documented to interact with the minor groove but, in most cases, indirectly reads out DNA sequences by binding narrow minor grooves adopted by AT-rich sequences (Rohs et al. 2009).
A single small CXC domain offers only limited binding specificity and affinity. Our data show that the MRE motif harbors tandem sites on opposite strands to cooperatively recruit two CXC domains, hence overcoming this problem. Such a binding mode is consistent with and would be facilitated by the dimeric organization of the MSL1/MSL2 complex (Hallacli et al. 2012). The tesmin/TSO1 family of proteins often contain two clustered homologous CXC domains (Cvitanich et al. 2000; Marin 2003; Schmit et al. 2009), which are distantly related to the CXC of MSL2 and probably recognize DNA by forming an intramolecular dimer.
We observed three distinct DNA-binding modes in the two determined DNA complex structures of the CXC domain. In mode I, molecules 1 and 2 in the S15 complex bind as a dimer and recognize two dinucleotide sequences on opposite DNA strands. This cooperative binding mode appears to be the key determinant of high-affinity recognition of the MRE motif. The S15 complex also shows that molecules 3 and 4 each bind DNA as an isolated domain and recognize dinucleotide sequences at the minor groove. This binding, termed mode II here, should be of lower affinity compared with mode I due to a lack of protein cooperativity. Finally, the (GC)6 DNA complex illustrates another low-affinity binding mode (mode III) in which individual CXC domains recognize a G at the major groove but not dinucleotide sequences at the minor groove. It is unclear at present whether there is an additional binding mode; for instance, one involving both R543 and R526 in sequence recognition.
The CXC domain may assume different binding modes in various functional contexts. Interaction of DNA-binding domains with specific sequences is often preceded by interaction with nonspecific or less specific sequences (Kalodimos et al. 2004). MSL2 may initially bind DNA in a low-affinity noncooperative manner (mode II or III), which would facilitate diffusion along DNA to search for target sites. Once an MRE is encountered, the interaction transforms into the high-affinity mode involving synergistic interactions between two CXC domains. In addition, when the MSL-DCC spreads to nearby active genes after binding to HASs, MLS2 could bind non-MRE regions with low-affinity modes. The plasticity in DNA binding would enable the CXC domain to bind various sequences encountered at different stages of X targeting.
Our structure shows that the S15 sequence could bind simultaneously with four CXC domains. Given the dimeric assembly of the MSL complex (Hallacli et al. 2012), molecules 1 and 2 that interact with each other likely belong to one dimeric MSL complex, whereas molecules 3 and 4 belong to another complex. In this case, the MRE motif is recognized by two MSL dimers. However, whether sites 3 and 4 are actually bound in vivo is more uncertain because of the lack of protein contact between the bound CXC domains. The two MSL dimers might be further connected by, for instance, the roX RNA, as shown for the polycomb-repressive complex-2 dimers tethered by long noncoding RNAs (Davidovich et al. 2014). This would enhance the recognition of the MRE motif and spreading of the MSL complex.
Mammals contain a similar MSL complex that is composed of orthologs of MSL1, MSL2, MSL3, and MOF and conducts H4K16 acetylation of all chromosomes (Smith et al. 2005; Taipale et al. 2005; Mendjan et al. 2006; Wu et al. 2014). Because of the high level of conservation in the sequences of CXC domain (a multiple sequence alignment can be found in Zheng et al. 2012), its DNA-binding mode and even sequence specificity are likely conserved at the single-domain level. In particular, the critical R543 residue is invariant and may recognize similar dinucleotide sequences at the minor groove. Although R526 is often replaced by lysine, lysine can still make electrostatic interaction with DNA. In addition, the mammalian MSL complex is also dimeric (Hallacli et al. 2012), which would favor cooperative binding of two CXC domains to the target sequence. However, the loop (residues 532–537) that mediates intermolecular contact of the CXC domain is conserved only in closely related Drosophila species. Consequently, the mammalian CXC domain might form an alternative dimer during combinatory recognition of DNA sequences. The binding consensus sequence of mammalian MSL2, which has not been reported yet, might also differ from the MRE motif, since the overall binding specificity is determined by both the sequence specificity of the single CXC domain and the higher-order structure of the CXC–DNA complex. The CXC domain of human MSL2 cannot substitute for that of Drosophila MSL2 in HAS targeting (Fauth et al. 2010), suggesting that they have different binding specificities.
Although earlier studies implicated the CXC domain in HAS interaction (Fauth et al. 2010), this domain alone does not suffice to direct the MSL-DCC to the X chromosome but requires two additional domains. First, complex formation with MSL1 is an obligatory requirement for HAS binding, and the MSL1 interaction is mediated by the RING domain of MSL2 (Lyman et al. 1997; Copps et al. 1998; Li et al. 2008). Biochemical data indicate that MSL1 does not contribute another DNA-binding surface (Fauth et al. 2010) but serves as a scaffold for dimerization of the complex (Hallacli et al. 2012). Second, work from Scott and colleagues (Li et al. 2008) highlighted the importance of a region rich in prolines and basic residues (Pro/Bas, between residues 684 and 725) C-terminal of the CXC domain for correct X chromosome targeting. These investigators suggested that a motif within the Pro/Bas region may be important for incorporating roX RNA into the MSL-DCC. However, one limitation of their approach was that mapping of MSL2-Flag derivatives on polytene chromosomes did not reliably distinguish HASs from generic sequences.
Where in the nucleus the MSL-DCC assembles is unclear. In one scenario, the assembly of the active MSL-DCC (involving dimerization) happens at the HASs. Protein factors that bind to the HASs may facilitate assembly of the MSL-DCC or its structural reorganization into the high-affinity binding mode. One such factor is the CLAMP protein, which cohabitates with MSLs at virtually all HAS elements (Soruco et al. 2013). However, the presence of the CLAMP protein alone at a GAGA-rich sequence that conforms to the MRE consensus does not determine whether the element actually functions as a HAS.
Can MSL2 and CLAMP bind simultaneously at the MRE motif from a structural point of view? The CLAMP protein contains six tandem C2H2-type Zn fingers at the C terminus that are a well-characterized DNA-binding domain and employs an α helix to recognize a triplet in the major groove (Pavletich and Pabo 1991; Wolfe et al. 2000). Structural modeling suggests that the MRE DNA with two CXC domains bound at sites 1 and 2 could accommodate three Zn fingers at the major groove, but additional Zn fingers would clash with the R526 loop in the CXC domain (Supplemental Fig. S5). The cooperative binding mode of the CXC dimer seems to be incompatible with binding of all six Zn fingers of CLAMP. Nevertheless, in low-affinity binding modes (II and III), a single CXC domain could bind at a common region with a peripheral Zn finger of CLAMP.
Our recent observations that the RNA helicase MLE can be directly cross-linked to all HASs (Straub et al. 2013) and that MLE remodels the noncoding roX RNA to facilitate specific interaction of MSL2 (Maenner et al. 2013) may point to a role for MLE/roX RNA in HAS definition or in modulating the nature of the CXC arrangements in response to its local microenvironment.
The process of dosage compensation in Drosophila illustrates fundamental principles of target gene selection and coregulation. Variations of these fundamental principles are also at work in other broad domains of chromatin-mediated regulation, such as the repression of large groups of genes through the polycomb machinery (McElroy et al. 2014). Our study suggests that the interplay between instructive forces of DNA sequence and the conformational flexibility of binding domains can bring about the kind of high-affinity interactions that are the basis of stringent regulation in an intrinsically dynamic environment.
Materials and methods
Protein expression and purification
All proteins of the CXC domain used in this study consisted of MSL2 residues 520–570 and a glycine substitution of the nonconserved residue C560. Point mutations were introduced into the expression plasmid by the QuikChange method. Protein expression, 15N labeling, and purification were previously described (Zheng et al. 2012).
Crystallization, data collection, and structure determination
The purified CXC protein (15 mg/mL in 5 mM MES at pH 8.0, 50 mM KCl) was incubated with DNA duplex at a 1:1 molar ratio for 1 h on ice and crystallized at 20°C by the hanging-drop vapor diffusion method. A number of DNAs with different lengths and sequences were screened. The S15 complex was crystallized in 0.1 M HEPES-Na (pH 7.5), 10% (w/v) PEG3350, and 0.2 M proline. The (GC)6 complex was crystallized in 0.1 M HEPES-Na (pH 7.5) and 25% (w/v) PEG3350. For cryoprotection, all crystals were soaked in 20% glycerol made in the reservoir solution and flash-cooled in liquid nitrogen.
All data were collected at the Shanghai Synchrotron Radiation Facility (SSRF) beamline BL17U and processed by HKL2000 (Otwinowski and Minor 1997). The crystal structure of the CXC domain was first determined for a different kind of DNA complex with SHARP (Vonrhein et al. 2007) using the single-wavelength anomalous dispersion method based on a data set collected at 1.28174 Å, the anomalous peak wavelength of Zn (S Zheng, J Wang, and K Ye, unpubl.). This structure could not be properly refined but provided a reasonable model of the CXC domain for molecular replacement. Other structures were determined by molecular replacement using the crystal structure of the CXC domain and standard B-form DNA duplexes as a search model. The structures were built with Coot (Emsley and Cowtan 2004) and refined with Refmac and PHENIX (Murshudov et al. 1999; Adams et al. 2010). Residues 530–332 were not all modeled in some molecules due to weak electron density. RAMPAGE analysis of the S15 complex shows that 96.7% of the residues are in favored regions, and 3.3% are in allowed regions. Structure figures were prepared with PyMol (http://www.pymol.org).
NMR experiments
NMR data were recorded at 298° K on Bruker DMX600 spectrometers equipped with a triple resonance cryoprobe. All spectra were processed with Felix (Accelrys, Inc.) and analyzed with NMRViewJ (Johnson 2004).
The S12 and CES11D1 DNA samples (1 mM) were prepared in 100% 2H2O. The unlabeled protein of the CXC domain in 50 mM potassium phosphate (pH 6.0) was lyophilized and added to the DNA solution to a final concentration of 1 mM. 2D 1H–1H NOESY spectra were collected for the free and CXC-bound DNA with a mixing time of 200 msec at 278° K. Proton resonances were assigned based on 2D 1H–1H NOESY and TOCSY spectra. The NOE cross-peaks between sugar protons H1′ and base protons H6/8 were assigned by sequential NOE walk. The top strand of CES11D1 was assigned with the help of S12 assignments; the bottom strand of CES11D1 cannot be assigned due to severe spectral overlaps.
DNA titration
DNA oligonucleotides were purchased from Invitrogen. Complementary strands (5 mM) were annealed in 50 mM phosphate potassium buffer (pH 6.0) for 2 min at 95°C and then incubated for 1 h at room temperature. The titration starting sample (500 μL) contained 0.2 mM 15N-labeled protein of the wild-type or mutant CXC domain in 50 mM potassium phosphate (pH 6.0), 0.01% (w/v) sodium 2,2-dimethylsilapentane-5-sulfonate, and 10% (v/v) 2H2O. An 1H–15N HSQC spectrum was recorded after every aliquot (1–10 μL) of DNA duplex (5 mM) was added.
The chemical shift changes of backbone amide hydrogen (ΔδH) and nitrogen (ΔδN) were calculated in reference to the free protein and converted to the combined change Δδ:
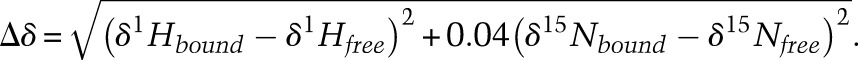 |
Assuming that the DNA has n equivalent and noninteracting binding sites for protein, Δδ is related to the DNA concentration according to the following equation (Fielding 2007):
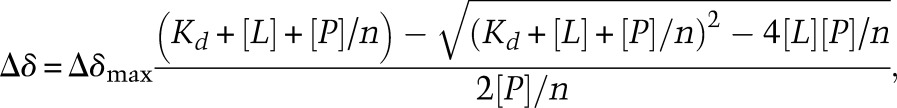 |
where [L] and [P] are the concentration of total DNA and protein, respectively; n is the number of equivalent and independent CXC-binding sites in DNA; Δδmax is the maximal chemical shift change for fully bound protein; and Kd is the apparent dissociation constant. The Δδ values for three significantly perturbed residues (R526 or A526, G528, and G531) were globally fit to the above equation. Δδmax and Kd were allowed to float in the fitting. The titration curve and the structural analysis suggest that the S12 DNA could simultaneously accommodate three CXC molecules. When n was allowed to float, a value of 3–4 was obtained for S12. To be consistent, n was fixed as 2 for 8-bp DNAs, 3 for 12-bp DNAs, and 3.5 for 13-bp DNAs. The fitting curves were displayed in Supplemental Figure S6. The real binding of the MRE motif by the CXC domain involves multiple binding sites of different affinities and binding cooperativity. The above model is an approximation, but analysis using a more complicated model is not possible due to the limited number and insufficient accuracy of titration data points.
Plasmids and antibodies
The expression vector for MSL2-GFP under the hsp70 promoter was previously described (Straub et al. 2005). Point mutations in MSL2 were introduced using the QuikChange site-directed mutagenesis kit (Stratagene). MSL2 and MSL3 antibodies were previously described (Gilfillan et al. 2006; Straub et al. 2008), the lamin antibody was a mouse monoclonal antibody (T40) kindly provided by Professor H. Saumweber, and the anti-GFP antibody was from Roche (118144600001).
Cells and immunostaining
Culture of Drosophila male S2 cells and generation of stable cell lines were performed as previously described (Straub et al. 2005) with few modifications: The GFP fusion proteins were cotransfected with a plasmid expressing a hygromycin resistance gene. Seventy-two hours after transfection, cells were put under selection with 300 μg/mL hygromycin for 3 wk. Immunostaining was done as described (Morales et al. 2004). Images were acquired on a Zeiss Axiovert 200M equipped with a 100× objective.
ChIP
ChIP experiments were performed as described (Straub et al. 2008) with minor modifications: Sonication was done with the Covaris AFA system (peak incident power of 60 W, duty factor 20%, and 200 cycles per burst for 90 min), and the immunoprecipitation was performed overnight with the appropriate amount of GFP-trap coupled to agarose beads (ChromoTek) (Rothbauer et al. 2008). Real-time PCRs were performed using SYBR Green dye (Applied Biosystems). Sequences of the primers are available on request.
Accession code
The atomic coordinates and experimental data have been deposited in the Protein Data Bank under accession codes 4RKG for the (GC)6 complex and 4RKH for the S15 complex.
Supplementary Material
Acknowledgments
We thank Hongwei Li, Xiaogang Niu, Bin Xia, and Changwen Jin at the Peking University NMR Center for generous help in NMR experiments, and the staff at the Shanghai Synchrotron Radiation Facility (SSRF) beamline BL17U for assistance in data collection. This research was supported by the Ministry of Science and Technology of China (2012CB910900 and 2010CB835402 to K.Y.), Strategic Priority Research Program of the Chinese Academy of Sciences (XDB08010203 to K.Y.), Beijing Municipal Government (to K.Y.), National Natural Science Foundation of China (31325007 to K.Y. and 31170701 to Y.F.), and the European Research Council (ERC) under the European Union’s Seventh Framework Programme (FP7/2007-2013)/ERC grant agreement number 293948 to P.B.B.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.250936.114.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyenko AA, Peng S, Larschan E, Gorchakov AA, Lee OK, Kharchenko P, McGrath SD, Wang CI, Mardis ER, Park PJ, et al. 2008. A sequence motif within chromatin entry sites directs MSL establishment on the Drosophila X chromosome. Cell 134: 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyenko AA, Ho JW, Peng S, Gelbart M, Tolstorukov MY, Plachetka A, Kharchenko PV, Jung YL, Gorchakov AA, Larschan E, et al. 2012. Sequence-specific targeting of dosage compensation in Drosophila favors an active chromatin context. PLoS Genet 8: e1002646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyenko AA, Ellison CE, Gorchakov AA, Zhou Q, Kaiser VB, Toda N, Walton Z, Peng S, Park PJ, Bachtrog D, et al. 2013. Conservation and de novo acquisition of dosage compensation on newly evolved sex chromosomes in Drosophila. Genes Dev 27: 853–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copps K, Richman R, Lyman LM, Chang KA, Rampersad-Ammons J, Kuroda MI. 1998. Complex formation by the Drosophila MSL proteins: role of the MSL2 RING finger in protein complex assembly. EMBO J 17: 5409–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvitanich C, Pallisgaard N, Nielsen KA, Hansen AC, Larsen K, Pihakaski-Maunsbach K, Marcker KA, Jensen EO. 2000. CPP1, a DNA-binding protein involved in the expression of a soybean leghemoglobin c3 gene. Proc Natl Acad Sci 97: 8163–8168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidovich C, Goodrich KJ, Gooding AR, Cech TR. 2014. A dimeric state for PRC2. Nucleic Acids Res 42: 9236–9248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Berletch JB, Ma W, Nguyen DK, Hiatt JB, Noble WS, Shendure J, Disteche CM. 2013. Mammalian X upregulation is associated with enhanced transcription initiation, RNA half-life, and MOF-mediated H4K16 acetylation. Dev Cell 25: 55–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disteche CM. 2012. Dosage compensation of the sex chromosomes. Annu Rev Genet 46: 537–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison CE, Bachtrog D. 2013. Dosage compensation via transposable element mediated rewiring of a regulatory network. Science 342: 846–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132. [DOI] [PubMed] [Google Scholar]
- Fauth T, Muller-Planitz F, Konig C, Straub T, Becker PB. 2010. The DNA binding CXC domain of MSL2 is required for faithful targeting the dosage compensation complex to the X chromosome. Nucleic Acids Res 38: 3209–3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielding L. 2007. NMR methods for the determination of protein–ligand dissociation constants. Prog Nucl Magn Reson Spectrosc 51: 219–242. [Google Scholar]
- Freemont PS, Lane AN, Sanderson MR. 1991. Structural aspects of protein-DNA recognition. Biochem J 278: 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelbart ME, Kuroda MI. 2009. Drosophila dosage compensation: a complex voyage to the X chromosome. Development 136: 1399–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilfillan GD, Straub T, de Wit E, Greil F, Lamm R, van Steensel B, Becker PB. 2006. Chromosome-wide gene-specific targeting of the Drosophila dosage compensation complex. Genes Dev 20: 858–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorchakov AA, Alekseyenko AA, Kharchenko P, Park PJ, Kuroda MI. 2009. Long-range spreading of dosage compensation in Drosophila captures transcribed autosomal genes inserted on X. Genes Dev 23: 2266–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Szauter P, Lucchesi JC. 1998. Targeting of MOF, a putative histone acetyl transferase, to the X chromosome of Drosophila melanogaster. Dev Genet 22: 56–64. [DOI] [PubMed] [Google Scholar]
- Hallacli E, Lipp M, Georgiev P, Spielman C, Cusack S, Akhtar A, Kadlec J. 2012. Msl1-mediated dimerization of the dosage compensation complex is essential for male X-chromosome regulation in Drosophila. Mol Cell 48: 587–600. [DOI] [PubMed] [Google Scholar]
- Johnson BA. 2004. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol Biol 278: 313–352. [DOI] [PubMed] [Google Scholar]
- Kadlec J, Hallacli E, Lipp M, Holz H, Sanchez-Weatherby J, Cusack S, Akhtar A. 2011. Structural basis for MOF and MSL3 recruitment into the dosage compensation complex by MSL1. Nat Struct Mol Biol 18: 142–149. [DOI] [PubMed] [Google Scholar]
- Kageyama Y, Mengus G, Gilfillan G, Kennedy HG, Stuckenholz C, Kelley RL, Becker PB, Kuroda MI. 2001. Association and spreading of the Drosophila dosage compensation complex from a discrete roX1 chromatin entry site. EMBO J 20: 2236–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalodimos CG, Biris N, Bonvin AM, Levandoski MM, Guennuegues M, Boelens R, Kaptein R. 2004. Structure and flexibility adaptation in nonspecific and specific protein-DNA complexes. Science 305: 386–389. [DOI] [PubMed] [Google Scholar]
- Larschan E, Soruco MM, Lee OK, Peng S, Bishop E, Chery J, Goebel K, Feng J, Park PJ, Kuroda MI. 2012. Identification of chromatin-associated regulators of MSL complex targeting in Drosophila dosage compensation. PLoS Genet 8: e1002830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Schiemann AH, Scott MJ. 2008. Incorporation of the noncoding roX RNAs alters the chromatin-binding specificity of the Drosophila MSL1/MSL2 complex. Mol Cell Biol 28: 1252–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyman LM, Copps K, Rastelli L, Kelley RL, Kuroda MI. 1997. Drosophila male-specific lethal-2 protein: structure/function analysis and dependence on MSL-1 for chromosome association. Genetics 147: 1743–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maenner S, Muller M, Frohlich J, Langer D, Becker PB. 2013. ATP-dependent roX RNA remodeling by the helicase maleless enables specific association of MSL proteins. Mol Cell 51: 174–184. [DOI] [PubMed] [Google Scholar]
- Marin I. 2003. Evolution of chromatin-remodeling complexes: comparative genomics reveals the ancient origin of ‘novel’ compensasome genes. J Mol Evol 56: 527–539. [DOI] [PubMed] [Google Scholar]
- McElroy KA, Kang H, Kuroda MI. 2014. Are we there yet? Initial targeting of the Male-Specific Lethal and Polycomb group chromatin complexes in Drosophila. Open Biol 4: 140006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meller VH, Gordadze PR, Park Y, Chu X, Stuckenholz C, Kelley RL, Kuroda MI. 2000. Ordered assembly of roX RNAs into MSL complexes on the dosage-compensated X chromosome in Drosophila. Curr Biol 10: 136–143. [DOI] [PubMed] [Google Scholar]
- Mendjan S, Taipale M, Kind J, Holz H, Gebhardt P, Schelder M, Vermeulen M, Buscaino A, Duncan K, Mueller J, et al. 2006. Nuclear pore components are involved in the transcriptional regulation of dosage compensation in Drosophila. Mol Cell 21: 811–823. [DOI] [PubMed] [Google Scholar]
- Morales V, Straub T, Neumann MF, Mengus G, Akhtar A, Becker PB. 2004. Functional integration of the histone acetyltransferase MOF into the dosage compensation complex. EMBO J 23: 2258–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ. 1999. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr D Biol Crystallogr 55: 247–255. [DOI] [PubMed] [Google Scholar]
- Oh H, Park Y, Kuroda MI. 2003. Local spreading of MSL complexes from roX genes on the Drosophila X chromosome. Genes & Dev 17: 1334–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Park Y, Kelley RL, Oh H, Kuroda MI, Meller VH. 2002. Extent of chromatin spreading determined by roX RNA recruitment of MSL proteins. Science 298: 1620–1623. [DOI] [PubMed] [Google Scholar]
- Park SW, Oh H, Lin YR, Park Y. 2010. MSL cis-spreading from roX gene up-regulates the neighboring genes. Biochem Biophys Res Commun 399: 227–231. [DOI] [PubMed] [Google Scholar]
- Pavletich NP, Pabo CO. 1991. Zinc finger–DNA recognition: crystal structure of a Zif268–DNA complex at 2.1 Å. Science 252: 809–817. [DOI] [PubMed] [Google Scholar]
- Rohs R, West SM, Sosinsky A, Liu P, Mann RS, Honig B. 2009. The role of DNA shape in protein-DNA recognition. Nature 461: 1248–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbauer U, Zolghadr K, Muyldermans S, Schepers A, Cardoso MC, Leonhardt H. 2008. A versatile nanotrap for biochemical and functional studies with fluorescent fusion proteins. Mol Cell Proteomics 7: 282–289. [DOI] [PubMed] [Google Scholar]
- Schmit F, Cremer S, Gaubatz S. 2009. LIN54 is an essential core subunit of the DREAM/LINC complex that binds to the cdc2 promoter in a sequence-specific manner. FEBS J 276: 5703–5716. [DOI] [PubMed] [Google Scholar]
- Smith ER, Cayrou C, Huang R, Lane WS, Cote J, Lucchesi JC. 2005. A human protein complex homologous to the Drosophila MSL complex is responsible for the majority of histone H4 acetylation at lysine 16. Mol Cell Biol 25: 9175–9188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soruco MM, Chery J, Bishop EP, Siggers T, Tolstorukov MY, Leydon AR, Sugden AU, Goebel K, Feng J, Xia P, et al. 2013. The CLAMP protein links the MSL complex to the X chromosome during Drosophila dosage compensation. Genes Dev 27: 1551–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub T, Becker PB. 2011. Transcription modulation chromosome-wide: universal features and principles of dosage compensation in worms and flies. Curr Opin Genet Dev 21: 147–153. [DOI] [PubMed] [Google Scholar]
- Straub T, Neumann MF, Prestel M, Kremmer E, Kaether C, Haass C, Becker PB. 2005. Stable chromosomal association of MSL2 defines a dosage-compensated nuclear compartment. Chromosoma 114: 352–364. [DOI] [PubMed] [Google Scholar]
- Straub T, Grimaud C, Gilfillan GD, Mitterweger A, Becker PB. 2008. The chromosomal high-affinity binding sites for the Drosophila dosage compensation complex. PLoS Genet 4: e1000302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub T, Zabel A, Gilfillan GD, Feller C, Becker PB. 2013. Different chromatin interfaces of the Drosophila dosage compensation complex revealed by high-shear ChIP-seq. Genome Res 23: 473–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale M, Rea S, Richter K, Vilar A, Lichter P, Imhof A, Akhtar A. 2005. hMOF histone acetyltransferase is required for histone H4 lysine 16 acetylation in mammalian cells. Mol Cell Biol 25: 6798–6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa R, Forne I, Muller M, Imhof A, Straub T, Becker PB. 2012. MSL2 combines sensor and effector functions in homeostatic control of the Drosophila dosage compensation machinery. Mol Cell 48: 647–654. [DOI] [PubMed] [Google Scholar]
- Vonrhein C, Blanc E, Roversi P, Bricogne G. 2007. Automated structure solution with autoSHARP. Methods Mol Biol 364: 215–230. [DOI] [PubMed] [Google Scholar]
- Wolfe SA, Nekludova L, Pabo CO. 2000. DNA recognition by Cys2His2 zinc finger proteins. Annu Rev Biophys Biomol Struct 29: 183–212. [DOI] [PubMed] [Google Scholar]
- Wu L, Zee BM, Wang Y, Garcia BA, Dou Y. 2011. The RING finger protein MSL2 in the MOF complex is an E3 ubiquitin ligase for H2B K34 and is involved in crosstalk with H3 K4 and K79 methylation. Mol Cell 43: 132–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Li L, Zhou B, Qin Z, Dou Y. 2014. H2B ubiquitylation promotes RNA Pol II processivity via PAF1 and pTEFb. Mol Cell 54: 920–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Wang J, Feng Y, Wang J, Ye K. 2012. Solution structure of MSL2 CXC domain reveals an unusual Zn3Cys9 cluster and similarity to pre-SET domains of histone lysine methyltransferases. PLoS ONE 7: e45437. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.