

Abstract
We describe a method that exploits contiguity preserving transposase sequencing (CPT-seq) to facilitate the scaffolding of de novo genome assemblies. CPT-seq is an entirely in vitro means of generating libraries comprised of 9216 indexed pools, each of which contains thousands of sparsely sequenced long fragments ranging from 5 kilobases to >1 megabase. These pools are “subhaploid,” in that the lengths of fragments contained in each pool sums to ∼5% to 10% of the full genome. The scaffolding approach described here, termed fragScaff, leverages coincidences between the content of different pools as a source of contiguity information. Specifically, CPT-seq data is mapped to a de novo genome assembly, followed by the identification of pairs of contigs or scaffolds whose ends disproportionately co-occur in the same indexed pools, consistent with true adjacency in the genome. Such candidate “joins” are used to construct a graph, which is then resolved by a minimum spanning tree. As a proof-of-concept, we apply CPT-seq and fragScaff to substantially boost the contiguity of de novo assemblies of the human, mouse, and fly genomes, increasing the scaffold N50 of de novo assemblies by eight- to 57-fold with high accuracy. We also demonstrate that fragScaff is complementary to Hi-C-based contact probability maps, providing midrange contiguity to support robust, accurate chromosome-scale de novo genome assemblies without the need for laborious in vivo cloning steps. Finally, we demonstrate CPT-seq as a means of anchoring unplaced novel human contigs to the reference genome as well as for detecting misassembled sequences.
The broad adoption of next-generation sequencing (NGS) has resulted in a proliferation of de novo genome assemblies (Pagani et al. 2012). For the most part, these assemblies are of far lower quality than the reference genomes produced by the International Human Genome Consortium (The International Human Genome Consortium 2001, 2004; Blanco-Ulate et al. 2013; Rong and McSpadden Gardener 2013; Shemesh et al. 2013; Wang et al. 2013), largely secondary to a dearth of readily accessible NGS-based methods for generating midrange and long-range contiguity information. Conventional NGS-based assemblies rely on deep sequencing of shotgun fragments and ∼3-kbp mate-pair libraries to provide short-range contiguity, both of which are generated via straightforward in vitro protocols. Dilution pools of PCR amplicons can provide long virtual reads, but these are limited to 10 kbp by the use of PCR, and the majority are ∼8 kbp (Voskoboynik et al. 2013). Midrange contiguity information requires still-expensive (but rapidly evolving) long sequencing reads (e.g., PacBio at ∼18 kbp) (Koren et al. 2012) or nanopore sequencing (Laszlo et al. 2014) or labor-intensive fosmid or BAC clone libraries (Gnerre et al. 2011; Zhang et al. 2012). Alternatively, long-range contiguity information can be generated in vitro via contact probability maps, wherein Hi-C read-pairs are used for chromosome-scale scaffolding (Burton et al. 2013). However, the performance of Hi-C-based scaffolding depends heavily on input assembly scaffold size, with optimal results requiring an input N50 of ∼200 kbp or greater, a level of contiguity that can be challenging to achieve with shotgun fragment and 3-kbp mate-pair libraries alone (Blanco-Ulate et al. 2013; Rong and McSpadden Gardener 2013; Shemesh et al. 2013; Wang et al. 2013). As such, there remains a strong need for robust in vitro methods to capture midrange contiguity information for de novo genome assembly.
Transposase-mediated library construction, or “tagmentation,” utilizes a hyperactive Tn5 transposase to both fragment and append universal adaptors in a single enzymatic step (Goryshin and Reznikoff 1998; Adey et al. 2010). In recent years, tagmentation has been applied in diverse ways including DNA-seq (Adey et al. 2010), stranded RNA-seq (Gertz et al. 2012), whole-genome bisulfite sequencing (Adey and Shendure 2012), chromatin profiling (Buenrostro et al. 2013), and in situ mate-pair library preparation directly on a sequencing flowcell (Schwartz et al. 2012). We recently demonstrated a novel method, contiguity preserving transposase sequencing (CPT-seq) for haplotype-resolved genome sequencing (Amini et al. 2014). CPT-seq utilizes an inherent property of the Tn5 transposase in which the enzyme remains tightly bound to the target DNA after tagmentation, physically linking adjacent library molecules (Fig. 1A). Prior to PCR amplification, the high molecular weight products of linked templates are subjected to subhaploid dilution and compartmentalization, followed by protein denaturation in order to free the templates for amplification. To increase the effective number of compartments, a two-tiered indexing approach (Erlich et al. 2009) is applied (Fig. 1B). The initial tagmentation is performed using 96 uniquely indexed transposase-adaptor complexes. The high molecular weight linked templates are then pooled, followed by limiting dilution into 96 indexed PCR reactions such that each PCR well contains templates from 96 originating transposase reactions, thus producing 96 × 96 = 9216 distinct index combinations. Sequence reads corresponding to each of the 9216 “virtual compartments” (also referred to as indexed pools) provide sparse, shotgun representation of the high molecular weight genomic DNA fragments that were transposed within that indexed pool (Fig. 1C), analogous to fosmid (Kitzman et al. 2011) or in vitro dilution pools (Kaper et al. 2013) but with a ∼100× higher effective number of pools. With appropriate dilution of the input genomic DNA, these indexed pools are “subhaploid,” in that the lengths of fragments contained in each pool sums to ∼5% to 10% of full genome length. From an experimental perspective, the library preparation steps of CPT-seq are straightforward and scalable, with an overall processing time of <3 h, relying on readily available equipment and requiring minimal hands-on time (Amini et al. 2014).
Figure 1.

CPT-seq method and performance. (A) High molecular weight (HMW) genomic DNA reacted with hyperactive Tn5 transposase loaded with indexed adaptors. After the transposase complex fragments the DNA and appends the indexed adaptors, the enzyme remains tightly bound to the DNA, such that library molecules derived from the same HMW genomic DNA molecule remain physically linked. Once the transposase is removed by denaturation, PCR amplification of viable templates (gray boxes) can be performed. (B) Schematic of two tier indexing. A 96-plex indexed tagmentation is performed (but without removing the transposase), followed by pooling, mixing, and redistribution to 96 wells. These new pools are subjected to removal of the transposase, 96-plex indexed PCR and then pooling to a single sequencing library. Individual molecules within the final library have indices corresponding to both the pool in which their originating HMW genomic DNA fragment was present during tagmentation (96 indices) as well as during PCR (96 indices), such that there are effectively 96 × 96 = 9216 compartments. (C) Representation of coverage profiles for indexed fragment pools, i.e., compartments (top) and trimodal distribution of adjacently aligning reads within individual compartments. The first peak (∼100 bp; red) corresponds to simple read pairs; the second peak (∼3.2 kbp; green) corresponds to reads originating from the same HMW genomic DNA fragment; the third peak (∼1 Mbp; blue) corresponds to reads originating from different HMW genomic DNA fragments. (D) Distribution of estimated HMW genomic DNA fragment lengths for CPT-seq of GM12878. The mean fragment size is 33.9 kbp, but it is a broad distribution and nearly 1M fragments are >100 kbp.
Results
We speculated that the large number of effective dilution pools in CPT-seq (n = 9216), each of which contains thousands of long DNA fragments (Fig. 1D), might be an excellent source of midrange contiguity information for de novo genome assembly. Many contemporary midrange contiguity methods involve mate-pair sequencing, in which each read in a sequenced pair is derived from the end of a long genomic DNA fragment, e.g., 3–40 kbp in length. These methods can use existing software to perform scaffolding such as Bambus2 (Koren et al. 2011), GRASS (Gritsenko et al. 2012), SCARPA (Donmez and Brudno 2013), or SOAPdenovo2 (Luo et al. 2012), which were evaluated extensively against one another and several other tools by Hunt et al. (2014) as well as other de novo assembly algorithms evaluated as part of the Assemblathon project (Earl et al. 2011; Bradnam et al. 2013). However, the data produced by CPT-seq is very different from that of mate-pair sequencing, consisting instead of sparse, shotgun representation of genomic DNA fragments that were transposed within each of 9216 pools, wherein each pool derives from an essentially random mix of thousands of variably sized, high molecular weight genomic DNA fragments. We therefore developed a new algorithm, fragScaff, which leverages coincidences between the content of different pools as a source of contiguity information for linking proximally located sequences for de novo genome assembly (Fig. 2; Supplemental Fig. S1; Supplemental Note).
Figure 2.

fragScaff assembly method. (A) The ends of contigs in a de novo genome assembly (gray boxes) are defined as nodes, and the subsets of the 9216 CPT-seq compartments, i.e., indexed pools, containing reads that align to each node are identified. The fraction of shared compartments between every possible pair of nodes is calculated. Pairs of nodes that are truly adjacent to one another in the genome are expected to exhibit excess sharing with respect to CPT-seq compartments as a result of HMW genomic DNA fragments that bridge the gap in the de novo genome assembly. Nonadjacent pairs of nodes will co-occur in a small fraction of compartments by chance, as each contains HMW genomic fragments that cover ∼10% of the genome. (B) The fraction of shared compartments is calculated for all possible pairs of nodes, and distributions are generated for each node. Outlier nodes in each distribution are identified assuming normality and using a P-value cutoff. If a link is reciprocated, i.e., if two nodes are each outliers in the other’s distribution, it is stored as an edge. (C) Subgraphs are reduced to their minimum spanning tree (MST), and the longest path (Trunk) is found. Branches (light nodes) are then placed to produce the final output scaffold. (D) Size distribution of gaps between properly linked contigs. Boxes indicate joins spanning gaps just beyond the 2.5-kbp mate-pair library (red), ∼6 kbp L1 repeat elements (green), and joins longer than 35 kbp, which cannot be achieved via fosmid mate-pair libraries (blue; n = 664).
The input to fragScaff consists of an initial genome assembly (e.g., based on shotgun and mate-pair sequencing data) and data produced by CPT-seq. First, the input contigs’ or scaffolds’ (referred to here as input contigs) lengths are used to determine the “end nodes,” or sequence at the ends of each input contig to be utilized in the assembly process, thus producing an end node pair for each input contig. The alignment of CPT-seq reads to the input assembly is then used to determine which of the 9216 CPT-seq pools have quality alignments (Q ≥ 10) to each of the end nodes. The distribution of the number of groups hitting the end nodes (Supplemental Fig. S2) is then used to exclude those at the extremes that may contain repetitive sequence (high extreme) or misassembled sequence (low extreme).
In the next step, which makes up the core of the fragScaff algorithm, the shared fraction of pools between every possible node pair is calculated as the number of pools hitting both the nodes divided by the total number of unique pools hit by either node. This results in a distribution of shared fractions for each individual node (Fig. 2B) for which a mean and standard deviation can be calculated and used to estimate the probability that the reciprocal node is an outlier. A P-value threshold is then manually set or automatically determined based on the number of outliers that would produce a predefined target mean number of outliers per node (Supplemental Table S1). In cases in which two nodes are called as outliers with respect to the distribution of the other, it is considered a “reciprocated link” and forms an edge in the subsequent graph manipulation steps with a weight corresponding to the outlier score defined as the –log10(P-value).
The third and final stage of fragScaff is the ordering of end nodes based on the constructed graph (Fig. 2C; Supplemental Fig. S3). The maximum-weight minimum spanning tree (MST) is first identified for each connected component, and the longest path (diameter) through the MST is identified as the trunk. An MST approach was chosen because the majority of connected components are already very close to their MST, and only very few edges are removed for the purpose of trunk identification. After the trunk is established, the end nodes not present in the trunk (branches) are placed using an iterative approach in which the end node with the highest weighted edge to a trunk end node is inserted into the trunk path such that the new trunk path score is maximized. This method utilizes all edges as opposed to solely the MST edges and is iterated until every branch end node is incorporated.
Run times and memory requirements of fragScaff are highly dependent on both the size of the input assembly as well as the number of input contigs present (Supplemental Note). For efficiency in optimizing the scaffolding process, each stage of fragScaff can be run independently to allow for multiple iterations of the fast graph manipulation stage using varying cutoffs (Supplemental Table S1) without rerunning the more time consuming I/O steps or the calculation of pool overlaps. For the determination of end nodes and BAM file parsing, the number of reads is the primary constraint, with run times of 4 h for 1324 M reads (human) and 40 min for 334 M reads (fly) using a single core and requiring a <1 GB memory footprint. The all-by-all node calculation is generally the most computationally intensive step, requiring 22 min for the human assembly (N50 = 437 kbp, 18,922 input contigs, 50 threads executed via the Sun Grid Engine [SGE]). This increases sharply with higher contig counts due to the O(n2) complexity, reaching 16 h using 50 threads (via SGE) for the 25-kbp simulated human assembly, which contained 115,162 input contigs. Finally, the graph manipulation steps are minimally intensive, requiring 2 min on a single core for the human assembly (N50 = 437 kbp, 18,922 input contigs).
We applied fragScaff to a human (GM12878) de novo assembly with an N50 scaffold size of 437 kbp generated by ALLPATHS-LG using only shotgun and 3-kbp mate-pair libraries (Gnerre et al. 2011). CPT-seq data was generated on GM12878 DNA (Amini et al. 2014), yielding 9216 pools with a mean 2513 fragments ≥5 kbp per pool (23.2 M fragments in total with a mean fragment size of 33.9 kbp and 0.9 M fragments >100 kbp). The resulting fragScaff assembly using conservative parameters incorporated 97.1% of sum length of the input contigs, resulting in an N50 of 4.4 Mbp (10× increase) with a 97.1% join accuracy, a 92.4% orientation accuracy, and 98.5% of base pairs properly placed (Table 1; Supplemental Table S2; Supplemental Figs. S4, S5), including a perfectly ordered scaffold of 23.9 Mbp. Additionally, we performed several less stringent iterations of fragScaff, which resulted in N50 increases up to 42× (N50 = 18.2 Mbp) while still maintaining a 97.8% base pair placement accuracy.
Table 1.
fragScaff performance summary
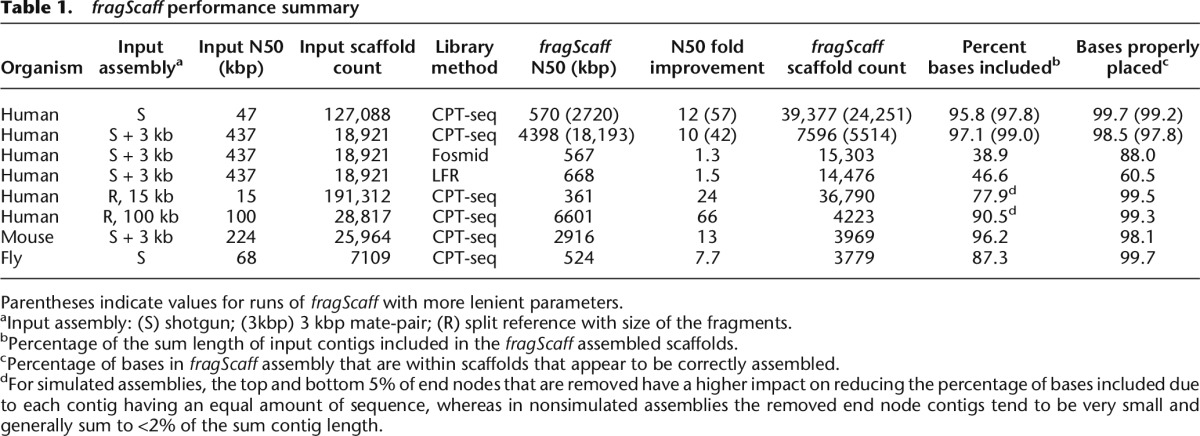
It is important to note that unlike mate-pair sequencing, in which orientation information is captured via read pair alignment orientation, fragScaff has no such information and relies on the link scores of each input contig end node. Therefore, as input contig size decreases, the spacing between the end nodes decreases and so does the ability to infer orientation, reaching zero when the end nodes of an input contig completely overlap (Supplemental Fig. S6). When excluding end nodes for which there is no orientation information (i.e., where end nodes completely overlap), the orientation accuracy increases to 96.8%. In situations in which orientation information is limited, the quality scores generated by fragScaff are of particular importance to inform whether or not the reverse orientation should be considered in downstream analyses.
We also attempted to apply fragScaff to data generated with fosmid (Kitzman et al. 2011) or long fragment read (LFR) (Kaper et al. 2013) dilution pools, rather than CPT-seq. However, this resulted in low link counts of poor accuracy (N50 improvement: 1.3× and 1.5×; join accuracy: 71.6% and 34.0%; sequence joined: 38.9% and 46.6% for fosmid and LFR, respectively), predominantly due to the reduced pool counts, which restrict the number of coincidences between pool content on which this method is based (fosmid: 288, LFR: 96 versus CPT-seq: 9216). Consistent with this, we observed markedly worse performance when down-sampling to a low number of CPT-seq pools (e.g., 47.9% [288 pools] versus 87.9% [9216 pools] of sequence scaffolded for fly; see below).
Because achieving a scaffold N50 of even 437 kbp with shotgun and 3-kbp libraries is quite challenging for complex genomes (Blanco-Ulate et al. 2013; Rong and McSpadden Gardener 2013; Shemesh et al. 2013; Wang et al. 2013), we sought to evaluate this approach on less contiguous input assemblies. As a first analysis, we fragmented the human input de novo assembly at every gap to produce a contig-only assembly (N50 reduced from 437 to 47 kbp) and then applied fragScaff with CPT-seq data. This resulted in an assembly that joined 97.7% of input contig sequence for a scaffold N50 of 2.7 Mbp (57× improvement), 97.9% join accuracy, and 99.2% of base pairs properly placed. However, the reduced contiguity of the input assembly and the effective exclusion of 3-kbp mate-pair data resulted in a substantial decrease in orientation accuracy to 71.4%. As a second analysis, we fragmented the human reference genome in silico to sizes ranging from 15 to 300 kbp. The scaffolding of these simulated input assemblies with fragScaff resulted in N50 improvements ranging from 24× to 75×, with >99.3% of base pairs properly placed and 96.0% properly oriented in every assembly (Table 1; Supplemental Table S2).
We also evaluated fragScaff and CPT-seq with Mus musculus (mouse) and Drosophila melanogaster (fruit fly), for which high-quality reference genomes are available for comparison. For both organisms, we used assemblies generated using ALLPATHS-LG from either shotgun and mate-pair sequencing (mouse, N50 = 224 kbp) or just shotgun sequencing (fly, N50 = 68 kbp) (Gnerre et al. 2011; Burton et al. 2013). CPT-seq data and fragScaff were then used to increase the N50 to 2.9 Mbp for mouse (13× improvement) and 524 kbp for fly (7.7× improvement) with 98.1% and 99.7% of base pairs properly placed for mouse and fruit fly, respectively (Table 1; Supplemental Table S2).
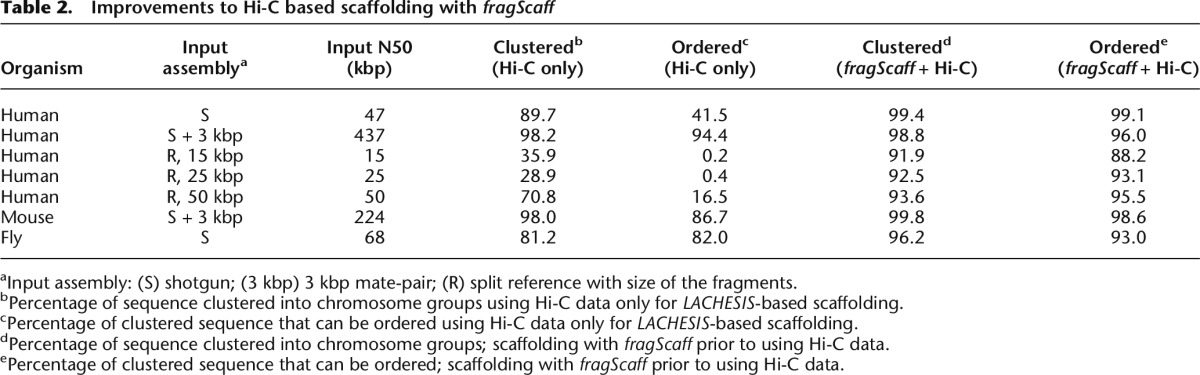
We recently reported the use of Hi-C data for chromosome-scale de novo assembly on human, mouse, and fruit fly genomes (Burton et al. 2013). For input assemblies with a large N50, Hi-C-based scaffolding produced high-quality scaffolds such as on human (input assembly with N50 = 437 kbp), where the resulting output clustered 98.2% of sequence into chromosome groups with 94.4% ordered. However, on the smaller fly input assembly (N50 = 68 kbp), Hi-C-based scaffolding was able to cluster only 81.2% and order 82.0% of sequence. Similarly, when we applied Hi-C-based scaffolding to our human contig assembly (N50 = 47 kbp), 89.7% of sequence was clustered with only 41.5% ordered and even less for the simulated 15 kbp contig assembly (35.9% clustered, 0.2% ordered). However, when we perform CPT-seq and fragScaff prior to Hi-C-based scaffolding, the completeness and quality of the resulting chromosome-scale de novo assembly markedly improves for the human contig assembly (Table 2; Supplemental Table S3) with 99.4% of sequence clustered (gain of 9.7%) and more than doubling the amount of sequence ordered to 99.1% (gain of 57.6%). Similar improvements were also observed for simulated contig assemblies of fly and human, most strikingly for the 15-kbp human simulated contig input, wherein the proportion of the input contig assembly that is ultimately clustered and ordered increased from 35.9% to 91.9% and from 0.2% to 88.2%, respectively. These results demonstrate that CPT-seq and fragScaff can improve the contiguity of assemblies generated from shotgun and short-range mate-pair libraries to a point suitable for chromosome-scale scaffolding (Supplemental Fig. S7).
Table 2.
Improvements to Hi-C based scaffolding with fragScaff
In Kitzman et al. (2011), we described a method that utilized the subhaploid content of each fosmid pool to anchor de novo assembled contigs from reads that did not align to the human reference genome, as well as a set of previously anchored sequences (hg18) described in Kidd et al. (2010) (Supplemental Fig. S8). This method worked on the premise that each window in the genome is hit by a discrete set of pools, as is each novel contig, and the window with maximum pool overlap with a novel contig is the most probable anchor location. We applied this approach to anchor the same set of contigs to hg18 (many of these have since been incorporated to the GRCh37 human reference assembly) but used CPT-seq data generated on GM12878, thus increasing the number of pools, and therefore anchoring power, from 115 to 9216. We were able to place 1816 of the 2363 contigs (76.9%), although a number of unplaced contigs are likely population-specific and not present in GM12878 (Kidd et al. 2010; Genovese et al. 2013). Kidd et al. (2010) previously placed 1226 of our placed contigs with 1154 (94.1%) in agreement with our calls.
We next evaluated whether the same method used for anchoring unplaced genomic content could be applied to detect misassemblies in a de novo genome assembly, i.e., by examining the pool overlap fractions of immediately adjacent windows and windows one apart (Fig. 3A). For example, using this method, 3081 suspicious windows from 926 scaffolds were identified in the de novo assembly based on shotgun, 3-kbp mate-pair and CPT-seq data (N50 = 4.4 Mbp), of which 400 scaffolds (43% of those flagged) contained bona fide misassemblies upon comparison with the GRCh37 reference assembly (Fig. 3B). These misassemblies resulted in reduced pool overlap fractions over a span of several windows as opposed to false positive flaggings, which were generally single window calls. Of course, the set of misassemblies identified in this example is biased toward those that were present in the input genome, and we have less power to detect misassemblies by fragScaff due to utilization of the same data that went into the scaffolding process.
Figure 3.

Misassembly detection using CPT-seq. (A) Three regions of assembled scaffolds representing various misassembly detections are shown. For each region, the set of CPT-seq indexed pools that cover each 5-kbp window (x-axis) was determined. The shared fraction of indexed pools between immediately adjacent windows (blue) and for windows one apart (purple) is plotted. Subregions for which both shared fraction values were in the bottom fifth percentile overall were called as potential misassemblies. False positive misassembly calls (i.e., no misassembly is actually present) overwhelmingly consisted of an isolated window (green shading), whereas multiple consecutive windows with low shared fraction values corresponded to misassemblies by fragScaff (yellow shading) or in the initial input assembly (red shading). (B) Breakdown of regions called as potentially misassembled by this approach (left) versus a randomly selected set of windows for comparison (right).
Lastly, we sought to take advantage of the haplotype-specific nature of CPT-seq data (Amini et al. 2014) by haplotype-resolving the GM12878 fragScaff assembly (input N50 = 437 kbp, fragScaff N50 4.4 Mbp), i.e., to produce a diploid de novo genome assembly. We first aligned shotgun reads back to the assembly (56×) and performed variant calling to identify 902,905 heterozygous sites, substantially fewer than the 2,180,767 high quality GM12878 heterozygous sites previously described (Zook and Salit 2013). Furthermore, only 327,828 (36.3%) of variants could be uniquely assigned to reference genome coordinates (GRCh37) that matched the coordinates of known GM12878 variants. Of the 575,077 unassigned variant calls, 50.6% aligned to repeat elements and 34.8% aligned to segmental duplications, indicating that the majority are false positive variants secondary to collapsed regions in the original assembly.
We then endeavored to perform haplotype resolution of either the full (n = 902,905) or validated (n = 327,828) sets of heterozygous sites using CPT-seq data and ReFHap (Duitama et al. 2010). For the full set, the high burden of false positive calls confounded the graph-based algorithm and was abandoned after 164 h of run time. On a smaller subset of scaffolds containing only 8437 variants, ReFHap completed but produced an essentially random result with a pairwise phasing accuracy of 50.1%. For the validated set, haplotype resolution was achieved in <1 h, with 82.4% of variants phased with a haplotype block N50 of 356 kbp (max 4.4 Mbp) and a pairwise phasing accuracy of 95.0%. Thus, although we were able to successfully achieve haplotype resolution concurrent with de novo genome assembly, this was only possible by restricting the analysis to variants validated by alignments to the human reference genome. This result underscores the need for experimental methods and/or algorithms that improve repeat disambiguation in de novo genome assemblies.
Discussion
We demonstrate that a new, entirely in vitro, method of transposase-mediated library construction, CPT-seq, which preserves the contiguity of high molecular weight transposed molecules, can be used to provide midrange sequence information. This library construction method results in 9216 distinct groups of reads, each of which contains sparse sequence reads over a set of thousands of long 5-kbp to 1-Mbp fragments that sum up to ∼5%–10% of the target genome.
We emphasize that the quality of input genomic DNA is a critical parameter in the performance of this method (and more generally, methods based on subhaploid complexity reduction) (Lo et al. 2013). The decreasing abundance of fragments at longer lengths (Fig. 1D) is largely due to fragmentation occurring at the transposase incorporation step and subsequent handling prior to PCR; however, the maximum size of fragments is also inherently limited by the quality of the input material. As such, CPT-seq is best performed on DNA freshly isolated using protocols that minimize fragmentation (Methods).
In order to use this new form of sequence data to aid in de novo genome assembly, we designed and implemented a novel algorithm, fragScaff, which produces midrange scaffolding comparable to 40-kbp fosmid-based mate-pair libraries, yet uses the entirely in vitro CPT-seq method. Furthermore, the algorithm can be tuned to allow for longer assemblies at a minor cost to accuracy, depending on downstream requirements, and provides detailed quality score information on the joins and sequence orientations present in the final assembly (Supplemental Note). Beyond the scaffolding of contigs, we were also able to utilize CPT-seq data to haplotype-resolve a de novo genome assembly, thus producing a diploid assembly. However, this was only possible by restricting the analysis to validated heterozygous variants, because the large number of false positive variant calls due to collapsed repeat elements confounds algorithms for haplotype resolution. In future work, this may be circumvented by improving repeat disambiguation during the genome assembly process or by improving methods for calling truly heterozygous variants within imperfect genome assemblies.
The primary limitation of CPT-seq and fragScaff at present is that short input contigs (<3 kbp) are difficult to scaffold confidently due to a reduced number of pools covering the contig. In these situations, removing the shorter contigs prior to fragScaff followed by placement using the described anchoring method may be preferred to avoid potential misassemblies during the scaffolding process. A second limitation of the method is that CPT-seq does not provide orientation information that is inherent in more traditional mate-pair sequencing approaches. As such, the only means of orientation is the difference in signal between the end node pairs of each input scaffold or contig. For longer input contigs, in which the end nodes do not overlap, orientation is possible with high accuracy; however, input contigs shorter than twice the end node size suffer from an increase in shared pools due to the overlapping end nodes, and therefore orientation accuracy rapidly decreases to random where the end nodes overlap completely. An additional challenge that is shared by fragScaff and traditional mate pair–based scaffolding is that unrecognized repeat elements will lead to false joins. This will be particularly problematic for genomes that contain large numbers of closely related sequences. Indeed, the accuracies demonstrated here are dependent on robust repeat masking prior to scaffolding. Given that the termini of many or most contig/scaffold ends after the primary de novo assembly are repetitive, an advantage of fragScaff, relative to traditional mate pair–based scaffolding, is that the end node can effectively be defined as the unique sequence just internal to this.
A key component of CPT-seq and its ability to generate useful data for either haplotype phasing as described in Amini et al. (2014) or for use in de novo assembly scaffolding via fragScaff is the partitioning of fragments such that each pool contains 1%–10% of the organism’s haploid genome. The distribution of fragments occurs after the indexed tagmentation of the high molecular weight DNA via dilution, such that each of the 96 PCR reactions contains ∼96 times the amount of desired long fragments per pool (e.g., a sum contiguous transposed template length of 32–320 Mbp per pool for roughly 0.3–3 Gbp per PCR reaction for human). On larger genomes, such as human and mouse, the size of the haploid genome allows for only modest dilution factors, with substantially more dilution required for fly. Although CPT-seq performed well on the modest, ∼170 Mbp fly genome, it is likely that the dilution factor required for genomes in the sub-10 Mbp range may be challenging or prohibitive. However, the pooling of genomes prior to the CPT-seq workflow may allow for a workaround with the added advantage of increased parallelization as long as special care is taken to insure the pooled genomes are sufficiently diverged (e.g., spiking in a bacterial genome into a primate CPT-seq prep). On a related note, we recently described the use of Hi-C data for the purposes of metagenome deconvolution and assembly (Burton et al. 2014). In principle, the contiguity provided by CPT-seq could be used in a similar fashion; however, the varying abundances of species within a metagenome would likely render titration to 1%–10% representation of each target genome very challenging or impossible for most samples. Nonetheless, this approach could be fruitful for subsets of species within a metagenome that presently are at approximately equal abundances.
The ease of CPT-seq library preparation and the amount of information obtained per base of sequencing performed makes it a very appealing tool for routine use in de novo genome assembly. These methods bridge the gap between short-range assemblies and the scaffold sizes needed to perform chromosome-scale scaffolding with Hi-C-based methods. We envision that the combination of four types of sequencing libraries—shotgun fragment, 3-kbp mate-pair, CPT-seq, and Hi-C—will provide a robust strategy to de novo assemble complex genomes to chromosome-scale contiguity using entirely in vitro methods that can be carried out cheaply and rapidly.
Methods
Library construction
Genomic DNA was prepared using the Qiagen Gentra Puregene Kit on 10 mg fresh liver (mus musculus, BL6) and 10 flies (Drosophila melanogaster, Canton S.) and analyzed on a pulsed field gel (Supplemental Fig. S10). CPT-seq libraries for human (GM12878) were those used in Amini et al. (2014) and are available from the Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under project accession PRJNA241346. For mouse and fly, CPT-seq libraries were prepared and sequenced using custom chemistry as described in Amini et al. (2014) with a slight modification to PCR template amounts for the fly sample to account for substantially decreased genome size (1, 2, or 3 pg inputs were used in each PCR reaction as opposed to 10 pg for Human and Mouse) (Supplemental Fig. S11). Fosmid libraries were previously generated for Adey et al. (2013). Long fragment read (LFR) libraries were generated on CEU gDNA using methods outlined in Kaper et al. (2013).
Input assemblies
The human, mouse, and fly input assemblies were the same input assemblies generated using ALLPATHS-LG as described in Burton et al. (2013) using reads accessed under Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) accessions SRA024407 (human, GM12878), SRA009956 (mouse), and SRR516038 and SRR516001 (fly). For an additional human assembly of lower quality, we broke up the original assembly into individual contigs by splitting scaffolds at any N base (N50 437 → 47 kbp). Simulated input assemblies were generated by splitting the human reference sequence at regular intervals. In cases in which repeat content was considered for the determination of input contig end nodes, the entire Repbase version 19.03 database FASTA (Jurka et al. 2005) was aligned to the input assembly using BLAST with default parameters followed by the merging of entries in the form of a bed file and including all segments ≥50 bp (half of the CPT-seq read length). End nodes were then adjusted to include additional unique sequence to compensate for the exclusion of repeat sequence. Reads in which ≥50% of the read aligned to a repeat element were excluded from any further analysis.
Contig scaffolding
Reads were aligned using BWA (Li and Durbin 2009) to references created from the input assemblies. The ends of each input contig (1 kbp fosmid and LFR, 5–10 kbp CPT-seq) were then used as end nodes, and the pools that had reads aligning to these end nodes were considered hits (reads that aligned to flagged repeats were excluded) (Supplemental Note). The distribution of the number of pools hitting each end node was then used to exclude the top and bottom 5% of end nodes. This exclusion does not necessarily remove the entire input contig, as the paired end node at the other end of the input contig may allow scaffolding; however, orientation information would be substantially weaker (Supplemental Fig. S2). Each end node was then analyzed individually by calculating the fraction of pools that overlap with all other end nodes (shared_pools/[total_node1_pools + total_node2_pools – shared_pools]) and subsequently calculating the mean and standard deviation for the end node. P-value cutoff thresholds were then determined by finding the –log10(P-value) score that would result in the desired mean number of outliers per end node. End nodes (node 1) that have an outlier end node (node 2), where node 1 is also an outlier in node 2, were then considered as potential reciprocated links with an edge weight of the combined –log10(P-value) score of each outlier and stored as an included edge if that weight was above a set threshold based on a multiple of the –log10(P-value) cutoff score (Supplemental Table S1; Supplemental Note). End nodes that were at the ends of the same input contig (end node pairs) were automatically given an edge score beyond the maximum score cutoff to prevent input contig splitting. Connected components in the graph were then determined followed by identification of the maximum-weight minimum spanning tree (MST) by implementing Prim’s algorithm. The true longest path (trunk) was then determined by identifying every path between degree one end nodes and taking the maximum length path. Since a typical MST in fragScaff has very few branches, the brute-force approach to finding the trunk can be implemented without excessive compute times. Nontrunk end nodes (branches) were then placed by inserting the branch into the trunk at a position that allows for the maximum trunk path edge weight based on all edges (not only the MST edges).
fragScaff parameters
fragScaff was run with varying parameters based on input assembly size and the desired output contiguity. The primary variable parameters are (1) end node size, (2) mean passing links per end node, and (3) link reciprocation factor (Supplemental Table S1); however, a number of other options can be modified, including the use of a repeat-masking bed file to exclude reads in identified repeats (Supplemental Note). For the end node size, the primary determining factor was the N90 of the input scaffolds. The end node size was set to a minimum of 5 kbp with an optimal size at approximately half the input N90, up to a maximum of 10 kbp. A small N50 generally requires an increase to the mean passing links per end node as well as modifications to the combined score cutoff. These parameters (j and u) are optimized in Supplemental Table S1 for each assembly. A detailed description of all parameters used in fragScaff is provided in the Supplemental Note.
Scaffolding accuracy measurement
Join accuracy was assessed only at locations where consecutive joined input scaffolds are properly mapped back to the reference assembly and were considered an accurate join if the scaffolds were within five of one another in the ordered rank. Orientation accuracy was also only assessed for consecutive, reference aligned scaffolds and considered accurate if the correct ends of the input scaffolds were joined. The fraction of base pairs properly placed was assessed by totaling the amount of correctly joined sequence in all scaffolds and then totaling the amount of sequence in all scaffolds that did not belong to the dominant locus of that scaffold. For example, if 3.5 Mbp of a scaffold was properly joined in correct order and was fused with another set of properly joined scaffolds that was 2 Mbp in length, we would consider the 2-Mbp set of sequence as improperly placed bases. Scaffolds were considered fused if >10% of the sequence in the scaffold was considered improperly placed. A graphical representation of accuracy measurements can be found in Supplemental Fig. S4.
Contig anchoring
CPT-seq reads were aligned to the human reference assembly using BWA (to hg18, as GRCh37 contains a number of the sequences we were anchoring). Fragments were then called in each pool using an alignment-distance cutoff of 15 kbp (Amini et al. 2014) on reads with a mapping quality ≥10. Unaligned reads were then aligned using BWA to a reference of all of the unanchored contigs. Pools with at least one read aligned with a mapping quality ≥10 were then considered hits. Pools with fragments spanning windows of 1–5 kbp in the reference genome were then identified (Supplemental Fig. S8). For each contig, the fraction of window pools shared with the contig pools and the fraction of contig pools shared with the window were calculated for each window, sorted, and ranked. The window with the top combined rank was then assigned as the anchor position. Agreement with published loci was determined by checking if our anchoring position was within 1 Mbp of the published position; however, 96% of the agreement loci were within 100 kbp.
Misassembly detection
CPT-seq reads were aligned to the resulting fragScaff assembly using BWA to assign a pool set for every 5-kbp window (excluding N’s) in the assembly. The shared pool fraction for immediately adjacent windows as well as for windows one apart was calculated. Window junctions that had shared pool fractions in the bottom fifth percentile for both immediately adjacent windows and windows one apart were flagged as putative misassemblies. Regions flanking the misassembled regions were aligned to the human reference via BLAST and analyzed whether or not they should be adjacent as well as whether or not the detected misassembly falls at a fragScaff join.
Haplotype resolution
GM12878 shotgun reads used in the original ALLPATHS-LG assembly (SRA024407, N50 = 437 kbp) were aligned onto the conservative fragScaff assembly (N50 = 4.4 Mbp) using BWA. Variants were then called using SAMtools (Li et al. 2009) and filtered to remove indels and require coverage of at least 10, at least one read for each allele on each strand, and an allele balance >0.25 and <0.75. The called heterozygous sites were then validated against a high quality reference set produced by the NIST (Zook and Salit 2013) on GM12878 by pulling the flanking 50 bp of assembled sequence around each called variant and aligning the 101-bp segments to the human reference genome (GRCh37) using BWA. The variants that were uniquely aligned and matched the position and alleles of the reference set were then utilized for haplotype resolution. CPT-seq reads were then aligned to the reference and used to call fragments by a thresholding approach by which a distance between subsequent aligned reads in a given read group must be ≤15,000 bp or else it is determined to be a break between fragments (Amini et al. 2014). During fragment calling, genotyping was performed on the fragments at the previously validated heterozygous sites. The genotyped fragments were then subjected to haplotype phasing using ReFHap (Duitama et al. 2010), and accuracy was determined by mapping the phasing of the validated sites back to the human reference genome positions and then comparing them to known haplotypes determined by pedigree information (Supplemental Fig. S9).
Software availability
The fragScaff software is available for download from Sourceforge (https://sourceforge.net/projects/fragscaff/files/).
Data access
Mouse and fly CPT-seq data generated for this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession numbers SRP041913 (mouse) and SRP041914 (fly).
Competing interest statement
Several authors of this manuscript are employed by Illumina Inc.
Supplementary Material
Acknowledgments
We thank R. Patwardhan for generating ALLPATHS-LG assemblies and other members of the Shendure laboratory for helpful discussions. We would also like to thank M. Wilken and the Reh laboratory for supplying a fresh mouse liver, and N. Peters and the Berg laboratory for supplying fly cultures. Our work was supported by grant HG006283 from the National Human Genome Research Institute (NHGRI; to J.S.); graduate research fellowship DGE-0718124 from the National Science Foundation (to A.A. and J.O.K.); and grant T32HG000035 from the NHGRI (to J.N.B.).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.178319.114.
References
- Adey A, Shendure J. 2012. Ultra-low-input, tagmentation-based whole-genome bisulfite sequencing. Genome Res 22: 1139–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adey A, Morrison HG, Asan, Xun X, Kitzman JO, Turner EH, Stackhouse B, MacKenzie AP, Caruccio NC, Zhang X, Shendure J. 2010. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol 11: R119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adey A, Burton JN, Kitzman JO, Hiatt JB, Lewis AP, Martin BK, Qiu R, Lee C, Shendure J. 2013. The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature 500: 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amini S, Pushkarev D, Christiansen L, Royce T, Turk C, Pignatelli N, Adey A, Kitzman JO, Ronaghi M, Shendure J, et al. . 2014. Haplotype-resolved whole-genome sequencing by contiguity-preserving transposition and combinatorial indexing. Nat Genet doi: 10.1038/ng.3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Ulate B, Rolshausen PE, Cantu D. 2013. Draft genome sequence of the grapevine dieback fungus Eutypa lata UCR-EL1. Genome Announc 1: e00228-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradnam KR, Fass JN, Alexandrov A, Baranay P, Bechner M, Birol I, Boisvert S, Chapman JA, Chapuis G, Chikhi R, et al. . 2013. Assemblathon 2: evaluating de novo methods of genome assembly in three vertebrate species. GigaScience 2: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton JN, Adey A, Patwardhan RP, Qiu R, Kitzman JO, Shendure J. 2013. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol 13: 1119–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton JN, Liachko I, Dunham MJ, Shendure J. 2014. Species-level deconvolution of metagenome assemblies with Hi-C-based contact probability maps. G3 (Bethesda) 4: 1339–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donmez N, Brudno M. 2013. SCARPA: scaffolding reads with practical algorithms. Bioinformatics 29: 428–434. [DOI] [PubMed] [Google Scholar]
- Duitama J, Gayle M, Eun-Kyung S, Hoehe MR. 2010. ReFHap: a reliable and fast algorithm for single individual haplotyping. http://dna.engr.uconn.edu/bibtexmngr/upload/Dal.10.pdf.
- Earl D, Bradnam K, John SJ, Darling A, Lin D, Fass J, Yu HO, Buffalo V, Zerbino DR, Diekhans M, et al. . 2011. Assemblathon 1: a competitive assessment of de novo short read assembly methods. Genome Res 21: 2224–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlich Y, Chang K, Gordon A, Ronen R, Navon O, Rooks M, Hannon GJ. 2009. DNA Sudoku—harnessing high-throughput sequencing for multiplexed specimen analysis. Genome Res 19: 1243–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G, Handsaker RE, Li H, Kenny EE, McCarroll SA. 2013. Mapping the human reference genome’s missing sequence by three-way admixture in Latino genomes. Am J Hum Genet 93: 411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz J, Varley KE, Davis NS, Baas BJ, Goryshin IY, Vaidyanathan R, Kuersten S, Myers RM. 2012. Transposase mediated construction of RNA-seq libraries. Genome Res 22: 134–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnerre S, Maccallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, Sharpe T, Hall G, Shea TP, Sykes S, et al. . 2011. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci 108: 1513–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryshin IY, Reznikoff WS. 1998. Tn5 in vitro transposition. J Biol Chem 273: 7367–7374. [DOI] [PubMed] [Google Scholar]
- Gritsenko AA, Nijkamp JF, Reinders MJ, de Ridder D. 2012. GRASS: a generic algorithm for scaffolding next-generation sequencing assemblies. Bioinformatics 28: 1429–1437. [DOI] [PubMed] [Google Scholar]
- Hunt M, Newbold C, Berriman M, Otto TD. 2014. A comprehensive evaluation of assembly scaffolding tools. Genome Biol 15: R42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International Human Genome Consortium 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921. [DOI] [PubMed] [Google Scholar]
- The International Human Genome Consortium 2004. Finishing the euchromatic sequence of the human genome. Nature 431: 931–945. [DOI] [PubMed] [Google Scholar]
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. 2005. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110: 462–467. [DOI] [PubMed] [Google Scholar]
- Kaper F, Swamy S, Klotzle B, Munchel S, Cottrell J, Bibikova M, Chuang H, Kruglyak S, Ronaghi M, Eberle MA, et al. . 2013. Whole-genome haplotyping by dilution, amplification, and sequencing. Proc Natl Acad Sci 110: 5552–5557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Sampas N, Antonacci F, Graves T, Fulton R, Hayden HS, Alkan C, Malig M, Ventura M, Giannuzzi G, et al. . 2010. Characterization of missing human genome sequences and copy-number polymorphic insertions. Nat Methods 7: 365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitzman JO, Mackenzie AP, Adey A, Hiatt JB, Patwardhan RP, Sudmant PH, Ng SB, Alkan C, Qiu R, Eichler EE, et al. . 2011. Haplotype-resolved genome sequencing of a Gujarati Indian individual. Nat Biotechnol 29: 59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Treangen TJ, Pop M. 2011. Bambus 2: scaffolding metagenomes. Bioinformatics 27: 2964–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Schatz MC, Walenz BP, Martin J, Howard JT, Ganapathy G, Wang Z, Rasko DA, McCombie WR, Jarvis ED, et al. . 2012. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat Biotechnol 30: 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laszlo AH, Derrington IM, Ross BC, Brinkerhoff H, Adey A, Nova IC, Craig JM, Langford KW, Samson JM, Daza R, et al. . 2014. Decoding long nanopore sequencing reads of natural DNA. Nat Biotechnol 32: 829–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26: 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup . 2009. The Sequence Alignment/Map (SAM) format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo C, Liu R, Lee J, Robasky K, Byrne S, Lucchesi C, Aach J, Church G, Bafna V, Zhang K. 2013. On the design of clone-based haplotyping. Genome Biol 14: R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, et al. . 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagani I, Liolios K, Jansson J, Chen IA, Smirnova T, Nosrat B, Markowitz VM, Kyrpides NC. 2012. The Genomes OnLine Database (GOLD) v.4: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 40: D571–D579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong X, McSpadden Gardener BB. 2013. Draft genome sequence of Cryptococcus flavescens strain OH182.9_3C, a biocontrol agent against fusarium head blight of wheat. Genome Announc 1: e00762-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JJ, Lee C, Hiatt JB, Adey A, Shendure J. 2012. Capturing native long-range contiguity by in situ library construction and optical sequencing. Proc Natl Acad Sci 109: 18749–18754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemesh M, Pasvolsky R, Sela N, Green SJ, Zakin V. 2013. Draft genome sequence of Alicyclobacillus acidoterrestris strain ATCC 49025. Genome Announc 1: e00638-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voskoboynik A, Neff NF, Sahoo D, Newman AM, Pushkarev D, Koh W, Passarelli B, Fan HC, Mantalas GL, Palmeri KJ, et al. . 2013. The genome sequence of the colonial chordate, Botryllus schlosseri. eLife 2: e00569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Wu R, Xu Y, Li M. 2013. Draft genome sequence of Rhizopus chinensis CCTCCM201021, used for brewing traditional Chinese alcoholic beverages. Genome Announc 1: e0019512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Fang X, Guo X, Li L, Luo R, Xu F, Yang P, Zhang L, Wang X, Qi H, et al. . 2012. The oyster genome reveals stress adaptation and complexity of shell formation. Nature 490: 49–54. [DOI] [PubMed] [Google Scholar]
- Zook J, Salit M. 2013. NIST NA12878 high-quality variant call set. ftp://ftp-trace.ncbi.nih.gov/giab/ftp/data/NA12878/variant_calls/NIST.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.