

Abstract
Owing to recent developments in computational algorithms and architectures, it is now computationally tractable to explore biologically relevant, equilibrium dynamics of realistically sized functional proteins using all-atom molecular dynamics simulations. Molecular dynamics simulations coupled with Markov state models is a nascent but rapidly growing technology that is enabling robust exploration of equilibrium dynamics. The objective of this work is to explore the challenges of coupling molecular dynamics simulations and Markov state models in the study of functional proteins. Using recent studies as a framework, we explore progress in sampling, model building, model selection, and coarse-grained analysis of models. Our goal is to highlight some of the current challenges in applying Markov state models to realistically sized proteins and spur discussion on advances in the field.
Introduction
Proteins dynamically explore their free energy landscape, giving rise to their form and function. Environmental changes can induce significant perturbations to a protein’s free energy landscape and modify its dynamics.1−4 Protein dynamics can be broadly sorted into equilibrium and nonequilibrium dynamics. Experimentally, established techniques in NMR and room temperature X-ray crystallography are able to quantify equilibrium dynamics at disparate time scales.5−8 From a computational standpoint, all-atom molecular dynamics (MD) simulations can survey the equilibrium dynamic of proteins; however, while a full accounting of equilibrium dynamics is theoretically accessible, in practice it is difficult to obtain.
MD is an N-body simulation scheme where atoms in the system of interest are treated classically and the Newtonian equations of motion are integrated numerically to propagate the system dynamics over time. Computational resources limit the system’s spatial scale, as well as achievable simulation time scale bound the applicable scope of MD simulations. As system size increases, the computational requirements to perform the simulation also increase, leading to a trade-off between system size and computationally tractable simulation length. Recent advances in computational hardware has expanded the scope and scale of MD simulations, owing to the deployment of multiple petascale national supercomputers (i.e., Stampede, Titan, Mira, BlueWaters), GPUs9−11 that have allowed routine access to continuous simulations of biologically relevant systems on the microsecond time scale, and specialized architectures such as Anton12,13 that have achieved millisecond resolution. Yet, accurately quantifying equilibrium properties from MD simulations remains a challenge not only because of limited sampling but also because of the still rather limited development of integration and analysis technologies that allow researchers to systematically derive information from multiple MD simulations in a rigorous and fully reproducible manner.
A recent approach to characterize equilibrium properties is to integrate MD simulations using Markov state models (MSM).14−16 A MSM is a stochastic model that assumes the Markov property that the system is memoryless (i.e., the conditional probability distribution of future states depend only upon the current state and not on prior states). Given a set of states Q, with Qt denoting the state at time t, if a process undergoes a transition i → j at time t, this is written as Qt = i and Qt+1 = j. The transition data of a Markov chain is given by a n × n transition matrix δ = pij, where pij = P(Qt+1 = j|Qt = i) is the probability of transitioning from state i to state j. Formally a process is Markovian if the following is true
There are various MSM packages that analyze MD trajectories and create clusters of microstates as well as to survey the transition state probabilities. Two of the most prominent are MSMBuilder217 and EMMA.18 From a practical standpoint, MSMBuilder2 appears to be designed for larger systems and with speed of data processing in mind. On the other hand, EMMA is designed for smaller MSMs that can be statistically validated and provides tools to quantify statistical uncertainty for quantities of interest.
To create a MSM, conformational space needs be explored, and subsequently discretized into microstates from which transition probabilities are calculated, and finally refined and validated.19 Classical MD simulations can be used to explore conformational space,12,13 but because the MSM is built from transition probabilities, enhanced sampling techniques such as replica exchange,20−22 simulated tempering,23 coarse graining,24,25 aMD,26 or using simplified force fields, which do not reproduce kinetic rates, can be used to provide an initial sampling of configuration space which is followed classical MD simulations to recapture the correct underlying thermodynamics. Microstates are defined through various clustering algorithms and state definitions depending on the biological question of interest (e.g., backbone RMSD for folding). After microstates are determined, transition rates between states are calculated from the MD trajectories and the MSM generated. Improvements on the initial MSM can be made through adaptive sampling, in which configurations with low sampling count (and thus high statistical error) become new starting configurations for subsequent MD runs. This allows one to obtain vastly improved statistics for more rarely sampled states.27 Finally, MSMs must be validated for self-consistency and that the Markov property is observed after some lag time.
While MSMs were initially used to couple Brownian dynamics simulations with MD simulations in order to quantify binding kinetics for biological systems in the early 1990s,28 the application of MSMs to reconstruct thermodynamics based solely on MD trajectory data is much more recent. In 2004 Swope, Pitera, and co-workers laid a theoretical framework and applied MSM coupled with MD simulations (MD-MSM) to the study of protein folding.29,30 MD-MSM development and application continues to be driven largely by the study of protein folding.31−37 The study of protein function has been more limited, however, within the past six months a number of new works have been published. Roux and co-workers used MSM in conjunction with their string method to study the activation of pathway of Scr-kinases.38,39 Kohlhoff and co-workers used MSM with trajectory data collected on Google’s exascale cloud computing resources to study agonism and inverse agonism in a GPCR.40 Recently, our lab completed our study of cAMP agonism of a cyclic-nucleotide binding domain (CBD) in the regulatory unit of protein kinase A (PKA).41 These studies each show the potential power of MD-MSM analysis in understanding protein function at an atomic scale and also highlight the computational difficulties of building these models as well as the challenges inherent in using an emerging analytical technique. The objective of the current work is to explore the challenges of using MD-MSM to study functional protein systems utilizing our recent study on PKA’s CBD; in other words, to explore the reality of moving from simple model systems to complex biological systems. After a brief introduction of the CBD studies, we discuss relevant challenges, including sampling conformational space, microstate definitions, model building, model comparison, and model analysis.
Biological Background of a Canonical Signal Transduction Domain
CBDs are a ubiquitous and ancient signaling domains.42,43 PKA’s regulatory subunit contains two tandem CBDs that cooperatively bind cAMP to regulate PKA’s enzymatic activity.44,45 Upon cAMP binding, the structure of the regulatory subunit of PKA changes from an extended or holo-enzyme (H) conformation that inhibits PKA’s catalytic subunit to a compact or cAMP-bound (B) conformation.46,47 The conformational changes in regulatory subunit are reflected in the motions of key structural motifs within the CBD that govern the ligand induced conformational changes in the regulatory subunit.48 Paralleling NMR studies on the same system,49 we explored the role of cAMP in the regulation of the conformational dynamics of the H-to-B conformational change in a single functional CBD domain using MD-MSM.41
Sampling
The sampling of rough energy landscapes is a computationally intensive task. The Arrhenius relationship suggests that the rate of reaction varies exponentially with the increase in barrier of activation. For energy landscapes with large barriers, using classical MD, a trajectory can become trapped in a local minima and never escape within the time scale of the simulation. Owing to the complexity of hyper-dimensional conformation space, it becomes computationally intractable to sample all configurations that contribute to the transition state probabilities with good statistics. While there have been many advances in computer hardware and MD software optimizations, computationally tractable time scales are currently only on the order of microseconds. Often, large domain motions of proteins have an intrinsic time scale on the order of several milliseconds. Even if one is lucky enough to sample a slow event, with n = 1 we cannot practically make any conclusions about the system and thus the main challenge in MD simulations is sampling.
A Markov State Model helps overcome sampling challenges by shifting the sampling focus from identifying local energy minima toward the practice of simulating more informative transition pathways. Many methods to improve sampling and statistical confidence in the MSMs have been developed. The most elementary method is to simply perform one (or a few) long time scale runs; however, there is a large computational cost associated with this approach.19 The computational burden of calculating extreme length trajectories (e.g., at the time of writing of this manuscript, we define “extreme length” by over 10 μs of total sampling in a single run) has been reduced by extraordinary progress in GPUs as well as specialized MD architectures such as Anton.50 Alternately, instead of calculating one or a few long trajectories, similar results can be obtained by performing multiple randomly seeded shorter runs. Instead of naively starting random trajectories that may be kinetically trapped in highly populated metastable states, initial states can also be selected near more rarely sampled configurations based upon the initial MSM.51 As additional statistics are gathered and integrated into the MSM, the sampling distribution will even out and new regions of less confidence can be used to pick new initial simulations seeds in a process called adaptive sampling.52 It is important to note that while simulations of initial configurations that do not lie on transition pathways will increase the total sampling time, the incorporated data will not bias the final results.19
The biology underlying the system of interest is a critical aspect of successful application of MD-MSM. Aware of the challenges of exploring the conformational ensemble, we selected to study the CBD. The CBD systems were sufficiently small, containing 126 amino acids and ∼30 000 atoms when fully solvated. Importantly, the critical CBD dynamics occur on the microsecond time scale or faster.53 Effectively accessing dynamics on the microsecond time scale allows us to leverage GPU and Anton-based MD for parallel multiple sampling runs seeded from the available crystal structures, thus avoiding the need to apply an alternate (e.g., enhanced or biased) sampling method or simulation methodology per se. We started MD simulations from the X-ray crystal structures of PKA’s regulatory subunit in both the H or B conformations and with and without cAMP thus creating four simulation families. Our objective was to explore the conformational space from two starting points hoping that the trajectories will overlap in conformational space allowing us to build a MSM. (Figure 1) We performed an initial sampling using long time scale, ∼13 μs, simulations on Anton12,13 with 4 parallel shorter 0.5 to 1.0 μs GPU enabled MD simulations using Amber9−11,41 Each of the parallel runs was started from the same equilibrated conformation but new initial velocities were assigned for each simulation.
Figure 1.
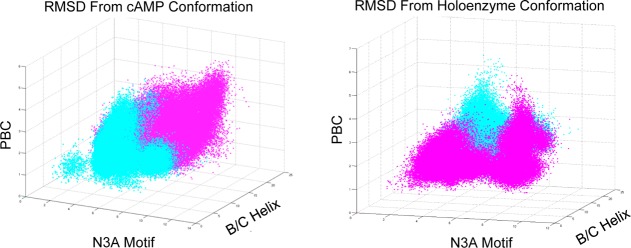
3D plots of conformational space exploration for two ∼13 μs simulations of apo CBD started at different conformations. MD simulations were started in either the cAMP bound conformation (cyan dots) or holoenzyme conformation (magenta dots). Each point represents a conformational snapshot (i.e., frame) from the MD trajectories plotted based on the RMSD from the crystallographic structure of three CBD structural motifs, the phosphate binding cassette (PBC), the N3A motif, and the B/C helix, that characterize the conformational change in the CBD. Each plot contains the same two trajectories with different reference structures.
In addition to our initial sampling, we performed multiple rounds of adaptive sampling guided by our primary MSM. In order to improve the quality of the MSM and enhance our conformational sampling we started short 10–15 ns MD simulations from microstates in the MSM with few, 1 or 2, conformations. Because the microstates are determined by clustering, conformations in these outlier clusters represented extreme conformations that could possibly be on transition pathways. Also, as the microstates had few members, the total number of transitions into and out of the microstate were extremely low decreasing our confidence in the rate of transitions to that state. For each of the conformations selected, 3 MD simulations were performed, assigning new starting velocities to explore the conformational space. The lengths of the simulations were sufficient to explore the local conformational and return to a local minimum. (Figure 2) Multiple rounds of adaptive sampling, composed of 68 MD simulations ranging from 10 to 200 ns in individual length, were performed until there was no noticeable change in the implied time scale plots, discussed below.
Figure 2.

Example conformational space exploration of apo CBD plotted relative to holoenzyme conformation of two 10 ns adaptive sampling runs. MD simulations were started in either the cAMP bound conformation (red dots) or holoenzyme conformation (blue dots). Adaptive sampling runs, indicated by black arrows, were started from frames form trajectory started in either cAMP bound conformation (magenta dots) or holoenzyme conformation (cyan dots) Each point represents a conformational snapshot (i.e., frame) from the MD trajectories plotted based on the RMSD from the crystallographic structure of three CBD structural motifs, the phosphate binding cassette (PBC), the N3A motif, and the B/C helix, that characterize the conformational change in the CBD.
Our approach is a hybrid of the two main sampling approaches in the literature: long time scale simulations, such as in the case of FiP35 WW domain folding a single MSM built from two 100 μs Anton generated trajectories stated from an unfolded state,32 and multiple short time scale simulations, such as in the case of GPCR MSMs simulations that were started from two structures, active and inactive, collecting multiple 100 ps trajectories for a total of 2.15 ms leveraging a grid-computing environment.54 A hybrid approach on our dynamic system allowed us to leverage the best of both approaches. The long time scale simulations enabled extensive sampling of the conformational ensemble followed by short time scale directed-sampling to improve the model. Using this hybrid method and starting with experimentally determined starting conformations allows for an unbiased exploration of conformational space. This sampling approach is similar to the work of Head–Gordon and co-workers who are able to identify key transition pathways combining long time scale initial sampling with directed sampling guided by instantaneous normal modes analysis.55,56 Together these methods establish the feasibility of unbiased, directed sampling of the conformational ensemble to detect important structural and dynamical transitions. Because our approach also allowed us to rely only on classical MD seeded from experimental structures, it is hoped that this data set will provide an effective test bed for comparing sampling methods. Our full trajectory data is available by request.
Of note, using our final MSM we determined the half-life of transitioning out each microstate by treating the transition out of a microstate into several microstates as a multipliable parallel reaction thus treating the transition as a first order reaction. Some of the half-lives for these microstates were >200 ns, a standard sampling duration for current classical MD studies. This illustrates the sampling limitations for single MD simulations; and suggests caution should be taken when selecting initial seed conformations of simulations with short time scale sampling lengths.
In addition to being able to effectively sample conformational space, it is also important to know how to track the progress of sampling and assess when it is complete. For our CBD models, we employed two approaches to assess sampling robustness. First, we developed a human readable metric that would allow us to track the progress of the MD simulations though conformational space and identify if the simulations have overlapped in conformational space. Because the CBD H-to-B conformational change is characterized by the orientation of key structural motifs in the CBD, we used the RMSD of each structural motif from the experimental structures to characterize a confirmation. This allowed us to plot a projection of the protein’s location in conformational space onto two 3D plots. (Figure 1) We employed both reference conformations to increase the likelihood of knowing the simulations overlapped in conformational space, as the distance between points becomes distorted in a projection of the hyper-dimensional conformational space. Our initial sampling, particularly with the Anton runs, overlapped in conformational space and allowed us to build a MSM of the CBD making the H-to-B transition with and without cAMP bound. Second, for adaptive sampling, we employed implied time scale plots, described below, to evaluate convergence of our model.
Microstate Definitions
In order to build a MSM the conformational space explored by the MD simulations needs to be discretized. Ideally the discretization occurs along kinetic boundaries between metastable states in conformational space minimizing approximation error.57 Since those boundaries are unknown a priori, the secondary objective is to make the division in conformational space as small as possible.57 Many approaches have been used and developed, from traditional root mean squared distance (RMSD) methods to independent component analysis.15,19 While some recent efforts have been able to recapture kinetic information within the microstate definitions, generally, any kinetic information associated with the conformations (e.g., velocities from MD trajectories) is discarded, and it is assumed that conformations that are structurally similar are kinetically similar.51,58 Fundamentally, the process of discretization involves defining a vector that describes a state, selecting a distance metric, and applying a clustering algorithm.
In our study of CBDs, we initially treated the problem CBD conformational dynamics as a protein folding problem, building on existing applications of MSMs to study proteins. We defined the conformation of a protein as spatial coordinates of the backbone Cα atoms. Corresponding microstates were based on RMSD clustering. For the sample MSM of the whole CBD presented here, we utilized the Theobald RMSD clustering algorithm implemented in MSMBuilder217 allowing alignment. For the MSM of the substructures of CBD we aligned the MD sampled conformations to a common reference frame, the crystallographic structures, in order to study the motions of the CBD subdomains relative to dynamically stable core of the CBD. Other studies used different microstate definitions. The Src kinase studies defined conformations by using subset of heavy atoms that exhibited the greatest conformational changes.39 For the GPCR studies, RMSD of active site atom and interdomain distances were used to define microstates.54 In all three approaches, microstates were determined according to the scientific question and structural domains of interest. These focused state definitions allow for efficient sampling with the potential neglect of portions of conformational space.
Model Selection
After microstates have been defined and sampling completed, the MSM needs to be built and its quality determined. In particular, the quality of a MSM can be quantified by whether the lag time is sufficiently long that the chosen microstate decomposition behaves according to the Markov property. The established best practice is to use methods from spectral theory such as the Swope–Pitera eigenvalue test, Chapman–Kolmogorov test, and Bayesian Model selection.59 Assuming that the system is in state q1 and progresses through state q2 to state q3, these methods are measures of whether or not the P(q3|q1q2) = P(q2|q1)P(q3|q2). While these methods are mathematically rigorous and informative, often a visual approach is easier to interpret. The best practice is to generate an implied time scale plot (ITP) of the model relaxation time scale versus the model lagtime of various coarse-grain methods or parameters. For a transition matrix T, we can calculate
where τk is the implied time scale, λk is an eigenvalue of the transition matrix with lag time τ. Ultimately we expect that the condition
is observed. This is visualized as an exponential decay in the ITP to system equilibrium.
In accordance with established best practices, we employed ITP to determine the quality of our MSM. Using the hybrid k-centers k-medoids clustering algorithm within MSMbuilder2,17 we carried out a parameter sweep of RMSD cut-offs producing variable divisions of conformational space and computed ITPs for each model. Interestingly, as the RMSD cutoff radius increased, decreasing the total number of clusters, the quality of the ITPs increased. (Figure 3) Based on the previous theoretical work, this finding would suggest that as the model error increased due to poorer division of the conformational ensemble, the MSM appears more Markovian. The apparent increase in model quality is likely due to the time exposure of dampening and thermal fluctuations manifested over the MD simulation. For the case of an extremely small RMSD cutoff, every conformation in the simulated trajectory may be considered its own cluster-state and the transition between one cluster-state to the next is extremely fast (the sampling rate). In this case, over dampening must be used to destroy memory in order to satisfy the Markovian criteria. For high friction (high dampening), the probability distribution of the conformational ensemble is temporally governed by the Fokker–Planck equation and this technique has been used determine reaction coordinates via diffusion maps.60
Figure 3.

Sample comparison between ITP plots and Chapman–Kolmogorov test plots for the apo CBD at different clustering cut-offs. In the left panel, implied time scale plots (ITPs) are presented. In the right panel, results from the Chapman–Kolmogorov (CK) tests are presented; MD data is blue; MSM results at different lag times are shown in green (lag 2.4 μs), magenta (lag 4.8 μs), and red (lag 9.6 μs).
In the case where the RMSD cutoff is large, clusters are often generated with hundreds of conformations and the transition between clusters is comparatively slower. The slower the transition is between clusters, the more the dynamics are exposed to dampening and thermal fluctuations when integrating the equations of motion for the simulation. It may be that the trajectories’ memory is semidestroyed by the amount of time it resides in a given cluster-state and that if the residence time is long enough, the trajectory will make an approximately memoryless transition to another cluster state increasing the apparent adherence to the Markov criteria.
In addition to ITPs, we performed a Chapman–Kolmogorov test to validate the models.57,61−63 The Chapman–Kolmogorov test compares the probability of remaining in a selected state at increasing time steps of the MD trajectories to that of MSM. A MSM is considered internally consistent with its source MD trajectories if the probability of remaining in given state falls within 1-σ standard error of the MD data.59 Consistent with theory, the Chapman–Kolmogorov test showed that increasing the number of microstates and increasing the lag time improved the consistency of the MSMs (Figure 3). However, as the quality of the ITP improved, with decreasing number of microstates, the quality of Chapman–Kolmogorov analyses decreased. The Chapman–Kolmogorov analyses can be thought of as a measure of how consistent the Markov State Model is with the actual MD simulation.57 Therefore, it seems that the choice of cutoff must balance the Makovian quality (as judged by the ITP) of the model with the internal consistency (Chapman–Kolmogorov analysis) of the model. A good MSM will minimize the error between both.
The error seems to arise from the memory inherent in the MD trajectory. We argue that a larger cutoff distance must be used in order to destroy memory but that this causes the continuity of the MD trajectory to be destroyed. The cluster states are often represented as crisp partitions in conformational space. However, a crisp partition implies that the same transition probabilities are assigned to all conformations associated within a given cluster-state. It may be likely that a conformation buried within a cluster-state does not have the same transition attributes as a conformation located on the peripheral boundary of that cluster-state. Anchoring a conformation to an assigned transition probability completely destroys the continuity of the dynamics and transitions between cluster-states become more like a jumping process rather than a smooth conformational transition. The Chapman–Kolmogorov test is likely sensitive to this discontinuity introduced by larger cluster sizes.
To counteract this issue in our work, we selected the MSM that had the largest number of clusters with the best-resolved ITPs at a long lag time, as described above. For the example CBD model, this resulted in models of ∼120 clusters—significantly fewer than the few thousand node models published for other systems.32,39,54 However, the RMSD cutoff was 3.5 Å, similar to folding models that resulted in thousands of nodes.32 The lag time for our models, 9.6 ns, was also similar to GPCR MSMs,54 7.5 ns, and the Scr kinase MSM,39 5 ns. The ITP for the CBD’s MSM (Figure 3) was better resolved than respective ITPs in either the GPCR MSMs or the Scr kinase MSM.39,54
Comparing Models
In the study of functional proteins, it is critical to be able to compare different MSMs to understand how ligands or mutations perturb the underlying free energy landscape and thus modify function. To our knowledge, only our CBD study41 and the GPCR study54 have attempted to compare MSM from perturbed systems, and in both cases, bound ligands were the source of perturbation. For the GPCR work, the same microstate definitions were used for each model and MSMs was generated independently with a unique selection of microstates. Comparison between MSMs of different systems was thus done using information derived from transition state matrix such as TPT or the generation of representative trajectories via Markov Chain Monte Carlo techniques and then comparing information derived from those trajectories. To compare the CBD MSMs, we employed a “unified clustering” approach. We started by clustering conformational states explored by both systems with and without cAMP-bound simulations together in order to produce a single shared set of microstates. Subsequently, separate MSMs were built for systems with and without cAMP-bound systems, using selected clustering parameters that produced high-quality ITPs and the same lag times for both systems. Unified clustering allowed for direct comparison between two MSMs on a microstate-by-microstate basis. We were able to observe the changes in equilibrium populations of microstates upon cAMP binding and furthermore, to identify unique microstates in each system. However, unified clustering may lead to a less optimal discretization of conformational space, as microstates are “forced” upon the model for the sampling of the other systems. We selected MSMs where the ITPs well resolved for both systems to minimize any distortion generated by unified clustering.
Coarse-Grained Models
Due to the inherent complexity of the conformational space a MSM for a well-sampled system may contain hundreds or thousands microstates. As a result, while high-resolution models may robustly describe the free energy landscape; it is arduous and time-consuming to extract distinct human readable kinetic states. In order to reduce the human burden of interpreting high-resolution MSMs, coarse-graining methods, which serve to aggregate similar microstates, are typically used. To understand the functional dynamics of the CBD in PKA, we employed two types of coarse-grained models: the first to study metastable states and the second to explore mapping of metastable states to putative functional conformations.41
First, we looked at the metastable states using the Robust Perron Cluster Analysis (PCCA+) method17,64,65 as implemented in MSMBuilder2 followed by the Markov-clustering algorithm (MCL).66,67 The PCCA+ algorithm performs a fuzzy spectral clustering based upon the eigenvalues of the stochastic transition matrix. PCCA+ assumes that there should be a separation of time scales between slow transitions across barriers and more rapid transitions within a single basin. This can be mathematically described by a distinction between the eigenvalues corresponding to the slowest dynamics (with values close to 1) and eigenvalues of faster transitions (with values less than one). Finally, the fuzzy clusters are obtained as a linear transformation of the slowest eigenvectors.65 Essentially, the conformations are grouped into clusters that are separated by the slow dynamics.
In contrast the MCL algorithm, designed originally for clustering of simple and weighted graphs, clusters based upon the propensity of a random walker leaving a particular set of states. This propensity is calculated by the alternate application of an expansion and inflation operator until the transition matrix converges. For a given stochastic transition matrix (A), the expansion operator computes the power of A using the normal matrix product (A*A). Successive applications of the expansion operator on A will eventually converge upon a stationary distribution. On the other hand, the inflation operator corresponds with taking the Hadamard power of a matrix (the elementwise power) followed by a scaling step to ensure the matrix is stochastic again. The inflation operator is formally written as,
where Γr is the inflation operator, Mpq is the value of stochastic transition matrix A at row p and column q, and r is a tunable parameter that determines the amount that strong neighbors strengthened and weak neighbors demoted. By analogy, parameter r correlates with how much the contrast ratio between barrier height and well depth is increased per iteration. Effectively, larger values of r will lead to enhanced sensitivity to small basins of attraction in the network, resulting in additional clusters and has been compared to the rate of temperature decrease in simulated annealing.68 In summary, MCL clusters states that are tightly connected by fast transitions.
As shown in Figure 4, both PCCA+ and MCL coarse-grained mostly share the same microstates. It is likely that PCCA+ and MCL coarse-grain to similar clusters because fundamentally both cluster based upon the propensity of a random walker to remain in a cluster with minimal jumps between clusters. MCL calculates this stationary distribution by strengthening fast transitions while weakening slow transitions, manipulating the transition matrix in a nonphysical way. While PCCA+ calculates this stationary distribution by using linear combinations of eigenvectors corresponding to the slow-transitions thus preserving the slow time scales of the transition matrix.
Figure 4.
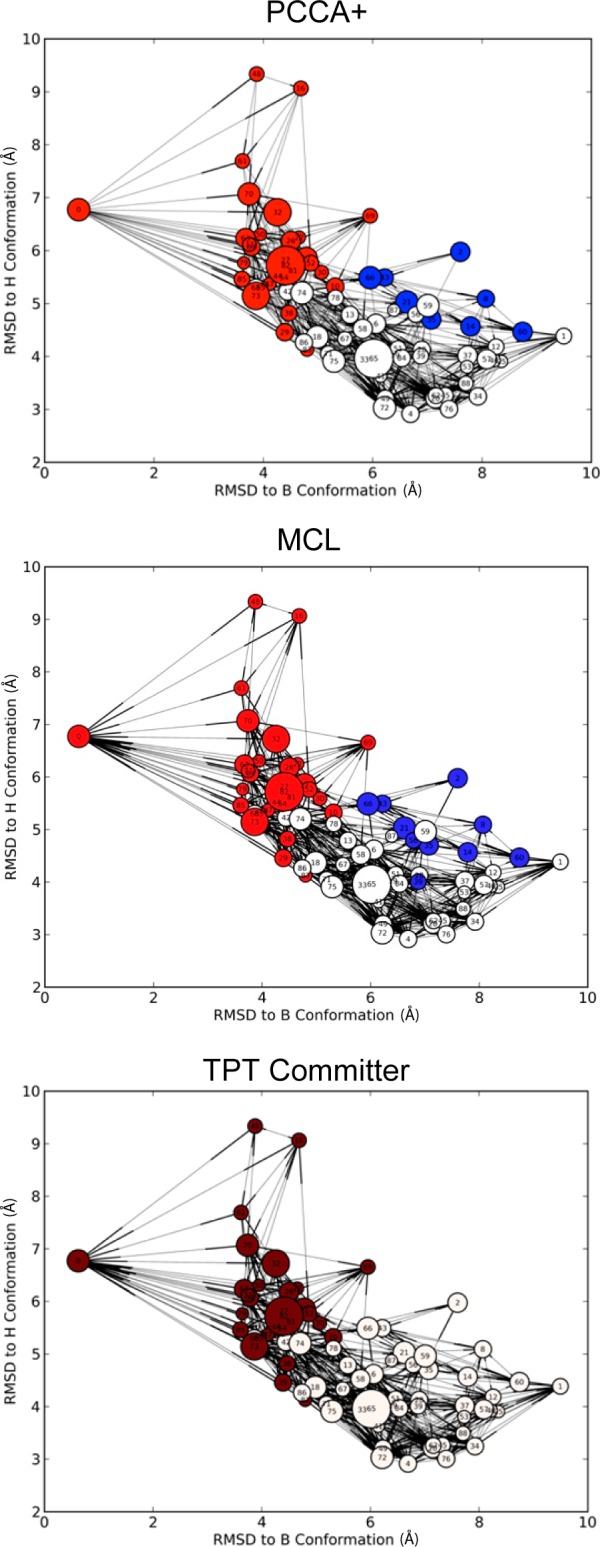
Sample comparison of PCCA+, MCL, and TPT committer macrostate models. Color-coding of the microstates identifies macrostate membership. Microstate nodes are plotted based on the RMSD of the cluster generator from two reference structures and the node diameters are proportional to the log of their proportional equilibrium population.
Due to its deterministic nature and r’s relationship to the barrier height, we used MCL for our analyses. For example, MCL facilitated the qualitative comparison of two energy landscapes of a structural motif of CBD. By comparing MCL at different r values clearly showed how the addition of cAMP changed the free energy landscape from a shallow surface with multiple metastable states to a defined two state system. (Figure 5).
Figure 5.

Comparison of MCL macrostate models for a subdomain of the CBD with and without cAMP-bound at different MCL-p values. Color-coding of the microstates identifies macro states membership. Microstate nodes are plotted based on the RMSD of the cluster generator from two reference structures and the node diameters are proportional to the log of their proportional equilibrium population.
Second, we used transition pathway theory (TPT) committer analysis69 to divide the conformational ensemble into “functional” macrostates. We used results of the committer analysis to divide the microstates into clusters with >50% of transitioning to microstates that contained one of the crystallographic conformations. This model assumes that the end-point crystal structures best represent the functional state of the protein. In effect, this method determines a functional “continental divide” in the free energy landscape. Encouragingly, the functional divisions corresponded with the kinetic divisions determined with the MCL analysis (Figure 4). The combinations of these two analysis techniques identified which of the kinetic transitions corresponded to functional transitions. While this approach was helpful in the analysis of the CBD, a robust study of this approach should be performed before it is more generally applied.
Conclusions Looking Forward
The advancements in computational resources and architectures have increased our MD sampling capabilities, and with the addition of MSMs as an emerging new general data analytics framework, it is now possible to characterize full equilibrium thermodynamics for functional protein systems with atomistic detail. In this work, we discussed issues surrounding sampling, microstate definitions, model selection, model comparison, and coarse-grained models, and attempt to highlight the challenges arising from more complicated systems.
In the area of sampling, ideally we would like to use experimental observables, for example NMR-derived order parameters, to determine if our models are converged and whether sampling is sufficient. Relationships between MSM and experimental observables are only beginning to be explored,70 but should be a major focus of research moving forward. Currently, it seems the general approach is to sample to the maximum capabilities of the employed computational resources. Going forward, the development of automated metrics to assess sampling convergence on-the-fly may improve utilization of available resources and make the process of creating MSMs more efficient, reproducible, and reliable.
To our knowledge, there has been no systemic evaluation of microstate definitions and their effect on sampling, MSM outcomes, and scientific insight. Unfortunately, at this point, there is likely insufficient publically available data to conduct a systematic analysis. However, as the implementations of MD-MSM increase, a well-described test set would be a desirable community outcome in order to facilitate meaningful comparison between definitions.
In the realm of model selection, the inconsistences with the Chapman–Kolmogorov test in our work and the lack of reporting of the Chapman–Kolmogorov test in the other published functional studies indicate the continued reliance on ITP to determine model quality. The relatively poor resolution of slow motions in the GPCR and Src studies and the inconsistency of the Chapman–Kolmogorov test results in our study suggest that there is more to be done in either improving the models or methods for evaluating the models.
As MSMs are constructed for an increasing number of biological systems, coarse-graining methods will need to continue to be developed, studied and evaluated. Coarse-graining models not only provide intuitive models of MSM, they can also form the basis for multiscale modeling approaches, allowing atomic scale MSM to be integrated in Markov models of macromolecular and subcellular processes. This integration requires development of transparent error analysis techniques that facilitate the assessment of error propagation between models and across scales.
Moving forward, methods for comparing MSMs need to be developed that are both qualitative and quantitative. While the scientific questions of each study generally guide the specific ways two MSMs are compared, a systematic and theoretically sound approach to assessing differences in the resulting two free energy landscapes needs to be addressed.
As more complicated systems are explored, researchers in the field will need to be continually critical of the employed approaches. We need to understand and continue to define limitations of the theoretical underpinnings of our work and where we are being guided by our biological intuition. We hope that the frank discussion of methods presented in this work will encourage discussion and continue methodological advancements related to the application of MSMs to larger-scale biological systems.
Acknowledgments
The authors would like to thank Prof. Susan Taylor and Dr. Alexandr Kornev for their collaboration and insight into the biological system. This work was funded in part by the National Institutes of Health (NIH) through the NIH Director’s New Innovator Award Program DP2-OD007237 and through the NSF XSEDE Supercomputer resources grant RAC CHE060073N to R.E.A. Additional funding from the National Biomedical Computation Resource, NIH P41 GM103426, is gratefully acknowledged. Anton computer time was provided by the Pittsburgh Supercomputing Center (PSC) and the National Center for Multiscale Modeling of Biological Systems (MM Bios) through Grant P41GM103712-S1 from the National Institutes of Health. D.E. Shaw Research generously made the Anton machine at PSC available.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Onuchic J. N.; Luthey-Schulten Z.; Wolynes P. G. Annu. Rev. Phys. Chem. 1997, 48, 545–600. [DOI] [PubMed] [Google Scholar]
- Ozenne V.; Schneider R.; Yao M.; Huang J.; Salmon L.; Zweckstetter M.; Jensen M. R.; Blackledge M. J. Am. Chem. Soc. 2012, 134, 15138–15148. [DOI] [PubMed] [Google Scholar]
- Levy Y.; Jortner J.; Becker O. M. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 2188–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y.; Nichols S. E.; McCammon J. A. Phys. Chem. Chem. Phys. 2014, 30, 6398–6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenwick R. B.; van den Bedem H.; Fraser J. S.; Wright P. E. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, E445–E454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar I.; Lezon T. R.; Yang L.-W.; Eyal E. Annu. Rev. Biophys. 2010, 39, 23–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin K.; Castañeda C. a; Schneidman-Duhovny D.; Sali A.; Nava-Tudela A.; Fushman D. J. Am. Chem. Soc. 2013, 135, 16595–16609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleckner I. R.; Foster M. P. Biochim. Biophys. Acta 2011, 1814, 942–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone J. E.; Phillips J. C.; Freddolino P. L.; Hardy D. J.; Trabuco L. G.; Schulten K. J. Comput. Chem. 2007, 28, 2618–2640. [DOI] [PubMed] [Google Scholar]
- Salomon-Ferrer R.; Götz A. W.; Poole D.; Le Grand S.; Walker R. C. J. Chem. Theory Comput. 2013, 9, 3878–3888. [DOI] [PubMed] [Google Scholar]
- Götz A. W.; Williamson M. J.; Xu D.; Poole D.; Le Grand S.; Walker R. C. J. Chem. Theory Comput. 2012, 8, 1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piana S.; Lindorff-Larsen K.; Shaw D. E. Proc. Natl. Acad. Sci. U.S.A. 2012, 1201811109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindorff-Larsen K.; Piana S.; Dror R. O.; Shaw D. E. Science 2011, 334, 517–520. [DOI] [PubMed] [Google Scholar]
- Pande V. S.; Beauchamp K.; Bowman G. R. Methods 2010, 52, 99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodera J. D. J.; Noé F. Curr. Opin. Struct. Biol. 2014, 25C, 135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz J. H.; Keller B.; Noe F. Phys. Chem. Chem. Phys. 2011, 13, 16912–16927. [DOI] [PubMed] [Google Scholar]
- Beauchamp K. A.; Bowman G. R.; Lane T. J.; Maibaum L.; Haque I. S.; Pande V. S. J. Chem. Theory Comput. 2011, 7, 3412–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senne M.; Trendelkamp-Schroer B.; Mey A. S. J. S.; Schutte C.; Noe F. J. Chem. Theory Comput. 2012, 8, 2223–2238. [DOI] [PubMed] [Google Scholar]
- Pande V. S.; Beauchamp K.; Bowman G. R. Methods 2010, 52, 99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swendsen R. H.; Wang J.-S. Phys. Rev. Lett. 1986, 57, 2607–2609. [DOI] [PubMed] [Google Scholar]
- Hansmann U. H. E. Chem. Phys. Lett. 1997, 281, 140–150. [Google Scholar]
- Zhou R. Methods Mol. Biol. 2007, 350, 205–223. [DOI] [PubMed] [Google Scholar]
- Prinz J.-H. H.; Chodera J. D.; Pande V. S.; Swope W. C.; Smith J. C.; Noé F.; Noe F. J. Chem. Phys. 2011, 134, 244108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders M. G.; Voth G. A. Annu. Rev. Biophys. 2013, 42, 73–93. [DOI] [PubMed] [Google Scholar]
- Tozzini V. Curr. Opin. Struct. Biol. 2005, 15, 144–150. [DOI] [PubMed] [Google Scholar]
- Xin Y.; Doshi U.; Hamelberg D. J. Chem. Phys. 2010, 132, 224101. [DOI] [PubMed] [Google Scholar]
- Huang X.; Bowman G. R.; Bacallado S.; Pande V. S. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 19765–19769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luty B. A.; Elamrani S.; Mccammon J. A. J. Am. Chem. Soc. 1993, 115, 11874–11877. [Google Scholar]
- Swope W. C.; Pitera J. W.; Suits F. J. Phys. Chem. B 2004, 108, 6571–6581. [Google Scholar]
- Swope W. C.; Pitera J. W.; Suits F.; Pitman M.; Eleftheriou M.; Fitch B. G.; Germain R. S.; Rayshubski A.; Ward T. J. C.; Zhestkov Y.; Zhou R. J. Phys. Chem. B 2004, 108, 6582–6594. [Google Scholar]
- Voelz V. A.; Bowman G. R.; Beauchamp K.; Pande V. S. J. Am. Chem. Soc. 2010, 132, 1526–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane T. J.; Bowman G. R.; Beauchamp K.; Voelz V. a; Pande V. S. J. Am. Chem. Soc. 2011, 133, 18413–18419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman G. R.; Pande V. S. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 10890–10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng N.; Dai W.; Levy R. M. J. Phys. Chem. B 2013, 117, 12787–12799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radford I. H.; Fersht A. R.; Settanni G. J. Phys. Chem. B 2011, 115, 7459–7471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodera J. D.; Singhal N.; Pande V. S.; Dill K. A.; Swope W. C. J. Chem. Phys. 2007, 126, 155101. [DOI] [PubMed] [Google Scholar]
- Chodera J. D.; Swope W. C.; Pitera J. W.; Seok C.; Dill K. a. J. Chem. Theory Comput. 2007, 3, 26–41. [DOI] [PubMed] [Google Scholar]
- Yang S.; Banavali N. K.; Roux B. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 3776–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla D.; Meng Y.; Roux B.; Pande V. S. Nat. Commun. 2014, 5, 3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlhoff K. J.; Shukla D.; Lawrenz M.; Bowman G. R.; Konerding D. E.; Belov D.; Altman R. B.; Pande V. S. Nat. Chem. 2014, 6, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmstrom R. D.; Kornev A. P.; Taylor S. S.; Amaro R. E.. 2014, Submitted. [Google Scholar]
- Berman H. M.; Ten Eyck L. F.; Goodsell D. S.; Haste N. M.; Kornev A.; Taylor S. S. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan N.; Wu J.; Anand G. S.; Yooseph S.; Neuwald A. F.; Venter J. C.; Taylor S. S. Genome Biol. 2007, 8, R264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor S. S.; Ilouz R.; Zhang P.; Kornev A. P. Nat. Rev. Mol. Cell Biol. 2012, 13, 646–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoberg T. J.; Kornev A. P.; Taylor S. S. Protein Sci. 2010, 19, 1213–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y.; Dostmann W. R.; Herberg F. W.; Durick K.; Xuong N. H.; Ten Eyck L.; Taylor S. S.; Varughese K. I. Science 1995, 269, 807–813. [DOI] [PubMed] [Google Scholar]
- Kim C.; Xuong N.-H. H.; Taylor S. S. Science 2005, 307, 690–696. [DOI] [PubMed] [Google Scholar]
- Sjoberg T. J.; Kornev A. P.; Taylor S. S. Protein Sci. 2010, 19, 1213–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R.; Esposito V.; Abu-Abed M.; Anand G. S.; Taylor S. S.; Melacini G. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw D. E.; Chao J. C.; Eastwood M. P.; Gagliardo J.; Grossman J. P.; Ho C. R.; Lerardi D. J.; Kolossváry I.; Klepeis J. L.; Layman T.; McLeavey C.; Deneroff M. M.; Moraes M. A.; Mueller R.; Priest E. C.; Shan Y.; Spengler J.; Theobald M.; Towles B.; Wang S. C.; Dror R. O.; Kuskin J. S.; Larson R. H.; Salmon J. K.; Young C.; Batson B.; Bowers K. J. Commun. ACM 2008, 51, 91. [Google Scholar]
- Schwantes C. R.; Pande V. S. J. Chem. Theory Comput. 2013, 9, 2000–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman G. R.; Ensign D. L.; Pande V. S. J. Chem. Theory Comput. 2010, 6, 787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R.; Esposito V.; Abu-Abed M.; Anand G. S.; Taylor S. S.; Melacini G. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlhoff K. J.; Shukla D.; Lawrenz M.; Bowman G. R.; Konerding D. E.; Belov D.; Altman R. B.; Pande V. S. Nat. Chem. 2014, 6, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C.; Head-Gordon T. PLoS Comput. Biol. 2011, 7, e1002082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C.; Zhang L.; Head-Gordon T. Biophys. J. 2010, 98, 2356–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz J.-H.; Wu H.; Sarich M.; Keller B.; Senne M.; Held M.; Chodera J. D.; Schütte C.; Noé F. J. Chem. Phys. 2011, 134, 174105. [DOI] [PubMed] [Google Scholar]
- McGibbon R. T.; Pande V. S. J. Chem. Theory Comput. 2013, 9, 2900–2906. [DOI] [PubMed] [Google Scholar]
- Prinz J.-H. H.; Wu H.; Sarich M.; Keller B.; Senne M.; Held M.; Chodera J. D.; Schütte C.; Noé F.; Schutte C.; Noe F. J. Chem. Phys. 2011, 134, 174105. [DOI] [PubMed] [Google Scholar]
- Rohrdanz M. a; Zheng W.; Maggioni M.; Clementi C. J. Chem. Phys. 2011, 134, 124116. [DOI] [PubMed] [Google Scholar]
- Chodera J. D.; Singhal N.; Pande V. S.; Dill K. A.; Swope W. C. J. Chem. Phys. 2007, 126, 155101. [DOI] [PubMed] [Google Scholar]
- Swope W. C.; Pitera J. W.; Suits F. J. Phys. Chem. B 2004, 108, 6571–6581. [Google Scholar]
- Chodera J. D.; Swope W. C.; Pitera J. W.; Dill K. a. Multiscale Model. Simul. 2006, 5, 1214–1226. [Google Scholar]
- Bowman G. R.; Huang X. H.; Pande V. S. Methods 2009, 49, 197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röblitz S.; Weber M. Adv. Data Anal. Classif. 2013, 7, 147–179. [Google Scholar]
- Berezovska G.; Prada-Gracia D.; Mostarda S.; Rao F. J. Chem. Phys. 2012, 137, 194101. [DOI] [PubMed] [Google Scholar]
- Enright A. J.; Van Dongen S.; Ouzounis C. A. Nucleic Acids Res. 2002, 30, 1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gfeller D.; De Los Rios P.; Caflisch A.; Rao F. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 1817–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzner P.; Schütte C.; Vanden-Eijnden E. Multiscale Model. Simul. 2009, 7, 1192–1219. [Google Scholar]
- Xia J. C.; Deng N. J.; Levy R. M. J. Phys. Chem. B 2013, 117, 6625–6634. [DOI] [PMC free article] [PubMed] [Google Scholar]