

Abstract
The major Vibrio cholerae virulence gene transcription activator, ToxT, is responsible for the production of the diarrhea-inducing cholera toxin (CT) and the major colonization factor, toxin coregulated pilus (TCP). In addition to the two primary virulence factors mentioned, ToxT is responsible for the activation of accessory virulence genes, such as aldA, tagA, acfA, acfD, tcpI, and tarAB. ToxT activity is negatively modulated by bile and unsaturated fatty acids found in the upper small intestine. Conversely, previous work identified another intestinal signal, bicarbonate, which enhances the ability of ToxT to activate production of CT and TCP. The work presented here further elucidates the mechanism for the enhancement of ToxT activity by bicarbonate. Bicarbonate was found to increase the activation of ToxT-dependent accessory virulence promoters in addition to those that produce CT and TCP. Bicarbonate is taken up into the V. cholerae cell, where it positively affects ToxT activity by increasing DNA binding affinity for the virulence gene promoters that ToxT activates regardless of toxbox configuration. The increase in ToxT binding affinity in the presence of bicarbonate explains the elevated level of virulence gene transcription.
INTRODUCTION
Vibrio cholerae has two distinct phases in its life cycle. In the planktonic state in the aquatic environment, V. cholerae represses the expression of pathogenesis genes while increasing the transcription of genes involved in environmental survival, such as those required for motility (1, 2). Upon entry into a human host via consumption of contaminated water or food, the bacteria encounter signals resulting in an inversion of their transcriptome profile. In this new, virulent state, the expression of genes involved in motility and environmental survival is repressed, while the expression of genes involved in host survival and pathogenesis is activated, resulting in the diarrheal disease cholera (1, 2). Virulence is controlled by a cascade of positive transcription regulators, known as the ToxR regulon (3). Many of the signals required to initiate change in the transcriptional profile have been identified in recent years (1, 2, 4).
The main signals that induce the transcription of V. cholerae pathogenesis genes in the host act on different steps in the virulence regulatory cascade. At the uppermost level of the cascade, AphA and AphB activate the transcription of tcpPH (5–7). AphB activity is enhanced by both low pH and anaerobic conditions (8, 9), such as those that would be found as V. cholerae transitions through the stomach and into the upper small intestine. The next level of the cascade includes the aforementioned TcpP, along with the constitutively produced ToxR, both of which are integral membrane proteins that work in tandem to activate toxT transcription (10). TcpP activity recently has been shown to be enhanced by taurocholate (11), a bile salt, and interaction between TcpP and ToxR is enhanced under oxygen-limiting conditions (12). The final level of the cascade consists of ToxT, which directly activates the production of the major virulence factors cholera toxin (CT) and toxin coregulated pilus (TCP) (13–16). ToxT also directly controls the transcription of other accessory virulence genes, including aldA, tagA, acfA, acfD, tcpI, tarA, and tarB (17–21). ToxT activity is reduced in the presence of the unsaturated fatty acid (UFA) components of bile (22, 23) and enhanced in the presence of bicarbonate (24), both of which are present in the upper small intestine where V. cholerae preferentially colonizes.
ToxT is a 276-amino-acid member of the AraC/XylS family of transcriptional regulators. The ToxT C-terminal domain contains two helix-turn-helix motifs responsible for DNA binding (25). ToxT activates virulence genes by binding to a 13-bp degenerate DNA sequence called a toxbox (19). All ToxT-dependent virulence genes have two toxboxes upstream of their transcriptional start site in either direct or inverted repeat configurations, with the exception of aldA, which has only a single toxbox (19). ToxT can bind toxboxes as a monomer (18, 19, 26) but is presumed to dimerize to fully activate at least some virulence genes (27–31).
The activation of the ToxT-dependent virulence genes can be altered in the presence of the positive and negative ToxT effectors. After egress from the stomach into the intestine, the bacteria encounter high concentrations of both bile and bicarbonate. As mentioned above, the UFA components of bile are negative effectors of ToxT activity (22, 27, 29, 30). The N terminus of ToxT is involved in the response to these effectors, presumably by decreasing ToxT dimerization (30). Bicarbonate is present in the upper small intestine to buffer stomach acid and also to protect the epithelial layer (32). Bicarbonate enhances the ability of ToxT to increase CT and TCP production (24). How bicarbonate enhances ToxT activity is not well understood.
Here, we report that bicarbonate increases transcription activation of other ToxT-dependent genes in addition to the genes encoding the two major virulence factors. We show that bicarbonate enters the bacterium, where it could interact with ToxT in the cytoplasm. Furthermore, we have determined that the mechanism for bicarbonate-mediated enhancement of ToxT activity is due to increased binding affinity of ToxT for the promoters it activates. This increase in binding occurs at promoters of each ToxT-dependent gene, regardless of toxbox configuration. This work establishes a direct mechanistic link between a signal from the host and increased transcription of pathogenesis genes that occurs in the host.
MATERIALS AND METHODS
Bacterial strains and plasmids.
Strains and plasmids used in this work are listed in Table 1. All strains were maintained at −70°C in LB containing 20% glycerol. Wild-type (WT) V. cholerae classical biotype strain O395 and an isogenic ΔtoxT mutant (15) with corresponding plasmids were used for β-galactosidase assays. The tcpA::lacZ and ctxAB::lacZ promoter fusions were constructed using pTL61t (19, 33) and contained 138 bp and 76 bp, respectively, upstream of the transcriptional start site. The pTL61t fusions aldA::lacZ and tagA::lacZ contained 158 bp and 92 bp, respectively, upstream of their transcriptional start sites (17). Fusions acfA::lacZ and acfD::lacZ were constructed in pTL61t (18) and contain 104 bp and 99 bp, respectively, upstream of the transcriptional start site. The tcpI::lacZ fusion contains 76 bp upstream of the translational start site (unpublished work). Strains and plasmids used in this study are listed in Table 1. The antibiotic concentration for strains with the pTL61t plasmid was 100 μg/ml ampicillin, and that for strains without plasmid was 100 μg/ml streptomycin.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Description | Source or reference |
|---|---|---|
| Strains | ||
| JW 467 | Escherichia coli BL21(DE3) | Laboratory collection |
| JW 1560 | JW 467 with pJW 407 (MBP-ToxT) | Laboratory collection |
| JW 9 | Vibrio cholerae classical strain O395 | Laboratory collection |
| JW 150 | O395 ΔtoxT | Laboratory collection |
| JW 18 | pJW 54 with JW 9 | 19 |
| JW 441 | pJW 211 with JW 9 | 33 |
| JW 87 | pJW 82 with JW 9 | 17 |
| JW 97 | pJW 89 with JW 9 | 17 |
| JW 86 | pJW 81 with JW 9 | 18 |
| JW 89 | pJW 84 with JW 9 | 18 |
| JW 99 | pJW 91 with JW 9 | Laboratory collection |
| JW 515 | pJW 54 with JW 150 | Laboratory collection |
| JW 1809 | pJW 211 with JW 150 | 33 |
| JW 167 | pJW 82 with JW 150 | Laboratory collection |
| JW 168 | pJW 89 with JW 150 | Laboratory collection |
| JW 165 | pJW 81 with JW 150 | Laboratory collection |
| JW 166 | pJW 84 with JW 150 | Laboratory collection |
| JW 169 | pJW 91 with JW 150 | Laboratory collection |
| Plasmids | ||
| pJW 407 | pMAL-c2e-ToxT | 33 |
| pJW 54 | pTL61t containing 138 bp upstream of tcpA | 19 |
| pJW 211 | pTL61t containing 76 bp upstream of ctxAB | 33 |
| pJW 82 | pTL61t containing 158 bp upstream of aldA | 17 |
| pJW 89 | pTL61t containing 92 bp upstream of tagA | 17 |
| pJW 81 | pTL61t containing 104 bp upstream of acfA | 18 |
| pJW 84 | pTL61t containing 99 bp upstream of acfD | 18 |
| pJW 91 | pTL61t containing 76 bp upstream of tcpI | Laboratory collection |
β-Galactosidase assay.
Bicarbonate virulence-inducing conditions were as previously described (24). Briefly, V. cholerae classical biotype strain O395 was grown overnight in LB at 37°C and subcultured 1:100 into AKI medium in the presence or absence of freshly prepared 0.3% sodium bicarbonate (36 mM) for 4 h. Cultures were grown statically for 4 h at 37°C and analyzed. The β-galactosidase assay was performed as previously described (34).
H14CO3 uptake assay.
V. cholerae classical biotype strain O395 was grown overnight in LB at 37°C and subcultured 1:100 into AKI medium in the absence of bicarbonate for 3 h. At 3 h, 1 μCi NaH14CO3 (54 mCi/mmol) (Perkin-Elmer) was added for each milliliter of V. cholerae subculture. Immediately, 1 ml of culture was centrifuged and supernatant was discarded. The cell pellet was washed 3 times with 1 ml of AKI medium and recentrifuged. Cell pellets were resuspended in 100 μl AKI medium and added to 5 ml scintillation cocktail (Fisher Scientific). The same procedure was followed in 15-min intervals for 2 h. After uptake, counts per minute (cpm) were measured for each time point using an LS6000IC liquid scintillation counting system (Beckman).
Cell fractionation.
V. cholerae strain O395 was grown overnight in LB at 37°C and subcultured 1:100 into AKI medium in the absence of bicarbonate for 3 h. At 3 h, 1 μCi NaH14CO3 (54 mCi/mmol) (Perkin-Elmer) was added to one milliliter of subculture and incubated at room temperature for 1 h. The bacteria were harvested by centrifugation and washed 3 times in 1× phosphate-buffered saline (PBS). Bacteria were resuspended in a 1-ml solution of 20% sucrose, 20 mM Tris-HCl (pH 8.0), and 1 mM Na-EDTA. Cells were lysed by sonication (10 s on/10 s off; amplitude setting, 60%; total time, 7 min; Branson Sonifier 450). Bacteria then were fractionated by centrifugation for 30 min at 10,000 × g to separate the periplasm/membranes and cytoplasm. The supernatant was taken as the cytoplasmic portion, and the pellet was resuspended in PBS. The cpm in each fraction subsequently were measured using an LS6000IC liquid scintillation counting system (Beckman).
Protein purification.
Maltose binding protein (MBP)-ToxT and MBP-AraC purification was performed as previously described (33). Briefly, fusion proteins were purified from Escherichia coli strain BL21(DE3) with plasmid pMAL-c2e containing MBP-ToxT or MBP-AraC. E. coli cells were subcultured until the optical density at 600 nm reached 0.5. The fusion protein then was induced with the addition of 0.25 mM isopropyl-β-d-thiogalactopyranoside (IPTG). Cells were lysed by a French press, and lysate was run over an amylose column (New England BioLabs). Fractions containing MBP-ToxT were dialyzed against buffer containing 50 mM Na2HPO4 (pH 8.0), 10 mM Tris-HCl (pH 8.0), and 100 mM NaCl and then dialyzed again against the same solution with 20% glycerol. The protein concentration was determined using a Qubit 2.0 Fluorometer (Invitrogen).
EMSA.
Electrophoretic mobility shift assay (EMSA) was performed as previously described (33). DNA probes were produced by PCR of pTL61T containing appropriate promoter sequence with one unlabeled primer and one primer end labeled with γ-32P (Perkin-Elmer) by T4 polynucleotide kinase (New England BioLabs). The binding reaction mixtures were prepared to a final volume of 30 μl and contained 10 μg/ml salmon sperm DNA, 10 mM Tris-acetate (pH 7.4), 1 mM potassium EDTA (pH 7.0), 100 mM KCl, 1 mM dithiothreitol (DTT), 0.3 mg/ml bovine serum albumin (BSA), and 10% glycerol. Binding reaction mixtures contained various concentrations of purified MBP-ToxT, and all reaction mixes had a constant concentration of labeled DNA probe. For assays using MBP-AraC, binding reaction mixes had a final concentration of 50 mM KCl, 5% glycerol, and 50 mM l-arabinose. Sodium bicarbonate, sodium biselenite, and sodium acetate were added to a final concentration of 36 mM in binding reaction mixtures containing the respective molecules. The binding reaction mixtures were incubated at 30°C for 30 min and immediately loaded into a 6% polyacrylamide gel and run at 4°C. Gels were dried and analyzed by autoradiography.
Binding curve analysis.
Autoradiographs were analyzed using ImageJ software (NIH). The percentage of labeled DNA bound (% bound) by protein was determined for each lane. GraphPad Prism 5 software was used for curve fitting to the following equation: % bound = Bmax × [protein]h/(KDh + [protein]h), where h is the Hill coefficient. Bmax is the amount of bound DNA where the curve plateaus, which we set to a constraint of 100%. The KD (equilibrium dissociation constant) for each condition was calculated, and significance was determined. Nonspecific binding was omitted due to the excess of nonspecific salmon sperm DNA added to binding reactions.
RESULTS
Bicarbonate increases activation of ToxT-dependent promoters.
Bicarbonate previously has been shown to increase the production of the major ToxT-dependent virulence factors TCP and CT (24); however, the effect of bicarbonate on other ToxT-activated promoters had not been assessed. Earlier work had demonstrated that the activation of genes aldA, tagA, acfA, acfD, and tcpI is dependent on the production of ToxT (17–19). We assessed the transcriptional activity of these ToxT-dependent promoters using plasmid-borne β-galactosidase promoter fusions in the classical biotype V. cholerae strain O395 and an isogenic toxT deletion (15). The location and orientation of the ToxT binding sites, or toxboxes, within each ToxT-dependent promoter is shown (Fig. 1A and B). Fusion construct activity from ctxAB::lacZ, aldA::lacZ, tagA::lacZ, acfA::lacZ, acfD::lacZ, and tcpI::lacZ was measured in the presence and absence of 36 mM sodium bicarbonate to determine if bicarbonate enhanced the ability of ToxT to activate the transcription of these genes, as it does with the major virulence genes tcpA and ctxAB. Culture conditions used to assess activity were adopted from previous work with bicarbonate (24). Our results confirm that bicarbonate activates each of these ToxT-dependent promoters in wild-type O395 while having no effect on a ΔtoxT strain (Fig. 1C). Therefore, bicarbonate has a global effect by enhancing ToxT activity at all ToxT-dependent promoters.
FIG 1.

Transcriptional effects of exogenous bicarbonate (Bicarb.) on ToxT-dependent virulence gene promoters. (A) Orientation and positioning of direct repeat toxbox virulence gene promoters, PtcpA and PctxAB, and the single toxbox promoter, PaldA. (B) Orientation and positioning of inverted repeat toxbox virulence gene promoters, PtagA, PacfA, PacfD, and PtcpI. Toxbox represents known ToxT binding sites. (C) β-Galactosidase activity produced from plasmid-borne virulence gene promoter fusion constructs in classical strain O395 and the isogenic ΔtoxT strain. Cultures were grown in AKI media in the absence or presence of 36 mM NaHCO3. Statistical significance was determined using Student's t test (*, P < 0.00025). Error bars represent standard deviations.
Radiolabeled bicarbonate is taken up by V. cholerae.
Previous work (24) and our findings (Fig. 1C) strongly suggested that the positive effect of bicarbonate on ToxT activity was the result of a direct interaction, but this had not been confirmed. ToxT protein levels are roughly equivalent in the presence or absence of bicarbonate (24); therefore, ToxT is thought to be in a less active state in the absence of bicarbonate. The simplest mechanism for bicarbonate activation of ToxT would be a direct interaction between bicarbonate and ToxT within the bacterial cytosol. To determine whether such a direct interaction between bicarbonate and ToxT is possible, we performed an NaH14CO3 uptake assay to measure bicarbonate import into the V. cholerae cell. First, classical biotype V. cholerae was subcultured statically in AKI medium for 2 h in the absence of bicarbonate. 14C-labeled bicarbonate then was added to the culture. Aliquots were centrifuged at 15-min intervals and washed with AKI medium three times, and 14C radiolabel in the cell pellet was quantified using a scintillation counter.
Results of the bicarbonate uptake assay displayed a linear increase of radiolabel within the cell pellet of the culture (Fig. 2). Linear regression of the data set revealed an equation of the best-fit line of y = 27.46x + 525, with an R2 value of 0.984. The results from the time course uptake show that bicarbonate is entering the bacteria at a linear rate. Further work involving the fractionation of the V. cholerae cytosol from the periplasm/membranes revealed that 84.2% ± 3.3% of the pellet-associated radiolabeled bicarbonate was located in the cytosol. This finding suggests that bicarbonate can interact directly with ToxT in the bacterial cytosol.
FIG 2.
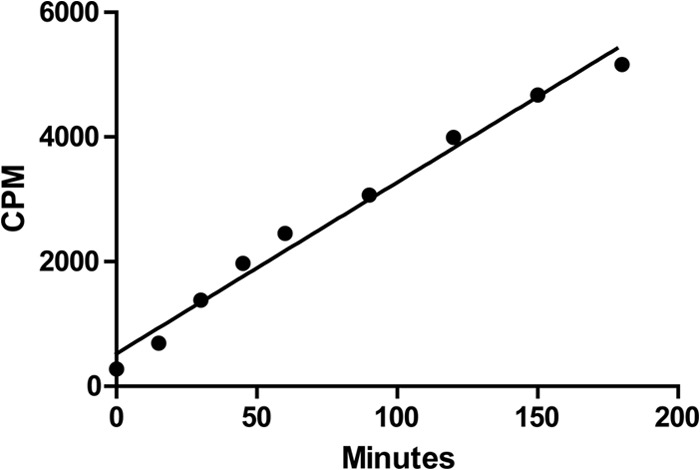
Radiolabeled bicarbonate is taken up by V. cholerae. Classical strain O395 cells were grown in AKI media in the absence of bicarbonate for 2 h. NaH14CO3 was added at time zero, and uptake was analyzed every 15 min by liquid scintillation counting. NaH14CO3 uptake was measured as cpm, reflecting the amount of radioactive 14C found within V. cholerae culture. Error bars represent standard deviations from three separate experiments.
Bicarbonate increases ToxT equilibrium binding affinity to PtcpA.
The simplest mechanism for a direct interaction between bicarbonate and ToxT that results in increased transcription activation by ToxT would be increased occupancy of toxboxes by ToxT, leading to enhanced interactions between ToxT and RNA polymerase. In the murine pathogen Citrobacter rodentium, an interaction between bicarbonate and another AraC/XylS protein family member, RegA, leads to the stabilization of RegA binding to the grlA promoter (35, 36). To determine whether bicarbonate directly affects ToxT binding to its specific DNA sites, we characterized the binding pattern of ToxT to virulence gene promoters using EMSA. The assays were designed to establish an estimation of the equilibrium dissociation constant, KD, to quantify the interaction of ToxT with the promoter of ToxT-dependent virulence genes in the absence and presence of bicarbonate. Here, KD represents the concentration of ToxT needed to bind 50% of the promoter DNA at equilibrium in the EMSA. Mathematically speaking, there is an inverse relationship between KD and binding affinity, meaning the lower the KD, the higher the binding affinity of ToxT to the DNA.
To determine the KD of the interaction between ToxT and PtcpA, we titrated purified MBP-ToxT, in the presence or absence of 36 mM bicarbonate, with a constant concentration of 32P-labeled PtcpA PCR product below the empirically estimated KD. MBP-ToxT was used because the independently folded MBP helps to solubilize ToxT and prevent aggregation during EMSA. The percentage of bound DNA at each concentration of MBP-ToxT was calculated using densitometry of the EMSA autoradiographs with ImageJ software. Curve fitting was performed with GraphPad software using the following equation: % bound = Bmax × [protein]h/(KDh + [protein]h), with the Bmax constraint set to 100%. This constraint represents all free DNA being bound by ToxT. A representative EMSA for PtcpA is shown in Fig. 3A. Binding reactions that correspond to lanes 1 to 7 were allowed to equilibrate in the absence of bicarbonate, while binding reactions shown in lanes 8 to 14 included bicarbonate. The percentage of bound DNA was calculated and curves were fit for both sets of reactions (Fig. 3B). With the addition of bicarbonate, the concentration of MBP-ToxT to bind 50% of PtcpA was 145.3 nM, significantly lower than the 287.9 nM KD without bicarbonate. The lower KD in the presence of bicarbonate indicates a higher binding affinity for the tcpA promoter. Interestingly, the Hill coefficient of the binding curves is greater than one, and the curves take on a sigmoidal shape. This is consistent with the possibility that there are multiple binding sites for ToxT and there is a degree of cooperativity in binding both sites. The presence of two toxboxes at PtcpA has been shown previously (19), but further work would be needed for the confirmation of cooperativity between ToxT monomers. The increase in binding affinity in the presence of bicarbonate in an in vitro EMSA indicates a proximal relationship between the bicarbonate ion and ToxT and likely a direct interaction.
FIG 3.

Bicarbonate increases binding affinity of MBP-ToxT to PtcpA. MBP-ToxT binding to PtcpA was analyzed using EMSA. Autoradiograph of EMSAs presented are representative of three or more independent experiments. (A) Binding reactions between MBP-ToxT and PtcpA in lanes 1 to 7 were conducted in the absence of NaHCO3. Lanes 8 to 14 were incubated in the presence of 36 mM NaHCO3. Lanes 1 and 8 contained PtcpA DNA in the absence of MBP-ToxT. Subsequent lanes contained a titration of MBP-ToxT with the indicated concentrations. (B) Binding curve for the autoradiograph shown in panel A. The densitometry of the autoradiograph was performed with ImageJ software. Circles represent percent PtcpA bound by MBP-ToxT in the absence of bicarbonate. The solid line corresponds to the binding curve for MBP-ToxT to PtcpA, determined by the equation % bound = Bmax × [protein]h/(KDh + [protein]h), with the Bmax constraint set to 100 using GraphPad Prism 5 software. Squares and the dashed line represent percent bound and binding curve, respectively, in the presence of bicarbonate. The KD for each condition is inset, and significant difference between the best-fit values of each data set is denoted by an asterisk (P < 0.00025). (C) Autoradiograph of EMSA showing titration of MBP-ToxT bound to PtcpA at pH 7.4 (lanes 1 to 7) and pH 8.6 (lanes 8 to 14). (D) Binding curves of each pH condition with KD. (E) Autoradiograph of EMSA showing titration of MBP-AraC bound to PBAD in the absence of NaHCO3 (lanes 1 to 7) and presence of 36 mM NaHCO3 (lanes 8 to 14). (F) Binding curves of MBP-AraC in the absence and presence of 36 mM NaHCO3.
The addition of bicarbonate to the binding reaction increases the pH from 7.4 to 8.6. To confirm that the increase in binding affinity is due to the bicarbonate ion itself and not an increase in pH, we performed an EMSA in which binding reactions at pH 7.4 (Fig. 3C, lanes 1 to 7) and 8.6 (Fig. 3C, lanes 8 to 14) were compared in the absence of bicarbonate. Densitometric analysis and nonlinear regression provided the binding curves and KD values for the EMSA (Fig. 3D). The binding curves at the two pH values overlapped, and the KD values for each curve were not statistically different. The KD of these two conditions were lower than those shown in Fig. 3A, because the concentration of active ToxT protein varies among batches and decreases over time. However, the overall response to the different binding conditions remains similar in different purified protein batches.
Both ToxT and RegA, transcriptional regulators of the AraC/XylS family, exhibit increased binding in the presence of bicarbonate (35, 36). We next analyzed whether another family member, AraC, showed signs of stabilized promoter architecture in the presence of bicarbonate to assess whether this was a common feature shared among the entire AraC family. Our results indicate that the addition of bicarbonate inhibited binding of AraC to the araBAD promoter (Fig. 3E and F). Therefore, not all AraC/XylS family members are positively modulated by bicarbonate.
Other small effector molecules do not increase ToxT binding to PtcpA.
The results described above suggest that the addition of bicarbonate to binding reactions increases the binding affinity of ToxT to PtcpA. However, it is possible that this phenomenon is not limited to bicarbonate specifically and also could be mediated by other small molecules similar to bicarbonate. We next tested the binding response of ToxT to molecules with structure or molecular weight similar to that of bicarbonate. Sodium biselenite has a structure similar to that of bicarbonate with a different central atom. ToxT had a greatly reduced binding affinity in the presence of biselenite (Fig. 4A and B). We also tested sodium acetate, which has a molecular weight similar to that of sodium bicarbonate. ToxT also showed decreased binding to PtcpA with the addition of this small molecule (Fig. 4C and D). The increase in binding affinity of ToxT to PtcpA appears to be bicarbonate specific, as other small molecules do not enhance ToxT binding or activity (data not shown).
FIG 4.

Small molecules, biselenite and acetate, do not enhance MBP-ToxT binding to PtcpA. The autoradiograph of EMSA shows binding reactions of MBP-ToxT to PtcpA with different small molecules similar to those of NaHCO3. Binding reactions in the absence and presence of 36 mM NaHSeO3 (A) with corresponding binding curves (B). Binding reactions in the absence and presence of 36 mM NaC2H3O2 (C) along with corresponding binding curves (D).
Bicarbonate increases binding to ToxT-dependent promoters regardless of toxbox configuration.
EMSA equilibrium binding experiments indicated that bicarbonate increases ToxT binding affinity for PtcpA. However, there is significant diversity in the configuration and spacing of toxboxes at different ToxT-activated promoters (19). ToxT directly controls the transcription of several other virulence genes besides tcpA, including ctxAB, aldA, tagA, acfA, acfD, and tcpI (17–19). The tcpA promoter contains two toxboxes in a direct repeat configuration. PctxAB, like PtcpA, also is configured as two direct repeat toxboxes (Fig. 1A). However, the orientation of the toxboxes is in the opposite direction relative to the promoter at PctxAB and the spacing between toxboxes differs between PtcpA and PctxAB (19, 33, 37). In contrast to PtcpA and PctxAB, PtagA, PacfA, PacfD, and PtcpI each contain a pair of toxboxes in an inverted repeat configuration and with variations in spacing between toxboxes (Fig. 1B). Finally, PaldA contains a single toxbox (Fig. 1A). As described earlier, the addition of bicarbonate to V. cholerae cultures increases activity from all of these promoters (Fig. 1C).
To determine whether bicarbonate affects the binding affinity of ToxT for each of these unique promoters, we performed EMSAs and calculated KD as previously described for PtcpA. First, we tested the ability of bicarbonate to increase the binding affinity of ToxT to PctxAB, which controls the production of CT and has two direct repeat toxboxes. Figure 5A shows the autoradiograph of ToxT binding to PctxAB in the absence and presence of bicarbonate. Densitometry and linear regression revealed that the KD is reduced from 328.7 nM MBP-ToxT to 231.4 nM with bicarbonate, indicating an increase in binding affinity. We next investigated if bicarbonate increased ToxT binding at the inverted repeat promoter of tagA. We used PtagA as an example of inverted repeat toxbox configurations that also occur in the promoters of acfA, acfD, and tcpI. As with the direct repeat promoters, bicarbonate increased the binding affinity of ToxT to PtagA (Fig. 5C and D). Finally, we performed EMSAs to determine if bicarbonate increased binding to the single toxbox promoter of aldA. The EMSA with binding reactions of MBP-ToxT and PaldA in the absence and presence of bicarbonate is shown in Fig. 5E. The binding curve was developed and revealed that bicarbonate decreased the KD for ToxT binding to PaldA from 173.7 nM to 113.9 nM. The increase in binding affinity to each of the promoters with bicarbonate agreed with the results of the analysis of each promoter in the β-galactosidase assays. Together, these data demonstrate that bicarbonate increases binding at each promoter regardless of toxbox orientation, including a single toxbox promoter, and reveals that the mechanism by which bicarbonate enhances ToxT activity most likely is a change in protein conformation, resulting in increased DNA binding affinity.
FIG 5.

Bicarbonate increases ToxT binding affinity to promoters having all known toxbox configurations. Each autoradiograph is a representative of three or more independent experiments. Binding reactions shown in lanes 1 to 7 in all autoradiographs were conducted in the absence of NaHCO3. Binding reactions shown in lanes 8 to 14 in all autoradiographs were conducted in the presence of 36 mM NaHCO3. All binding curves were generated using GraphPad Prism 5 software using the equation % bound = Bmax × [protein]h/(KDh + [protein]h), with the Bmax constraint set to 100. (A) Autoradiograph of EMSA showing MBP-ToxT binding to PctxAB, in which two toxboxes are oriented as direct repeats. (B) Representative binding curves. (C) Autoradiograph of EMSA showing MBP-ToxT binding to PtagA, in which two toxboxes are oriented as inverted repeats. (D) Binding curves corresponding to MBP-ToxT binding to PtagA. (E) Autoradiograph of EMSA showing MBP-ToxT binding to the single toxbox promoter, PaldA. (F) Binding curves of PaldA from EMSA results.
DISCUSSION
The regulatory network that controls the production of the major virulence factors responsible for disease symptoms of cholera culminates with the production of ToxT. ToxT is the transcription regulator directly responsible for the activation of tcpA and ctxAB transcription (13–16). Previous studies have shown that bicarbonate, a signal located in the upper small intestine where V. cholerae colonizes, enhances the ability of ToxT to activate these major virulence genes (24). In addition to its effect on these genes, we have shown that bicarbonate increases the transcription of other ToxT-dependent virulence genes, aldA, tagA, acfA, acfD, and tcpI.
ToxT activity is modulated by different effector molecules. It is negatively regulated by bile and its UFA components (22, 23), as well as the small molecule virstatin (28, 38). Bile is located at high concentrations in the lumen of the upper small intestine, and the interaction between the UFAs of bile and ToxT are thought to decrease ToxT's ability to dimerize effectively and bind to DNA (27, 29, 30). Our findings have shown that bicarbonate, which is also present at high concentrations in the upper small intestine, has a mechanism that operates conversely to that for bile. Our results indicate that bicarbonate-mediated activation of virulence gene transcription occurs as a result of an increase in ToxT binding affinity to virulence gene promoters. The increase in binding affinity in response to bicarbonate can be seen at each ToxT-dependent promoter regardless of toxbox configuration and positioning.
The binding curves generated from EMSAs performed on different ToxT-dependent promoters were sigmoidal. Sigmoidal binding curves generally relate to cooperative binding to multiple binding sites of a protein, similar to O2 binding by hemoglobin (39). Cooperativity occurs when one bound molecule increases the binding affinity of subsequent molecules. Positive cooperativity also can be seen in terms of transcription factors binding to promoters with multiple binding sites. One example of positive cooperativity of transcription factors is seen with the TtgR operator of Pseudomonas putida strain DOT-T1E (40). TtgR, which can form dimers in solution, exhibits biphasic cooperative binding at this promoter (40). ToxT is thought to have optimal activity when acting as a dimer (27–31). In contrast, it also has been shown that ToxT can bind DNA as a monomer (18, 19, 26). The sigmoidal ToxT binding curve at two toxbox promoters (Fig. 3B and 5B and D) suggests that a single ToxT monomer binds the DNA first, coinciding with previous findings. Additionally, the possibility of positive cooperativity derived from the sigmoidal binding curve and Hill coefficient implies that the binding of the first monomer increases the binding affinity of the second monomer. This can be explained by an increase in the incidence of association between ToxT monomers after one has already bound the promoter, increasing the binding affinity of the second ToxT monomer. Interestingly, the ToxT binding curve of the single toxbox promoter of aldA also exhibited a sigmoidal shape. Previous studies have shown that this promoter contained a single toxbox that was necessary and sufficient for full transcription activation (17); however, the ToxT binding curve at this promoter opens the possibility that another, atypical and low-affinity, ToxT binding site is present in the vicinity of the promoter. Further work is needed to determine the role of dimerization and cooperativity at each of these promoters.
Potential mechanisms for the increased ToxT DNA binding affinity in response to bicarbonate include a change in conformation of ToxT or increased frequency of ToxT dimerization. Indirect mechanisms that increase ToxT binding can be ruled out, as the increase in binding was seen in binding experiments using purified components. Work on an AraC/XylS family member of C. rodentium, RegA, revealed that bicarbonate increases RegA binding to promoter regions (35, 36). RegA can act as a monomer or dimer, similar to ToxT, and it was shown that bicarbonate did not increase the incidence of RegA dimerization (41). As a primary assessment of ToxT dimerization in the presence of bicarbonate, we analyzed the ability of bicarbonate to increase ToxT binding to the single toxbox of PaldA, in which dimerization should not be evident. However, as stated before, our findings revealed the possibility of more than one toxbox in or near the promoter of aldA. Consequently, further investigation into the precise mechanism of increased ToxT binding affinity in the presence of bicarbonate is needed.
Bicarbonate must first enter the bacterium to interact directly with ToxT. We report here that bicarbonate does get taken up into the bacterial cytosol. However, further investigation into the mechanism of uptake is needed. Previous work has shown that the addition of the carbonic anhydrase inhibitor ethoxzolamide (EZA) decreases activation of PtcpA in the presence of bicarbonate (24). V. cholerae contains genes encoding each of the three classes of bacterial carbonic anhydrase. A triple carbonic anhydrase deletion did not reveal a decrease in H14CO3 uptake compared to the level for WT O395 (data not shown). This suggests that the mechanism for uptake is not completely reliant on the carbonic anhydrase-dependent conversion of HCO3− to CO2.
Signals that V. cholerae encounters during passage to the intestine result in the production of ToxT (4), but due to the presence of a high concentration of bile and UFAs in the lumen of the intestine, ToxT initially is in an inactive form (22, 23, 27, 30). Bicarbonate also is present in the lumen where bile is present and could be activating some ToxT protein. The bacteria in the lumen still express flagella and undergo positive chemotaxis toward mucin proteins (1, 42). As the bacteria swim closer to the intestinal epithelium, the concentration of bile decreases, as the large molecules that comprise bile cannot enter the mucus layer. The concentration of bicarbonate increases as the bacteria get closer to the epithelium due to the direct secretion of bicarbonate from these cells (32), but more importantly, the ratio of bicarbonate to UFAs increases. ToxT protein becomes active as it reaches the high bicarbonate/low UFA breakpoint, and active ToxT increases transcription of genes involved in virulence, such as CT and TCP (24). In addition to these major virulence factors, bicarbonate activates accessory virulence factors important in disease progression. Our findings here reveal that the mechanism for bicarbonate-mediated enhancement of ToxT activity is due to an increase in ToxT binding affinity to virulence gene promoters. This provides a direct relationship between an intestinal signal and increased expression of genes required for pathogenesis.
ACKNOWLEDGMENTS
We thank the members of the Withey and Neely laboratories for helpful discussions.
This work was supported by PHS grants K22AI071011 and R56AI093622 (to J.H.W.) and by Wayne State University funds.
Footnotes
Published ahead of print 2 September 2014
REFERENCES
- 1.Butler SM, Camilli A. 2005. Going against the grain: chemotaxis and infection in Vibrio cholerae. Nat. Rev. Microbiol. 3:611–620. 10.1038/nrmicro1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krukonis ES, DiRita VJ. 2003. From motility to virulence: sensing and responding to environmental signals in Vibrio cholerae. Curr. Opin. Microbiol. 6:186–190. 10.1016/S1369-5274(03)00032-8. [DOI] [PubMed] [Google Scholar]
- 3.Peterson KM, Mekalanos JJ. 1988. Characterization of the Vibrio cholerae ToxR regulon: identification of novel genes involved in intestinal colonization. Infect. Immun. 56:2822–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matson JS, Withey JH, DiRita VJ. 2007. Regulatory networks controlling Vibrio cholerae virulence gene expression. Infect. Immun. 75:5542–5549. 10.1128/IAI.01094-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kovacikova G, Skorupski K. 2001. Overlapping binding sites for the virulence gene regulators AphA, AphB and cAMP-CRP at the Vibrio cholerae tcpPH promoter. Mol. Microbiol. 41:393–407. 10.1046/j.1365-2958.2001.02518.x. [DOI] [PubMed] [Google Scholar]
- 6.Kovacikova G, Lin W, Skorupski K. 2004. Vibrio cholerae AphA uses a novel mechanism for virulence gene activation that involves interaction with the LysR-type regulator AphB at the tcpPH promoter. Mol. Microbiol. 53:129–142. 10.1111/j.1365-2958.2004.04121.x. [DOI] [PubMed] [Google Scholar]
- 7.Kovacikova G, Skorupski K. 1999. A Vibrio cholerae LysR homolog, AphB, cooperates with AphA at the tcpPH promoter to activate expression of the ToxR virulence cascade. J. Bacteriol. 181:4250–4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Behari J, Stagon L, Calderwood SB. 2001. pepA, a gene mediating pH regulation of virulence genes in Vibrio cholerae. J. Bacteriol. 183:178–188. 10.1128/JB.183.1.178-188.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kovacikova G, Lin W, Skorupski K. 2010. The LysR-type virulence activator AphB regulates the expression of genes in Vibrio cholerae in response to low pH and anaerobiosis. J. Bacteriol. 192:4181–4191. 10.1128/JB.00193-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krukonis ES, Yu RR, Dirita VJ. 2000. The Vibrio cholerae ToxR/TcpP/ToxT virulence cascade: distinct roles for two membrane-localized transcriptional activators on a single promoter. Mol. Microbiol. 38:67–84. 10.1046/j.1365-2958.2000.02111.x. [DOI] [PubMed] [Google Scholar]
- 11.Yang M, Liu Z, Hughes C, Stern AM, Wang H, Zhong Z, Kan B, Fenical W, Zhu J. 2013. Bile salt-induced intermolecular disulfide bond formation activates Vibrio cholerae virulence. Proc. Natl. Acad. Sci. U. S. A. 110:2348–2353. 10.1073/pnas.1218039110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan F, Liu Z, Jabeen N, Birdwell LD, Zhu J, Kan B. 2014. Enhanced Interaction of Vibrio cholerae virulence regulators TcpP and ToxR under oxygen-limiting conditions. Infect. Immun. 82:1676–1682. 10.1128/IAI.01377-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hulbert RR, Taylor RK. 2002. Mechanism of ToxT-dependent transcriptional activation at the Vibrio cholerae tcpA promoter. J. Bacteriol. 184:5533–5544. 10.1128/JB.184.20.5533-5544.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu RR, DiRita VJ. 1999. Analysis of an autoregulatory loop controlling ToxT, cholera toxin, and toxin-coregulated pilus production in Vibrio cholerae. J. Bacteriol. 181:2584–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Champion GA, Neely MN, Brennan MA, DiRita VJ. 1997. A branch in the ToxR regulatory cascade of Vibrio cholerae revealed by characterization of toxT mutant strains. Mol. Microbiol. 23:323–331. 10.1046/j.1365-2958.1997.2191585.x. [DOI] [PubMed] [Google Scholar]
- 16.DiRita VJ, Parsot C, Jander G, Mekalanos JJ. 1991. Regulatory cascade controls virulence in Vibrio cholerae. Proc. Natl. Acad. Sci. U. S. A. 88:5403–5407. 10.1073/pnas.88.12.5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Withey JH, Dirita VJ. 2005. Vibrio cholerae ToxT independently activates the divergently transcribed aldA and tagA genes. J. Bacteriol. 187:7890–7900. 10.1128/JB.187.23.7890-7900.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Withey JH, DiRita VJ. 2005. Activation of both acfA and acfD transcription by Vibrio cholerae ToxT requires binding to two centrally located DNA sites in an inverted repeat conformation. Mol. Microbiol. 56:1062–1077. 10.1111/j.1365-2958.2005.04589.x. [DOI] [PubMed] [Google Scholar]
- 19.Withey JH, DiRita VJ. 2006. The toxbox: specific DNA sequence requirements for activation of Vibrio cholerae virulence genes by ToxT. Mol. Microbiol. 59:1779–1789. 10.1111/j.1365-2958.2006.05053.x. [DOI] [PubMed] [Google Scholar]
- 20.Bradley ES, Bodi K, Ismail AM, Camilli A. 2011. A genome-wide approach to discovery of small RNAs involved in regulation of virulence in Vibrio cholerae. PLoS Pathog. 7:e1002126. 10.1371/journal.ppat.1002126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Richard AL, Withey JH, Beyhan S, Yildiz F, DiRita VJ. 2010. The Vibrio cholerae virulence regulatory cascade controls glucose uptake through activation of TarA, a small regulatory RNA. Mol. Microbiol. 78:1171–1181. 10.1111/j.1365-2958.2010.07397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chatterjee A, Dutta PK, Chowdhury R. 2007. Effect of fatty acids and cholesterol present in bile on expression of virulence factors and motility of Vibrio cholerae. Infect. Immun. 75:1946–1953. 10.1128/IAI.01435-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schuhmacher DA, Klose KE. 1999. Environmental signals modulate ToxT-dependent virulence factor expression in Vibrio cholerae. J. Bacteriol. 181:1508–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abuaita BH, Withey JH. 2009. Bicarbonate induces Vibrio cholerae virulence gene expression by enhancing ToxT activity. Infect. Immun. 77:4111–4120. 10.1128/IAI.00409-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Higgins DE, Nazareno E, DiRita VJ. 1992. The virulence gene activator ToxT from Vibrio cholerae is a member of the AraC family of transcriptional activators. J. Bacteriol. 174:6974–6980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bellair M, Withey JH. 2008. Flexibility of Vibrio cholerae ToxT in transcription activation of genes having altered promoter spacing. J. Bacteriol. 190:7925–7931. 10.1128/JB.00512-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prouty MG, Osorio CR, Klose KE. 2005. Characterization of functional domains of the Vibrio cholerae virulence regulator ToxT. Mol. Microbiol. 58:1143–1156. 10.1111/j.1365-2958.2005.04897.x. [DOI] [PubMed] [Google Scholar]
- 28.Shakhnovich EA, Hung DT, Pierson E, Lee K, Mekalanos JJ. 2007. Virstatin inhibits dimerization of the transcriptional activator ToxT. Proc. Natl. Acad. Sci. U. S. A. 104:2372–2377. 10.1073/pnas.0611643104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lowden MJ, Skorupski K, Pellegrini M, Chiorazzo MG, Taylor RK, Kull FJ. 2010. Structure of Vibrio cholerae ToxT reveals a mechanism for fatty acid regulation of virulence genes. Proc. Natl. Acad. Sci. U. S. A. 107:2860–2865. 10.1073/pnas.0915021107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Childers BM, Cao X, Weber GG, Demeler B, Hart PJ, Klose KE. 2011. N-terminal residues of the Vibrio cholerae virulence regulatory protein ToxT involved in dimerization and modulation by fatty acids. J. Biol. Chem. 286:28644–28655. 10.1074/jbc.M111.258780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Childers BM, Weber GG, Prouty MG, Castaneda MM, Peng F, Klose KE. 2007. Identification of residues critical for the function of the Vibrio cholerae virulence regulator ToxT by scanning alanine mutagenesis. J. Mol. Biol. 367:1413–1430. 10.1016/j.jmb.2007.01.061. [DOI] [PubMed] [Google Scholar]
- 32.Hogan DL, Ainsworth MA, Isenberg JI. 1994. Review article: gastroduodenal bicarbonate secretion. Aliment. Pharmacol. Ther. 8:475–488. [DOI] [PubMed] [Google Scholar]
- 33.Dittmer JB, Withey JH. 2012. Identification and characterization of the functional toxboxes in the Vibrio cholerae cholera toxin promoter. J. Bacteriol. 194:5255–5263. 10.1128/JB.00952-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 35.Tauschek M, Yang J, Hocking D, Azzopardi K, Tan A, Hart E, Praszkier J, Robins-Browne RM. 2010. Transcriptional analysis of the grlRA virulence operon from Citrobacter rodentium. J. Bacteriol. 192:3722–3734. 10.1128/JB.01540-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang J, Hart E, Tauschek M, Price GD, Hartland EL, Strugnell RA, Robins-Browne RM. 2008. Bicarbonate-mediated transcriptional activation of divergent operons by the virulence regulatory protein, RegA, from Citrobacter rodentium. Mol. Microbiol. 68:314–327. 10.1111/j.1365-2958.2008.06171.x. [DOI] [PubMed] [Google Scholar]
- 37.Yu RR, DiRita VJ. 2002. Regulation of gene expression in Vibrio cholerae by ToxT involves both antirepression and RNA polymerase stimulation. Mol. Microbiol. 43:119–134. 10.1046/j.1365-2958.2002.02721.x. [DOI] [PubMed] [Google Scholar]
- 38.Hung DT, Shakhnovich EA, Pierson E, Mekalanos JJ. 2005. Small-molecule inhibitor of Vibrio cholerae virulence and intestinal colonization. Science 310:670–674. 10.1126/science.1116739. [DOI] [PubMed] [Google Scholar]
- 39.Goutelle S, Maurin M, Rougier F, Barbaut X, Bourguignon L, Ducher M, Maire P. 2008. The Hill equation: a review of its capabilities in pharmacological modelling. Fundam. Clin. Pharmacol. 22:633–648. 10.1111/j.1472-8206.2008.00633.x. [DOI] [PubMed] [Google Scholar]
- 40.Krell T, Teran W, Mayorga OL, Rivas G, Jimenez M, Daniels C, Molina-Henares AJ, Martinez-Bueno M, Gallegos MT, Ramos JL. 2007. Optimization of the palindromic order of the TtgR operator enhances binding cooperativity. J. Mol. Biol. 369:1188–1199. 10.1016/j.jmb.2007.04.025. [DOI] [PubMed] [Google Scholar]
- 41.Yang J, Dogovski C, Hocking D, Tauschek M, Perugini M, Robins-Browne RM. 2009. Bicarbonate-mediated stimulation of RegA, the global virulence regulator from Citrobacter rodentium. J. Mol. Biol. 394:591–599. 10.1016/j.jmb.2009.10.033. [DOI] [PubMed] [Google Scholar]
- 42.Allweiss B, Dostal J, Carey KE, Edwards TF, Freter R. 1977. The role of chemotaxis in the ecology of bacterial pathogens of mucosal surfaces. Nature 266:448–450. 10.1038/266448a0. [DOI] [PubMed] [Google Scholar]