

Abstract
Subway systems are indispensable for urban societies, but microbiological characteristics of subway aerosols are relatively unknown. Previous studies investigating microbial compositions in subways employed methodologies that underestimated the diversity of microbial exposure for commuters, with little focus on factors governing subway air microbiology, which may have public health implications. Here, a culture-independent approach unraveling the bacterial diversity within the urban subway network in Hong Kong is presented. Aerosol samples from multiple subway lines and outdoor locations were collected. Targeting the 16S rRNA gene V4 region, extensive taxonomic diversity was found, with the most common bacterial genera in the subway environment among those associated with skin. Overall, subway lines harbored different phylogenetic communities based on α- and β-diversity comparisons, and closer inspection suggests that each community within a line is dependent on architectural characteristics, nearby outdoor microbiomes, and connectedness with other lines. Microbial diversities and assemblages also varied depending on the day sampled, as well as the time of day, and changes in microbial communities between peak and nonpeak commuting hours were attributed largely to increases in skin-associated genera in peak samples. Microbial diversities within the subway were influenced by temperature and relative humidity, while carbon dioxide levels showed a positive correlation with abundances of commuter-associated genera. This Hong Kong data set and communities from previous studies conducted in the United States formed distinct community clusters, indicating that additional work is required to unravel the mechanisms that shape subway microbiomes around the globe.
INTRODUCTION
People in modern societies spend over 90% of their time indoors; thus, they are constantly exposed to contents present in this primary habitat (1). Indoor air consists of a myriad of solid aerosol particles, including inhalable bioaerosols, which recently have been the focus of scientific research because of their impacts on public health (2). Microbial causative agents of adverse health conditions have been documented in aerosols of different indoor built environments (3, 4), and such agents can be transmitted between individuals in close proximity. Therefore, characterizations of indoor microbial compositions will provide information regarding the nature and extent to which individuals in indoor settings are exposed to microbial life and the breadth of microbial life in which transmission can occur. Given its importance, microbial community assessments have been conducted in numerous indoor air and surface environments (3, 5–7).
Of the many indoor infrastructures, subways have become inseparable elements of urban centers. Global commuters spend sizable fractions of their times daily in this particular mode of transportation. Subway networks are present and are being built in many cities and are used regularly by an ever-increasing number of individuals. Therefore, the use of subways by commuters and their dependence on the subway are sure to be more widespread on a global scale for years to come. Unfortunately, the microbiological contents of the aerosol in this unique built environment are relatively unknown. The majority of current microbiological assessments of subway networks had been limited to conventional culture methods (8–17) or limited culture-independent methods (18, 19), which are likely to underestimate the true spectrum of microorganisms present. Culture-based studies are also inherently restrictive in the comparison of microbial communities between samples, as the organisms cultivated for analysis are dependent on the selective growth conditions chosen. As a result, these studies are limited to insensitive comparisons of microbial concentrations between samples, with crude analysis on the composition of microbial communities. While these cultivation-based studies demonstrated the roles ridership (10, 11, 17), time (18–20), and station location and design (8, 12, 13, 15) play in microbial concentrations and diversity in subway air, it is not known whether this difference is representative of the entire phylogenetic spectrum present given the methodologies employed. According to the only sequencing-based subway microbiome study conducted to date (21), subway air is made up predominantly of a small number of microbial phyla and families, and its community composition resembles that of outdoor air. While this study provides a more complete analysis of the subway microbiome, it is currently unknown whether the findings from the study can be extrapolated to subway networks of different regions around the globe, as different subway networks are architecturally distinct, and variations in geographic and demographic factors also may contribute to variations in microbiomes of built environments (22–24). Moreover, as all previous studies analyzed only air samples collected in stationary locations along platforms or concourses of a limited number of stations, the microbial communities described may not necessarily represent the repertoire of microorganisms a typical commuter is exposed to.
In this study, a comprehensive microbial community analysis of bioaerosols collected in numerous subway lines within the Hong Kong subway network, in addition to selected outdoor locations throughout the city, is described. Hong Kong is one of the most densely populated cities in the world (6,620 persons/km2), and the Hong Kong subway network is among the busiest in the world. To provide a more representative account of the breadth of microorganisms a subway passenger is exposed to, bioaerosols from subway rides rather than individual stations and concourse locations were collected to provide an integrated description of the microbial community of each sample. Using the Illumina sequencing technology targeting the 16S rRNA gene, communities from subway air collected in the different subway lines, times, and days are also compared.
MATERIALS AND METHODS
MTR characteristics and ventilation system.
The Hong Kong Mass Transit Railway (MTR) network is among the world's busiest subway system by patronage, with an annual ridership of approximately 1.5 billion (http://gia.info.gov.hk/general/201106/08/P201106080126_0126_79963.pdf; accessed January 2014). The network consists of 10 rail lines and 84 stations (at the time of manuscript preparation), and it spans 174.7 km of railroad tracks across the urban metropolis. Within the seven lines sampled, the stations and platforms of four lines (Tsuen Wan Line [TWL], Island Line [ILL], Kwun Tong Line [KTL], and Tseung Kwan O Line [TWOL]) are predominantly underground and indoor. The stations of these lines contain screen doors between the train tunnel and the platforms as security and safety measures (see Fig. S1 in the supplemental material). For the remaining three lines (East Rail Line [ERL], West Rail Line [WRL], Ma On Shan Line [MOSL]), the trains mostly travel outdoors and above ground. For these three outdoor lines, the station platforms in ERL and MOSL are open to ambient air, whereas screen doors are present in station platforms of the WRL. Trains are air conditioned, and a sophisticated ventilation system is required to ensure the efficient operation of the MTR in a subtropical climate, where heat-sink effects alone are not sufficient to cool down the subway environment (http://www.legco.gov.hk/yr10-11/chinese/panels/tp/tp_rdp/papers/tp_rdp0506cb1-2125-2-ec.pdf; accessed 10 March 2014). Briefly, the MTR ventilation system focuses on the underground tunnel and station areas. When piston effect is generated due to train movement across an underground tunnel, air is pushed out toward the forward ventilation shafts, while ambient air is drawn back into the tunnel at the rear of the train. When the train is stationary, the piston effect is absent and air is drawn in and out of the tunnel by intake and exhaust fans, respectively. Additional fans are present on platforms to replenish ambient air of the track area during prolonged stoppages at a station, where fresh air is supplied from below the platforms and air is extracted via ducts above the trains. Each station is installed with two (or more for stations with interchanges) such ventilation systems, one on each side of the platform. All underground lines are equipped with safety screen doors at station platforms, with the doors extending to the ceilings of the station platforms.
Experimental design and environmental parameters.
Seven lines on the Hong Kong subway network, ILL, TWL, KTL, ERL, WRL, TKOL, and MOSL, were included in this analysis. Sample collection was not performed on public holidays and days with extreme weather conditions, such as those with heavy rain and typhoon warnings (as issued by the Hong Kong Observatory), to minimize any possible influence these conditions have on microbial communities. For each line, four sampling times were selected: two during peak commute hours (07:45 to 09:45 and 17:45 to 19:45) and two during nonpeak hours (10:15 to 12:15 and 15:15 to 17:15). Samples also were classified as collected during morning (a.m.) hours (07:45 to 09:45 and 10:15 to 12:15) as well as afternoon and evening (p.m.) hours (15:15 to 17:15 and 17:45 to 19:45). Four replicates were performed for each line at the times indicated above, with each replicate performed over a span of an average of 15 days. Each sampling event on a line included air collected during entry into the paid zone, the platform environment, and inside the carriage. For each MTR sampling session, sampling began in the paid zone of the station concourse. Samplers boarded the first train arriving at the platform, disembarked after three stops, and waited by the same platform until the third train arrived onto the platform. The same procedure was repeated in one direction until the terminus station, and the samplers would board trains operating at the opposite direction and continue the sampling. After 2 h, the samplers exited the paid zone, and this marked the end of a sampling session. Thus, a sample represents a time-controlled and integrated assessment of potential microorganism exposure to simulate the typical commuting event of a passenger, combining air collected on concourse, train, and platform of a given line. In addition, outdoor air samples were collected at seven different locations on the ground level adjacent to one of the stations along the representative subway line. These locations were chosen so that each location is accessible from at least one of the seven sampled lines, and these locations are accessible regardless of the directionality of the train. A set of four replicates, each on a different day, was performed for each outdoor location (12:15 to 14:15) during the sampling of the respective lines. For all samples, environmental parameters, including temperature, relative humidity, and carbon dioxide (CO2) concentrations, were measured, logged, and recorded at 2-s intervals using the Q-Trak indoor air quality monitor (TSI Inc., Shoreview, MN, USA) over the course of samplings. Instruments were calibrated prior to use.
Sample collection.
Air samples were collected using Leland Legacy portable sample pumps (SKC Inc., Eighty Four, PA, USA), each at a flow rate of 9 liters/min for 2 h. Autoclaved cellulose nitrate filters (diameter, 25 mm; pore size, 0.2 μm; Whatman, Maidstone, United Kingdom) were used to collect air samples by impaction using a Sioutas Cascade Impactor (SKC Inc., Eighty Four, PA, USA) with a D-plate accelerator (collects particles with a diameter size of ≥0.25 μm). At any given sampling time, four pumps were in operation simultaneously, and a total of 4.32 m3 of air was collected for each sample. Prior to sampling, impactors were disassembled and irradiated with UV, rubber tubings were soaked in 3% sodium hypochlorite, and all openings were sealed to prevent air entering into the sampling system when apparatuses were not in use. Between sampling, filters were replaced using aseptic techniques, and the sampling apparatus, including impactors and other equipment used for handling the sampling apparatus, were sterilized with 70% ethanol and 3% sodium hypochlorite. Following sampling, filters were immediately stored at −80°C until genomic DNA (gDNA) extraction. Filters were processed within 1 month of storage at −80°C. During extraction, the four filters were combined into one DNA extraction reaction mixture in order to acquire sufficient biomass for downstream applications and analyses, as microbial loads in the atmosphere tend to be lower than those of samples obtained from other environments (25). Negative controls, including sterile filters not exposed to MTR/outdoor aerosols, and a no-filter control was included for extraction as well as PCR and sequencing.
Genomic DNA extraction.
Following sampling, cellulose nitrate filters were subjected to gDNA extraction using the PowerSoil DNA isolation kit (MO BIO Laboratories, Carlsbad, CA, USA) (25), with slight methodological modifications. Briefly, tubes containing filters and C1 lysis solution were incubated at 70°C for 10 min, followed by mechanical beating using a Mini-Bead Beater 16 (Biospec Products, Bartlesville, OK, USA) for 10 min. The remaining steps were performed according to the manufacturer's instructions. Eluted gDNA was sent to Health GeneTech Corporation (Taipei, Taiwan) for 16S rRNA gene amplification, sequence library construction, and sequencing.
PCR, library preparation, and 16S rRNA gene sequencing.
The 515f/806r primer pair was used to target and amplify the V4 hypervariable region of the 16S rRNA gene (26). The 16S region was selected because it provides a better resolution of bacterial phyla (27). PCR amplification was performed in a 20-μl reaction volume containing 10 μl 2× Phusion HF master mix (New England BioLabs, Ipswich, MA, USA), 0.5 μM each forward and reverse primer, consisting of customized barcodes present on both primers for multiplex sequencing, and 50 to 150 ng DNA template. The PCR conditions consisted of an initial 98°C for 30 s, followed by 30 cycles of 98°C for 10 s, 54°C for 30 s, and 72°C for 30 s, as well as a final extension of 72°C for 5 min. Positive amplification was verified next by agarose gel electrophoresis. Amplicons in triplicates were pooled and purified using AMPure XP beads (Agencourt, Brea, CA, USA) and quantified using a Qubit double-stranded DNA HS assay kit on a Qubit fluorometer (Invitrogen, Carlsbad, CA, USA), all according to the respective manufacturers' instructions. For sequencing the library preparation, Illumina adapters were attached to amplicons using the Illumina TruSeq DNA sample preparation kit, v3. Purified libraries were applied for cluster generation and sequencing on the Illumina MiSeq platform using paired-end 300-bp reads.
Sequence analysis.
FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit) and the QIIME pipeline (v.1.8.0) (28) were used to process the raw sequences, and sequence chimera filtering was performed with ChimeraSlayer via the QIIME “parallel_identify_chimeric_seqs.py” script. Nonchimeric sequences with a minimum acceptable Phred quality score of 20 in terminal bases and ≥20 for 70% of their length were retained for downstream analysis. Following quality filtering and trimming, sequences shorter than 100 bp were removed. The forward and reverse reads gave similar results, and the reverse reads were used for analysis. Unless otherwise described, data and statistical analyses were performed using R and Perl scripts. High-quality sequences were clustered into operational taxonomic units (OTUs) against the Greengenes rRNA gene sequence database (ftp://greengenes.microbio.me/greengenes_release/gg_13_5/gg_13_8_otus.tar.gz; 97% rep set, filtered to remove sequences without taxonomic information down to the genus level, total of 35,435 sequences retained in the database; retrieved 27 December 2013) using the UCLUST-based open-reference OTU clustering pipeline implemented in QIIME's “pick_open_reference_otus.py” script, with a 97% sequence identity cutoff (28–30). Sequences with <97% identity to database sequences were allowed to form de novo clusters with no taxonomic classification. As part of the QIIME pipeline, a preclustering filtering step is used such that data sequences below 60% identity to the reference data set are removed. Global singleton OTUs were removed, and in downstream steps requiring relative abundances, OTU proportions were standardized to the total number of high-quality reads. For downstream steps requiring phylogenetic tree construction (Faith's phylogenetic diversity [FPD], UniFrac distances), representative sequences for OTU clusters were selected based on the most abundant sequence within each cluster and aligned against the Greengenes reference alignment using PyNAST (31), as implemented in the QIIME script “align_seqs.py”.
To generate rarefaction curves for each of the three α-diversity matrices (observed OTUs, FPD, and Chao1) per sample, 10 increments of sampling depth between 10 and 35,640 (corresponding to the median depth across 139 samples) were selected, and for each increment, the averages of 10 repeated richness measurements were plotted, as implemented in the QIIME script “alpha_rarefaction.py”. As the sample with the fewest sequences had 7,210 reads (the third a.m. peak sample on the West Rail Line, WRLAMP3), samples were normalized at this sequence depth for subsequent α- and β-diversity measurements. For α-diversity, taxonomic (based on the number of OTUs present), phylogenetic (FPD) (32), and singleton-based (Chao1) (33) richness measurements were computed. For β-diversity, both the unweighted and abundance-weighted UniFrac distances were computed to compare phylogenetic dissimilarities of detected communities between sample types (34, 35). UniFrac distance matrices were used to construct principal coordinate analysis (PCoA) plots using the R package vegan (http://vegan.r-forge.r-project.org/). The analysis of similarities (ANOSIM) test was performed in PRIMER (v.6; PRIMER-E, Plymouth, United Kingdom) to determine whether the major sample factors (MTR line, MTR line type, indoors/outdoors, time, peak/nonpeak, sampling day) had significantly different microbial communities. UniFrac distance calculations, PCoA, and ANOSIM were also performed to compare microbial communities from our study to indoor samples collected from air and surface samples from previous reports (5, 36). These reports were selected because they targeted the same V4 16S rRNA gene region using the Illumina sequencing platform. Sequences from all three studies were clustered against the filtered Greengenes database in addition to de novo sequences from the current study, based on 97% sequence identity. Sequences from the other studies which did not cluster were placed into singleton clusters. The similarity percentage (SIMPER) algorithm was run in PRIMER to identify major bacterial genera contributing to any community compositional differences detected. To examine potential relationships between communities in the MTR lines and corresponding outdoor locations, the Bayesian SourceTracker algorithm (37) was employed, with MTR samples designated sinks and outdoor samples designated sources.
Statistical analysis.
Comparisons of environmental measurements between two sample types were performed using the nonparametric Mann-Whitney test, and a Kruskal-Wallis test was employed for comparisons of more than two sample types. Pearson's correlation coefficient was computed to determine significant correlations between environmental conditions (temperature, relative humidity, and CO2 levels) and relative abundance of bacterial taxa (only OTUs with average relative abundance of >1% were considered; a significance threshold of P < 0.05 with Bonferroni correction was done during comparison). The Mann-Whitney test was used for comparisons for significant differences in OTU richness between two sample types (e.g., a.m. versus p.m., peak versus nonpeak, underground versus above ground, etc.), and the Kruskal-Wallis one-way analysis of variance was used to detect significant OTU richness differences between at least three sample types (e.g., subway lines and outdoor locations). Line connectedness was calculated using a distance matrix, where distance between two lines sharing interchange stations (i.e., connected lines) is represented by 1/x, where x is the number of interchange stations shared and an integer for distance in the smallest number of stops and/or line change between nonconnected lines with no interchange station. A Mantel test (significance determined with 999 random permutations) was performed to examine correlations between the connectedness of subway lines and abundance-weighted UniFrac distances of their communities.
Sequence read accession numbers.
A total of 6,591,522 raw sequences have been deposited in the NCBI Sequence Read Archive under BioProject accession number PRJNA230428 and study accession number SRP039009.
RESULTS
Overview of the subway bioaerosol microbiome.
A total of 140 samples were collected on the MTR network and selected corresponding outdoor locations (Fig. 1). One sample (ILLAMP2) yielded a low number of reads (<1,000) and was removed from downstream analysis. Following quality filtering of the remaining 139 samples, a total of 5,456,084 high-quality sequences were clustered into 55,703 unique operational taxonomic units (OTUs). On average, 39,252 reads (from 7,210 to 113,729 reads) contributed to 2,473 OTUs (from 696 to 6,955 OTUs) in each sample. To account for differences in sequencing depth, rarefaction of 7,210 reads per sample was performed for subsequent analysis. The numbers of sequences and OTUs for each sample are listed in Table S1 in the supplemental material. Approximately 48% of sequences shared over 97% identity to microbial sequences in the Greengenes database (Fig. 2), and over 45% of the sequences formed de novo OTU clusters, a proportion similar to that of an urban outdoor aerosol study (38). The remaining 7% of the sequences were singletons removed from subsequent analysis. As documented in other urban environments (39, 40), archaeal sequences consisted of the Euryarchaeota and Crenarchaeota phyla and constituted a minimal portion (<0.1%) of the assigned reads. Proteobacteria (average prevalence of 23.4% across 139 samples), Actinobacteria (15.6%), Firmicutes (6.3%), and Deinococcus-Thermus (1.6%) were the most prevalent bacterial phyla detected, similar to the findings of Robertson et al. (21), with the exception of Bacteroidetes, seen in New York City (NYC), instead of Deinococcus-Thermus. These bacterial phyla also are among the most commonly detected in aerosols of other indoor built environments (39, 41). Skin-associated genera Micrococcus (4.9%), Enhydrobacter (3.1%), Propionibacterium (2.9%), Staphylococcus (1.8%), and Corynebacterium (1.5%) were among the most commonly detected genera in our data set (see Table S2 in the supplemental material). In addition, the soil-associated Sphingobium and freshwater-dwelling Blastomonas also were among the most common genera detected, with generally higher abundances in outdoor samples for both genera. Despite the general trends described, distinctive and unique communities could be observed for some samples, similar to observations reported by Robertson et al. (21). The fourth sample taken in the Po Lam outdoor location (PL4) contained high proportions of sequences belonging to the plant-associated Clostridium cellulovorans (45.2% versus and average of 0.28% for the other samples) and the marine genus Synechococcus (6.5% versus an average of 0.056% for other samples), and the first p.m. nonpeak sample on the Ma On Shan Line (MOSLPMNP1) contained a high proportion of Leuconostoc sequences (54.4% versus an average of 0.43% for other samples).
FIG 1.

Hong Kong MTR network map and information, including ridership information and connectedness of lines included in this study. (A) Schematic representation of MTR lines sampled in this study. The network map is not drawn to scale. Lines are color coded, and corresponding outdoor locations are indicated with the same color as the lines; the station is shaded yellow. (B) MTR line and ridership information are based on 2010 figures (http://gia.info.gov.hk/general/201106/08/P201106080126_0126_79963.pdf). (C) Pairwise comparisons between the lines are represented as a matrix. To systematically represent connectedness between lines, a system was devised considering the number of interchange stations (stations with oval shapes) between lines and the shortest distance between two lines if no interchange station is shared. Matrix of line-to-line connectedness for the line pair with interchange stations are represented as 1/x, where x is the number of shared interchange stations. For example, the Island (blue) and Tsuen Wan (red) Lines share two stations; hence, their connectedness is 1/2. The interchange stations allow one to go from one line to another in both directions. For lines with no sharing station, the distance is represented by the shortest unit distance separating the two lines, where one step is traveling from one station to the next or the change of a line. For example, as illustrated, from Kwun Tong Line (green) to Island Line (blue), a minimum of three steps are required, with a change from the Kwun Tong to the Tseung Kwan O (purple) Line (step 1), followed by traveling one station (step 2), and then changing from the Tseung Kwan O to the Island Line (step 3). The dotted line adjacent to the station Tsim Sha Tsui East can be treated as an interchange station for the study.
FIG 2.

Major phyla detected in MTR and outdoor samples. Each group represents the collection of samples within a given sample type, either a particular line or an outdoor location. The plot is arranged by similarities in proportions of phyla between samples. The corresponding outdoor samples are placed to the right of a set of train line samples. Taxonomic assignment was performed using the open-reference method, based on 97% sequence identity cutoff against a filtered Greengenes database to include only sequences with taxonomic information down to the genus level. The top five phyla present across the data set are indicated. Minor/unclassified includes sequences assigned to the remaining phyla according to the Greengenes database and de novo sequences with no Greengenes sequences sharing ≥97% identity.
The effects of spatial, temporal, and architectural attributes on α- and β-diversities.
We compared the within-sample diversities (α-diversities) between samples using taxonomic-based, phylogenetic-based (FPD), and singleton-based (Chao1) approaches (see Table S1 and Fig. S2 in the supplemental material). All three α-diversity indices provided consistent results for a given comparison. Collectively, outdoor locations had a higher taxonomic and phylogenetic diversity than the MTR environment (P < 0.05 for all three approaches) (see Fig. S3A). In addition, the 10 samples with the most OTUs following normalization all belonged to outdoor samples or samples from MTR lines predominantly outdoors and above ground. MTR lines did not show significantly different α-diversity, and when MTR samples were grouped by their architectural design, lines that were predominantly indoors and underground were similar in diversity to those outdoors and above ground. Diversity differences also were not observed between different outdoor samples. Temporal characteristics appeared to play roles in bacterial richness on the MTR, where afternoons and evenings (p.m.) exhibited a higher diversity on the MTR than the morning (a.m.) (P < 0.05 for all three approaches) (see Fig. S3B), which was consistent in all three α-diversity indices. In contrast, commuter traffic, as measured by comparing peak and nonpeak commute hour samples, did not appear to influence α-diversity.
As we collected a total of four replicates per line, with each replicate collected within an average span of 15 days, we analyzed whether α-diversities differ significantly on different days within the same line. For a particular line, samples collected on the same day, consisting of the four a.m. and p.m. peak and nonpeak samples, were combined, and comparisons were made between different days for a single line. Of the seven lines analyzed, two of the lines, both above ground (East Rail and Ma On Shan Lines), showed significant α-diversity changes on different days based on taxonomic richness and FPD (see Fig. S3C in the supplemental material). None of the lines showed day-to-day changes in Chao1 diversity.
The phylogenetic dissimilarity of communities (β-diversity) observed for each sample type was determined using both unweighted and abundance-weighted UniFrac distances, and analysis of similarity (ANOSIM) was computed to determine significance between sample types (Table 1). With the exception of the outdoor versus outdoor and peak versus nonpeak comparisons, both unweighted and weighted analyses gave consistent results for a given comparison. In contrast to what was observed for α-diversity indices, the bacterial community in MTR air as a whole was not significantly different from that of outdoor locations, consistent with observations seen in NYC (21). When considering only MTR samples, however, community phylogenetic variations were observed between different lines, and communities in MTR lines above ground and outdoors showed significantly different phylogenetic communities compared to those below ground. In contrast, significant phylogenetic dissimilarity was not observed between the different outdoor locations based on weighted UniFrac phylogenetic distances. Within-day temporal characteristics also contributed to community compositions, where a.m. and p.m. samples had marginal changes in communities. The effect of commuter traffic (peak versus nonpeak), a quasitemporal variable possibly better defined as a function of commuter traffic, showed significant differences only when abundance-weighted UniFrac was considered. Similarity percentage (SIMPER) analysis comparison based on Bray-Curtis measure of dissimilarity revealed that the increased abundances of skin-associated OTUs, such as Micrococcus spp., Enhydrobacter spp., and Staphylococcus spp., on the MTR explained the community differences between MTR and the outdoors (see Table S3 in the supplemental material). Within the MTR, higher abundances of these skin-associated OTUs in the underground lines during peak and p.m. hours also explained assemblage differences between lines, peak/nonpeak, and a.m./p.m. hours.
TABLE 1.
Comparison of β-diversity between sample types according to unweighted and weighted UniFrac distances
| Comparison by structure and time | Global R (unweighted) | P valuea | Global R (weighted) | P valuea |
|---|---|---|---|---|
| Spatial/architectural | ||||
| MTR vs outdoor | −0.023 | NS | 0.046 | NS |
| Outdoor vs outdoor | 0.161 | 0.021 | −0.003 | NS |
| Line vs line | 0.207 | 0.001 | 0.082 | 0.001 |
| Underground vs above ground | 0.232 | 0.001 | 0.064 | 0.004 |
| Temporal | ||||
| a.m. vs p.m. | 0.062 | 0.001 | 0.029 | 0.03 |
| Peak vs nonpeak | −0.005 | NS | 0.109 | 0.001 |
NS, not significant (P > 0.05). Boldfaced values indicate statistical significance.
Relationships between the MTR and outdoor microbiomes.
Analysis of the most common microbial members detected in the air of MTR lines revealed that these genera included not only normal inhabitants of the skin but also soil, water, and leaf-associated organisms, such as OTUs of the Sphingobium, Blastomonas, Acinetobacter, and Xanthomonas genera (see Table S2 in the supplemental material), reflecting the potential roles of these specific outdoor locations in shaping the MTR microbiome. We investigated whether the microbiomes from outdoor locations along the subway lines also can act as sources of the MTR microbiome. We selected representative outdoor locations where they can be accessed by at least one of the MTR lines sampled, and we employed the Bayesian SourceTracker approach to determine the proportions of contributions of the different outdoor sources, assuming that the bacterial community observed within a line (sink community) originated from the sampled outdoor locations (source communities), each contributing to a particular proportion of the line microbiome (Fig. 3). The strength of this model is that it provides a quantitative account of the potential contribution of each source, as supposed to the qualitative description seen in principal coordinate analysis (PCoA) plots derived from UniFrac distances. All outdoor locations could act as sources for the MTR microbiome, but different sources contributed different proportions within a given line, and each source contributed differently between different lines. Each outdoor location contributed a range of 0.41% to 50% of a given line's microbiome, and with the exceptions of the East Rail Line and Tseung Kwan O Line, the corresponding outdoor locations contributed the greatest to the microbial community of each line. It should be noted that both Admiralty and Causeway Bay are accessible by the Island Line (Fig. 1), and these two outdoor locations together contributed over 55% of the community seen on this line. Thus, adjacent outdoor locations play prominent roles in shaping the microbial communities of corresponding MTR lines.
FIG 3.
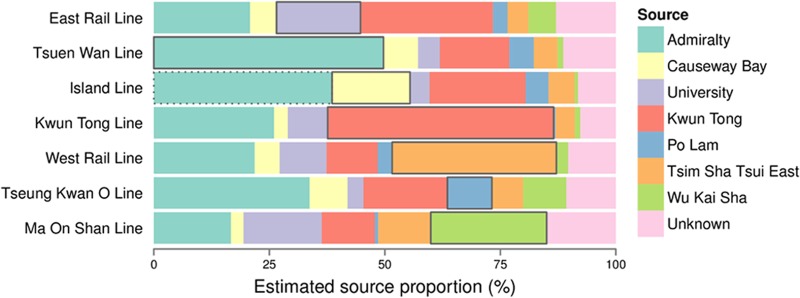
Relationship between MTR line community composition and corresponding outdoor locations. A Bayesian source-tracking approach (37) was used to represent the mean proportions of contributions of each outdoor-location bacterial community on each line. An overlap between line (sink) and outdoor (source) microbiomes is represented by ≥97% identity between sequences of the two sample environments. Sequences in which SourceTracker could not confidently assign a source environment are placed in the unknown category. The outdoor location corresponding to a specific line is indicated with a black bracket for each bar. While Causeway Bay was chosen as the corresponding location for the Island Line, Admiralty (dotted bracket) is also along the Island Line; hence, it also should be considered a source for this line.
The effects of line connectedness on the similarity of microbiomes.
In addition to the effects of nearby outdoor environments, we hypothesized that the microbiome of an MTR line is shaped in part by adjacent lines, where lines that are connected by interchanging stations would share more similar communities than those without interchanges. Specifically, we examined if the community differences observed for individual MTR lines follow a distance- (or connectedness-) dependent trend, similar to what was observed in microbial communities of residential air (42, 43). To address this, we determined the correlation between the connectedness of MTR lines to the phylogenetic similarities of their bacterial communities. Connectedness between lines was represented as a geographical distance matrix where the distance between two lines sharing interchange stations is represented by 1/x, with x being the number of interchange stations shared between two lines. If the two lines are not connected (no interchange station shared), an integer is given for the shortest number of steps between two lines, where a step is defined as either changing a line or going from one station to the next (Fig. 1A). A pairwise comparison matrix of line-to-line connectedness is present in Fig. 1C. A Mantel test of MTR line connectedness and weighted UniFrac distances indicated that closely connected MTR lines shared more similar microbial communities than pairs that are further apart (R = 0.47, P = 0.03), consistent with the suggestion that microbiome exchanges are more likely to occur between closely connected lines, possibly by distance-dependent dispersal and transferring commuters.
The effects of environmental conditions on microbiomes and relative abundances of genera.
Temperature, carbon dioxide (CO2), and relative humidity measurements were logged over 2-s intervals and averaged over the 2-h sampling time for each sample type (see Table S4 in the supplemental material). MTR samples showed significantly lower average temperatures (P = 4.1 × 10−10 by Mann-Whitney test) (see Fig. S4A) and higher average levels of CO2 (P = 3.5 × 10−15 by Mann-Whitney test) (see Fig. S4B) compared to those of outdoor samples. CO2 is likely to be a reflection of commuter density, as peak samples also showed significantly higher levels of CO2 than samples taken during off-peak hours (see Fig. S4C), and that the two lines with the lowest ridership figures (Tseung Kwan O and Ma On Shan Lines) (Fig. 1B) showed lower average CO2 levels (see Table S4). A Kruskal-Wallis test showed overall significant variations in all three environmental parameters between lines (see Fig. S4D to F); however, the variations in the three parameters could not be explained by whether the lines were underground or above ground (P > 0.05 by Mann-Whitney test for all three environmental comparisons). Furthermore, environmental conditions were similar between a.m. and p.m. samples, as well as between different outdoor locations.
We investigated whether the environmental conditions were correlated with changes in sample diversity within the MTR. For both taxonomic (based on rarefied sample OTU numbers) and phylogenetic (based on rarefied FPD) diversities, temperature (see Fig. S5A and B in the supplemental material) and relative humidity (see Fig. S5C and D) were inversely and proportionally correlated with diversities, respectively, while significant correlations between CO2 levels and diversity were not detected. However, the removal of outliers based on normalized numbers of OTUs (all outliers belong to samples taken on outdoor/aboveground MTR lines) rendered these correlations nonsignificant, suggesting that these environmental parameters have a greater impact on diversity in outdoor lines than on those indoors. Environmental conditions also affected the relative abundances of selected genera, with the Pearson's test revealing significant positive correlations between CO2 concentrations and abundances of members of Enhydrobacter spp. (ρ = 0.41, P = 9.8 × 10−7) and Micrococcus spp. (ρ = 0.46, P = 3.2 × 10−8) across the entire data set. However, as the genus comparisons were performed with both MTR and non-MTR outdoor samples, the correlations observed could be governed by sampling locations. However, more modest but similar CO2-associated correlations were observed for Enhydrobacter (ρ = 0.33, P = 0.0004) and Micrococcus (ρ = 0.35, P = 0.0001) within the MTR. In addition, inverse correlations between temperature on the MTR and abundances of soil-dwelling Sphingobium (ρ = −0.29, P = 0.002) and Blastomonas (ρ = −0.30, P = 0.002) were detected. Therefore, we provided evidence that humidity, temperature, and CO2 play roles in sample diversity and/or abundances of certain genera in the MTR.
Comparison of Hong Kong subway microbial community to those in other microbiome studies.
As one of the few large-scale studies investigating aerosol microbial community composition in Asia, we were interested in comparing our subway and outdoor data sets to those of indoor microbiome studies conducted elsewhere. We have chosen studies investigating the bioaerosols of a university building in Oregon (36) and the household surface microbial compositions in North Carolina homes (5). PCoA plots based on weighted UniFrac distances clearly showed that the Hong Kong cluster is distinct from the American groups (global R = 0.34, P = 0.001 by ANOSIM) (Fig. 4).
FIG 4.
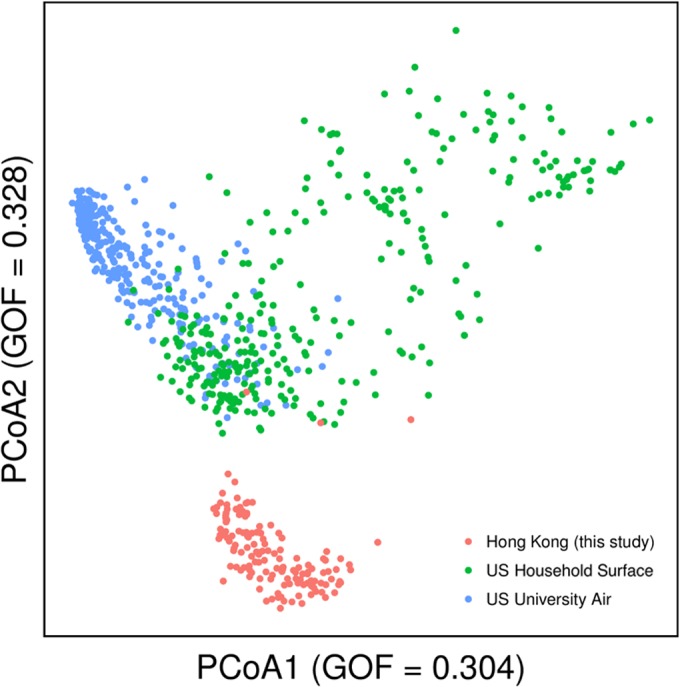
PCoA plot of phylogenetic dissimilarity between samples of different geographical regions. Comparison was performed between Hong Kong samples collected in this study and previous works investigating urban environments in the United States (5, 36). The plot was constructed based on abundance-weighted UniFrac phylogenetic distance. GOF represents the goodness of fit, an indication of the representation of the PCoA plot to the UniFrac distance data. PCoA dimensions 1 and 2 show 30% and 33% of the variances, respectively.
A UniFrac-based comparison between the Hong Kong and the NYC subway (21) aerosol microbial community was not feasible, as the latter study utilized Roche 454 pyrosequencing technology with lower sequence depth. Thus, a much lower rarefied read depth would be necessary, and the representation of the rarefied community in our sample would be questionable. However, taxonomic-based comparison of these communities revealed that some common genera were not shared between the studies. For example, soil-dwelling genera, such as Arthrobacter and Psychrobacter, which were among the common genera documented in NYC, all were detected at <0.5% across the Hong Kong data set. Conversely, Enhydrobacter, a genus previously detected in high proportions in Chinese young adults (44), were among the top skin-associated genera on the MTR but not considered to be a common genus in the NYC subway. Such differences call for the additional metagenomic analysis of subway systems around the world to determine whether additional geographical and/or cultural/ethnic factors play roles in driving differences in aerosol microbiomes.
DISCUSSION
Urbanization has led to dependence on public transportation, including subway networks. Despite its role in modern societies, little is known pertaining to the microbial exposure of commuters during subway use. The characterization of subway bioaerosols using high-throughput, culture-independent techniques is still in its nascency, conducted with the intention of improving one's understanding of public health risks and in biodefense preparation (19–21). Instead, the main focus of this study was to link patterns of observed microbial communities with intrinsic characteristics of subway usage patterns and architectural designs. We believe that the works presented in this study will pave the way for future bioaerosol investigations of subways to better understand microbiological profiles of this unique built environment.
As the first study unraveling the microbial diversity of an urban subway network in Asia using sequencing technology, we describe the Hong Kong subway bioaerosols as taxonomically diverse, more so than was seen in previous culture-independent reports of subway microbial communities (19, 21). We detected residents of various ecological habitats, including soil (Sphingobium and Acinetobacter), water (Blastomonas), and leaf (Xanthomonas). In addition, Micrococcus, Propionibacterium, and Staphylococcus were among the most commonly detected host-associated genera in our study, an observation also recorded in other indoor built-environment studies (4, 41, 45). While these members constitute the normal human skin and oral flora (46), recent reports of antibiotic-resistant microorganisms in subways underscore the need for the active surveillance of microbial communities in this environment (19, 47), as a crowded and enclosed MTR environment encourages the horizontal transmission of microorganisms between large numbers of commuters.
The effects of architecture, namely, indoor ventilation, has been demonstrated to play roles in shaping the extent to which microbial communities from outdoor air influence various indoor spaces (4, 36, 41, 42). We hypothesize that similar phenomena apply to the subway. A recent study employing conventional Sanger and pyrosequencing on the NYC subway indicated a lack of difference in bacterial communities between subway and outdoor air (21). The lack of mechanical ventilation in the NYC subway and the mixing of outdoor and subway air was proposed to contribute to the lack of distinct microbial profiles detected. In Hong Kong, the MTR microbiome also had no significant variation compared to the outdoor microbiome, suggesting equilibrium and complete air mixing of the microbial communities between the two environments. However, multiple observations in this study suggest that complete outdoor and MTR air mixing does not take place: (i) the greater α-diversity observed outdoors compared to inside the MTR in this study, (ii) the presence of community assemblage variations between MTR lines, (iii) the differences in contributions on MTR microbial assemblages depending on the outdoor locations, and (iv) the correlation between community phylogenetic distances and connectedness between MTR lines.
Thus, we postulate that while some air mixing occurs between outdoor locations and the MTR, other factors contributed to shaping the MTR microbial community. Specifically, the presence of mechanical ventilation may play an important role in shaping microbial communities in the MTR. Of the seven MTR lines sampled, three (East Rail, West Rail, and Ma On Shan Lines) are operated predominantly outdoors and are predominated by natural ventilation on the station platforms, and these aboveground lines showed community differences compared to the mechanically ventilated, indoor underground lines. The presence of screen doors along the platforms of the indoor and underground stations would restrict air exchanges between the tunnel track and the passenger platform areas in these lines (48). Outdoor air taken from the intake ventilation systems would fill up the tunnel area predominantly, but only small volumes of outdoor air will be introduced into the platform area during train stoppage because of the screen doors. This restricted influence of outdoor air is less so for aboveground lines, as most of the aboveground platforms are open to ambient air. As our integrated sampling approach included air taken on the platforms, the presence of screen doors may further restrict outdoor air entering into the underground platform areas. In addition, although additional information, such as station depths, was not obtained, stations deeper below ground also have been shown to contain more varied communities (8, 9, 12, 13). Thus, we believe that understanding microbial communities of the subway requires a thorough assessment of various features of its architecture.
Analysis of β-diversity using weighted UniFrac distances detected no significant difference in compositions between different outdoor locations. It was previously documented that different terrain types may reveal different microbial communities (49). However, our selection of outdoor locations may vary less in terms of land use types than in the degree of urbanization. Indeed, previous investigations of the effects of urbanization on microbial communities in Hong Kong revealed no variation between urban and rural areas (50). Alternatively, geographical variations that may explain differences in outdoor and/or indoor microbial communities may need to be on a regional rather than a local scale (22, 23, 51), mediated by great variations in climates in the atmosphere (52). In agreement, the microbial communities in the Hong Kong MTR were distinct from those of other studies in the United States (5, 36), revealing clustering based on continental geography. While it is possible that different indoor building functions and types selected played roles in such observed community variations, taxonomy-based comparison between MTR and the NYC subways revealed differences in some of the common environmental and human-associated genera detected, favoring some geography-governing community differences. Also, because occupants have a great impact on the indoor air microbiomes, variations in microbiomes of different population groups may influence air microbial assemblages (24), a phenomenon that has been reported in studies investigating indoor surfaces (22, 53). One of the common genera detected in this study, Enhydrobacter, is a documented resident on the human skin but may be present in higher proportions in Chinese individuals (44). Such observations underscore the need to take the occupants' microbiomes into account when examining indoor microbial assemblages.
Marginal intraday microbial differences were observed on the MTR overall, with p.m. hours showing higher α-diversity than a.m. hours. Each of the a.m. and p.m. groups is an accumulation of 4 h within a line, which may dampen any greater variations seen within shorter time frames (54). While meteorological factors may explain intraday differences in communities with low human activity (55, 56), in urban settings such as that of samples collected in this study, human factors are likely to take greater part in shaping day-to-day assemblage variations (19). The effect of commuters on MTR microbial communities is further exemplified by our observation that increases in skin-associated genera during peak hours contributed to differences in the peak and nonpeak communities. The increased number of commuters naturally will result in the increased shedding of skin-associated organisms into the subway environment (19, 57). In addition, daily variations in α-diversity were observed in aboveground lines, and interday community variations were observed in both MTR and outdoor environments. Interday distinct communities within the same locations are consistent with the day-to-day idiosyncrasy observed by Robertson et al. (21), as well as those in other urban (51) and rural (25) locations. These unique communities, usually dominated by a small number of genera, may be explained by sporadic anthropogenic factors (organism release via skin shedding, sneezing, talking, and coughing) and other source factors in the vicinity rather than environmental factors associated with seasonal community differences (54, 58). Therefore, commuters using the subway on a regular basis at different times and days are more likely to be exposed to a greater breadth of microbial life.
Decreasing temperature (within a range of ∼30°C to 24°C) and increasing relative humidity (within a range of ∼50% to 90%) within the MTR were associated with an increase in taxonomic and phylogenetic diversities. Similarly, other studies investigating effects of temperature on microbial communities in residential aerosols showed negative correlations between indoor temperature and bacterial survival (59). Tang (60) proposes that an increase in temperature from 24°C decreases the survival of different airborne bacterial species. Whether this observation can be applied and generalized to bacteria in aerosols and to those of different indoor environments is unknown. Nonetheless, one should use caution in such generalizations and comparisons, as environmental effects such as temperature and humidity on microbial community assemblages are likely to be influenced by sample locations, method of sampling, and the microbial members and communities in question (17, 59, 60). When outlier samples based on their high taxonomic diversity were removed, the effects of temperature and humidity on MTR α-diversity no longer were significant. Thus, it is possible that fluctuations in environmental properties would be more influential in the observed microbial community on outdoor rather than indoor MTR lines, where commuters and anthropogenic factors are likely to play more important roles. The observation that only outdoor lines showed significant interday α-diversity variations appear to support this. We found that temperatures on the MTR were inversely correlated with relative abundances of Sphingobium and Blastomonas, both of which are within the Sphingomonadales order. The relationship between temperature and these genera commonly detected in soil (27) and aquatic (61) environments is unknown; however, their presence in residential and other urban environments and their abilities to acquire antibiotic resistance (62) deserve further analysis of how environmental parameters in urban environments affect their survival. We believe that CO2 levels within the MTR is a function of commuter density, as the CO2 level was higher on the MTR than outdoors and higher during peak than nonpeak hours, concomitant with positive correlations between CO2 levels and the abundance of skin-associated bacteria. The changes observed in these environmental attributes took place on relatively short time scales (two hours between peak and nonpeak times). Therefore, further investigations on the relationships between such parameters and microbial assemblage changes in indoor environments will shed light on the microbial community dynamics of this unique indoor environment.
In conclusion, we have performed an integrated characterization of the microbial community of an urban subway network using a high-throughput culture-independent method, demonstrating that the air in MTR contained extensive microbial diversity. The detected differences in assemblages between space and time are likely to be due to a complex interplay between architectural, meteorological, and anthropogenic factors. Overall, we now have greater insights into the microbial life in the air of this unique built environment that serves millions of passengers daily.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the Research Grants Council of Hong Kong through project 124412.
We thank Wai Shan Chow, Catherine Chung, Ka Yan Ng, Yuet Ying Wong, and Flora Yeh for sampling assistance and Zhi Ning and Fenhuan Yang for their support with instruments. We thank Wei Chi Wang and the members of Health GeneTech Corporation for sample processing and sequence analysis.
Footnotes
Published ahead of print 29 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02244-14.
REFERENCES
- 1.Klepeis NE, Nelson WC, Ott WR, Robinson JP, Tsang AM, Switzer P, Behar JV, Hern SC, Engelmann WH. 2001. The National Human Activity Pattern Survey (NHAPS): a resource for assessing exposure to environmental pollutants. J. Expo. Anal. Environ. Epidemiol. 11:231–252. 10.1038/sj.jea.7500165. [DOI] [PubMed] [Google Scholar]
- 2.Douwes J, Thorne P, Pearce N, Heederik D. 2003. Bioaerosol health effects and exposure assessment: progress and prospects. Ann. Occup. Hyg. 47:187–200. 10.1093/annhyg/meg032. [DOI] [PubMed] [Google Scholar]
- 3.Kettleson E, Kumar S, Reponen T, Vesper S, Méheust D, Grinshpun SA, Adhikari A. 2013. Stenotrophomonas, Mycobacterium, and Streptomyces in home dust and air: associations with moldiness and other home/family characteristics. Indoor Air 23:387–396. 10.1111/ina.12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kembel SW, Jones E, Kline J, Northcutt D, Stenson J, Womack AM, Bohannan BJ, Brown GZ, Green JL. 2012. Architectural design influences the diversity and structure of the built environment microbiome. ISME J. 6:1469–1479. 10.1038/ismej.2011.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dunn RR, Fierer N, Henley JB, Leff JW, Menninger HL. 2013. Home life: factors structuring the bacterial diversity found within and between homes. PLoS One 8:e64133. 10.1371/journal.pone.0064133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hospodsky D, Qian J, Nazaroff W, Yamamoto N, Bibby K, Rismani-Yazdi H, Peccia J. 2012. Human occupancy as a source of indoor airborne bacteria. PLoS One 7:e34867. 10.1371/journal.pone.0034867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonetta S, Bonetta S, Mosso S, Sampò S, Carraro E. 2010. Assessment of microbiological indoor air quality in an Italian office building equipped with an HVAC system. Environ. Monit Assess. 161:473–483. 10.1007/s10661-009-0761-8. [DOI] [PubMed] [Google Scholar]
- 8.Gilleberg S, Faull J, Graeme-Cook K. 1998. A preliminary survey of aerial biocontaminants at six London Underground stations. Int. Biodeterior. Biodegrad. 41:149–152. 10.1016/S0964-8305(98)00005-5. [DOI] [Google Scholar]
- 9.Awad AHA. 2002. Environmental study in subway metro stations in Cairo, Egypt. J. Occup. Health 44:112–118. 10.1539/joh.44.112. [DOI] [Google Scholar]
- 10.Seino K, Takano T, Nakamura K, Watanabe M. 2005. An evidential example of airborne bacteria in a crowded, underground public concourse in Tokyo. Atmos. Environ. 39:337–341. 10.1016/j.atmosenv.2004.09.030. [DOI] [Google Scholar]
- 11.Cho J, Min K, Paik N. 2006. Temporal variation of airborne fungi concentrations and related factors in subway stations in Seoul, Korea. Int. J. Hyg. Environ. Health 209:249–255. 10.1016/j.ijheh.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 12.Bogomolova E, Kirtsideli I. 2009. Airborne fungi in four stations of the St. Petersburg underground railway system. Int. Biodeter. Biodegr. 63:156–160. 10.1016/j.ijgo.2012.03.027. [DOI] [Google Scholar]
- 13.Hwang S, Yoon C, Ryu K, Paik S, Cho J. 2010. Assessment of airborne environmental bacteria and related factors in 25 underground railway stations in Seoul, Korea. Atmos. Environ. 44:1658–1662. 10.1016/j.atmosenv.2010.01.047. [DOI] [Google Scholar]
- 14.Dong S, Yao M. 2010. Exposure assessment in Beijing, China: biological agents, ultrafine particles, and lead. Environ. Monit. Assess. 170:331–343. 10.1007/s10661-009-1236-7. [DOI] [PubMed] [Google Scholar]
- 15.Kawasaki T, Kyotani T, Ushiogi T, Izumi Y, Lee H, Hayakawa T. 2010. Distribution and identification of airborne fungi in railway stations in Tokyo, Japan. J. Occup. Health 52:186–193. 10.1539/joh.O9022. [DOI] [PubMed] [Google Scholar]
- 16.Kim K, Kim Y, Kim D, Kim H. 2010. Exposure level and distribution characteristics of airborne bacteria and fungi in Seoul metropolitan subway stations. Ind. Health 49:242–248. 10.2486/indhealth.MS1199. [DOI] [PubMed] [Google Scholar]
- 17.Kawasaki T, Kyotani T, Ushiogi T, Lee H. 2013. Distribution of airborne bacteria in railway stations in Tokyo, Japan. J. Occup. Health 55:495–502. 10.1539/joh.13-0055-FS. [DOI] [PubMed] [Google Scholar]
- 18.Birenzvige A, Eversole J, Seaver M, Francesconi S, Valdes E, Kulaga H. 2003. Aerosol characteristics in a subway environment. Aerosol Sci. Technol. 37:210–220. 10.1080/02786820300941. [DOI] [Google Scholar]
- 19.Dybwad M, Granum P, Bruheim P, Blatny J. 2012. Characterization of airborne bacteria at an underground subway station. Appl. Environ. Microbiol. 78:1917–1929. 10.1128/AEM.07212-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dybwad M, Skogan G, Blatny JM. 2014. Temporal variability of the bioaerosol background at a subway station: concentration level, size distribution, and diversity of airborne bacteria. Appl. Environ. Microbiol. 80:257–270. 10.1128/AEM.02849-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robertson C, Baumgartner L, Harris J, Peterson K, Stevens M, Frank D, Pace N. 2013. Culture-independent analysis of aerosol microbiology in a metropolitan subway system. Appl. Environ. Microbiol. 79:3485–3493. 10.1128/AEM.00331-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hewitt KM, Gerba CP, Maxwell SL, Kelley ST. 2012. Office space bacterial abundance and diversity in three metropolitan areas. PLoS One 7:e37849. 10.1371/journal.pone.0037849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amend A, Seifert K, Samson R. 2010. Indoor fungal composition is geographically patterned and more diverse in temperate zones than in the tropics. Proc. Natl. Acad. Sci. U. S. A. 107:13748–13753. 10.1073/pnas.1000454107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou Y, Gao H, Mihindukulasuriya K, Rosa P, Wylie K, Vishnivetskaya T, Podar M, Warner B, Tarr P, Nelson D, Fortenberry J, Holland M, Burr S, Shannon W, Sodergren E, Weinstock G. 2013. Biogeography of the ecosystems of the healthy human body. Genome Biol. 14:R1. 10.1186/gb-2013-14-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bowers R, McCubbin I, Hallar A, Fierer N. 2012. Seasonal variability in airborne bacterial communities at a high-elevation site. Atmos. Environ. 50:41–49. 10.1016/j.atmosenv.2012.01.005. [DOI] [Google Scholar]
- 26.Caporaso J, Lauber C, Walters W, Berg-Lyons D, Huntley J, Fierer N, Owens S, Betley J, Fraser L, Bauer M, Gormley N, Gilbert J, Smith G, Knight R. 2012. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6:1621–1624. 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peiffer J, Spor A, Koren O, Jin Z, Tringe S, Dangl J, Buckler E, Ley R. 2013. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. U. S. A. 110:6548–6553. 10.1073/pnas.1302837110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caporaso J, Kuczynski J, Stombaugh J, Bittinger K, Bushman F, Costello E, Fierer N, Peña A, Goodrich J, Gordon J, Huttley G, Kelley S, Knights D, Koenig J, Ley R, Lozupone C, McDonald D, Muegge B, Pirrung M, Reeder J, Sevinsky J, Turnbaugh P, Walters W, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:335–336. 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 30.Conlan S, Kong H, Segre J. 2012. Species-level analysis of DNA sequence data from the NIH Human Microbiome Project. PLoS One 7:e47075. 10.1371/journal.pone.0047075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. 2010. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26:266–267. 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Faith DP. 1992. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 61:1–10. 10.1016/0006-3207(92)91201-3. [DOI] [Google Scholar]
- 33.Chao A. 1984. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 11:265–270. [Google Scholar]
- 34.Lozupone C, Knight R. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71:8228–8235. 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lozupone C, Hamady M, Kelley S, Knight R. 2007. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 73:1576–1585. 10.1128/AEM.01996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meadow J, Altrichter A, Kembel S, Kline J, Mhuireach G, Moriyama M, Northcutt D, O'Connor T, Womack A, Brown G, Green J, Bohannan B. 2014. Indoor airborne bacterial communities are influenced by ventilation, occupancy, and outdoor air source. Indoor Air 24:41–48. 10.1111/ina.12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knights D, Kuczynski J, Charlson E, Zaneveld J, Mozer M, Collman R, Bushman F, Knight R, Kelley S. 2011. Bayesian community-wide culture-independent microbial source tracking. Nat. Methods 8:761–763. 10.1038/nmeth.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bertolini V, Gandolfi I, Ambrosini R, Bestetti G, Innocente E, Rampazzo G, Franzetti A. 2013. Temporal variability and effect of environmental variables on airborne bacterial communities in an urban area of northern Italy. Appl. Microbiol. Biotechnol. 97:6561–6570. 10.1007/s00253-012-4450-0. [DOI] [PubMed] [Google Scholar]
- 39.Flores G, Bates S, Caporaso J, Lauber C, Leff J, Knight R, Fierer N. 2013. Diversity, distribution and sources of bacteria in residential kitchens. Environ. Microbiol. 15:588–596. 10.1111/1462-2920.12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feazel LM, Baumgartner LK, Peterson KL, Frank DN, Harris JK, Pace NR. 2009. Opportunistic pathogens enriched in showerhead biofilms. Proc. Natl. Acad. Sci. U. S. A. 106:16393–16399. 10.1073/pnas.0908446106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kembel SW, Meadow JF, O'Connor TK, Mhuireach G, Northcutt D, Kline J, Moriyama M, Brown GZ, Bohannan BJM, Green JL. 2014. Architectural design drives the biogeography of indoor bacterial communities. PLoS One 9:e87093. 10.1371/journal.pone.0087093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams RI, Miletto M, Taylor JW, Bruns TD. 2013. Dispersal in microbes: fungi in indoor air are dominated by outdoor air and show dispersal limitation at short distances. ISME J. 7:1262–1273. 10.1038/ismej.2013.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adams RI, Miletto M, Lindow SE, Taylor JW, Bruns TD. 2014. Airborne bacterial communities in residences: similarities and differences with fungi. PLoS One 9:e91283. 10.1371/journal.pone.0091283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ling Z, Liu X, Luo Y, Yuan L, Nelson KE, Wang Y, Xiang C, Li L. 2013. Pyrosequencing analysis of the human microbiota of healthy Chinese undergraduates. BMC Genomics 14:390. 10.1186/1471-2164-14-390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rintala H, Pitkaranta M, Toivola M, Paulin L, Nevalainen A. 2008. Diversity and seasonal dynamics of bacterial community in indoor environment. BMC Microbiol. 8:56. 10.1186/1471-2180-8-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Human Microbiome Project Consortium. 2012. Structure, function and diversity of the healthy human microbiome. Nature 486:207–214. 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou F, Wang Y. 2013. Characteristics of antibiotic resistance of airborne Staphylococcus isolated from metro stations. Int. J. Environ. Res. Public Health 10:2412–2426. 10.3390/ijerph10062412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ke M-T, Cheng T-C, Wang W-P. 2002. Numerical simulation for optimizing the design of subway environmental control system. Build. Environ. 37:1139–1152. 10.1016/S0360-1323(01)00105-6. [DOI] [Google Scholar]
- 49.Bowers RM, McLetchie S, Knight R, Fierer N. 2011. Spatial variability in airborne bacterial communities across land-use types and their relationship to the bacterial communities of potential source environments. ISME J. 5:601–612. 10.1038/ismej.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Woo AC, Brar MS, Chan Y, Lau MCY, Leung FCC, Scott JA, Vrijmoed LLP, Zawar-Reza P, Pointing SB. 2013. Temporal variation in airborne microbial populations and microbially-derived allergens in a tropical urban landscape. Atmos. Environ. 74:291–300. 10.1016/j.atmosenv.2013.03.047. [DOI] [Google Scholar]
- 51.Brodie E, DeSantis T, Parker J, Zubietta I, Piceno Y, Andersen G. 2007. Urban aerosols harbor diverse and dynamic bacterial populations. Proc. Natl. Acad. Sci. U. S. A. 104:299–304. 10.1073/pnas.0608255104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Womack AM, Bohannan BJM, Green JL. 2010. Biodiversity and biogeography of the atmosphere. Philos. Trans. R. Soc. Lond. B Biol. Sci. 365:3645–3653. 10.1098/rstb.2010.0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Flores G, Bates S, Knights D, Lauber C, Stombaugh J, Knight R, Fierer N. 2011. Microbial biogeography of public restroom surfaces. PLoS One 6:e28132. 10.1371/journal.pone.0028132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maron P-A, Lejon D, Carvalho E, Bizet K, Lemanceau P, Ranjard L, Mougel C. 2005. Assessing genetic structure and diversity of airborne bacterial communities by DNA fingerprinting and 16S rDNA clone library. Atmos. Environ. 39:3687–3695. 10.1016/j.atmosenv.2005.03.002. [DOI] [Google Scholar]
- 55.Lighthart B, Shaffer BT. 1995. Airborne bacteria in the atmospheric surface layer: temporal distribution above a grass seed field. Appl. Environ. Microbiol. 61:1492–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lighthart B. 1999. An hypothesis describing the general temporal and spatial distribution of alfresco bacteria in the earth's atmospheric surface layer. Atmos. Environ. 33:611–615. 10.1016/S1352-2310(98)00215-5. [DOI] [Google Scholar]
- 57.Qian J, Hospodsky D, Yamamoto N, Nazaroff W, Peccia J. 2012. Size-resolved emission rates of airborne bacteria and fungi in an occupied classroom. Indoor Air 22:339–351. 10.1111/j.1600-0668.2012.00769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fierer N, Liu Z, Rodriguez-Hernandez M, Knight R, Henn M, Hernandez MT. 2008. Short-term temporal variability in airborne bacterial and fungal populations. Appl. Environ. Microbiol. 74:200–207. 10.1128/AEM.01467-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Frankel M, Beko G, Timm M, Gustavsen S, Hansen EW, Madsen AM. 2012. Seasonal variations of indoor microbial exposures and their relation to temperature, relative humidity, and air exchange rate. Appl. Environ. Microbiol. 78:8289–8297. 10.1128/AEM.02069-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang J. 2009. The effect of environmental parameters on the survival of airborne infectious agents. J. R. Soc. Interface 6:S737–S746. 10.1098/rsif.2009.0227.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zeng Y, Jiao N. 2007. Source environment feature related phylogenetic distribution pattern of anoxygenic photosynthetic bacteria as revealed by pufM analysis. J. Microbiol. 45:205–212. [PubMed] [Google Scholar]
- 62.Vaz-Moreira I, Egas C, Nunes OC, Manaia CM. 2013. Bacterial diversity from the source to the tap: a comparative study based on 16S rRNA gene-DGGE and culture-dependent methods. FEMS Microbiol. Ecol. 83:361–374. 10.1111/1574-6941.12002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.