

ABSTRACT
Human parainfluenza viruses (HPIVs) cause widespread respiratory infections, with no vaccines or effective treatments. We show that the molecular determinants for HPIV3 growth in vitro are fundamentally different from those required in vivo and that these differences impact inhibitor susceptibility. HPIV infects its target cells by coordinated action of the hemagglutinin-neuraminidase receptor-binding protein (HN) and the fusion envelope glycoprotein (F), which together comprise the molecular fusion machinery; upon receptor engagement by HN, the prefusion F undergoes a structural transition, extending and inserting into the target cell membrane and then refolding into a postfusion structure that fuses the viral and cell membranes. Peptides derived from key regions of F can potently inhibit HPIV infection at the entry stage, by interfering with the structural transition of F. We show that clinically circulating viruses have fusion machinery that is more stable and less readily activated than viruses adapted to growth in culture. Fusion machinery that is advantageous for growth in human airway epithelia and in vivo confers susceptibility to peptide fusion inhibitors in the host lung tissue or animal, but the same fusion inhibitors have no effect on viruses whose fusion glycoproteins are suited for growth in vitro. We propose that for potential clinical efficacy, antivirals should be evaluated using clinical isolates in natural host tissue rather than lab strains of virus in cultured cells. The unique susceptibility of clinical strains in human tissues reflects viral inhibition in vivo.
IMPORTANCE Acute respiratory infection is the leading cause of mortality in young children under 5 years of age, causing nearly 20% of childhood deaths worldwide each year. The paramyxoviruses, including human parainfluenza viruses (HPIVs), cause a large share of these illnesses. There are no vaccines or drugs for the HPIVs. Inhibiting entry of viruses into the human cell is a promising drug strategy that blocks the first step in infection. To develop antivirals that inhibit entry, it is critical to understand the first steps of infection. We found that clinical viruses isolated from patients have very different entry properties from those of the viruses generally studied in laboratories. The viral entry mechanism is less active and more sensitive to fusion inhibitory molecules. We propose that to interfere with viral infection, we test clinically circulating viruses in natural tissues, to develop antivirals against respiratory disease caused by HPIVs.
INTRODUCTION
Acute respiratory infection is the leading cause of mortality in young children under 5 years of age and accounts for nearly 20% of childhood deaths worldwide each year. Paramyxoviruses, particularly respiratory syncytial virus (RSV), human metapneumovirus, and the human parainfluenza viruses (HPIVs), cause the majority of childhood croup, bronchiolitis, and pneumonia (1). In adults, these viruses cause about two-thirds of respiratory illnesses, with high mortality in immunocompromised persons (2); for example, HPIV3 accounts for 90% of the respiratory illnesses in hematopoietic stem cell transplant patients (3) and carries high mortality (3–8). There are no effective vaccines or treatments for the HPIVs. Remarkably, while strategies of passive immunoprophylaxis for RSV protect infants at greatest risk (9), and effective antiviral drugs and vaccines are available for influenza (10, 11), there are no vaccines or drugs for the HPIVs (1, 12).
The first step of infection by HPIV, entry of virus into the target cell, is initiated by attachment of the hemagglutinin-neuraminidase receptor-binding protein (HN) to sialic acid-containing receptor molecules on the cell. Attachment starts with engagement of the primary sialic acid binding site (site I), which also possesses neuraminidase, or receptor cleaving, activity. Once the virus is receptor-bound, HN activates the viral fusion protein (F) to a fusion-ready state, permitting its hydrophobic fusion peptide to insert into the target membrane. After F has inserted, it undergoes a regulated sequence of structural transitions during which the heptad repeats (HR) at opposite ends of the molecule meet, eventually culminating in the formation of a six-helix bundle (6HB) and merger of the viral and cellular membranes. At the start of this process, F assumes a transient extended intermediate form. The N-terminal coiled coil is formed, relocating the fusion peptide to interact with the target cell's bilayer, and the C-terminal HR segments (HRC) separate from each other. At this transient stage, the fusion process can be halted using peptides derived from the HRC region that bind to the N-terminal HR segment (HRN) regions of the activated F and prevent formation of the 6HB (13, 14).
The efficacy of peptide fusion inhibitors for paramyxoviruses is determined in part by the strength of the HRC-derived peptides association with the target HRN sequence (14, 15). The kinetics of fusion activation by the attachment protein also modulates inhibitory efficacy, by varying the time window for a peptide to interact with the transient intermediate of F (16). A faster activation process results in less time in the vulnerable transient state. Enhanced fusion activation can be achieved by HN with higher receptor avidity, which permits more receptor-engaged time for activation, or with enhanced F-triggering activity to more efficiently activate fusion. HN provides ongoing activation to F, as fusion proceeds beyond insertion of the fusion peptide into the target (17); this continuous activation of F by HN can oppose the effect of peptide inhibitors. However, paradoxically, HNs that possess higher receptor avidity or enhanced activation of F during entry confer resistance to fusion peptide inhibition, and the viruses bearing such HN molecules are remarkably unfit for growth in vivo (18).
We proposed that specific functions of the viral fusion machinery—receptor avidity, HN activation efficiency, F activation kinetics—differ between laboratory reference strains and clinically relevant viruses and modulate the virus's susceptibility to fusion inhibitors. Previously, we had compared a series of HPIV3 laboratory reference strains that have fusion features affecting growth in human airway epithelial tissue culture (HAE) and in vivo. The comparisons included (i) the HPIV3 laboratory reference strain, (ii) a highly fusogenic strain bearing an HN mutated at the putative secondary sialic acid binding site (site II) (HN Q552), and (iii) a strain (S-18) derived from adaptation of the HPIV3 HN Q552 to growth in human airway, bearing two sequential mutations in F (G396D) and HN (Q559R). The laboratory reference strain and the HN Q552 strain, with an HN that interacts with F to trigger fusion more efficiently, are fusogenic in monolayer cell culture. However, these strains grow poorly or not at all in human airway or in vivo (18–20). The HAE-adapted strain (HN Q552–R559/F D396) has reduced receptor binding, interaction with F, and fusion promotion and grows poorly in monolayer cell culture but well in vivo (20), indicating that the optimal balance of binding and fusion properties is different in vivo and in cultured cell monolayers and that virus in HAE reflects infection in vivo.
We proposed that viruses with less active HN/F fusion machinery will be at a selective advantage in vivo. In contrast, viruses that grow well in monolayer cell cultures have a higher binding avidity than their neuraminidase activity, allowing for more receptor contact, and they have more fusogenic HN/F fusion machineries and thus would be at a disadvantage in vivo (20).
In the present study, we found that the fusion machinery of an HPIV3 clinical isolate (CI) strain is remarkably similar in its properties to the HAE-adapted strain, in terms of characteristics that contribute to viral entry: receptor avidity, HN′s activation efficiency, and F's activation readiness. Infection and growth in vivo correlate with the HAE-adapted/CI constellation of fusion properties. In fact, the growth of the HPIV3 strains in vivo ranked in the opposite order of their fusogenicity in monolayer culture cells (19) and in the opposite order of the activation readiness of their fusion machineries. None of the strains with proficient fusion activities that grow efficiently in cultured monolayer cells succeed in vivo. We observed that the HPIV3 CI-1 collected directly from a patient grew efficiently in cotton rats, producing 2 to 3 logs more virus than the laboratory reference strain at the peak of infection, similarly to the HAE-adapted strain, which we had previously found to be viable in vivo (19, 20).
The efficiency of a virus' fusion machinery correlated inversely to sensitivity to fusion peptide inhibition and growth in vivo (16–18, 20). We hypothesized that the HPIV3 CI's fusion characteristics would resemble viruses derived from adaptation to growth in HAE, rendering it sensitive to peptide inhibitors. Given that the reference strain is inhibited only by the most potent peptide (14), we asked whether the fusion machinery of HPIV3 CI-1 would, conversely, show sensitivity to less potent inhibitors, in HAE and in vivo. We found that HPIV3 CI-1 was highly sensitive in both infection models. These findings suggest that the balance of fusion/entry properties that correlate with growth in vivo (receptor avidity, activation efficiency) leads to less fusion than observed in laboratory-adapted strains, rendering CI strains sensitive to inhibitors in vivo. The results suggest improved strategies for antiviral screening and development.
MATERIALS AND METHODS
Peptide synthesis.
All peptides were produced by standard Fmoc solid-phase methods. Conjugation to a bromoacetyl derivative of cholesterol to produce the monomeric cholesterol-tagged inhibitor (see Fig. 4A) has also been described (14, 21–23).
FIG 4.
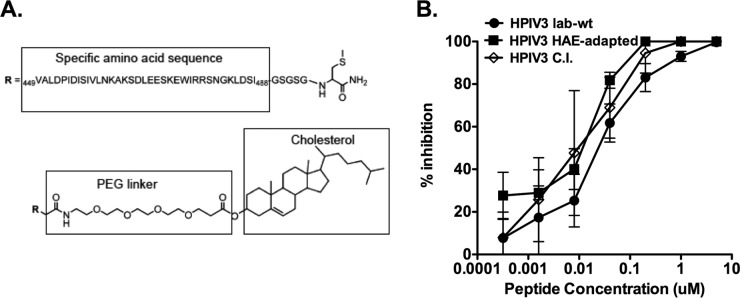
Inhibition of HPIV3 reference strain, HAE-adapted strain, and CI-1 infection by the cholesterol-tagged HPIV3 F HRC peptides in vitro. (A) Schematic of the monomeric cholesterol-tagged HPIV3 F HRC peptide, including its sequence derived from HPIV3 F amino acids 449 to 488, and the cholesterol tag attached to the C terminus by a maleimide (MAL) 4-unit polyethylene glycol (PEG 4) linker. (B) CV1 cell monolayers were infected with HPIV3 reference strain (circle), HAE-adapted strain (square), or CI-1 (open diamond) at a multiplicity of infection (MOI) of 6.7 in the presence of increasing concentrations of the PEG 4-cholesterol peptide. After a 90-min incubation at 37°C, cells were overlaid with Avicel, and plaques were stained and counted after 24 h. The percent inhibition of viral entry (compared to results for control cells infected in the absence of inhibitors) is shown as a function of the (log scale) concentration of peptide. Data points are means (±SD) from triplicate samples. These data are representative of results from three experiments.
Assay of neuraminidase activity.
Monolayers of 293T (human kidney epithelial) cells transiently expressing lab- or human airway epithelium-adapted or CI HNs were washed once with phosphate-buffered saline (PBS) and then incubated for 10 min in 5 mM EDTA in 1× PBS placed in pH 5.0 CO2-independent medium (GIBCO). Cells (150,000) were transferred to 96-well plates and incubated at 37°C for 10 min. 2′-(4-methylumbelliferyl)-a-d-N-acetylneuraminic acid sodium salt (MUNANA; 20 mM; Toronto Research Chemical) was added 1:1, vol/vol, for a final concentration of 10 mM MUNANA. A kinetic reading at 37°C was done using a Spectromax M5 enzyme-linked immunosorbent assay (ELISA) reader every 2 min for 1 h.
Use of receptor-depleted RBCs to assess HN receptor-binding avidity.
Partial receptor depletion of erythrocytes (RBCs) was done as previously described in reference 24. RBCs that were partially depleted of their surface sialic acid receptors were used to determine the relative receptor-binding avidities of variant HN molecules as described previously in references 24 and 25.
β-Galactosidase complementation-based fusion assay.
We adapted (26) an assay that detects early stages of fusion activation, performed as previously described in references 24 and 25.
Cells and viruses.
293T and CV1 cells were grown in Dulbecco's modified Eagle's medium (DMEM) (Mediatech; Cellgro) supplemented with 10% fetal bovine serum (FBS) and antibiotics in 5% CO2. The effect of peptides on HPIV3 plaque number was assessed by a plaque reduction assay performed as described previously in references 14 and 27. Briefly, CV-1 cell monolayers were inoculated with 100 PFU of HPIV3 in the presence of various concentrations of peptides. After 90 min, 2× minimal essential medium containing 10% FBS was mixed with 1% methylcellulose (or Avicel) and added to the dishes. The plates were then incubated at 37°C for 24 h. After the medium overlay was removed, the cells were immunostained for plaque detection. The number of plaques in the control (no peptide) and experimental wells were counted under a dissecting stereoscope. The CI viruses were obtained from deidentified clinical samples isolated in the clinical microbiology laboratory (directed by S.G.J.).
HAE.
The EpiAirway AIR-100 system (MatTek Corporation) consists of normal human-derived tracheal/bronchial epithelial cells that have been cultured to form a pseudostratified, highly differentiated mucociliary epithelium closely resembling that of epithelial tissue in vivo. Upon receipt from the manufacturer, HAE were transferred to 6-well plates (containing 0.9 ml medium per well), with the apical surface remaining exposed to air, and incubated at 37°C in 5% CO2 overnight.
Infection and treatment of HAE and measurement of viral titers from infected HAE.
HAE were infected by applying 200 μl of EpiAirway medium containing 4,000 PFU of reference or another strain of HPIV3 to the apical surface for 90 min at 37°C. At 90 min, the medium containing the inoculum was removed, and cultures were placed at 37°C and fed each day via the basolateral surface with 0.9 ml medium. Viruses were harvested by adding 200 μl medium per well to the HAE apical surface and allowed to equilibrate for 30 min at 37°C. The suspension was then collected, and viral titers were determined as previously described (18). This viral collection was performed sequentially on the same wells of cells on each day postinfection. Treatments were performed by adding medium containing 10, 1, or 0.1 μM of the indicated peptide at the time of infection only. The control wells were infected with no peptide treatment. Harvesting by the apical surface washes was done sequentially on the same HAE, at the indicated time points.
Animals, infection, and virus titration.
Inbred cotton rats were obtained from Harlan (Indianapolis, IN). Female animals, 6 to 10 weeks of age, were infected with 106 PFU intranasally (i.n.) as previously described in references 18 and 28. Groups of 4 animals were infected with the indicated virus and treated subcutaneously with 4 mg/kg of body weight/day of peptide with 2 doses per day for 3 days beginning 1 h after infection. Viral titers were determined as previously described in reference 18. The animal experiments were approved by the Institutional Animal Care and Use Committee of The Ohio State University.
RESULTS
HN/F machineries: receptor avidity and receptor cleaving activities differ in clinical isolate and laboratory-adapted strains.
HN performs four critical functions that are important for viral entry and spread: protection of F, receptor binding, activation of F, and receptor cleavage (1, 24, 29, 30). Prior to receptor engagement, HN stabilizes F and prevents premature activation of the fusion function; upon receptor engagement, HN activates F to fold into its fusion-ready conformation, thereby facilitating the merger of the viral envelope with the host cell membrane. After viral replication, HN′s neuraminidase activity is essential for release of newly budding viruses from the infected cell. To begin to understand these functional characteristics for the HN of the CI-1 strain, the three HNs—derived from the HPIV3 laboratory reference strain, the strain adapted to growth in HAE, and an HPIV3 clinical isolate (CI-1)—were first compared for their ability to bind and release sialic acid receptors. For measurement of HN receptor-binding avidity, cells transiently expressing each of the HN variants were pretreated with neuraminidase to deplete receptors on the expressing cells' surfaces (24, 25). Receptor-bearing cells, in this case erythrocytes (RBCs) with different degrees of receptor depletion, were then added and used to quantify binding to the HNs. HN molecules with higher avidity can bind RBCs that have lower receptor density, so the level of RBC receptor depletion that still binds provides a measure for avidity. The reference HN showed the highest avidity for receptor of the 3 HNs, with 50% of binding to RBCs treated with 40 mU of neuraminidase, compared to 50% binding to RBCs treated with 15 mU and 20 mU for HAE-adapted HN and CI-1, respectively (Fig. 1A).
FIG 1.
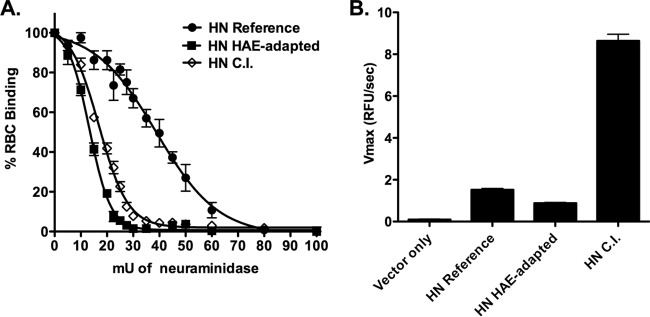
The HPIV3 clinical isolate HN has the highest neuraminidase/avidity ratio, indicating the least engagement with receptor. (A) To measure relative avidity for receptor, cell monolayers transiently expressing reference strain HN (circles), HAE-adapted strain HN (squares), or CI-1 HN (open diamonds) were assayed by hemadsorption (HAD) (at 4°C) using a series of receptor-depleted RBCs, as described in Materials and Methods. The binding (y axis) at the indicated degree of receptor depletion, expressed as milliunits of neuraminidase treatment (x axis), is expressed as the percentage of binding obtained with undepleted RBCs. The results are means ± standard errors from triplicate experiments. (B) To measure relative neuraminidase activity, cell monolayers transiently expressing reference strain HN, HAE-adapted strain HN, CI-1 HN, or empty vector were assayed at 37°C at pH 5. These results are means ± standard errors from triplicate experiments and expressed as relative fluorescent units (RFU)/s (y axis).
To compare receptor cleavage, each HN′s enzymatic activity was analyzed. HN CI-1 showed almost 6-fold-higher neuraminidase activity than the reference HN and approximately 9-fold more neuraminidase activity than the HAE-adapted HN (Fig. 1B). Thus, the HN of CI-1 has a high neuraminidase/avidity ratio (0.41) compared to that of the reference strain (0.05), indicating that CI-1 HN is less likely to engage receptor.
HN/F machineries: fusion promotion differs between clinical isolate and laboratory-adapted strains.
The HPIV3 HAE-adapted strain and the CI-1 strain attain viral titers of up to 107 and 108 PFU/ml, respectively, in HAE, and greater than 105 PFU/g of lung tissue in cotton rats compared to those of the HPIV3 laboratory reference strain, which we previously found to peak at 105 PFU/ml in HAE and 104 PFU/g of lung tissue in cotton rats (20). Viruses collected from HAE infected with the HAE-adapted HPIV3 produced small plaques (average diameter of 0.1 mm) relative to those of the reference strain (average diameter of 0.6 mm) (19), and the CI-1 virus produced plaques similar in size to those of the HAE-adapted strain (data not shown). CI-1 HN/F, like the HAE-adapted strain HN/F, fuses cultured cells little or not at all. The fusion promotion capacity of individual HN/F pairs was measured using a beta-galactosidase complementation fusion assay (24, 25). The HN from each virus was tested in a pair with its own F as well as paired with each of the other Fs; the 9 possible combinations are shown in the bars in Fig. 2. In each set of three bars, the solid black bar shows the HN specified on the x axis paired with the reference F, the gray bar shows the HN specified on the x axis paired with the HAE-adapted F, and the white bar shows the HN specified on the x axis paired with CI-1 F. Fusion mediated by the HAE-adapted strain HN/F (1491 RLU) and CI-1 HN/F (313 RLU) is minimal compared to that of the reference strain HN/F (29286 RLU), indicating that the HAE-adapted strain and CI-1 viruses that grow well in vivo carry a less efficient fusion machinery. As shown for the HAE-adapted strain (19, 20), both HN and F of the CI-1 strain contribute to the reduced fusion properties of the CI-1 HN/F pair, as is revealed by examining each of the pairings. CI-1 F permits less fusion than the reference F, even when paired with the reference HN. CI-1 HN promotes far less fusion than the reference HN, even when paired with the reference F.
FIG 2.
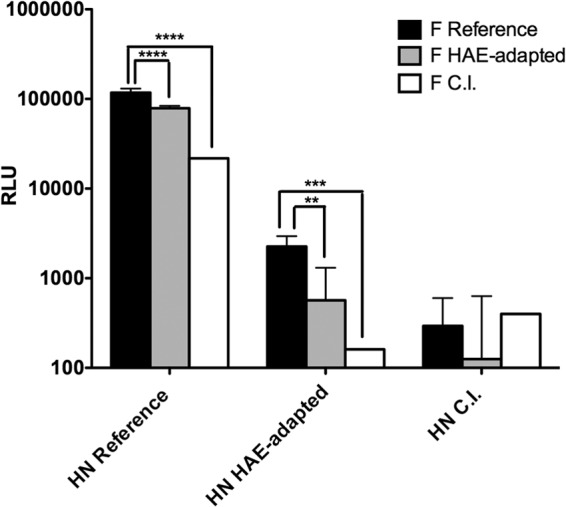
The HN/F pairs of the lung fit variants are less efficient at promoting cell-cell fusion than the proteins of the laboratory reference virus, and the difference is mediated by both HN and F partners in the pair. 293T cells coexpressing the indicated HN and F pair were allowed to fuse with receptor-bearing cells at 37°C for 3 h. Cell fusion was measured as RLU (y axis; log scale) in the presence of each HN (as indicated on the x axis) coexpressed with reference strain F (dark bars), HAE-adapted strain F (gray bars), or CI-1 F (white bars). The bars represent means ± standard errors of results from three different wells in triplicate experiments. ****, P < 0.0001; ***, P < 0.001; **, P < 0.01.
F activation is reduced in the clinical isolate strain compared to that in laboratory strains, and CI-1 F is not triggered by heat.
To study F activation for the CI-1 and laboratory-adapted strains in the absence of HN, influenza hemagglutinin (HA) protein (uncleaved) was used as a nonspecific binding protein to tether F-expressing cells to target cells (17, 31), and heat was used to activate F. The HA is not cleaved when produced in these cells and is not exposed to low pH, both requirements for the HA to express its own membrane fusing activities. We have shown previously that in the presence of uncleaved HA, heat can be used to activate F of the reference strain; at temperatures above 37°C, the reference strain F mediates fusion with RBC membranes (17). To assess F activation by CI-1 F (Fig. 3), we determined whether in the presence of uncleaved HA the reference F, F from the HAE-adapted strain, and F from CI-1 are activated by incubation for 10, 30, or 60 min at a temperature of 45°C (Fig. 3A, B, C). These conditions, as expected, provided sufficient energy to trigger the Fs of both the reference strain and the HAE-adapted strain (19) but failed to activate CI-1 F to its fusion competent state (black bars). In the control experiment shown in Fig. 3B, to address whether CI-1 F is capable of experimental activation, we coexpressed an HN molecule containing a mutation in the globular head (D216R), previously shown to eliminate neuraminidase activity while retaining binding and F activation properties and thereby conferring constitutive receptor engagement (25). In the presence of this HN, under the same conditions at 10, 30, or 60 min (Fig. 3D, E, F), while delayed at the early time points, over time CI-1 F was activated, confirming that despite being less prone to activation in the absence of a receptor-binding protein, it is capable of activation when paired with a constitutively engaged HN.
FIG 3.

Activation of F over time at 45°C either alone or with HN (reference strain, HAE adapted, or CI-1). 293T cells transiently expressing influenza virus HA (A, B, C) or HN D216R (D, E, F) and either F from the reference strain, F from the HAE-adapted strain, or F from the clinical isolate (CI-1) were overlaid with RBCs at 45°C for 10, 30, or 60 min as indicated. The percentage of RBCs released (white bars), bound (gray bars), or fused (black bars) was quantified. The values are means ± standard deviations (SD) from triplicate samples from a representative experiment.
Clinical isolate viruses are inhibited by fusion inhibitory peptides in human airway epithelium cultures, a model of the natural host.
We have described a 36-amino-acid fusion-inhibitory peptide corresponding to the HRC domain of HPIV3 F, linked at its C terminus to a cholesterol moiety by a 4-unit polyethylene glycol (PEG 4) (peptide VGPEG4-chol) (14) (Fig. 4A). This peptide inhibited infection by the HPIV3 reference strain in monolayer cell culture (14). We have also found that HN/F pairs that have enhanced kinetics of fusion activation render a virus less susceptible to peptide inhibition. Faster fusion activation decreases the time window for peptide activity (16), and HN′s ongoing activation of F after fusion peptide insertion also opposes the effect of peptide inhibitors (17). We compared this peptide's inhibitory activity against the HPIV3 reference strain, the HAE-adapted strain, and CI-1, hypothesizing that the less efficient fusion machinery of the HAE-adapted strain and CI strains might render them more sensitive to inhibition by VGPEG4-chol.
Cultured cell monolayers were infected with the HPIV3 reference strain (circles), the HAE-adapted strain (squares), or CI-1 (open diamonds) in the presence of increasing concentrations of peptide (Fig. 4B). After a 90-min adsorption period, cells were overlaid with Avicel (cellulose overlay material; FMC BioPolymer), and viral plaques were counted at 24 h. A single dose of inhibitor at 0.01 μM (10 nM) decreased the viral titer of HPIV3 CI-1 by approximately 50%, the HAE-adapted strain by approximately 40%, and the HPIV3 reference strain by only 20%. While all three strains were inhibited under these conditions, the CI and the HAE-adapted strain were more sensitive to the inhibitors than was the reference HPIV3 strain; the 50% inhibitory concentrations (IC50s) were 0.008 μM for CI-1, 0.01 μM for the HAE-adapted strain, and 0.03 μM for the reference strain HPIV3. Despite these relatively minor differences in sensitivity, all three strains are inhibited by the peptides in CV-1 cells.
We previously found that the VGPEG4-chol peptide did not curtail the replication of the HPIV3 reference strain virus in HAE (14) or in cotton rats (data not shown). We now assessed the VGPEG4-chol peptide's inhibitory activity against CI-1 in HAE, hypothesizing that the less efficient fusion machinery of the CI strain might render it sensitive to inhibition by VGPEG4-chol.
The pseudostratified epithelium was infected at the apical surface with 4,000 PFU of CI-1 in the presence of 10, 1, or 0.1 μM of VGPEG4-chol peptide (Fig. 5A). After a 90-min adsorption period, the liquid from the apical surface, containing virus and peptide, was aspirated, and the titer of virus emerging from the apical surface was measured at 1 day postinfection (Fig. 5A). A single dose of 1 μM of inhibitor decreased the viral titer of HPIV3 CI-1 by approximately 5-fold, and a single dose of 10 μM of inhibitor decreased the viral titer of CI-1 by more than 2 logs in HAE. However, note that a dose of 0.1 μM of inhibitor failed to inhibit viral growth in HAE cells, while the same dose inhibited viral entry by 90% in cultured monolayer cells (Fig. 4); HAE provides a more stringent condition for the inhibitors.
FIG 5.
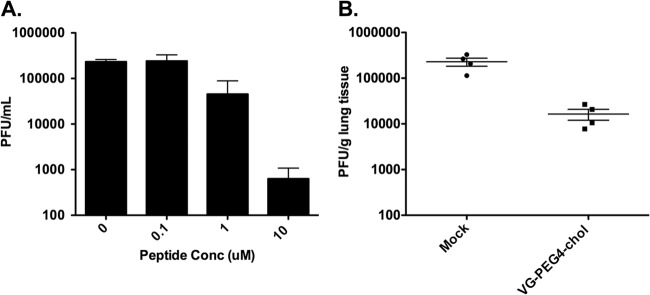
Inhibition of CI-1 infection by lipid-tagged HPIV3 F HRC peptides in HAE cells and cotton rats. (A) HAE were infected with 4,000 PFU of HPIV3 CI-1 in the absence or presence of 0.1, 1, or 10 μM of PEG 4-cholesterol peptide. After a 90-min incubation at 37°C, the inoculum was removed and the HAE were incubated at 37°C. The virus released from the apical surface was collected at 1 day after infection. The viral titer (PFU/ml, log scale) on the y axis is shown as a function of the concentration of HPIV3 HRC peptide. Data points are means (±standard errors) of results from three separate experiments. (B) Groups of 4 cotton rats were infected with HPIV3 CI-1 and treated with 2 mg/kg/day of monomeric peptide and then sacrificed 3 days postinfection. Control animals were infected with the respective virus but not treated with peptide. Viral titer (PFU/g lung tissue) (y axis) was determined by plaque assay.
Clinical isolate viruses are inhibited by cholesterol-conjugated fusion inhibitory peptides in vivo.
We evaluated the therapeutic efficacy of VGPEG4-chol in vivo. The peptides were administered to cotton rats subcutaneously (s.q.), at a dose of 4 mg/kg, twice a day for 3 days beginning 1 h after inoculation with HPIV3 CI-1. After 3 days, the animals were sacrificed and viral titers in the lungs were determined. Treatment of animals infected with HPIV3 CI-1 resulted in a 90% inhibition of viral growth in vivo (Fig. 5B). The inhibitory peptide is a potent in vivo inhibitor of clinically relevant strains of HPIV3.
Since patients rarely present at early times after infection, it is of interest to determine whether peptide inhibitors can be effective when administered later into the course. To this end, we treated cotton rats 48 h after infection with the CI and obtained a reduction of almost 1 log in viral titer (data not shown).
DISCUSSION
Fusion of enveloped viruses with receptor-bearing host cells is a critical step in viral infectivity and spread. Human parainfluenza viruses possess a paired receptor-binding protein (HN) and fusion protein (F) that in concert mediate fusion between the viral and host cell membranes. Inhibitory peptides interact with specific intermediate stages of F during fusion activation and inhibit only once F has begun its structural rearrangement and has extended, before refolding into the ultimate 6HB (16, 17, 32–34). The period of availability of this extended state—the peptide's window of time for inhibition—depends on factors such as the fusion promotion phenotype of the HN partner as well as properties intrinsic to F.
HPIV3 strains with a less active fusion machinery, with briefer receptor engagement due either to lower receptor avidity or higher neuraminidase activity, are more successful in the natural host than viruses with more avid or longer receptor engagement. As shown in Fig. 1, the clinical isolate and HAE-adapted strains have lower receptor avidity than the laboratory reference strain, suggesting that lower receptor avidity is advantageous in an environment likely to more closely resemble the lung. The HN of CI-1 also showed 600% higher receptor cleavage activity than the reference HN and thus a high neuraminidase/avidity ratio compared to that of the reference strain. This balance between receptor binding and cleavage found in the clinical strain favors short-term receptor engagement, versus the longer-term engagement favored by reference HPIV3 virus strains grown in monolayer cell culture. The higher barrier to activation that is therefore required for fusion in these strains may serve to protect F from premature activation, delaying the triggering of F until the conditions are right for fusion in the lung. CI-1 F is less readily activated by temperature than either the reference strain F or the HAE-adapted strain F, as shown in Fig. 3. Highly active fusion mediated by HN/F is not an advantage, but actually a detriment, in the natural host (19). Analysis of a series of additional clinical isolates is under way, from a collection of over 100 isolates. We have obtained the complete genome sequence and fusion experimental data for two isolates in addition to the one discussed here. The second isolate was identical in sequence to the first despite being from a different patient (and also shared fusion features); the third isolate diverged in sequence but demonstrated shared fusion features, supporting the generality of the hypothesis (data not shown).
We contend that viral fitness, defined as the ability to produce infective particles in vivo, correlates directly with peptide sensitivity and that the features of the HN/F machinery that regulate fusion simultaneously drive infectivity in vivo and sensitivity to peptide inhibition. The HN/F pairs from clinical strains, with a less active fusion machinery, are infectious in the natural host tissue and are sensitive to peptide inhibitors, suggesting that mutations in the HN/F fusion machinery that confer peptide resistance are disadvantageous to the virus. The HN/F pairs that confer resistance to peptide inhibitors in monolayer cell culture are growth-handicapped in HAE and in the cotton rat. The HPIV3 CI-1 is more sensitive than any of our lab-adapted or variant strains to peptide inhibition in HAE. We propose an approach to testing early candidate inhibitory molecules in which antivirals are tested against the circulating viral fusion machinery—clinical isolates or similar strains—in the natural host tissues as the prelude to in vivo studies. Antivirals would first be optimized using the HPIV3 laboratory reference strain in HAE, providing the most stringent conditions for inhibitors. While artificial, this condition (rapid fusion promotion and short window of opportunity for HRN binding) sets the highest bar. With the stringent requirement for efficacy, modifications to address the active fusion promotion can be tested. The most potent inhibitors would then be moved to CI assays. Using inhibitor-insensitive HPIV3 HN/F pairs (e.g., reference strain glycoprotein pairs) as a primary testing platform will aid in the design of better peptides against the clinical HPIV3 strains, which carry less efficient fusion machineries.
The results here provide an example of the pressure for infectious viruses to suit their hosts: circulating HPIV3 viruses bear HN/F fusion machineries that are well suited to their environment. CI HN and F carry several amino acid variations compared to reference strains; these likely combine to confer the observed fusion properties, and the genotype/phenotype correlation is under active investigation. However, there are no mutations in the HRN or HRC regions that might contribute to differential sensitivity to inhibitory peptides (data not shown). To interfere with viral infection, we confront the mechanisms that affect virus-cell interplay in the context of a particular host. Therefore, to dysregulate the HN/F fusion machinery, we need to move beyond cultured monolayer cells and lab strains of virus and test clinically circulating viruses in natural tissues.
Clinically useful peptide inhibitors include enfuvirtide (Fuzeon) for HIV-1 infection (35–38). While enfuvirtide provides a proof of concept for the use of fusion-inhibitory peptides, it has remained a drug of last resort, largely because it is a parenteral drug and oral HIV drugs are available; since HIV disease requires long-term treatment, parenteral administration is therefore less attractive. In the case of HPIV3, the disease is acute, and therefore even parenteral therapy will be of short duration. We are developing a formulation suitable for inhalation, to deliver the antiviral agent directly to the target host tissue. We contend that a combination of an agent like Fludase, a sialidase fusion protein which interferes with receptor binding and is currently in clinical trials for adults with HPIV (28), and fusion inhibitor peptides may be a feasible combination treatment for respiratory viruses and would reduce the likelihood of eliciting resistance.
RNA viruses rapidly evolve resistance to drugs that attack a single target. By substantially increasing the potency of peptide inhibitors through various methods of optimization, including lipid conjugation, we expect to create a barrier to resistance (23, 39, 40). We postulate that viruses that develop peptide resistance by the mechanisms identified here will simultaneously lose fitness for growth in the lung and that both properties (resistance and fitness in the natural host) are modulated by the HN/F fusion machinery. The promise of the antiviral approach we propose lies in the fact that the peptide inhibitors play against the virus' delicate balance between resistance and fitness.
Drug candidates that are considered to be promising after initial studies often fail in clinical trials, suggesting that the early development strategies failed to identify which drugs are most likely to work in humans. Early phase, even in vitro, models that set antiviral compounds on the track to clinical relevance are sorely needed. We propose first screening for molecules that pass a high bar for efficacy against fusogenic strains and then testing those molecules against HPIV3 clinical isolates in natural host tissues. This shift in strategy led us to an effective antiviral strategy for HPIV3, a strategy that may be adaptable to other pathogens.
ACKNOWLEDGMENTS
We thank Antonello Pessi for previous contributions to designing the antiviral peptide.
This work was supported by grant number R01AI31971 from the National Institutes of Health (NIAID) to A.M.
Footnotes
Published ahead of print 10 September 2014
REFERENCES
- 1.Moscona A. 2005. Entry of parainfluenza virus into cells as a target for interrupting childhood respiratory disease. J. Clin. Invest. 115:1688–1698. 10.1172/JCI25669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roghmann M, Ball K, Erdman D, Lovchik J, Anderson LJ, Edelman R. 2003. Active surveillance for respiratory virus infections in adults who have undergone bone marrow and peripheral blood stem cell transplantation. Bone Marrow Transplant. 32:1085–1088. 10.1038/sj.bmt.1704257. [DOI] [PubMed] [Google Scholar]
- 3.Nichols W, Corey L, Gooley T, Davis C, Boeckh M. 2001. Parainfluenza virus infections after hematopoietic stem cell transplantation: risk factors, response to antiviral therapy, and effect on transplant outcome. Blood 98:573–578. 10.1182/blood.V98.3.573. [DOI] [PubMed] [Google Scholar]
- 4.Maziarz RT, Sridharan P, Slater S, Meyers G, Post M, Erdman DD, Peret TC, Taplitz RA. 2010. Control of an outbreak of human parainfluenza virus 3 in hematopoietic stem cell transplant recipients. Biol. Blood Marrow Transplant 16:192–198. 10.1016/j.bbmt.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen YB, Driscoll JP, McAfee SL, Spitzer TR, Rosenberg ES, Sanders R, Moss RB, Fang F, Marty FM. 2011. Treatment of parainfluenza 3 infection with DAS181 in a patient after allogeneic stem cell transplantation. Clin. Infect. Dis. 53:e77–e80. 10.1093/cid/cir501. [DOI] [PubMed] [Google Scholar]
- 6.Cunha BA, Corbett M, Mickail N. 2011. Human parainfluenza virus type 3 (HPIV 3) viral community-acquired pneumonia (CAP) mimicking swine influenza (H1N1) during the swine flu pandemic. Heart Lung 40:76–80. 10.1016/j.hrtlng.2010.05.060. [DOI] [PubMed] [Google Scholar]
- 7.Hodson A, Kasliwal M, Streetly M, Macmahon E, Raj K. 2011. A parainfluenza-3 outbreak in a SCT unit: sepsis with multi-organ failure and multiple co-pathogens are associated with increased mortality. Bone Marrow Transplant. 46:1545–1550. 10.1038/bmt.2010.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Srinivasan A, Wang C, Yang J, Shenep JL, Leung WH, Hayden RT. 2011. Symptomatic parainfluenza virus infections in children undergoing hematopoietic stem cell transplantation. Biol. Blood Marrow Transplant. 17:1520–1527. 10.1016/j.bbmt.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loughlin GM, Moscona A. 2006. The cell biology of acute childhood respiratory disease: therapeutic implications. Pediatr. Clin. North Am. 53:929–959. 10.1016/j.pcl.2006.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Preziosi P. 2011. Influenza pharmacotherapy: present situation, strategies and hopes. Expert Opin. Pharmacother 12:1523–1549. 10.1517/14656566.2011.566557. [DOI] [PubMed] [Google Scholar]
- 11.Moscona A. 2005. Oseltamivir resistance—disabling our influenza defenses. N. Engl. J. Med. 353:2633–2636. 10.1056/NEJMp058291. [DOI] [PubMed] [Google Scholar]
- 12.DeLaMora P, Moscona A. 2007. A daring treatment and a successful outcome: the need for targeted therapies for pediatric respiratory viruses. Pediatr. Transplant. 11:121–123. 10.1111/j.1399-3046.2006.00654.x. [DOI] [PubMed] [Google Scholar]
- 13.Lambert DM, Barney S, Lambert AL, Guthrie K, Medinas R, Davis DE, Bucy T, Erickson J, Merutka G, Petteway SR., Jr 1996. Peptides from conserved regions of paramyxovirus fusion (F) proteins are potent inhibitors of viral fusion. Proc. Natl. Acad. Sci. U. S. A. 93:2186–2191. 10.1073/pnas.93.5.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Porotto M, Rockx B, Yokoyama C, Talekar A, DeVito I, Palermo L, Liu J, Cortese R, Lu M, Feldmann H, Pessi A, Moscona A. 2010. Inhibition of Nipah virus infection in vivo: targeting an early stage of paramyxovirus fusion activation during viral entry. PLoS Pathog. 6(10):e1001168. 10.1371/journal.ppat.1001168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Porotto M, Carta P, Deng Y, Kellogg G, Whitt M, Lu M, Mungall B, Moscona A. 2007. Molecular determinants of antiviral potency of paramyxovirus entry inhibitors. J. Virol. 81:10567–10574. 10.1128/JVI.01181-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Porotto M, Yokoyama C, Orefice G, Kim H-S, Moscona A. 2009. Kinetic dependence of paramyxovirus entry inhibition. J. Virol. 83:6947–6951. 10.1128/JVI.00416-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Porotto M, Devito I, Palmer SG, Jurgens EM, Yee JL, Yokoyama CC, Pessi A, Moscona A. 2011. Spring-loaded model revisited: paramyxovirus fusion requires engagement of a receptor binding protein beyond initial triggering of the fusion protein. J. Virol. 85:12867–12880. 10.1128/JVI.05873-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palermo L, Porotto M, Yokoyama C, Palmer S, Mungall B, Greengard O, Niewiesk S, Moscona A. 2009. Human parainfluenza virus infection of the airway epithelium: the viral hemagglutinin-neuraminidase regulates fusion protein activation and modulates infectivity. J. Virol. 83:6900–6908. 10.1128/JVI.00475-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palmer SG, Porotto M, Palermo LM, Cunha LF, Greengard O, Moscona A. 2012. Adaptation of human parainfluenza virus to airway epithelium reveals fusion properties required for growth in host tissue. mBio 3(3):e00137-12. 10.1128/mBio.00137-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu R, Palmer SG, Porotto M, Palermo LM, Niewiesk S, Wilson IA, Moscona A. 2013. Interaction between the hemagglutinin-neuraminidase and fusion glycoproteins of human parainfluenza virus type III regulates viral growth in vivo. mBio 4(5):e00803-13. 10.1128/mBio.00803-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ingallinella P, Bianchi E, Ladwa NA, Wang YJ, Hrin R, Veneziano M, Bonelli F, Ketas TJ, Moore JP, Miller MD, Pessi A. 2009. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc. Natl. Acad. Sci. U. S. A. 106:5801–5806. 10.1073/pnas.0901007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Porotto M, Yokoyama C, Palermo LM, Mungall B, Aljofan M, Cortese R, Pessi A, Moscona A. 2010. Viral entry inhibitors targeted to the membrane site of action. J. Virol. 84:6760–6768. 10.1128/JVI.00135-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kahle KM, Steger HK, Root MJ. 2009. Asymmetric deactivation of HIV-1 gp41 following fusion inhibitor binding. PLoS Pathog. 5:e1000674. 10.1371/journal.ppat.1000674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Porotto M, Fornabaio M, Kellogg GE, Moscona A. 2007. A second receptor binding site on human parainfluenza virus type 3 hemagglutinin-neuraminidase contributes to activation of the fusion mechanism. J. Virol. 81:3216–3228. 10.1128/JVI.02617-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palermo LM, Porotto M, Greengard O, Moscona A. 2007. Fusion promotion by a paramyxovirus hemagglutinin-neuraminidase protein: pH modulation of receptor avidity of binding sites I and II. J. Virol. 81:9152–9161. 10.1128/JVI.00888-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moosmann P, Rusconi S. 1996. Alpha complementation of LacZ in mammalian cells. Nucleic Acids Res. 24:1171–1172. 10.1093/nar/24.6.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levin Perlman S, Jordan M, Brossmer R, Greengard O, Moscona A. 1999. The use of a quantitative fusion assay to evaluate HN-receptor interaction for human parainfluenza virus type 3. Virology 265:57–65. 10.1006/viro.1999.0024. [DOI] [PubMed] [Google Scholar]
- 28.Moscona A, Porotto M, Palmer S, Tai C, Aschenbrenner L, Triana-Baltzer G, Li QX, Wurtman D, Niewiesk S, Fang F. 2010. A recombinant sialidase fusion protein effectively inhibits human parainfluenza viral infection in vitro and in vivo. J. Infect. Dis. 202:234–241. 10.1086/653621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Porotto M, Palmer SG, Palermo LM, Moscona A. 2012. Mechanism of fusion triggering by human parainfluenza virus type III: communication between viral glycoproteins during entry. J. Biol. Chem. 287:778–793. 10.1074/jbc.M111.298059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Porotto M, Salah ZW, Gui L, Devito I, Jurgens EM, Lu H, Yokoyama CC, Palermo LM, Lee KK, Moscona A. 2012. Regulation of paramyxovirus fusion activation: the hemagglutinin-neuraminidase protein stabilizes the fusion protein in a pretriggered state. J. Virol. 86:12838–12848. 10.1128/JVI.01965-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Russell CJ, Kantor KL, Jardetzky TS, Lamb RA. 2003. A dual-functional paramyxovirus F protein regulatory switch segment: activation and membrane fusion. J. Cell Biol. 163:363–374. 10.1083/jcb.200305130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harrison SC. 2008. Viral membrane fusion. Nat. Struct. Mol. Biol. 15:690–698. 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aguilar HC, Aspericueta V, Robinson LR, Aanensen KE, Lee B. 2010. A quantitative and kinetic fusion protein-triggering assay can discern distinct steps in the Nipah virus membrane fusion cascade. J. Virol. 84:8033–8041. 10.1128/JVI.00469-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Russell CJ, Jardetzky TS, Lamb RA. 2001. Membrane fusion machines of paramyxoviruses: capture of intermediates of fusion. EMBO J. 20:4024–4034. 10.1093/emboj/20.15.4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wild CT, Shugars DC, Greenwell TK, McDanal CB, Matthews TJ. 1994. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. U. S. A. 91:9770–9774. 10.1073/pnas.91.21.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wild C, Oas T, McDanal C, Bolognesi D, Matthews T. 1992. A synthetic peptide inhibitor of human immunodeficiency virus replication: correlation between solution structure and viral inhibition. Proc. Natl. Acad. Sci. U. S. A. 89:10537–10541. 10.1073/pnas.89.21.10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eckert DM, Kim PS. 2001. Design of potent inhibitors of HIV-1 entry from the gp41 N-peptide region. Proc. Natl. Acad. Sci. U. S. A. 98:11187–11192. 10.1073/pnas.201392898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kilby JM, Hopkins S, Venetta TM, DiMassimo B, Cloud GA, Lee JY, Alldredge L, Hunter E, Lambert D, Bolognesi D, Matthews T, Johnson MR, Nowak MA, Shaw GM, Saag MS. 1998. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat. Med. 4:1302–1307. 10.1038/3293. [DOI] [PubMed] [Google Scholar]
- 39.Welch BD, Francis JN, Redman JS, Paul S, Weinstock MT, Reeves JD, Lie YS, Whitby FG, Eckert DM, Hill CP, Root MJ, Kay MS. 2010. Design of a potent D-peptide HIV-1 entry inhibitor with a strong barrier to resistance. J. Virol. 84:11235–11244. 10.1128/JVI.01339-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Welch BD, VanDemark AP, Heroux A, Hill CP, Kay MS. 2007. Potent D-peptide inhibitors of HIV-1 entry. Proc. Natl. Acad. Sci. U. S. A. 104:16828–16833. 10.1073/pnas.0708109104. [DOI] [PMC free article] [PubMed] [Google Scholar]