

ABSTRACT
The leader (L) and 2A proteins of cardioviruses are the primary antihost agents produced during infection. For encephalomyocarditis virus (EMCV), the prototype of the genus Cardiovirus, these proteins interact independently with key cellular partners to bring about inhibition of active nucleocytoplasmic trafficking and cap-dependent translation, respectively. L and 2A also bind each other and require this cooperation to achieve their effects during infection. Recombinant L and 2A interact with 1:1 stoichiometry at a KD (equilibrium dissociation constant) of 1.5 μM. The mapped contact domains include the amino-proximal third of 2A (first 50 amino acids) and the central hinge region of L. This contact partially overlaps the L segment that makes subsequent contact with Ran GTPase in the nucleus, and Ran can displace 2A from L. The equivalent proteins from Theiler's murine encephalomyelitis virus (TMEV; BeAn) and Saffold virus interact similarly in any subtype combination, with various affinities. The data suggest a mechanism whereby L takes advantage of the nuclear localization signal in the COOH region of 2A to enhance its trafficking to the nucleus. Once there, it exchanges partners in favor of Ran. This required cooperation during infection explains many observed codependent phenotypes of L and 2A mutations.
IMPORTANCE Cardiovirus pathogenesis phenotypes vary dramatically, from asymptomatic, to mild gastrointestinal (GI) distress, to persistent demyelination and even encephalitic death. Leader and 2A are the primary viral determinants of pathogenesis, so understanding how these proteins cooperate to induce such a wide variety of outcomes for the host is of great important and interest to the field of virology, especially to those who use TMEV as a murine model for multiple sclerosis.
INTRODUCTION
The Cardiovirus genus of the Picornaviridae family is divided into several species and subtypes. Among the important members are encephalomyocarditis virus (EMCV), Theiler's murine encephalomyelitis virus (TMEV), and Saffold virus (1, 2). All have single-stranded, positive-sense RNA genomes containing single open reading frames. The polyproteins are cleaved co- and posttranslationally by an endogenous 3C protease (3Cpro) (3). Unique to this genus, the polyprotein begins with an amino-terminal leader protein (L) and a centrally located 2A protein that are without a homolog or analog in other viruses or cells. Together, they are primarily responsible for almost all cardiovirus antihost activities (4–8).
The EMCV L protein (LE) is 67 amino acids (aa). The Saffold (LS [71 aa]) and TMEV (LT [76 aa]) L proteins are slightly longer. The solution structure of the L protein (LM) of mengovirus (a subtype of EMCV) has been determined in free form and as bound to Ran GTPase, a key cellular participant in L-dependent activities (9). The conformation is primarily random coil, except for an amino-proximal CHCC zinc finger motif (aa 10 to 22). The structure of the remainder relies on induced-fit contacts dependent upon a specific binding partner or partners. The mapped functional units, in addition to the zinc finger, include a central “hinge” region (aa 35 to 44), essential to Ran interactions, and an acidic domain (aa 37 to 52) that confers an overall pI of 3.8 to the protein (10). The LS and LT homologs are similar, with equivalent low pIs, except they also have short, characteristic theilovirus domain (13-aa) and Ser/Thr domain (12-aa) insertions, configured putatively as linked helices, near their respective C termini (11).
For any LX to function in cells, it must be phosphorylated. The required sites include Tyr41 and Thr47 for LE, Ser57 for LT, and Thr58 for LS. Kinases CK2, SYK, and AMPK participate in these modifications, but the precise timing of the reactions and stepwise requirements during infection are not yet clearly understood (11, 12). It is clear, however, that during infection or in recombinant form, the introduction of phosphorylation-competent LE, LS, or LT into cells induces a rapid inhibition of active nucleocytoplasmic trafficking (NCT) (8, 12). The mechanism requires, in addition to L phosphorylation, specific L interactions with Ran GTPase, a key cellular trafficking regulator. When aided by catalytic amounts of nuclear Ran guanine-nucleotide exchange factor (RCC1), LM binds tightly to Ran (equilibrium dissociation constant [KD] of 3 nM), diverting its normal activities into antihost events (13). The consequence is induced hyperphosphorylation of Phe/Gly-containing nuclear pore proteins (nucleoporins [Nups]) by cellular kinases in the p38 and Erk1/2 pathways (8, 14, 15) and subsequent cessation of active NCT. It has been proposed that LM:Ran complexes achieve this by trapping exportin-bound activated kinases within the nuclear pores (nuclear pore complexes [NPC]) (9). Since Ran-dependent NCT is essentially shut down, the movement of cellular proteins and RNA through the NPC is reduced to that permitted by diffusion alone. Recombinant LM, LE, LT, or LS alone is necessary and sufficient for observing these effects when their genes are transfected into cells (11, 14). During infection, however, cardiovirus L proteins are not the exclusive antihost activators.
The functions of protein 2A are not as well characterized. EMCV 2A (143 aa) is translated between the P1 (capsid) and P2/3 (replication) regions of the polyprotein. The protein has a distinctive C-terminal 13- to 16-aa “scission cassette” (Fig. 1) ending with an Asn-Pro-Gly-Pro motif (NPG/P). The unit functions in viral or exogenous contexts, through a cotranslational ribosome-skipping mechanism, separating otherwise cojoined proteins between the Gly and Pro residues (16). The NPG/P event provides primary scission of cardiovirus polyproteins. The N-terminal release, as with the C-terminal release of L, from an L-P1-2A precursor, is subsequently catalyzed by viral 3Cpro. Antibodies specific to EMCV 2A track the dominant cellular localization to nucleoli during infection, although there is also significant cytoplasmic accumulation (4). The protein has a very basic pI of 9.67, which presumably allows it to remain nucleolar through rRNA binding contacts. Mutagenic mapping has identified a ribosome protein-like nuclear localization signal (NLS) and a C-proximal eIF4E binding site, which partially overlaps the scission cassette sequences and are common to all known cardioviruses (17). Similar mutations, tested during infection, link the activities of 2A (EMCV) to virus-induced shutdown of cap-dependent translation (4, 18). The protein influences 4EBP1 pathways in certain cell types, and moreover, 2A-deficient viruses can be rescued by chemical inhibitors of mTOR and phosphatidylinositol 3-kinase (PI3K), elements required for cap-dependent but not virus-dependent translation (19). During infection, a portion of 2A is found in association with 40S, but not 60S or 80S ribosomal subunits, though no determined mechanism yet links these observations (18).
FIG 1.
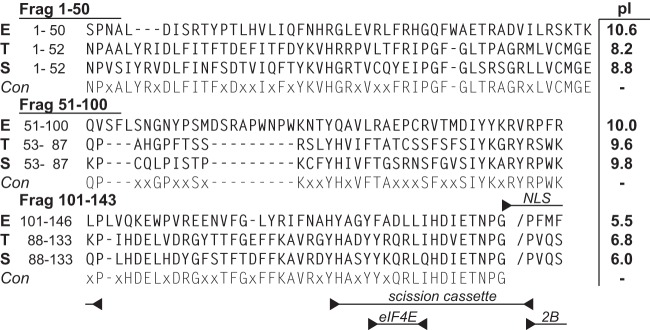
2A protein sequences of EMCV-R (sequence E; GenBank accession no. ABC25550), TMEV BeAn (sequence T; Swiss-Prot accession no. P08544), and Saffold-2 (sequence S; GenBank accession no. AFP86294) were aligned with (Lasergene 9) MegAlign software using the Jotun Hein method. The consensus (Con) required 2 or more identities. Tested protein fragments (LE), their pI values, and functional motifs are indicated.
Cardiovirus L and 2A interactions with various cellular partners have been the subject of much study and speculation (13, 14, 17, 18). As part of this process, we employed yeast two-hybrid systems to fish out unknown, potential reaction candidates (unpublished). Given their reciprocal pIs, perhaps it should not have been a surprise that both came back as preferred partners of each other. The specificity and required elements for these reactions have now been documented by mutagenesis and biochemistry. Within the virus life cycle, including those of theilovirus and Saffold virus, the mutual L:2A pathways may explain why these proteins' antihost activities should probably not be considered independently. Phenotypes attributed to one protein are, in some steps, codependent upon the other.
MATERIALS AND METHODS
Recombinant constructions.
The N-terminal His-tagged GB1 gene for parental plasmid pT-hGB1 originated from a pET30-GBFusion1 vector (a kind gift from John Markley), as excised by PCR using appropriate primers. After digestion with NcoI and HindIII, the amplicon was gel purified and then ligated into pTri-Ex 1.1 (Novagen) using the same restriction sequences. The EMCV 2A gene from pEC9 (20) was amplified in parallel and then digested with HindIII and XhoI. Plasmid pT-hGB1-2A substituted this fragment into the corresponding sites of parental pT-hGB1. Bacterial expression produces an in-frame His-tagged GB1-2A fusion protein (hGB1-2A). Derivative plasmids, using different primer sets, were equivalent but included only those EMCV 2A sequences encoding amino acids 1 to 50, 51 to 100, or 101 to 143. Expression plasmids for Saffold (SafV-2) and TMEV (BeAn) 2A were of similar configuration and founded on amplicons generated from infectious cDNAs (generous gifts from Howard Lipton). Leader–glutathione S-transferase (GST) fusion plasmids for EMCV (LE-GST), Saf-2, (LS-GST), and BeAn (LT-GST) have been described previously (12), as have GST-LE proteins with substitution mutations, GST-LK35Q, GST-LD37A, and GST-LW40A (21). The sequences of all materials were verified by restriction mapping and Sanger sequencing.
Protein purification.
For hGB1-2A synthesis, plasmids were transformed into Rosetta BL21(DE3)/pLac I cells (Novagen). Single colonies were picked then grown overnight in 2XYT (1% glucose, 34 μg/ml chloramphenicol, 50 μg/ml ampicillin) at 30°C. The stocks primed larger cultures, which at an optical density at 600 nm (OD600) of 0.6 were treated with 1 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG). Growth continued (30°C) until harvest at an OD600 of 2.4 to 3.2. The cells were collected (6,000 × g, 15 min, 4°C) and frozen at −80°C. Expressed proteins were extracted after resuspending the pellets in His-2A buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, 25% vol/vol glycerol) containing 1 mM phenylmethylsulfonyl fluoride (PMSF). After incubation with lysozyme (1 mg/ml, 30 min, 4°C), the DNA was sheared by sonication. The soluble fraction (20,000 × g, 45 min, 4°C) was filtered (0.2-μm-pore filter; GE Healthcare) and then loaded onto a HisTrap high-performance (HP) column (GE Healthcare). Bound proteins were eluted with an imidazole step gradient (20, 60, 120, 250, and 500 mM). Relevant fractions were pooled and concentrated and then applied to Sephacryl S-100 columns (GE Healthcare). Separation was by size exclusion (50 mM NaH2PO4, 300 mM NaCl, 25% vol/vol glycerol [pH 7.4]). The proteins were collected, dialyzed (same buffer), concentrated, and then stored at −80°C. The expression and purification of C-terminal GST-tagged leader proteins, LE-GST, LT-GST, and LS-GST were described previously (11), as were protocols for human Ran GTPase (N-terminal His tagged) and human guanine nucleotide exchange factor, RCC1 (N-terminal GST tagged) (13).
GST-L-2A interactions within infected lysates.
HeLa cells were infected with vEC9 at a multiplicity of infection (MOI) of 30. After 6 h, the cells were washed three times with phosphate-buffered saline (PBS) followed by incubation with Promega passive lysis buffer (PLB). The resulting whole-cell lysates were precleared with glutathione-Sepharose beads (GE Life Sciences) overnight at 4°C. They were then incubated with prebound GST or GST-L (10 μg) glutathione-Sepharose beads for 2 h at 37°C. Beads were washed (5× in PLB) and then boiled before the proteins were fractionated by SDS-PAGE and detected by Western analyses using murine anti-GST (product 71097; Novagen) or anti-2A (22) with an anti-murine secondary antibody (product A2554; Sigma-Aldrich).
Recombinant protein interactions.
Protein interaction assays took advantage of the respective GST and hGB1 tags on the LX and 2A recombinant panels. When GST proteins were the baits, they were prebound to glutathione-Sepharose 4B beads (10-μl reaction mixtures with 50 nM protein, 50 mM HEPES, 125 mM NaCl, 0.5% NP-40 [pH 7.4] at 4°C overnight). The beads were collected (500 × g), washed with the same buffer (2 times), and then incubated (1 h at 25°C) with increasing amounts of prey protein (e.g., hGB1-2A at 5 to 100 nmol/sample). For competition experiments between 2A and Ran, the bait protein (GST-LE or mutated variants) was prebound to beads as described above, before the incubation (2 h) with various prey combinations (at 50 nM). All reaction mixtures with Ran also included catalytic amounts of RCC1 (at 1 nM). Values for competition experiments were normalized to the amount of hGB1-2A pulled down by wild-type GST-LE. Reciprocal experiments used hGB1 protein baits bound to Ni2+-charged chelating-Sepharose beads in buffer (50 nM protein in 50 mM HEPES, 400 mM NaCl, 50 mM imidazole [pH 7.4]) for the capture of GST-LE preys (5 to 100 nmol/sample). Binding affinity reactions were similar, except for the variable salt concentrations (125 to 500 mM NaCl). Interspecies Lx-GST (on beads) and hGB1-2A reactions were performed as described above. For all reactions, after extensive washing (3 times) to reduce background signals, the bead-bound proteins were released by boiling in SDS buffer, fractionated by SDS-PAGE, detected by Coomassie staining, quantitated, and compared to input levels of GST-L or hGB1-2A (ImageQuant software). Alternatively, after transfer to polyvinylidene difluoride (PVDF) membranes, the proteins were detected by Western analyses. The antibodies included murine anti-GST, goat anti-Ran (product sc-1156; Santa Cruz Biotechnologies), and anti-murine secondary and anti-goat secondary antibody (A5420; Sigma). The GB1 tag is a derivative of the IgG binding B1 domain of streptococcal protein G (23). Assays to detect this protein (anti-GB1) need only the murine secondary antibody.
GST-LE phosphorylation.
Bait (GST-LE) and prey (hGB1-2A) complexes bound to glutathione-Sepharose 4B beads were established and collected as described above, except during the protein capture (1 h, 20°C) the prey concentration varied (2.5, 10, or 40 nM). Once the beads were collected, they were resuspended in the manufacturers' buffers supplemented with 5.0 μCi [γ-32P]ATP (3,000 Ci/mmol [10 mCi/ml]), 10 U of CK2 (New England BioLabs), 10.3 U of SYK (SignalChem), or 10 U of both CK2 and SYK. After the reaction (37°C, 60 min), the beads were washed (3 times) with PBS buffer (plus 500 mM NaCl and 0.02% Triton X-100) and then boiled in SDS, before protein fractionation by SDS-PAGE. Detection was by silver stain or phosphor screen, as visualized with a Typhoon scanner (GE Healthcare).
SPR.
Equilibrium binding studies used a BIAcore 2000 instrument (BIAcore AB, Uppsala, Sweden) loaded with CM5 research-grade sensor chips (GE Healthcare). Anti-GST (described above) was covalently attached to the chips with amine-coupling chemistry. GST-LE (5 μg/ml [120 nM]) diluted in surface plasmon resonance (SPR) buffer (10 mM Bis-Tris propane, 100 mM NaCl, 0.005% Tween 20 [pH 7.4]) flowed over individual chip cells at a rate of 10 μl/min (75 μg total, 25°C). The buffer was then changed to include hGB1-2A (or iterations) in various concentrations (10, 20, or 50 μg/ml at 20 μl/min). The total injection time was 450/600 s (120/150 μl total), with a dissociation time of 120 s. Chip surfaces were regenerated using 20 mM piperazine (pH 9.0) with 2 M KCl. Automatic parallel reference subtractions were performed with an antibody-only lane to account for nonspecific and bulk interactions. BIA evaluation software (version 4.1) calculated the normalized binding constants specific to LE and 2A. Association and dissociation rates were determined independently from best-fit curves, using Langmuir calculations at steady-state levels. The slope and y-intercept values, plotted in Excel, recording the concentration of analyte (hGB1-2A) against the observed rate constant (kobs), were used to determine the final KD.
RESULTS
LE-2A interactions.
The small size and high charge of cardiovirus LX proteins make them difficult to detect in experiments involving Western blot assays unless they are fused to tags like GST (220 aa). Such tags, whether C linked or N linked, do not affect the structure or biological activity of LE constructs (14). Likewise, the cardiovirus 2A proteins are relatively insoluble, unless they too are coupled to tags like hGB1 (56 aa) or maltose-binding protein (MBP [396 aa]) (unpublished). The combined tags make protein purification easier, and binding studies can take advantage of high-specificity commercial reagents. When GST-LE was reacted with infected HeLa cell lysates, the extracted binding partners included 2A (Fig. 2), as had been suggested by previous yeast two-hybrid assays (unpublished). The converse experiment with hGB1-2A baits was uninformative because native LE cannot be detected with Western blots. Instead, recombinant GST-LE and hGB1-2A were tested together in reciprocal pulldown assays, dependent upon their respective tags, and were shown to interact with each other (Fig. 3A). In multiple experiments, the “bait” protein captured “prey” in approximate proportion to its solution concentration, reaching saturation at about a 1:2 molar ratio (100 nmol/reaction of prey), regardless of the bead-bound protein. The interactions were not due to either protein's tag, as these alone were unable to capture cognates. Formation of such complexes withstood the presence of 250 to 500 mM salt (Fig. 3B), indicating a reasonably specific affinity between the LE and 2A proteins with a strength that could not be due to simple charge-charge interactions (i.e., pI 3.8 versus 9.7).
FIG 2.
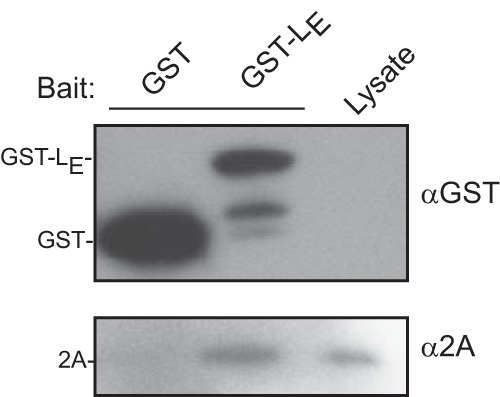
EMCV-infected HeLa lysates (100 μl) were incubated with either GST or GST-LE (10 μg/reaction). Beads were washed, boiled, and analyzed by Western blot analysis with anti-GST and anti-2A.
FIG 3.
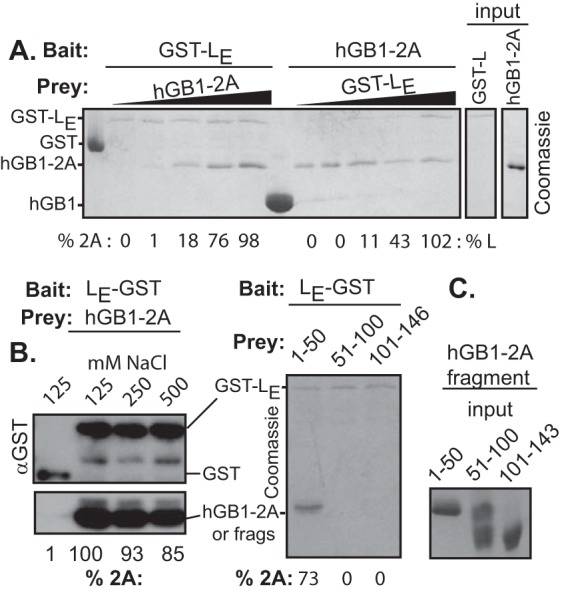
Pull-down assays. (A) The indicated recombinant “baits” and “preys” were used in reciprocal pulldown assays. The baits were held at 50 nM/reaction, while the prey concentrations varied (5 to 100 nM/reaction) as described in Materials and Methods. Band quantitation is relative to the highest value input (50 nM). (B) Similar to panel A, the association reactions and wash reactions used the indicated salt concentrations. Band detection was by Western blot analysis with anti-GST. hGB1(-2A) is recognized by the secondary anti-murine antibody. (C) Similar to panel A, EMCV 2A fragments were reacted with bead-bound LE-GST. Captured prey was detected with Coomassie staining. The right-hand marker panel shows the sizes of input 2A fragments were they to be detected.
As a better assessment of this complex, the binding constant was determined by surface plasmon resonance. SPR is essentially a pulldown assay using a mass-sensitive chip. In this case, three concentrations of hGB1-2A analyte were reacted over an antibody-fixed GST-LE surface, and the increased mass over 450 or 600 s of exposure was recorded in a sensorgram. A plot series is shown in Fig. 4A. From these curves, including the decay phase after the analyte is flushed, normalized values for kobs can be calculated for each concentration (Fig. 4B). These in turn extrapolate to absolute on/off rates, where kon = (1.4 ± 0.1) × 10−3 M−1 s−1 and koff = (2.1 ± 0.1) × 10−6 s−1, and a KD for the LE-2A reaction, determined here as 1.5 ± 0.1 μM. The shapes of the sensorgram curves are consistent with 1:1 stoichiometry. Higher-order cooperative interactions would have different plots (Fig. 4A) and nonlinear extrapolated slopes (Fig. 4B).
FIG 4.
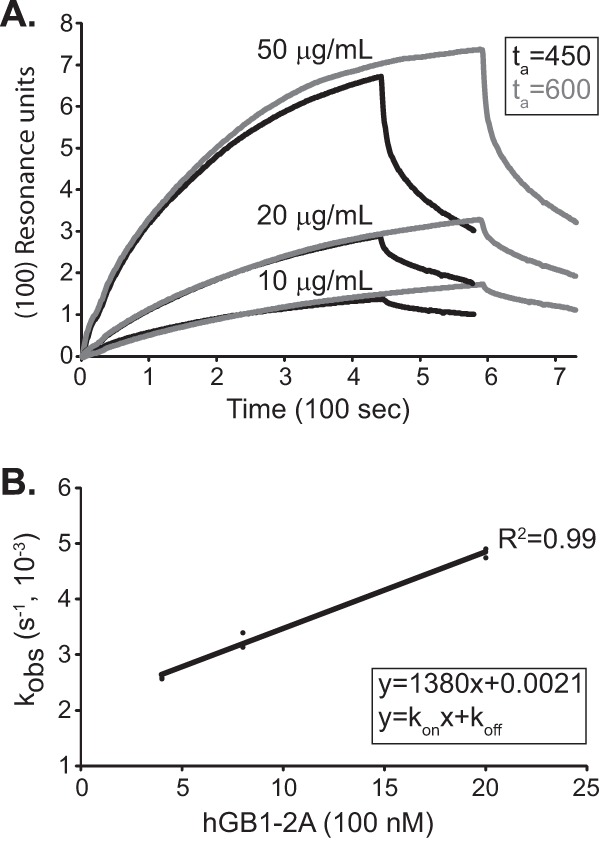
SPR of LE-2A binding. (A) SPR sensorgram curves for hGB1-2A flowed over GST-LE-bound anti-GST surfaces on CM5 chips at the indicated concentrations. The association phase (ta)was either 450 or 600 s with a dissociation phase of 120 s. Reference subtraction used a lane containing only anti-GST (not shown). (B) BIAevaluation software calculated association and dissociation rates based on the sensorgram curves in panel A, using Langmuir fitting. kobs was plotted against the concentration of hGB1-2A to extract the normalized KD using a best-fit line in Excel.
Homolog interactions.
Among LE, LS, and LT sequences (67 to 71 amino acids [aa]) for which there are cDNAs, there is about 29% shared amino acid identity and 42% amino acid similarity (11). The equivalent 2A proteins vary in length from 133 to 143 aa and share 14% identity (Fig. 1) with 39% similarity. If properly controlled, capture experiments can provide a measure of relative affinity for panels of similar proteins. In this case, a C-terminally tagged LX-GST panel was chosen as baits because Saffold virus and theilovirus leader proteins become biologically inactive if the tag is attached N terminally (11). These and cognate hGB1-2A proteins were isolated, quantitated, and then reacted in matched samples (Fig. 5). In repeated experiments (all data not shown), there was cross-reactivity with every combination, but surprisingly, the 2A from EMCV was always the most reactive with each of the leaders regardless of species. LS-GST and LT-GST bound nearly twice as much of this protein as they did their homologous counterparts. The EMCV 2A was clearly the preferred binding partner.
FIG 5.
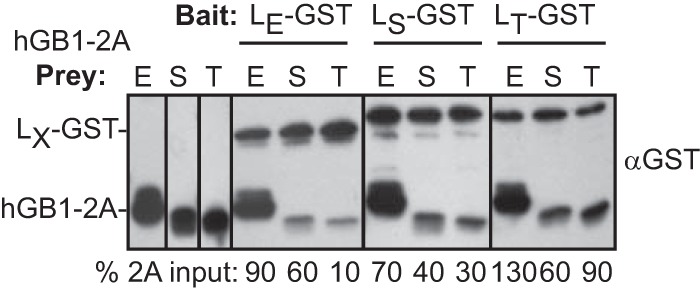
Intra- and interspecies reactions. LE-GST, LS-GST, and LT-GST baits were reacted with hGB1-2A prey from EMCV (E), SafV (S), or TMEV (T). Bead-bound protein was fractionated and then detected by Western blot analysis with anti-GST. The secondary (anti-murine) monoclonal antibody is also reactive with GB1 sequences. Band intensity values (hGB1-2A [ImageQuant]) were normalized to each respective input lane.
Required 2A elements.
No 2A structure is available for any cardiovirus. For all of these sequences though, the C-terminal third of the protein maintains characteristics of an extended alpha helix (17). The first and second portions are not responsive to structure predictions. These regions are more variable in sequence between species. Most of the basic residues contributing to the pI map in these upstream regions, imparting the clearest pI differential to the first two-thirds of the protein (Fig. 1). Among 2As, this portion of the EMCV protein is the most basic. The EMCV 2A binding segments making contact with LE were approximated by dividing the gene into fragments encoding residues from positions 1 to 50 (fragment 1–50), 51 to 100 (fragment 51–100), and 101 to 143 (fragment 101–143). The peptides were then expressed with hGB1 tags for solubility. In turn, these served as prey in LE-GST capture experiments (Fig. 3C). GST-LE was able to pull down a significant portion of fragment 1–50 (73% compared to input), but neither of the other fragments was reactive in this context. Therefore, fragment 1–50 probably contains the dominant 2A determinants for LE interactions, at least as measured in the absence of an intact 2A conformation. Follow-up SPR experiments with the hGB1-2A1–50 fragment and GST-LE were inconclusive because the much smaller mass change of the prey did not give reproducible signals, especially at low concentrations.
LE partner competition.
In the presence of catalytic amounts (1 nmol/reaction) of RCC1, LE binds Ran GTPase at 1:1 stoichiometry with a KD of 3 nM (13). The KD for LE:2A, as determined above by SPR, is much higher (1.5 μM), so in theory, Ran should be able to outcompete 2A if the preferred LE bait sites overlap. Ran interacts with the central hinge region of the LE protein (9), within which mutations at K35, D37, and W40 mark the most significant sites (21). 2A (50 nmol), Ran (50 nmol), or a mixture of both preys (50 nmol each) was added to GST-LE bait, allowed to reach equilibrium (2 h), and then assessed for relative 2A binding. All reactions with Ran included catalytic amounts (1 nM) of GST-RCC1. A representative gel series is shown in Fig. 6A. The same experiment was repeated (n = 4) and averaged to produce the values indicated in Fig. 6B. They show that hGB1-2A binding was reduced by 16 to 26% when the bait GST-LE had any of the key mutations in the hinge region, even in the absence of Ran (white bars). Ran, when present, bound simultaneously to the same GST-LE beads with essentially 1:1 stoichiometry (13). The combined preys reduced hGB1-2A binding to the wild-type GST-LE by 22%, and clearly that binding was further weakened with mutant LE sequences, because Ran then displaced even more hGB1-2A (29 to 60% reduced binding [gray bars]). Still, that Ran did not entirely displace hGB1-2A from the bound GST-LE suggests these proteins have partially overlapping but not mutually exclusive preferences for LE sites. While these mutations were previously shown to reduce L-Ran binding (21) in the absence or RCC1, as shown here, addition of this Ran-activating factor can help overcome some of the mutational inhibition.
FIG 6.
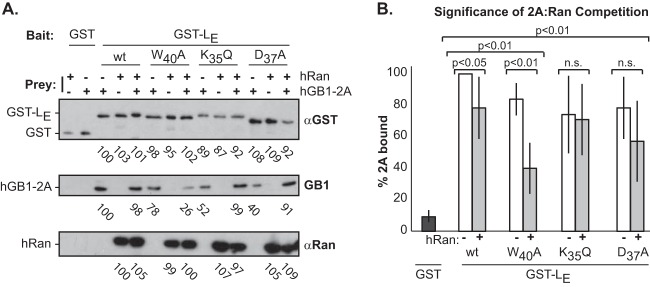
Ran and 2A competitions. (A) Recombinant GST or GST-LE with substitution mutation K35Q, D37A, or W40A was prebound to beads as baits and then reacted with equimolar (50 nM) hGB1-2A, hRan (with 1 nM GST-RCC1), or a combination of hGB1-2A and hRan. Values for the captured proteins were detected by ImageQuant software and normalized to levels of hGB1-2A pulled down by wild-type LE. (B) The experiment in panel A was repeated (n = 4), values were averaged over all experiments and plotted to show the variance. Standard t test significance for values ± Ran is indicated. n.s., not significant.
2A impedes LE phosphorylation.
During infection, LE is sequentially phosphorylated at T47 and Y41 by the CK2 and SYK enzymes, respectively (12). The addition process, even with recombinant proteins, is obligatory, because mutations that block the CK2 reaction (e.g., T47A) also prevent the SYK reaction at Y41 unless the mutation is a phosphomimetic (e.g., T47E) (12). When added to GST-LE, neither hGB1 nor hGB1-2A prevented the incorporation of 32P, as long as the bait protein had a wild-type CK2 site at T47 (Fig. 7A). However, when treated with CK2 and then SYK, the Y41 site became masked in the presence of hGB1-2A (Fig. 7B). The control hGB1 alone did not allow this masking, either on the wild-type GST-LE or with the phosphomimetic bait, T47E. Therefore, Y41, which lies to the C-terminal side of the LE hinge domain, is among the likely contact sites for 2A binding. This site and T47 were found to be solvent exposed when LE binds Ran (9).
FIG 7.

Phosphorylation of LE:2A complexes. (A) GST-LE and mutant derivatives (bait) and hGB1-2A or hGB1-2A (prey) bead-bound complexes were reacted with CK2 in the presence of [32P]ATP. (B) Similar to panel A, the SYK plus [32P]ATP reactions were preceded by incubation with CK2 and cold ATP as described in reference 12. Fractionated proteins were detected by phosphorimaging.
DISCUSSION
At the earliest stages of a cardiovirus infection, viral proteins are in low abundance, and yet the virus must take swift action to combat innate host antiviral defenses. The LE protein of EMCV achieves this by leveraging a cell kinase-based phosphorylation cascade directed against Phe/Gly-containing nuclear pore proteins (15). The effect is a rapid shutdown of active transport of macromolecules across the NPC (8). Addition of LE to permeabilized cells or transfection of LE-encoding cDNA into intact cells can readily demonstrate this effect (8, 15). However, in both cases, the viral protein concentrations are effectively much higher than the scant few molecules initially translated from an infecting genome. We previously hypothesized that the viral protein 2A gene, which encodes an active nuclear localization signal, may help shuttle LE to the nuclear rim, thereby placing it directly into contact with Ran GTPase, the required LE activation partner (13). This would, however, require a physical interaction between LE and 2A, either directly or indirectly.
These proteins, encoded at opposite ends of the L-P1-2A precursor, are released by tandem cleavages with 3Cpro almost as soon as the protease is available (24–26). Their respective pIs as the most basic (2A) and acidic (LX) units in the polyprotein should make obvious the potential for interaction. Indeed, we demonstrated here that LX and 2A from three different cardioviruses can bind directly in vitro and in any combination from any virus (Fig. 5). The binding is stoichiometric. For EMCV 2A, it can be almost entirely recapitulated with a shorter fragment containing only the first 50 amino acids (Fig. 3C). Theilovirus and Saffold virus LX cognates reacted with all homologous 2A proteins but preferred the sequence from EMCV, presumably because that particular 1–50 segment is almost 2 logs more basic than their normal partners (Fig. 1). When measured by SPR, the EMCV proteins had a KD of 1.5 μM that was partially responsive to salt, but the majority of complexes were still able to form at concentrations up to 500 mM. Therefore, these proteins must have a degree of specificity in addition to simple charge-charge interactions.
The preferred partner for LE, Ran, binds with a much lower KD (3 nM). Competitions between 2A and Ran for bead-bound LE, however, suggest that both proteins can be accommodated simultaneously, implying only partially overlapping binding sites. Mutated LE sequences with weaker binding affinities for 2A (e.g., W40A) were more readily displaced by Ran (Fig. 6A). Interestingly, LE-2A interactions also clearly masked LE residue Y41, one of two crucial phosphorylation sites for the activity of LE. Phosphorylation is not required for LE interactions with Ran, but without these modifications, the subsequent complex cannot proceed to ternary or quaternary reactions required to trigger the Nup phosphorylation cascade (9). Therefore, logically, 2A cannot remain perpetually bound to LE during the normal course of events during infection. For complete LE phosphorylation after it is bound to Ran, the 2A protein must be released. Since LE-Ran interactions are facilitated by the conformational morphing of Ran, as catalyzed by RCC1 tethered to chromatin just inside the nuclear rim, the results are consistent with a 2A-dependent trafficking pathway of LE to nuclear RCC1 sites, where it is displaced by Ran. Subsequently, LE bound to Ran can be dual phosphorylated and primed for Nup inhibition activities. The freed 2A then presumably proceeds to nucleoli and initiates its independent cellular translation inhibition activities.
If this scenario is true, it can explain some previously observed experimental anomalies in 2A and LE mutational studies. For example, mutants with deletions in LE (27) or 2A (17) or chimeric viruses exchanging EMCV and theilovirus LX or their 2A (25) typically have incomplete or improperly processed L-P1-2A regions. Presumably, the L-2A interaction, even in this precursor stage, could facilitate proper P1 folding, creating the requisite conformational substrates for sequential reactions with 3Cpro. Without this interaction, disrupted by the deletion or mutation of either protein, 3Cpro would not efficiently process protomers into functional assembly intermediates. The LX-2A binding reactions we tested with EMCV, Saffold virus, and theilovirus proteins showed that some chimeric combinations had poorer affinities. For example (Fig. 5), LE and theilovirus 2A bind to only 10% saturation compared to LE and EMCV 2A. When tested in a virus context, it has been reported the same homologous swaps have, as expected, concordant processing and replication defects (25, 28). The results also imply that studies aimed at mutagenesis of LT domains (29, 30), with regard to its assigning nuclear pore activities or effects on cytokine trafficking, could easily cause unintended disruption of 2A-dependent trafficking or reduced LT:2A affinities that would manifest as phenotypes with impeded LT localization to the nuclear pore and subsequent shutoff of NCT. All told, our findings show that the antihost activities of cardiovirus LX and 2A proteins should not be considered independent of one another. Some phenomena previously ascribed solely to 2A or to LX may result from their affinity for each other.
ACKNOWLEDGMENTS
This work was supported by NIH grant AI017331 to A.C.P. and NIH training grants GM07215 to R.V.P. and CVA347300 to V.R.B.-D. Additional support was from a University of Wisconsin Science and Medicine Graduate Research Scholars Fellowship (V.R.B.-D.) and University of Wisconsin Department of Biochemistry Wharton Fellowship (R.V.P.). Instrument data were obtained at the University of Wisconsin Biophysics Instrumentation Facility, which was established with support from the University of Wisconsin and grants BIR-9512577 (NSF) and S10 RR13790 (NIH).
We thank Howard Lipton for the generous gift of SafV-2 and TMEV (BeAn) cDNAs, Lukasz Ochyl for cloning help, and Darrell McCaslin for advice and expertise.
Footnotes
Published ahead of print 10 September 2014
REFERENCES
- 1.Pevear DC, Calenoff MA, Rozhon EJ, Lipton HL. 1987. Analysis of the complete nucleotide sequence of the picornavirus Theiler's murine encephalomyelitis virus indicates that it is closely related to cardioviruses. J. Virol. 61:1507–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zoll J, Hulshof SE, Lanke K, Lunel FV, Melchers WJG, Schoondermark-van de Ven E, Roivainen M, Galama JMD, van Kuppeveld FJM. 2009. Saffold virus, a human Theiler's-like cardiovirus, is ubiquitous and causes infection early in life. PLoS Pathog. 5:e1000416. 10.1371/journal.ppat.1000416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martinez-Salas E, Ryan MD. 2010. Translation and protein processing, p 141–161 InEhrenfeld E, Domingo E, Roos RP. (ed), The picornaviruses. ASM Press, Washington, DC. [Google Scholar]
- 4.Aminev AG, Amineva SP, Palmenberg AC. 2003. Encephalomyocarditis viral protein 2A localizes to nucleoli and inhibits cap-dependent mRNA translation. Virus Res. 95:45–57. 10.1016/S0168-1702(03)00162-X. [DOI] [PubMed] [Google Scholar]
- 5.Zoll J, Galama JMD, van Kuppeveld FJM, Melchers WJG. 1996. Mengovirus leader is involved in the inhibition of host cell protein synthesis. J. Virol. 70:4948–4958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Pesch V, van Eyll O, Michiels T. 2001. The leader protein of Theiler's virus inhibits immediate-early alpha/beta interferon production J. Virol. 75:7811–7817. 10.1128/JVI.75.17.7811-7817.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zoll J, Melchers WJ, Galama JM, van Kuppeveld FJ. 2002. The mengovirus leader protein suppresses alpha/beta interferon production by inhibition of the iron/ferritin-mediated activation of NF-kappa B. J. Virol. 76:9664–9672. 10.1128/JVI.76.19.9664-9672.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Porter FW, Palmenberg AC. 2009. Leader-induced phosphorylation of nucleoporins correlates with nuclear trafficking inhibition of cardioviruses. J. Virol. 83:1941–1951. 10.1128/JVI.01752-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bacot-Davis VR, Ciomperlik JJ, Cornilescu CC, Basta HA, Palmenberg AC. Solution structures of Mengovirus leader protein, its phosphorylated derivatives, and in complex with RanGTPase. Proc. Nat. Acad. Sci. U. S. A., in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cornilescu CC, Porter FW, Zhao Q, Palmenberg AC, Markley JL. 2008. NMR structure of the Mengovirus leader protein zinc-finger domain. FEBS Lett. 582:896–900. 10.1016/j.febslet.2008.02.023. [DOI] [PubMed] [Google Scholar]
- 11.Basta HA, Palmenberg AC. 2014. AMP-activated protein kinase phosphorylates EMCV, TMEV, and SafV leader proteins at different sites. Virology 462–463:236–240. 10.1016/j.virol.2014.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Basta HA, Bacot-Davis VR, Ciomperlik JJ, Palmenberg AC. 2014. Encephalomyocarditis virus leader is phosphorylated by CK2 and Syk as a requirement for subsequent phosphorylation of cellular nucleoporins. J. Virol. 88:2219–2226. 10.1128/JVI.03150-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petty RV, Palmenberg AC. 2013. Guanine-nucleotide exchange factor RCC1 facilitates a tight binding between encephalomyocarditis virus leader and cellular Ran GTPase. J. Virol. 87:6517–6520. 10.1128/JVI.02493-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Porter FW, Bochkov YA, Albee AJ, Wiese C, Palmenberg AC. 2006. A picornavirus protein interacts with Ran-GTPase and disrupts nucleocytoplasmic transport. Proc. Natl. Acad. Sci. U. S. A. 103:12417–12422. 10.1073/pnas.0605375103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Porter FW, Brown B, Palmenberg A. 2010. Nucleoporin phosphorylation triggered by the encephalomyocarditis virus leader protein is mediated by mitogen-activated protein kinases. J. Virol. 84:12538–12548. 10.1128/JVI.01484-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Donnelly ML, Luke G, Mehrotra A, Li X, Hughes LE, Gani D, Ryan MD. 2001. Analysis of the aphthovirus 2A/2B polyprotein ‘cleavage' mechanism indicates not a proteolytic reaction, but a novel translational effect: a putative ribosomal ‘skip.' J. Gen. Virol. 82:1013–1025. [DOI] [PubMed] [Google Scholar]
- 17.Groppo R, Brown BA, Palmenberg AC. 2011. Mutational analysis of EMCV 2A protein identifies a nuclear localization signal and an eIF4E binding site. Virology 410:257–267. 10.1016/j.virol.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Groppo R, Palmenberg A. 2007. Cardiovirus 2A protein associates with 40S but not 80S ribosome subunits during infection. J. Virol. 81:13067–13074. 10.1128/JVI.00185-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Svitkin YV, Hahn H, Gingras AC, Palmenberg AC, Sonenberg N. 1998. Rapamycin and wortmannin enhance replication of a defective encephalomyocarditis virus. J. Virol. 72:5811–5819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin LR, Neal ZC, McBride MS, Palmenberg AC. 2000. Mengovirus and encephalomyocarditis virus poly(C) tract lengths can affect virus growth in murine cell culture. J. Virol. 74:3074–3081. 10.1128/JVI.74.7.3074-3081.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bacot-Davis VR, Palmenberg AC. 2013. Encephalomyocarditis virus leader protein hinge domain is responsible for interactions with Ran GTPase. Virology 443:177–185. 10.1016/j.virol.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aminev AG, Amineva SP, Palmenberg AC. 2003. Encephalomyocarditis virus (EMCV) proteins 2A and 3BCD localize to nuclei and inhibit cellular mRNA transcription but not rRNA transcription. Virus Res. 95:59–73. 10.1016/S0168-1702(03)00163-1. [DOI] [PubMed] [Google Scholar]
- 23.Cheng Y, Patel DJ. 2004. An efficient system for small protein expression and refolding. Biochem. Biophys. Res. Commun. 317:401–405. 10.1016/j.bbrc.2004.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hall DJ, Palmenberg AC. 1996. Mengo virus 3C proteinase: recombinant expression, intergenus substrate cleavage and localization in vivo. Virus Genes 13:99–110. 10.1007/BF00568903. [DOI] [PubMed] [Google Scholar]
- 25.Zoll J, van Kuppeveld FJM, Galama JMD, Melchers WJG. 1998. Genetic analysis of mengovirus protein 2A: its function in polyprotein processing and virus reproduction. J. Gen. Virol. 79:17–25. [DOI] [PubMed] [Google Scholar]
- 26.Butterworth BE, Rueckert RR. 1972. Kinetics of synthesis and cleavage of encephalomyocarditis virus-specific polypeptides. Virology 50:535–549. 10.1016/0042-6822(72)90405-9. [DOI] [PubMed] [Google Scholar]
- 27.Dvorak CMT, Hall DJ, Hill M, Riddle M, Pranter A, Dillman J, Deibel M, Palmenberg AC. 2001. Leader protein of encephalomyocarditis virus binds zinc, is phosphorylated during viral infection and affects the efficiency of genome translation. Virology 290:261–271. 10.1006/viro.2001.1193. [DOI] [PubMed] [Google Scholar]
- 28.Paul S, Michiels T. 2006. Cardiovirus leader proteins are functionally interchangeable and have evolved to adapt to virus replication fitness. J. Gen. Virol. 87:1237–1246. 10.1099/vir.0.81642-0. [DOI] [PubMed] [Google Scholar]
- 29.Ricour C, Delhaye S, Hato SV, Olenyik TD, Michel B, van Kuppeveld FJ, Gustin KE, Michiels T. 2009. Inhibition of mRNA export and dimerization of interferon regulatory factor 3 by Theiler's virus leader protein. J. Gen. Virol. 90:177–186. 10.1099/vir.0.005678-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ricour C, Borghese F, Sorgeloos F, Hato SV, van Kuppeveld FJ, Michiels T. 2009. Random mutagenesis defines a domain of Theiler's virus leader protein that is essential for antagonism of nucleocytoplasmic trafficking and cytokine gene expression. J. Virol. 83:11223–11232. 10.1128/JVI.00829-09. [DOI] [PMC free article] [PubMed] [Google Scholar]