

Abstract
Autophagy and apoptosis play critical roles in cellular homeostasis and survival. Subtilase cytotoxin (SubAB), produced by non-O157 type Shiga-toxigenic Escherichia coli (STEC), is an important virulence factor in disease. SubAB, a protease, cleaves a specific site on the endoplasmic reticulum (ER) chaperone protein BiP/GRP78, leading to ER stress, and induces apoptosis. Here we report that in HeLa cells, activation of a PERK (RNA-dependent protein kinase [PKR]-like ER kinase)-eIF2α (α subunit of eukaryotic initiation factor 2)-dependent pathway by SubAB-mediated BiP cleavage negatively regulates autophagy and induces apoptosis through death-associated protein 1 (DAP1). We found that SubAB treatment decreased the amounts of autophagy markers LC3-II and p62 as well as those of mTOR (mammalian target of rapamycin) signaling proteins ULK1 and S6K. These proteins showed increased expression levels in PERK knockdown or DAP1 knockdown cells. In addition, depletion of DAP1 in HeLa cells dramatically inhibited the SubAB-stimulated apoptotic pathway: SubAB-induced Bax/Bak conformational changes, Bax/Bak oligomerization, cytochrome c release, activation of caspases, and poly(ADP-ribose) polymerase (PARP) cleavage. These results show that DAP1 is a key regulator, through PERK-eIF2α-dependent pathways, of the induction of apoptosis and reduction of autophagy by SubAB.
INTRODUCTION
Shiga-toxigenic Escherichia coli (STEC) infection causes gastrointestinal disease, including diarrhea, hemorrhagic colitis (1), and hemolytic-uremic syndrome (HUS) (1–3). The bacterial products Shiga toxins 1 and 2 are important virulence factors in the pathogenesis of disease (4). In addition, subtilase cytotoxin (SubAB) was discovered in STEC O113:H21 strain 98NK2, which was responsible for an outbreak of HUS (5). SubAB is produced primarily by a variety of non-O157 serotypes of STEC; STEC O157:H7, the most common serogroup implicated in hemorrhagic colitis and HUS, almost never produces SubAB (6–9). SubAB has an enzymatically active subunit, which is a subtilase-like serine protease, and five receptor recognition domains, which play important roles in binding to the receptor on the target cell surface (5).
In order to understand SubAB cytotoxicity, it was investigated in cultured cells. First, it was observed that SubAB bound to surface receptors (e.g., Neu5Gc [10]), shown to be terminally sialic acid modified membrane proteins (11, 12), and was translocated into target cells. After being endocytosed, SubAB was transported to the Golgi apparatus, which was confirmed by its colocalization with golgin-97, a marker protein of the Golgi apparatus. SubAB was delivered to the endoplasmic reticulum (ER) via a COG (conserved oligomeric Golgi)/Rab6/COPI (coat protein I)-dependent pathway (13). In the ER, SubAB cleaves a specific site at Leu416 on endoplasmic reticulum chaperone BiP/GRP78 (14). SubAB-dependent BiP cleavage is inhibited by brefeldin A (BFA), a Golgi complex-disrupting agent (15, 16). SubAB-induced ER stress due to BiP cleavage causes activation of stress sensor proteins, followed by the induction of various cellular events leading to cell damage, e.g., transient inhibition of protein synthesis (17), G0/G1 cell cycle arrest (15, 17), caspase-dependent apoptosis via mitochondrial membrane damage (18), activation of Akt-NF-κB signaling (19), downregulation of gap junction expression (20), activation of RNA-dependent protein kinase (PKR)-like ER kinase (PERK) followed by caspase-dependent apoptosis (12), and inhibition of lipopolysaccharide (LPS)-stimulated NO production through inhibition of NF-κB nuclear translocation and inducible nitric oxide synthase (iNOS) expression (21).
Macroautophagy (referred to below as autophagy) is mediated by autophagosomes, double-membrane vesicles that enclose a portion of the cytoplasm for delivery to the lysosome. Autophagosome formation is dynamically regulated by starvation and other stresses and involves complicated membrane reorganization (22). Recent studies have shown that autophagy is an important component of the innate defense against a variety of infectious agents. Microorganisms, however, have evolved strategies for evading or subverting host autophagy so as to survive and establish persistent infections (23, 24). In addition, there are negative regulators of autophagy (e.g., HO-1, Nrf2) (25–27). Death-associated protein 1 (DAP1) has been identified as a novel substrate of mammalian target of rapamycin (mTOR) that negatively regulates autophagy (28). DAP1 (15 kDa) was initially identified for its role in programmed cell death (29) and was shown to be ubiquitously expressed in many types of cells and tissues (30). We show here the molecular mechanisms involved in SubAB-mediated suppression of the generation of autophagy marker LC3-II, including reduced expression levels of factors of autophagy. We observed that DAP1 was a key factor in the regulation of SubAB-induced apoptosis and autophagy.
MATERIALS AND METHODS
Subtilase cytotoxin preparation.
Escherichia coli producing recombinant His-tagged wild-type (wt) subtilase cytotoxin (SubAB) or catalytically inactivated mutant (mt) SubAB (with an S272A alteration in SubA) was used as the source of toxins for purification according to a published procedure (17).
Antibodies and other reagents.
Antibodies against Atg5, Atg7, Atg12, Atg16L1, Beclin 1, DAP1, eIF2α, phospho-eIF2α(Ser51), LC3B, ULK1, phospho-ULK1(Ser757), p70 S6 kinase (S6K), phospho-S6K(Thr389), cleaved caspase-7 (cCas7), cleaved poly(ADP-ribose) polymerase (cPARP), PERK, SQTMT1/p62, mTOR, and phospho-mTOR(S2448) were purchased from Cell Signaling. Mouse monoclonal antibodies against BiP/GRP78 were from BD Biosciences, and the antibody against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was from GeneTex. The anti-LC3 monoclonal antibody (clone 1703) was from Cosmo Bio; Z-Val-Ala-Asp-fluoromethylketone (Z-VAD-FMK) was from R&D Systems; Necrostatin-1 and the anti-α-tubulin antibody were from Sigma-Aldrich; 3-methyladenine (3-MA) was from MP Biomedicals; and bafilomycin A1 (Baf A1) and cycloheximide were from Wako.
Cell culture and gene transfection.
HeLa cells were cultured in Earle's minimal essential medium (Sigma) containing 10% fetal calf serum (FCS). Cells were plated into 24-well dishes (5 × 104 cells/well) or 12-well dishes (1 × 105 cells/well) with medium containing 10% FCS. RNA interference-mediated gene knockdown was performed using validated Qiagen HP small interfering RNAs (siRNAs) for Atg12 (SI02655289) and PERK (SI02223718). The Atg16L1 siRNA (5′-CAGGACAATGTGGATACTCAT-3′) was designed and validated as described previously (31). SQSTM1/p62 siRNAs (5′-GUAAGCCUAGGUGUUGUCATT-3′) were designed and validated as described previously (32). DAP1 siRNAs were purchased from Dharmacon. Negative-control siRNAs were purchased from Sigma-Aldrich. Cells were transfected with siRNAs at 50 to 100 nM for 48 h by using Lipofectamine RNAiMax transfection reagent (Invitrogen) according to the manufacturer's protocol. The knockdown of the target proteins was confirmed by immunoblotting. The Myc-tagged hULK1 plasmid (33) was obtained from Addgene (plasmid 31961). HeLa cells were transfected with X-tremeGENE HP DNA transfection reagent (Roche) according to the manufacturer's instruction manual.
Cell viability assay.
HeLa cells (1 × 104/well) were incubated with wild-type or catalytically inactive mutant SubAB (200 ng/ml) for the indicated times. Cell viability was measured by a Cell Counting kit (Dojindo) according to the manufacturer's instructions.
Immunoprecipitation.
Conformationally changed Bax or Bak was coimmunoprecipitated as described previously (34). Briefly, the indicated siRNA-transfected HeLa cells were treated with wt or mt SubAB for 3 h. After a wash with ice-cold phosphate-buffered saline (PBS), cells were solubilized with lysis buffer {10 mM HEPES, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 2% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate [CHAPS] [pH 7.4]} containing protease inhibitor cocktail (Roche Diagnostics) and were incubated for 30 min on ice. After centrifugation at 17,400 × g for 15 min at 4°C, solubilized extracts (100 μg/200 μl) were collected and were incubated with a conformation-specific anti-Bax (clone 3; BD Bioscience) or anti-Bak (Ab-2; Calbiochem) antibody at 4°C for 3 h. Immunoprecipitates were collected by incubation with protein G-Sepharose (Invitrogen) for 1 h, followed by centrifugation for 1 min at 4°C. After immunocomplexes were washed three times with lysis buffer, proteins were dissolved in SDS sample buffer, subjected to SDS-PAGE in 15% gels, and transferred to polyvinylidene difluoride (PVDF) membranes, which were then analyzed by Western blotting using anti-Bax or anti-Bak antibodies (Cell Signaling).
Immunoblot analysis.
Cell lysis and immunoblotting were performed as described previously (12). Briefly, proteins were separated by SDS-PAGE and were transferred to PVDF membranes, which were incubated with the indicated primary antibodies. Detection was performed with horseradish peroxidase-labeled goat anti-mouse or anti-rabbit secondary antibodies, followed by enhanced chemiluminescence (SuperSignal; Pierce). Bands were visualized using a LAS-1000 system (Fujifilm). Densitometric analysis was performed by Image Gauge software (Fujifilm) on the scanned blots, and protein levels were normalized to those of α-tubulin or GAPDH.
Immunofluorescence confocal microscopy.
For immunofluorescence analysis of LC3B or SQSTM1/p62, 1 × 105 HeLa cells on the microcoverglass (Matsunami) were incubated with 200 ng/ml wt or mt SubAB for the indicated times. Cells were fixed with 4% paraformaldehyde (PFA) at room temperature for 15 min, washed twice with PBS, and then immediately permeabilized with ice-cold 100% methanol for 10 min at −20°C. The cells were then rinsed three times with PBS and incubated with blocking buffer (5% goat serum, 0.3% Triton X-100 in PBS) at room temperature for 1 h. To visualize LC3B (D11; dilution, 1:200) or SQSTM1/p62 (D10E10; dilution, 1:400), cells were further incubated with primary antibodies in 0.4% bovine serum albumin (BSA)-PBS buffer at 4°C overnight, washed twice with PBS, and incubated with a DyLight 488-conjugated anti-mouse antibody (Rockland) or a Cy3-conjugated anti-rabbit antibody (Sigma) at room temperature for 1 h in the dark. After three washes with PBS, cells were mounted on glass slides using Prolong Gold Antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI). The stained cells were visualized by FV10i-LIV confocal microscopy (Olympus). The images were arranged with Adobe Photoshop CS4.
Statistics.
The P values for densitometric analysis and vacuolating assays were determined by Student's t test with GraphPad Prism software (GraphPad, San Diego, CA). P values of <0.05 were considered statistically significant.
RESULTS
SubAB negatively regulates autophagy in HeLa cells.
To understand the relationship between autophagy and SubAB-induced apoptosis in HeLa cells, we examined the effect of SubAB on the conversion of LC3 and the expression level of p62. Studies have shown that during autophagosome formation, the level of autophagy marker LC3-II was increased and it accumulated in autophagosomal membranes, while the level of p62 was decreased in autophagosomes (35, 36). We first investigated the effect of SubAB on HeLa cell viability, which was significantly decreased after 48 h of incubation with wild-type (wt) SubAB, but not with catalytically inactive mutant (mt) SubAB (Fig. 1a). Although SubAB-induced caspase-7 activation was observed after 3 h of incubation, the amounts of LC3-II and p62 were decreased in a time-dependent manner (Fig. 1b). We next investigated the effects of SubAB on the classical mTOR pathway in HeLa cells. As shown in Fig. 1c, wt SubAB, but not mt SubAB, induced BiP cleavage within 1 h. SubAB treatment for 3 h suppressed ULK1 and S6K expression. Previous studies have shown that ULK1 is a regulator of autophagy (33). Therefore, we examined the effect of transient expression of ULK1 in HeLa cells on the inhibition of LC3-II generation by SubAB. Compared with control plasmid-transfected cells, ULK1 overexpression did not inhibit the suppression of LC3-II and p62 by SubAB (Fig. 1d). Further, we also investigated the effect of ULK1 overexpression on SubAB-induced apoptosis. As shown in Fig. 1e, overexpression of ULK1 did not affect SubAB-induced PERK phosphorylation, eIF2α phosphorylation, caspase-7 activation, or PARP cleavage. These data suggest that SubAB functions independently of ULK1.
FIG 1.

Effect of SubAB on autophagy in HeLa cells. (a) HeLa cells were incubated with catalytically inactivated (mt) or wild-type (wt) SubAB (200 ng/ml) for 0, 3, 6, 24, and 48 h. Cell viability with SubAB was determined with a Cell Counting kit as described in Materials and Methods. Data are means ± standard deviations of values from three experiments, with three duplicates per experiment (n = 6). *, P < 0.05. (b) HeLa cells were incubated with mt or wt SubAB (200 ng/ml) for 0, 0.5, 1, and 3 h. (Top) Cell lysates were analyzed by Western blotting using the indicated antibodies. (Bottom) The amounts of LC3-II and p62 after incubation with mt or wt SubAB were quantified by densitometry. Data are means ± standard deviations of values from three experiments. *, P < 0.05. (c) HeLa cells were treated with mt or wt SubAB for the indicated times. (Top) Cell lysates were analyzed by Western blotting using the indicated antibodies. (Bottom) The amounts of ULK1 and S6K after incubation with mt or wt SubAB were quantified by densitometry. Data are means ± standard deviations of values from three experiments. *, P < 0.05. (d) Cells transiently expressing Myc-tagged ULK1 were incubated with mt or wt SubAB for 3 h at 37°C. (Top) The amounts of LC3-II, p62, and ULK1 were determined by immunoblot analysis. (Bottom) The amounts of LC3-II and p62 after incubation with mt or wt SubAB for 3 h were quantified by densitometry. Data are means ± standard deviations of values from three experiments. *, P < 0.05. (e) Cells transiently expressing ULK1 cDNAs were treated with mt or wt SubAB for 0, 1, or 3 h at 37°C. (Left) Cell lysates were analyzed by Western blotting using specific antibodies as indicated. All experiments were repeated three times with similar results. (Right) The amounts of cCas7 and cPARP after incubation for 3 h with mt or wt SubAB were quantified by densitometry. Data are means ± standard deviations of values from three experiments. *, P < 0.05.
SubAB suppression of autophagy is required for PERK-mediated signaling.
Previously, we demonstrated that SubAB induction of apoptosis by BiP cleavage was mediated by the ER stress sensor protein PERK (12). Therefore, we next investigated if PERK-related signaling pathways were associated with SubAB suppression of autophagy in HeLa cells. We determined the effects of SubAB on the mTOR-signaling pathway in PERK knockdown cells. In control siRNA-transfected cells, the amounts of phospho-mTOR, ULK1, phospho-ULK1, S6K, phospho-S6K, p62, and LC3-II were decreased by SubAB. In contrast, SubAB increased the levels of activation of caspase-7. In PERK knockdown cells, the expression level of PERK was significantly suppressed, and the amounts of ULK1, S6K, p62, and LC3-II were not decreased in the presence of SubAB. SubAB-induced caspase-7 activation was significantly suppressed in PERK knockdown cells, as described previously (12). The catalytically inactive SubAB had no effect (Fig. 2a).
FIG 2.

PERK controls apoptosis and autophagy by SubAB in HeLa cells. (a) Nontargeting control (NC) and PERK siRNA-transfected HeLa cells were incubated with mt or wt SubAB for 3 h. NC, nontargeting control siRNA. (Left) Cell lysates were analyzed by Western blotting using specific antibodies as indicated. (Right) The amounts of PERK, LC3-II, S6K, phospho-S6K (p-S6K), phospho-mTOR (p-mTOR), and p62 after incubation with mt or wt SubAB in control (NC) or PERK knockdown cells were quantified by densitometry. All experiments were repeated three times with similar results. Data are means ± standard deviations of values from three experiments. *, P < 0.05. (b) The siRNA-transfected HeLa cells were incubated with mt or wt SubAB in the presence or absence of 100 nM bafilomycin A1 (Baf A1) for 3 h. (Top) Cell lysates were analyzed by Western blotting using anti-LC3B, anti-p62, and anti-BiP antibodies. Tubulin was used as a loading control. (Bottom) The amounts of LC3-II and p62 were quantified by densitometry. All experiments were repeated three times with similar results. Data are means ± standard deviations of values from three experiments. *, P < 0.05. (c) (Top) The indicated siRNA-transfected HeLa cells were incubated with mt or wt SubAB in the presence or absence of 100 nM bafilomycin A1 (Baf A1) for 3 h, followed by fixation and then reaction with the indicated antibodies as described in Materials and Methods. (Bottom) The LC3 puncta in a single cell were manually counted under a confocal microscope (*, P < 0.05). For each group, 30 cells were randomly selected for averaging the number of LC3 puncta per cell.
We next investigated the effect of bafilomycin A1 (Baf A1) on the expression levels of LC3-II and p62 in the presence of SubAB in PERK knockdown cells. In control siRNA-transfected cells, the decreases in LC3-II and p62 levels by SubAB were not inhibited by Baf A1, which prevents the maturation of autophagic vacuoles (37). In PERK knockdown cells, the amounts of LC3-II and p62 were not decreased in the presence of mt or wt SubAB; they were, however, significantly increased in the presence of Baf A1 (Fig. 2b). As observed by confocal microscopy, SubAB decreased the number of cells showing LC3-II, even in the presence of Baf A1 (Fig. 2c, top). In contrast, in PERK knockdown cells, basal amounts of LC3-II were increased and were not significantly suppressed by SubAB with or without Baf A1 (Fig. 2c, bottom).
Previously, we demonstrated that SubAB-induced apoptosis is caspase dependent (18). Recent studies have shown that apoptosis-mediated cleavage of Beclin 1 and Atg5 inhibits autophagy (38, 39). We next investigated the effect of SubAB on the expression of Atg5 and Beclin 1 in HeLa cells. SubAB treatment did not alter the amounts of Atg5 and Beclin 1 (Fig. 3a). We next examined if SubAB-induced caspase activation had an effect on the suppression of LC3-II and p62. A general caspase inhibitor, Z-VAD-FMK, completely suppressed SubAB-induced caspase-7 activation; however, SubAB action resulting in the reduction of LC3-II and p62 expression was not affected. Further, SubAB did not affect the amounts of Atg5 and Beclin 1. A necrosis inhibitor, Necrostatin, did not affect SubAB-induced reduction of LC3-II and p62 expression (Fig. 3b).
FIG 3.

Effects of SubAB on autophagy-related proteins in HeLa cells. (a) HeLa cells were incubated with mt or wt SubAB for 3 h. Cell lysates were analyzed by Western blotting using anti-Beclin1 and anti-Atg5 antibodies. GAPDH was used as a loading control. N.S., not significant. (b) HeLa cells were pretreated with 10 μM Z-VAD-FMK (VAD) or 20 μM Necrostatin 1 (Necro) for 30 min and were then incubated with mt or wt SubAB for 3 h. Cell lysates were analyzed by Western blotting using specific antibodies as indicated. Cont, control. (c) HeLa cells were preincubated with 5 mM 3-methyladenine (3-MA) for 30 min and were then incubated with mt or wt SubAB for 3 h. (Left) Cell lysates were analyzed by Western blotting using specific antibodies as indicated. (Right) The amounts of LC3-II, p62, and p-S6K were quantified by densitometry. All experiments were repeated three times with similar results. Data are means ± standard deviations of values from three experiments. *, P < 0.05. (d and e) The indicated siRNA-transfected HeLa cells were incubated with mt or wt SubAB for 3 h. Cell lysates were analyzed by Western blotting using the indicated antibodies. The amounts of cPARP and cCas7 were quantified by densitometry. All experiments were repeated three times with similar results. Data are means ± standard deviations of values from three experiments. *, P < 0.05. (f) NC, Atg12, and Atg7 siRNA-transfected HeLa cells were incubated with mt or wt SubAB for 3 h. (Left) Cell lysates were analyzed by Western blotting using specific antibodies as indicated. All experiments were repeated three times with similar results. (Right) Atg7, Atg12, cCas7, and cPARP obtained with mt and wt SubAB were quantified by densitometry. Data are means ± standard deviations of values from three experiments. *, P < 0.05.
Furthermore, we investigated the effect of 3-methyladenine (3-MA), an autophagy inhibitor, on SubAB-suppressed LC3-II and p62. Although treatment with 3-MA enhanced SubAB-induced S6K dephosphorylation, 3-MA incubation did not alter SubAB-mediated BiP cleavage or SubAB-suppressed LC3-II and p62 expression (Fig. 3c, left). To examine whether autophagosome formation is involved in SubAB-induced apoptosis, we tested the effect of silencing of the Atg16L1 gene on SubAB-induced caspase activation. PERK knockdown dramatically suppressed SubAB-induced activation of caspase-7 and cleavage of PARP. The amounts of LC3-II and p62 were not decreased by SubAB in PERK knockdown cells (Fig. 2). Knockdown of Atg16L1 reduced the basal levels of LC3-II and Atg5 proteins. However, SubAB-induced PARP cleavage and caspase-7 activation were not affected by Atg16L1 knockdown. In PERK and Atg16L1 double-knockdown cells, SubAB-induced PARP cleavage and caspase-7 activation were not significantly increased over levels in mt SubAB-treated cells (Fig. 3d). We next investigated the effects of p62 knockdown on SubAB-induced apoptosis. p62, a substrate of autophagy, is accumulated under autophagy-defective conditions and perturbs signal transduction pathways (36). As shown in Fig. 3e, p62 knockdown did not affect SubAB-induced PARP cleavage or caspase-7 activation. Taken together, these findings suggest that autophagosome-related proteins (e.g., Atg16L1, p62) do not affect SubAB-induced apoptosis. Further, we investigated the effect of other types of Atg proteins, such as Atg12 and Atg7, on SubAB-induced apoptosis. Knockdown of Atg12, which is essential for autophagosome formation, did not affect SubAB-induced PARP cleavage or caspase activation. However, Atg7 knockdown significantly enhanced SubAB-induced apoptotic activity (Fig. 3f), as reported previously (40). These results suggest that Atg7 not only participates in autophagosome formation but also is involved in apoptosis.
Death-associated protein 1 (DAP1) plays an important role in SubAB-regulated apoptosis and autophagy.
Since Atg family proteins were not associated with SubAB-regulated autophagy and apoptosis, as shown above, we next examined the role of DAP1, which has been reported to be an mTOR substrate and a negative regulator of autophagy (28, 41), in SubAB-regulated autophagy and apoptosis. First, we investigated the effect of siRNA-mediated knockdown of DAP1 on SubAB-associated apoptosis and autophagy. DAP1 expression was significantly suppressed by the siRNA. Depletion of DAP1 did not inhibit SubAB-mediated BiP cleavage. In control siRNA-transfected cells, SubAB treatment for 3 h significantly decreased LC3-II and p62 protein levels, while it increased caspase-7 activation and PARP cleavage. In contrast, in DAP1 knockdown cells, the basal level of DAP1 was decreased and SubAB treatment slightly decreased LC3-II and p62 levels, while SubAB-induced caspase-7 activation and PARP cleavage were significantly suppressed (Fig. 4a and b). To explore whether DAP1 regulates the PERK-eIF2α signaling pathway, we investigated, in DAP1 knockdown cells, the effect of SubAB on phospho-eIF2α. As shown in Fig. 4c, in control cells, SubAB increased eIF2α phosphorylation, which was not inhibited in DAP1 siRNA-transfected cells. As a positive control, in PERK knockdown cells, SubAB-induced eIF2α phosphorylation was suppressed. Thus, DAP1 acts downstream of SubAB-mediated BiP cleavage and PERK-eIF2α and controls both apoptosis and autophagy.
FIG 4.

Knockdown of DAP1 rescues SubAB-mediated autophagy and apoptosis. (a) Nontargeting control (NC) and DAP1 siRNA-transfected HeLa cells (1 × 105/well) were incubated with mt or wt SubAB (0.2 μg/ml) for 3 to 4 h at 37°C. (Left) Cell lysates were analyzed by Western blotting using the indicated antibodies. (Right) The amounts of DAP1, p62, and LC3-II after incubation with mt or wt SubAB in NC or DAP1 knockdown cells were quantified by densitometry. All data are representative of the results of at least three separate experiments. Data are means ± standard deviations of values from three experiments. #, P < 0.03; *, P < 0.05. (b) NC and DAP1 siRNA-transfected cells were incubated with mt or wt SubAB (0.2 μg/ml) for 3 to 4 h at 37°C. (Left) Cell lysates were analyzed by Western blotting using anti-cCas7 and anti-cPARP antibodies. GAPDH was used as a loading control. (Right) The amounts of cCas7 and cPARP were quantified by densitometry. All data are representative of the results of at least three separate experiments. Data are means ± standard deviations of values from three experiments. #, P < 0.03; *, P < 0.05. (c) NC and DAP1 siRNA-transfected cells were incubated with wt or mt SubAB at 37°C for 2 h, followed by immunoblotting with anti-p-eIF2α(Ser51), anti-eIF2α, anti-DAP1, and anti-PERK antibodies. All data are representative of the results of at least three separate experiments. (d) NC and DAP1 siRNA-transfected cells were incubated with wt or mt SubAB at 37°C for 3 to 4 h, followed by immunoblotting with specific antibodies as indicated. (e) (Left) The siRNA-transfected cells were incubated and immunoblotted as described for panel d. (Right) The amounts of Atg5, Atg12, and Atg16L1 after incubation with mt or wt SubAB in NC or DAP1 knockdown cells were quantified by densitometry. All data are representative of the results of at least three separate experiments. Data are means ± standard deviations of values from three experiments. *, P < 0.05. (f) Cells were pretreated with the indicated concentrations of rapamycin (Rapa) for 1 h and were then incubated with mt or wt SubAB for 4 h, followed by immunoblotting with the indicated antibodies. All data are representative of the results of at least three separate experiments.
We next explored the effect of DAP1 depletion on mTOR signaling by SubAB. A previous study showed that mTOR activity in HeLa cells was not affected by depletion of DAP1 (28). As shown in Fig. 4d, SubAB treatment significantly decreased phospho-ULK1 and phospho-S6K levels in DAP1 knockdown cells, effects similar to those seen in control siRNA-transfected cells, suggesting that these effects were independent of DAP1.
We also assessed the effect of DAP1 knockdown on the levels of Atg proteins. As shown in Fig. 4e, in control siRNA-transfected cells, SubAB treatment slightly decreased the amount of Atg16L1. Depletion of DAP1 increased the amounts of Atg5, Atg12, and Atg16L1, which were not decreased by SubAB.
Next, we investigated the effect of rapamycin, an mTOR complex 1 (mTORC1) inhibitor (42), on caspase activation by SubAB. It is known that rapamycin has protective effects against proapoptotic insults (40). S6K phosphorylation was suppressed by rapamycin in a dose-dependent manner. SubAB treatment decreased the amounts of LC3-II, p62, and S6K; rapamycin did not prevent caspase-7 activation or PARP cleavage by SubAB (Fig. 4f). Thus, rapamycin did not prevent the proapoptotic effects of SubAB.
DAP1 regulates Bax/Bak conformational changes, Bax/Bak oligomerization, and cytochrome c release.
DAP1 affected SubAB-induced apoptosis and negatively regulated autophagy as shown above. Our previous studies and those of others showed that SubAB-induced apoptosis in HeLa cells was caused by Bax/Bak conformational changes and cytochrome c release, followed by caspase activation (12, 43, 44). We next examined the effect of the transfection of HeLa cells with DAP1 siRNA on SubAB-induced Bax/Bak conformational changes and Bax/Bak oligomerization by using conformation-specific anti-Bax (cBax) or anti-Bak (cBak) monoclonal antibodies. We also monitored the release of cytochrome c from mitochondria to the cytosol. Silencing of the DAP1 gene did not affect Bax and Bak levels. Densitometric analysis showed that SubAB-induced Bax/Bak conformational changes, Bax/Bak oligomerization, and cytochrome c (Cyt c) release were dramatically reduced in DAP1 knockdown cells from those in control siRNA-transfected cells (Fig. 5a and b).
FIG 5.
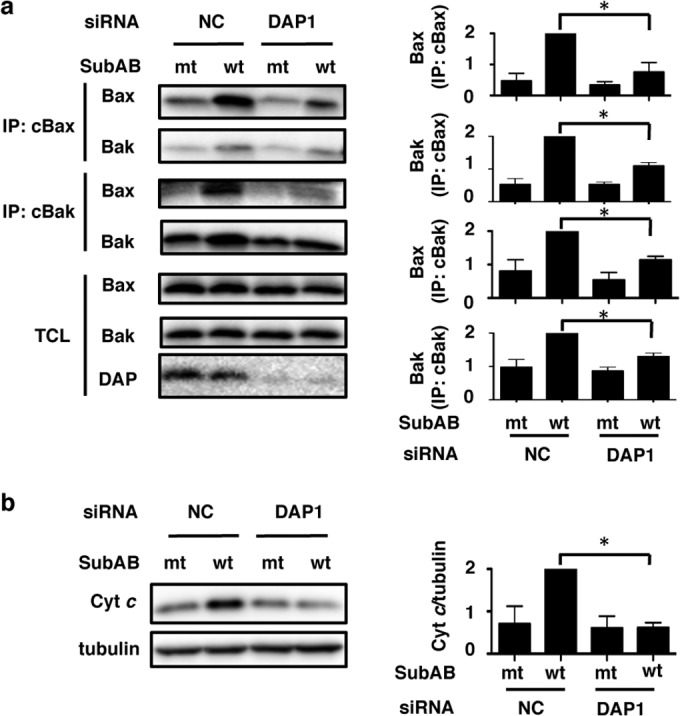
Depletion of DAP1 inhibits SubAB-induced Bak/Bax conformational changes. (a) NC and DAP1 siRNA-transfected cells were incubated with mt or wt SubAB (0.2 μg/ml) for 3 h at 37°C. Then cells were lysed, and proteins were immunoprecipitated with conformation-specific anti-Bax (cBax) or anti-Bak (cBak) monoclonal antibodies as described in Materials and Methods. The immunocomplexes (IP) or total-cell lysates (TCL) were analyzed by SDS-PAGE, followed by immunoblotting with anti-Bax and anti-Bak antibodies. (b) The level of cytochrome c (Cyt c) release into the cytoplasmic fraction was determined as described in Materials and Methods. Cytochrome c and tubulin were detected by Western blotting. Conformationally changed Bax, conformationally changed Bak, Bax/Bak complexes, and released Cyt c obtained with mt and wt SubAB were quantified by densitometry. Data are means ± standard deviations of values from three experiments. *, P < 0.05.
DISCUSSION
Recent studies have shown that several bacterial cytotoxins induced autophagy, which was associated with cell death (45–49). In contrast, cyclic AMP (cAMP)-elevating toxins from Bacillus anthracis and Vibrio cholerae suppressed host autophagy and thereby suppressed immune responses and modulated host cell physiology (50). It was also suggested that these toxins might disrupt a downstream step in autophagosome formation (50). Furthermore, it was demonstrated that cAMP-elevating toxins contribute to increased bacterial replication (50). In STEC infection, it is not known whether autophagy contributes to pathogenesis. SubAB was found mainly in non-O157, LEE-negative Stx2-producing STEC strains (51, 52). A recent study showed that Shiga toxins (Stxs), which are major virulence factors, induced autophagy in Stx-sensitive and Stx-insensitive cells (46). It showed that Stx-induced LC3-II generation depended on Stx B subunits, indicating that toxin enzymatic activity was not required for autophagosome formation; rather, toxin activity in sensitive cells was involved in caspase activation, leading to Atg5 and Beclin 1 cleavage (46). After treatment with SubAB, the amounts of Atg5 and Beclin 1 were unaltered. Pretreatment with a general caspase inhibitor (Z-VAD-FMK) or a necrosis inhibitor (Necrostatin) did not prevent SubAB-induced reduction of LC3-II and p62 levels, suggesting that SubAB-mediated inhibition of autophagy is not associated with inactivation or reduction of Atg proteins and Beclin 1 due to activation of caspase.
Here we show that SubAB toxin appears to suppress basal levels of autophagy in HeLa cells. Our results indicate that SubAB caused ER stress by BiP cleavage, resulting in the activation of a PERK-dependent pathway, leading to cell damage and a cytotoxic response, with suppression of autophagosome formation. This inhibition was not observed in PERK knockdown cells, where the basal level of LC3-II was significantly increased. Thus, a PERK-dependent pathway was involved in the SubAB-dependent decrease in autophagy.
Previous studies showed activation of PERK/eIF2α/ATF4 by the unfolded protein response, leading to inhibition of mTORC1–phospho-S6K signaling and the subsequent induction of cytoprotective autophagy (53, 54). We also investigated the effect of SubAB on mTORC1 signaling in HeLa cells. SubAB treatment decreased the levels of ULK1, S6K, phospho-ULK1, and phospho-S6K. Knockdown of PERK led to increases in the basal levels of ULK1 and S6K proteins, which were slightly reduced by SubAB treatment. In addition, we observed that SubAB decreased LC3-II and p62 protein levels even in ULK1-overexpressing cells. Thus, inhibition of autophagosome formation by SubAB is independent of ULK1-signaling pathways.
Several studies have identified factors responsible for the negative regulation of autophagy (55). We looked for candidate proteins that could serve as negative regulators of autophagy in SubAB-treated cells. Interestingly, we found that DAP1, which is expressed ubiquitously in many types of cells and tissues (28), is involved both in the suppression of autophagy by SubAB and in SubAB-induced apoptosis. DAP1 was identified as a novel substrate of mTOR that negatively regulates autophagy (28). Moreover, DAP1 has been shown to be a positive mediator of gamma interferon (IFN-γ)- and tumor necrosis factor alpha (TNF-α)-induced programmed cell death (30, 56). Silencing of the DAP1 gene in HeLa cells significantly inhibited the reduction in LC3-II and p62 expression seen with SubAB, effects similar to those of PERK knockdown. SubAB-induced phosphorylation of eIF2α was not altered by DAP1 depletion, suggesting that DAP1 was downstream of PERK-eIF2α. DAP1 knockdown did not affect the reduction of mTOR-dependent S6K phosphorylation by SubAB, suggesting that DAP1 may negatively regulate autophagy via an mTOR-independent pathway. Recent studies have shown that various factors, e.g., intracellular Ca2+, cAMP, extracellular signal-regulated kinase (ERK), p38 mitogen-activated protein kinase (MAPK), reactive oxygen species (ROS), FoxO proteins, and transcription factor EB (TFEB), are involved in the induction of autophagy (57, 58). We did not observe any effect of an ERK inhibitor (U0126) on the protein levels of LC3-II and p62 or on the activation of caspase-7 by SubAB in DAP1 knockdown cells (data not shown). These results indicated that ERK phosphorylation did not participate either in the suppression of autophagy by SubAB or in SubAB-induced apoptosis through DAP1. We found that the levels of some proteins (e.g., ULK1, S6K) that were decreased by SubAB were significantly increased in DAP1 knockdown cells. The proteins whose levels were increased may be associated with mTOR-independent control of autophagy by DAP1 and may play an essential role in the reduction of autophagy by SubAB.
A previous report showed that DAP1 is functionally silenced in growing cells through mTOR-dependent phosphorylation of Ser3 and Ser51, with the dephosphorylated form of DAP1 acting as the active form that suppresses autophagy (28). To investigate whether the dephosphorylated form of DAP1 activates or inactivates the effects of SubAB, either FLAG-tagged DAP1, a nonphosphorylated (S3A S51A) mutant of DAP1, or the phosphomimetic (S3D S51D) mutant of DAP1 was transiently transfected into HeLa cells. However, these overexpressed DAP1 mutants did not alter SubAB-induced PARP cleavage from that with wild-type DAP1 (data not shown). Thus, our findings suggest that these DAP1 phosphorylation sites do not participate in SubAB-induced apoptosis. DAP1 may require other phosphorylation sites or an unknown interaction factor to participate in SubAB-induced apoptosis.
Further, we found that DAP1 was required for the stimulation of Bak/Bax-dependent apoptosis by SubAB. In SubAB-induced apoptosis, Bak/Bax conformational changes, Bak/Bax oligomerization, and cytochrome c release, leading to activation of caspases, were regulated by the ubiquitin-proteasome pathway (43, 44). Interestingly, sequence analysis of DAP1 showed that it is identical to promyelocytic leukemia (PML)-interacting clone 1 (PIC1) and sentrin, a Fas receptor-associated protein. These proteins share a significant degree of homology to ubiquitin and other ubiquitin homology proteins (56). In conclusion, we found that SubAB suppression of autophagy proceeded through a PERK-eIF2α-dependent pathway, which might promote STEC infection and participate in the pathogenesis of severe disease. Further, we showed here that DAP1 is a key regulator of both autophagy and apoptosis in cells intoxicated by SubAB. Recent studies have shown that DAP1 plays a proapoptotic role in human breast cancer (59). Loss of DAP1 in colorectal cancer cells resulted in a gain in cellular migration and a loss of sensitivity to apoptosis induced by a chemotherapeutic agent, 5-fluorouracil (5-FU) (60). Thus, DAP1 plays critical roles in cellular homeostasis and survival. Finally, understanding of DAP1-related signaling and its component proteins may help better characterize mTOR-independent autophagy and apoptosis.
ACKNOWLEDGMENTS
This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science and Culture of Japan and from the Improvement of Research Environment for Young Researchers program of the Japan Science and Technology Agency, as well as by grants-in-aid from the Ministry of Health, Labour, Welfare of Japan (H24-Shinkou-Ippan-012) and Takeda Science Foundation. Joel Moss was supported by the Intramural Research Program, National Institutes of Health, National Heart, Lung, and Blood Institute.
We acknowledge the expert technical assistance of K. Hirano.
Footnotes
Published ahead of print 2 September 2014
REFERENCES
- 1.Riley LW, Remis RS, Helgerson SD, McGee HB, Wells JG, Davis BR, Hebert RJ, Olcott ES, Johnson LM, Hargrett NT, Blake PA, Cohen ML. 1983. Hemorrhagic colitis associated with a rare Escherichia coli serotype. N. Engl. J. Med. 308:681–685. 10.1056/NEJM198303243081203. [DOI] [PubMed] [Google Scholar]
- 2.Latorre-Martinez JC, Garcia-Lozano T, Blanco J, Buesa J. 2007. Characterization of Escherichia coli O157:H7 strains isolated from sporadic cases of hemolytic-uremic syndrome in children. Enferm. Infecc. Microbiol. Clin. 25:603–604 (In Spanish.) 10.1157/13111190. [DOI] [PubMed] [Google Scholar]
- 3.Shiomi M, Togawa M. 1997. Sporadic cases of hemolytic uremic syndrome and hemorrhagic colitis with serum IgM antibodies to lipopolysaccharides of enterohemorrhagic Escherichia coli O157. Nihon Rinsho 55:686–692 (In Japanese.) [PubMed] [Google Scholar]
- 4.Karmali MA. 2004. Prospects for preventing serious systemic toxemic complications of Shiga toxin-producing Escherichia coli infections using Shiga toxin receptor analogues. J. Infect. Dis. 189:355–359. 10.1086/381130. [DOI] [PubMed] [Google Scholar]
- 5.Paton AW, Srimanote P, Talbot UM, Wang H, Paton JC. 2004. A new family of potent AB5 cytotoxins produced by Shiga toxigenic Escherichia coli. J. Exp. Med. 200:35–46. 10.1084/jem.20040392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cergole-Novella MC, Nishimura LS, Dos Santos LF, Irino K, Vaz TM, Bergamini AM, Guth BE. 2007. Distribution of virulence profiles related to new toxins and putative adhesins in Shiga toxin-producing Escherichia coli isolated from diverse sources in Brazil. FEMS Microbiol. Lett. 274:329–334. 10.1111/j.1574-6968.2007.00856.x. [DOI] [PubMed] [Google Scholar]
- 7.Tozzoli R, Caprioli A, Cappannella S, Michelacci V, Marziano ML, Morabito S. 2010. Production of the subtilase AB5 cytotoxin by Shiga toxin-negative Escherichia coli. J. Clin. Microbiol. 48:178–183. 10.1128/JCM.01648-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu Y, Hinenoya A, Taguchi T, Nagita A, Shima K, Tsukamoto T, Sugimoto N, Asakura M, Yamasaki S. 2010. Distribution of virulence genes related to adhesins and toxins in Shiga toxin-producing Escherichia coli strains isolated from healthy cattle and diarrheal patients in Japan. J. Vet. Med. Sci. 72:589–597. 10.1292/jvms.09-0557. [DOI] [PubMed] [Google Scholar]
- 9.Buvens G, Lauwers S, Pierard D. 2010. Prevalence of subtilase cytotoxin in verocytotoxin-producing Escherichia coli isolated from humans and raw meats in Belgium. Eur. J. Clin. Microbiol. Infect. Dis. 29:1395–1399. 10.1007/s10096-010-1014-z. [DOI] [PubMed] [Google Scholar]
- 10.Byres E, Paton AW, Paton JC, Lofling JC, Smith DF, Wilce MC, Talbot UM, Chong DC, Yu H, Huang S, Chen X, Varki NM, Varki A, Rossjohn J, Beddoe T. 2008. Incorporation of a non-human glycan mediates human susceptibility to a bacterial toxin. Nature 456:648–652. 10.1038/nature07428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yahiro K, Satoh M, Morinaga N, Tsutsuki H, Ogura K, Nagasawa S, Nomura F, Moss J, Noda M. 2011. Identification of subtilase cytotoxin (SubAB) receptors whose signaling, in association with SubAB-induced BiP cleavage, is responsible for apoptosis in HeLa cells. Infect. Immun. 79:617–627. 10.1128/IAI.01020-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yahiro K, Tsutsuki H, Ogura K, Nagasawa S, Moss J, Noda M. 2012. Regulation of subtilase cytotoxin-induced cell death by an RNA-dependent protein kinase-like endoplasmic reticulum kinase-dependent proteasome pathway in HeLa cells. Infect. Immun. 80:1803–1814. 10.1128/IAI.06164-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith RD, Willett R, Kudlyk T, Pokrovskaya I, Paton AW, Paton JC, Lupashin VV. 2009. The COG complex, Rab6 and COPI define a novel Golgi retrograde trafficking pathway that is exploited by SubAB toxin. Traffic 10:1502–1517. 10.1111/j.1600-0854.2009.00965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paton AW, Beddoe T, Thorpe CM, Whisstock JC, Wilce MC, Rossjohn J, Talbot UM, Paton JC. 2006. AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature 443:548–552. 10.1038/nature05124. [DOI] [PubMed] [Google Scholar]
- 15.Morinaga N, Yahiro K, Matsuura G, Moss J, Noda M. 2008. Subtilase cytotoxin, produced by Shiga-toxigenic Escherichia coli, transiently inhibits protein synthesis of Vero cells via degradation of BiP and induces cell cycle arrest at G1 by downregulation of cyclin D1. Cell. Microbiol. 10:921–929. 10.1111/j.1462-5822.2007.01094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chong DC, Paton JC, Thorpe CM, Paton AW. 2008. Clathrin-dependent trafficking of subtilase cytotoxin, a novel AB5 toxin that targets the endoplasmic reticulum chaperone BiP. Cell. Microbiol. 10:795–806. 10.1111/j.1462-5822.2007.01085.x. [DOI] [PubMed] [Google Scholar]
- 17.Morinaga N, Yahiro K, Matsuura G, Watanabe M, Nomura F, Moss J, Noda M. 2007. Two distinct cytotoxic activities of subtilase cytotoxin produced by Shiga-toxigenic Escherichia coli. Infect. Immun. 75:488–496. 10.1128/IAI.01336-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsuura G, Morinaga N, Yahiro K, Komine R, Moss J, Yoshida H, Noda M. 2009. Novel subtilase cytotoxin produced by Shiga-toxigenic Escherichia coli induces apoptosis in Vero cells via mitochondrial membrane damage. Infect. Immun. 77:2919–2924. 10.1128/IAI.01510-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamazaki H, Hiramatsu N, Hayakawa K, Tagawa Y, Okamura M, Ogata R, Huang T, Nakajima S, Yao J, Paton AW, Paton JC, Kitamura M. 2009. Activation of the Akt-NF-κB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response. J. Immunol. 183:1480–1487. 10.4049/jimmunol.0900017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang T, Wan Y, Zhu Y, Fang X, Hiramatsu N, Hayakawa K, Paton AW, Paton JC, Kitamura M, Yao J. 2009. Downregulation of gap junction expression and function by endoplasmic reticulum stress. J. Cell. Biochem. 107:973–983. 10.1002/jcb.22202. [DOI] [PubMed] [Google Scholar]
- 21.Tsutsuki H, Yahiro K, Suzuki K, Suto A, Ogura K, Nagasawa S, Ihara H, Shimizu T, Nakajima H, Moss J, Noda M. 2012. Subtilase cytotoxin enhances Escherichia coli survival in macrophages by suppression of nitric oxide production through the inhibition of NF-κB activation. Infect. Immun. 80:3939–3951. 10.1128/IAI.00581-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mizushima N, Yoshimori T, Ohsumi Y. 2011. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 27:107–132. 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 23.Yuk JM, Yoshimori T, Jo EK. 2012. Autophagy and bacterial infectious diseases. Exp. Mol. Med. 44:99–108. 10.3858/emm.2012.44.2.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patel KK, Stappenbeck TS. 2013. Autophagy and intestinal homeostasis. Annu. Rev. Physiol. 75:241–262. 10.1146/annurev-physiol-030212-183658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rao VA, Klein SR, Bonar SJ, Zielonka J, Mizuno N, Dickey JS, Keller PW, Joseph J, Kalyanaraman B, Shacter E. 2010. The antioxidant transcription factor Nrf2 negatively regulates autophagy and growth arrest induced by the anticancer redox agent mitoquinone. J. Biol. Chem. 285:34447–34459. 10.1074/jbc.M110.133579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim HP, Wang X, Chen ZH, Lee SJ, Huang MH, Wang Y, Ryter SW, Choi AM. 2008. Autophagic proteins regulate cigarette smoke-induced apoptosis: protective role of heme oxygenase-1. Autophagy 4:887–895. 10.4161/auto.6767. [DOI] [PubMed] [Google Scholar]
- 27.Bolisetty S, Traylor AM, Kim J, Joseph R, Ricart K, Landar A, Agarwal A. 2010. Heme oxygenase-1 inhibits renal tubular macroautophagy in acute kidney injury. J. Am. Soc. Nephrol. 21:1702–1712. 10.1681/ASN.2010030238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koren I, Reem E, Kimchi A. 2010. DAP1, a novel substrate of mTOR, negatively regulates autophagy. Curr. Biol. 20:1093–1098. 10.1016/j.cub.2010.04.041. [DOI] [PubMed] [Google Scholar]
- 29.Levy-Strumpf N, Kimchi A. 1998. Death associated proteins (DAPs): from gene identification to the analysis of their apoptotic and tumor suppressive functions. Oncogene 17:3331–3340. [DOI] [PubMed] [Google Scholar]
- 30.Deiss LP, Feinstein E, Berissi H, Cohen O, Kimchi A. 1995. Identification of a novel serine/threonine kinase and a novel 15-kD protein as potential mediators of the gamma interferon-induced cell death. Genes Dev. 9:15–30. 10.1101/gad.9.1.15. [DOI] [PubMed] [Google Scholar]
- 31.Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJ, Campbell BJ, Jewell D, Simmons A. 2010. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 16:90–97. 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 32.Pursiheimo JP, Rantanen K, Heikkinen PT, Johansen T, Jaakkola PM. 2009. Hypoxia-activated autophagy accelerates degradation of SQSTM1/p62. Oncogene 28:334–344. 10.1038/onc.2008.392. [DOI] [PubMed] [Google Scholar]
- 33.Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. 2009. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 20:1992–2003. 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mikhailov V, Mikhailova M, Degenhardt K, Venkatachalam MA, White E, Saikumar P. 2003. Association of Bax and Bak homo-oligomers in mitochondria. Bax requirement for Bak reorganization and cytochrome c release. J. Biol. Chem. 278:5367–5376. 10.1074/jbc.M203392200. [DOI] [PubMed] [Google Scholar]
- 35.Mizushima N, Yoshimori T. 2007. How to interpret LC3 immunoblotting. Autophagy 3:542–545. 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 36.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. 2007. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282:24131–24145. 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 37.Klionsky DJ, Elazar Z, Seglen PO, Rubinsztein DC. 2008. Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy 4:849–950. 10.4161/auto.6845. [DOI] [PubMed] [Google Scholar]
- 38.Wirawan E, Vande Walle L, Kersse K, Cornelis S, Claerhout S, Vanoverberghe I, Roelandt R, De Rycke R, Verspurten J, Declercq W, Agostinis P, Vanden Berghe T, Lippens S, Vandenabeele P. 2010. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 1:e18. 10.1038/cddis.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kang R, Zeh HJ, Lotze MT, Tang D. 2011. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 18:571–580. 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ravikumar B, Berger Z, Vacher C, O'Kane CJ, Rubinsztein DC. 2006. Rapamycin pre-treatment protects against apoptosis. Hum. Mol. Genet. 15:1209–1216. 10.1093/hmg/ddl036. [DOI] [PubMed] [Google Scholar]
- 41.Koren I, Reem E, Kimchi A. 2010. Autophagy gets a brake: DAP1, a novel mTOR substrate, is activated to suppress the autophagic process. Autophagy 6:1179–1180. 10.4161/auto.6.8.13338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sabers CJ, Martin MM, Brunn GJ, Williams JM, Dumont FJ, Wiederrecht G, Abraham RT. 1995. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J. Biol. Chem. 270:815–822. 10.1074/jbc.270.2.815. [DOI] [PubMed] [Google Scholar]
- 43.May KL, Paton JC, Paton AW. 2010. Escherichia coli subtilase cytotoxin induces apoptosis regulated by host Bcl-2 family proteins Bax/Bak. Infect. Immun. 78:4691–4696. 10.1128/IAI.00801-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yahiro K, Morinaga N, Moss J, Noda M. 2010. Subtilase cytotoxin induces apoptosis in HeLa cells by mitochondrial permeabilization via activation of Bax/Bak, independent of C/EBF-homologue protein (CHOP), Ire1α or JNK signaling. Microb. Pathog. 49:153–163. 10.1016/j.micpath.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liévin-Le Moal V, Comenge Y, Ruby V, Amsellem R, Nicolas V, Servin AL. 2011. Secreted autotransporter toxin (Sat) triggers autophagy in epithelial cells that relies on cell detachment. Cell. Microbiol. 13:992–1013. 10.1111/j.1462-5822.2011.01595.x. [DOI] [PubMed] [Google Scholar]
- 46.Lee MS, Cherla RP, Jenson MH, Leyva-Illades D, Martinez-Moczygemba M, Tesh VL. 2011. Shiga toxins induce autophagy leading to differential signalling pathways in toxin-sensitive and toxin-resistant human cells. Cell. Microbiol. 13:1479–1496. 10.1111/j.1462-5822.2011.01634.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gutierrez MG, Saka HA, Chinen I, Zoppino FC, Yoshimori T, Bocco JL, Colombo MI. 2007. Protective role of autophagy against Vibrio cholerae cytolysin, a pore-forming toxin from V. cholerae. Proc. Natl. Acad. Sci. U. S. A. 104:1829–1834. 10.1073/pnas.0601437104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Di Venanzio G, Stepanenko TM, García Véscovi E. 2014. Serratia marcescens ShlA pore-forming toxin is responsible for early induction of autophagy in host cells and is transcriptionally regulated by RcsB. Infect. Immun. 82:3542–3554. 10.1128/IAI.01682-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Terebiznik MR, Raju D, Vazquez CL, Torbricki K, Kulkarni R, Blanke SR, Yoshimori T, Colombo MI, Jones NL. 2009. Effect of Helicobacter pylori's vacuolating cytotoxin on the autophagy pathway in gastric epithelial cells. Autophagy 5:370–379. 10.4161/auto.5.3.7663. [DOI] [PubMed] [Google Scholar]
- 50.Shahnazari S, Namolovan A, Mogridge J, Kim PK, Brumell JH. 2011. Bacterial toxins can inhibit host cell autophagy through cAMP generation. Autophagy 7:957–965. 10.4161/auto.7.9.16435. [DOI] [PubMed] [Google Scholar]
- 51.Wolfson JJ, Jandhyala DM, Gorczyca LA, Qadeer Z, Manning SD, Hadler J, Rudrik JT, Thorpe CM. 2009. Prevalence of the operon encoding subtilase cytotoxin in non-O157 Shiga toxin-producing Escherichia coli isolated from humans in the United States. J. Clin. Microbiol. 47:3058–3059. 10.1128/JCM.00706-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paton AW, Paton JC. 2005. Multiplex PCR for direct detection of Shiga toxigenic Escherichia coli strains producing the novel subtilase cytotoxin. J. Clin. Microbiol. 43:2944–2947. 10.1128/JCM.43.6.2944-2947.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Avivar-Valderas A, Bobrovnikova-Marjon E, Alan Diehl J, Bardeesy N, Debnath J, Aguirre-Ghiso JA. 2013. Regulation of autophagy during ECM detachment is linked to a selective inhibition of mTORC1 by PERK. Oncogene 32:4932–4940. 10.1038/onc.2012.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hart LS, Cunningham JT, Datta T, Dey S, Tameire F, Lehman SL, Qiu B, Zhang H, Cerniglia G, Bi M, Li Y, Gao Y, Liu H, Li C, Maity A, Thomas-Tikhonenko A, Perl AE, Koong A, Fuchs SY, Diehl JA, Mills IG, Ruggero D, Koumenis C. 2012. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J. Clin. Invest. 122:4621–4634. 10.1172/JCI62973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liang C. 2010. Negative regulation of autophagy. Cell Death Differ. 17:1807–1815. 10.1038/cdd.2010.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liou ML, Liou HC. 1999. The ubiquitin-homology protein, DAP-1, associates with tumor necrosis factor receptor (p60) death domain and induces apoptosis. J. Biol. Chem. 274:10145–10153. 10.1074/jbc.274.15.10145. [DOI] [PubMed] [Google Scholar]
- 57.Lavallard VJ, Meijer AJ, Codogno P, Gual P. 2012. Autophagy, signaling and obesity. Pharmacol. Res. 66:513–525. 10.1016/j.phrs.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 58.Ravikumar B, Futter M, Jahreiss L, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Narayanan U, Renna M, Jimenez-Sanchez M, Sarkar S, Underwood B, Winslow A, Rubinsztein DC. 2009. Mammalian macroautophagy at a glance. J. Cell Sci. 122:1707–1711. 10.1242/jcs.031773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wazir U, Jiang WG, Sharma AK, Mokbel K. 2012. The mRNA expression of DAP1 in human breast cancer: correlation with clinicopathological parameters. Cancer Genomics Proteomics 9:199–201. [PubMed] [Google Scholar]
- 60.Jia Y, Ye L, Ji K, Toms AM, Davies ML, Ruge F, Ji J, Hargest R, Jiang WG. 2014. Death associated protein 1 is correlated with the clinical outcome of patients with colorectal cancer and has a role in the regulation of cell death. Oncol. Rep. 31:175–182. 10.3892/or.2013.2866. [DOI] [PubMed] [Google Scholar]