

Abstract
The mycobacterial Esx-1 (ESAT-6 system 1) exporter translocates virulence factors across the cytoplasmic membrane to the cell wall, cell surface, and the bacteriological medium in vitro. The mechanisms underlying substrate targeting to distinct locations are unknown. Several Esx-1 substrates are N-α-terminally acetylated. The role of this rare modification in bacteria is unclear. We sought to identify genes required for Esx-1 substrate modification, transport, and localization. Pathogenic mycobacteria lyse Acanthamoeba castellanii in an Esx-1-dependent manner. We conducted a genetic screen to identify Mycobacterium marinum strains which failed to lyse amoebae. We identified a noncytotoxic M. marinum strain with a transposon insertion in a predicted N-α-terminal acetyltransferase not previously linked to mycobacterial pathogenesis. Disruption of this gene led to attenuation of virulence, failure to induce a type I interferon response during macrophage infection, and loss of hemolytic activity. The major Esx-1 substrates, EsxA and EsxB, were exported to the cell surface, but only low levels were released into the bacteriological medium. The balance of EsxA N-α-terminal acetylation was disrupted, resulting in a mycobacterial strain in which surface-associated EsxA was hyperacetylated. Genetic complementation completely restored Esx-1 function and the levels of N-α-terminally acetylated EsxA on the surface but restored only low levels of Esx-1 substrates in the bacteriological medium. Our results reveal a novel gene required for mycobacterial Esx-1 export. Our findings indicate that maintaining the homeostasis of Esx-1 substrate N-α-terminal acetylation is essential for Esx-1-mediated virulence. We propose an inverse correlation between EsxA acetylation and virulence.
INTRODUCTION
The ESAT-6 system 1 (Esx-1) exporter/WXG-100 secretion system (WSS) promotes the virulence of both mycobacterial and Gram-positive pathogens (1–7). In pathogenic mycobacteria, Esx-1 mediates the interaction between the bacteria and the macrophage cytosol by permeabilizing the phagosomal membrane (8–13). The Esx-1/WSS are conserved in several nonpathogenic bacterial species where they have diverse roles (14–20).
Mycobacterial protein “substrates” are transported across the cytoplasmic membrane to extracytoplasmic locations in vitro in an Esx-1-dependent manner (4, 6, 21–33). The major Esx-1 substrates, EsxA (ESAT-6, for early secreted antigenic target, 6 kDa) and EsxB (CFP-10, for culture filtrate protein, 10 kDa) have been localized to the cell wall, the cell surface, the capsule, and the bacteriological medium (culture supernatant) in vitro (21, 28, 29, 31, 33). It is unclear if substrates are in the bacteriological medium because they are extrinsically associated with the cell wall and passively shed into the medium or true exoproteins that are actively secreted (34, 35). Recently, we demonstrated that surface association of EsxA and EsxB, rather than secretion into the culture supernatant, correlated with virulence in Mycobacterium marinum (27).
Mycobacterium marinum is a pathogenic mycobacterial species that serves as an established model for Esx-1 export in Mycobacterium tuberculosis (2, 36–38). In M. marinum, the esx-1 locus (MMAR_5439 through MMAR_5459) includes the genes required for Esx-1 export (2, 23, 24, 30, 39). We recently reported a second genomic region, MMAR_1663 to MMAR_1668, disruption of which also abrogates Esx-1 substrate export to the cell surface and into the culture supernatant (27). Several individual genes are required for substrate export, disruption of which generally results in a loss of substrate export into the bacteriological medium and Esx-1-mediated virulence. However, several recent studies have indicated that the relationship between Esx-mediated export in vitro and virulence is not straightforward (65, 66).
Based on the predicted localizations of Esx-1-associated proteins, the Esx-1 exporter is thought to span the cytoplasmic membrane (34, 40, 41). How the Esx-1 substrates traverse the mycolate outer membrane (MOM) to distinct locations or if the known Esx-1 genes promote this process are unknown (34, 41).
EsxA in the M. tuberculosis culture supernatant is N-α-terminally acetylated (42, 43). N-α-terminal acetylation is the acetylation of the α-amino group of the N-terminal amino acid of a protein substrate and is completely distinct from N-ε lysine acetylation. Although N-α-terminal acetylation is a nearly ubiquitous modification of human and Saccharomyces cerevisiae proteins, there are very few specific examples of N-α terminally acetylated proteins in bacteria (44, 67). EsxA is the only bacterial virulence factor known to be N-α-terminally acetylated (43, 44). The mechanism behind the N-α-terminal acetylation of EsxA, and how this modification affects Esx-1-mediated secretion and virulence are unknown.
Because we do not know how Esx-1 substrates cross the MOM, how they are targeted to distinct extracytoplasmic locations, or how they are modified, we hypothesize that there are several additional loci required for Esx-1 export that are undefined.
The uptake and growth of pathogenic mycobacteria in both macrophages and amoebae result in Esx-1-mediated cytolysis (8, 9, 45, 46). We generated an M. marinum M strain transposon (Tn) insertion library including ∼25,000 independent strains (47). Using this near-saturating library, we conducted a pilot genetic screen to identify novel genes required for Esx-1-mediated cytolysis. Thus far, we have screened ∼1,100 mycobacterial strains and identified 7 strains with decreased cytolytic activity with defects in the Esx-1 export system. Here, we describe one strain isolated from our screen and characterize a novel gene in M. marinum that is required for Esx-1-mediated export and virulence. We demonstrate that loss of this gene disrupts the homeostasis of EsxA acetylation on the mycobacterial cell surface, which can be restored using genetic complementation. We propose a model for Esx-1 export in which the unacetylated form of EsxA on the cell surface correlates with mycobacterial virulence. Understanding how the Esx-1 system functions to promote protein translocation at the molecular level will advance both our understanding of mycobacterial virulence mechanisms and how to counteract them.
MATERIALS AND METHODS
Microbiological strains and growth conditions.
All of the Mycobacterium marinum strains used in this study were derived from the M strain (ATCC BAA-535) and are listed in Table S1 in the supplemental material. We follow the nomenclature rules for Esx-1-associated genes that were proposed by Bitter et al. (48). Briefly, Ecc is Esx conserved components, Esp is Esx secretion-associated protein, etc.; subscript 1 designates that the genes are found at the esx-1 locus. The 120A3 strain is part of a Tn insertion library maintained in the laboratory (47). In the 120A3 M. marinum strain, the Tn inserted between the T/A dinucleotide at bases 42194 and 42195.
M. marinum strains were grown in Middlebrook 7H9 defined liquid broth (Sigma-Aldrich) with 0.005% glycerol and 0.1% Tween 80 (Fisher Scientific) or on Middlebrook 7H11 agar as previously described (24). Strains were supplemented with kanamycin (20 μg/ml; IBI) or hygromycin (50 μg/ml; EMD Millipore) when appropriate. For in vitro growth experiments, mycobacterial strains were grown to an optical density at 600 nm (OD600) of ∼1.8 in Middlebrook 7H9 broth. Each strain was inoculated into 25 ml of fresh 7H9 broth to an OD600 of 0.4 and grown at 30°C under agitation. The OD600 was measured at the time points shown in Figure S1 in the supplemental material.
Identification of the Tn insertion in the 120A3 strain.
The Tn insertion site in the 120A3 strain was determined by generating a plasmid library using 120A3 genomic DNA and selecting for the plasmid bearing the DNA with the Tn insertion on kanamycin, exactly as described previously (27, 47). Plasmid preparations were performed using an AccuPrep Plasmid Miniprep DNA extraction kit (Bioneer). The Tn insertion site was identified by Sanger sequencing analysis performed by the Genomics Facility at the University of Notre Dame using the 821A and 822A primers (see Table S1 in the supplemental material), which anneal to the Tn and allow sequencing of the Tn/genome junction (49). All primers in this study were synthesized by Integrated DNA Technologies (IDT).
Plasmid construction.
The PMOPMMAR_0039 (where MOP is mycobacterial optimal promoter) complementation plasmid was constructed by amplifying the MMAR_0039 gene from M. marinum genomic DNA as annotated on MycoBrowser (50) using the opc242 and opc244 oligonucleotide primers (see Table S1). The resulting PCR product was purified using an AccuPrep PCR purification kit (Bioneer) and digested with HpaI and SnaB1 (New England BioLabs). The pMH406 plasmid includes the esxBA genes from M. tuberculosis behind the constitutive MOP (4, 51). The hygromycin (Hyg) variant of this plasmid was a gift from Jeffery S. Cox. The esxBA genes were excised from the pMH406 Hyg plasmid by digestion with PvuII and HpaI. The MMAR_0039 gene was introduced into the digested and purified pMH406 Hyg vector lacking the esxBA genes. The resulting complementation plasmid was confirmed by Sanger sequencing analysis and was introduced into the 120A3 strain by electroporation. The resulting complementation strain was confirmed by amplifying the MMAR_0039 gene using ofm19 and ofm20 oligonucleotide primers (Table S1).
Protozoan strains and growth conditions.
Acanthamoeba castellanii (Douglas) Page was obtained from the American Type Culture Collection (ATCC 30234) and was grown and maintained as previously described (45).
Genetic screen to identify genes required for mycobacterial virulence.
A total of 2 × 104 A. castellanii amoebae were seeded in each well of a 96-well plate (Sarstedt). M. marinum strains from the Tn insertion library (47) were delivered from colonies on agar plates using a 48-pin replicator tool to Page's modified Neff's amoeba saline (PAS) medium in a fresh 96-well plate. A multichannel pipette was used to mix cells in suspension, and 10 μl of each M. marinum suspension was delivered to the amoebae, resulting in various multiplicities of infection (MOIs). The plate was centrifuged to promote interaction between the amoebae and bacteria (1,000 rpm for 30 min) and incubated for 2 h at room temperature (RT). The infected monolayers were washed three times with phosphate-buffered saline (PBS) to remove external bacteria. The amoebae were incubated in fresh PAS medium, and the monolayers were visualized under a light microscope every day for up to 5 days postinfection. Strains that failed to significantly disrupt the monolayer, indicating a loss of cytotoxicity against the amoebae similar to the M. marinum control strain with a deletion of region of difference 1 (ΔRD1) were selected for future study. We screened 13 96-well plates for the pilot screen.
Cytotoxicity assays.
M. marinum strains were incubated with A. castellanii at an MOI of 10 as described by Kennedy et al. (45). Following 2 h of incubation with M. marinum, the amoebae were washed three times with PBS, fresh peptone-yeast-glucose (PYG) medium was added, and the cells were incubated for 24 h at RT. RAW 264.7 macrophages were coincubated with M. marinum at an MOI of 25 for 2 h. The macrophages were washed three times with PBS and incubated at 37°C and 5% CO2 in fresh Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) for 3 h. Amoebae and macrophages were stained using either ethidium homodimer 1 (EthD-1; Invitrogen/Molecular Probes) or a Live/Dead viability/cytotoxicity kit for mammalian cells (Life Technologies) and imaged using an AxioObserver inverted microscope (Zeiss) exactly as described by Kennedy et al. (27).
Gentamicin protection assays.
Amoebae and RAW macrophages were infected with M. marinum at an MOI of 1 using the same procedure as described above. After phagocytosis, 100 μg/ml of gentamicin was added to the medium, and the cells were incubated for an additional 2 h to kill extracellular bacteria. The amoebae and macrophages were washed three times with PBS. Amoebae were incubated at RT in fresh PAS medium. The infected RAW macrophages were maintained in fresh DMEM supplemented with 10% FBS at 37°C and 5% CO2. Bacteria were harvested, and CFU were counted at specific time points as described previously (52). Statistically significant differences between the wild-type (WT) and either the 120A3 or ΔRD1 CFU count at each time point were determined by performing a two-tailed Student's t test using the TTEST function in Microsoft Excel. P values of <0.05 were considered significant.
Hemolysis assay.
Sheep red blood cell (sRBC) (defibrinated sheep RBCs; BD) lysis assays were performed by incubating M. marinum with washed sRBCs for 2 h at 30°C exactly as described previously (47). Each assay was performed on at least three biological replicates. The data provided here are representative of the three replicates. Error bars represent standard deviations.
Esx-1 secretion assays.
M. marinum bacteria were grown in Sauton's liquid medium to induce Esx-1 export exactly as described previously (27). Cell lysate (CL) and culture filtrate (CF) fractions were prepared and quantitated exactly as previously described (27). Substrate export was detected by either Western blot analysis, matrix-assisted laser desorption ionization–mass spectrometry (MALDI-MS), or quantitative mass spectrometry (see below). For the Western blot analysis, 12.5 μg of cell lysates and culture filtrates was separated on a 4 to 20% Criterion Tris-HCl polyacrylamide gel (Bio-Rad) and transferred to nitrocellulose. Nitrocellulose membranes were incubated with antibodies against ESAT-6 (EsxA) (1:3,000 [ab26246; Abcam]), CFP-10 (EsxB) (1:5,000 [NR-13801; BEI Resources]), MPT-32 (1:5,000 [NR-13807; BEI Resources]), or RNA polymerase subunit β (RNAP-β) (1:5,000 [ab12087; Abcam]) and detected using either chemiluminescence or a Li-Cor Odyssey Imager as described previously (47). Surface protein (SP) fractions were generated by washing M. marinum colonies grown on Sauton's agar with water, as described previously (27, 47). MALDI-MS analysis of SP fractions was performed identically as described by M. M. Champion et al. (47). Briefly 1 μl of SPs from the WT, eccCb::Tn, 120A3, and complemented (MMAR_0039) strains was spotted and dried on a 384-well MALDI target, overlaid with 1 μl of a saturated solution of sinapinic acid in 0.1% trifluoroacetic acid (TFA; Sigma, St. Louis, MO), and dried. MALDI spectra were acquired on a Bruker AutoFlex tandem time of flight (TOF/TOF) MS in linear mode. External (adjacent-spot) calibration was performed using myoglobin (16,952.3 m/z, 8,476.6 m/z) and insulin (5,734.5 m/z). Spectra were acquired at 100 Hz; 2,000 spectra were summed per spot. Remaining SP fractions were digested and analyzed as described below.
Mycobacterial RNA extraction and qualitative RT-PCR.
Total RNA was extracted from 25 ml of exponential-phase cultures of M. marinum using an RNeasy minikit (Qiagen) according to the manufacturer's recommendation with the following modifications. After bacterial cells were harvested, they were resuspended in 1 ml of RLT buffer containing 0.1% 2-mercaptoethanol and transferred to a 2-ml screw-cap tube containing glass beads. The mixture was homogenized in a mini-bead beater (BioSpec Products) with three 30-s pulses. RNA samples were treated with DNase I. Reverse transcriptase PCR (RT-PCR) was performed using a SuperScript III One-Step RT-PCR kit (Life Technologies) according to the instructions. The thermal cycler program was as follows: cDNA synthesis step at 45°C for 30 min; denaturation step 94°C for 2 min, followed by 30 cycles of amplification (94°C for 15 s, 58°C for 30 s, and 68°C for 1 min) and a final extension of 68°C for 5 min.
Macrophage infection and RNA extraction.
The RAW 264.7 mouse macrophage cell line was maintained at 37°C and 5% CO2 in DMEM supplemented with 10% FBS. A total of 5 × 105 cells were preseeded in a 24-well plate (Greiner Bio-one) for 24 h. The cells were incubated with 5 × 106 bacteria for 2 h at 37°C. The nonphagocytized bacteria were removed by washing, and fresh medium was added. RNA was extracted at 4 h postinfection using a Direct-zoL RNA MiniPrep kit (Zymo Research) according to the manufacturer's instructions. Eluted RNA was digested with RNase-free DNase I and repurified using a Direct-zoL column. The RNA was quantified using a Nanodrop 2000 instrument (Thermo Fisher Scientific).
qRT-PCR.
Quantitative real-time PCR (qRT-PCR) was performed using a Power SYBR Green 1-Step kit (Life Technologies) and an ABI Prism 7500 Fast real-time PCR system (Life Technologies). The reaction was performed in 20 μl using 100 nM primers and 50 ng of RNA as the template. The thermal cycling parameters were as follows: step 1, 48°C for 30 min; step 2, 95°C for 10 min; step 3, 95°C for 15 s; step 4, 60°C for 15 s. Steps 3 and 4 were repeated for 40 cycles, followed by melt curve analysis. The mRNA level of beta interferon (IFN-β) was normalized to that of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The IFN-β and GAPDH primers used here are listed in Table S1 in the supplemental material. Statistical analysis was performed with GraphPad Prism, version 5, software using a Mann-Whitney U nonparametric test.
Mass spectrometry proteomics.
Targeted mass spectrometry (multiple-reaction monitoring [MRM]) with stable isotope dilution mass spectrometry were performed essentially as described in Kennedy et al. (27) and as briefly described below.
Protein digestion.
Digestion and cleanup were performed as described previously (27, 47). Fifty micrograms of cell lysate or culture filtrate or 100 μl of surface protein (ca. 20 μg) was desalted and delipidated by two rounds of acetone precipitation and washing. Samples were resuspended in 25 μl of 2,2,2-trifluoro-ethanol (TFE; Sigma), 25 μl of 50 mM ammonium bicarbonate (ABC; Sigma), and 10 mM dithiothreitol (DTT; Sigma) and reduced at 56°C for 1 h. Following this, samples were alkylated with 15 mM iodoacetic acid (Sigma) for 15 min at RT in the dark. Next, samples were diluted to ∼500 μl with 50 mM ABC, and 500 ng of sequencing grade trypsin (Promega) was added in two additions 1.5 h apart; samples were allowed to digest for 8 h at 37°C. Following digestion, samples were quenched by the addition of 5 μl of TFA (Sigma), dried to <20 μl, and then resuspended to 100 μl total in 0.3% formic acid (Fisher). Samples were desalted using a C18 spin column (Protea) according to the manufacturer's instructions. To 10 μg of each sample, stable heavy-isotope peptides with 13C15N labeling of Lys and Arg were added as described by Kennedy et al. (27) from GroES, EsxA, MMAR_2929, and the N-terminal peptide of EsxA (NT-EsxA) with and without acetylation (New England Peptide). All peptides were added as 500× dilutions from the following stocks: GroES, 130 μM stock, 130 fmol on-column with each sample; MMAR_2929, 116 μM; EsxA (internal), 142 μM. NT-EsxA (TEQQWNFAGIEAASSAIQGNVTSISHLLDEGK) and EsxA (Ac-TEQQWNFAGIEAASSAIQGNVTSISHLLDEGK, where Ac is acetylated) were added at 1:50 dilutions due to poor recovery (used as absolute internal standards[IS] only, normalized to GroES and EsxA internal peptide sequence). All stable heavy-isotope peptides were tested for purity, and transitions were determined using MRM as described below for liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. Samples were then dried to dryness in a SpeedVac and resuspended in 25 μl of 0.5% formic acid prior to LC-MS/MS analysis.
LC-MS/MS (MRM) targeted quantitative mass spectrometry was performed nearly identically as described by Kennedy et al. (27). Two microliters of the above-digested culture filtrate, cell lysate, or cell surface proteins plus stable isotope standards (800-ng total) was loaded onto a 100-μm by 100-mm C18 BEH column (Waters) running at 600 nl/min on a two-dimensional (2D) nano-Ultra high-performance liquid chromatography (HPLC) system (Eskigent). A 90-min gradient running from 2 to 50% buffer B (buffer A consists of 0.1% formic acid in water; buffer B consists of 0.1% formic acid in acetonitrile) (Burdick and Jackson) was used for separation (100-min total). MRM mass spectrometry was performed on a QTrap 6500 (AB Sciex) running in triple-quadrupole mode. Source was set as curtain (CUR) 25, voltage at 2.3 kV (ion spray voltage [ISV]), and sheath gas (GS1) at 12 lb/in2. MRM transition parameters are provided as Fig. S3 in the supplemental material. A total of 111 transitions were monitored including stable heavy isotopes and quantifier qualifier. MRM transitions were determined as published previously (24, 27, 53, 54). All samples were analyzed in triplicate with biological duplicates.
Peak area integration.
The top transition (quantifier) from each set of peptide transitions was integrated using MultiQuant (version 2.1) (AB Sciex) using the MQ4 integration algorithm with 3-point Gaussian smoothing. Area ratios were taken versus the peak area of the constant stable heavy-isotope area for each peptide. The absolute quantification for each peptide/protein was taken as the absolute ratio of the known stable heavy-isotope peak area to the endogenous area, averaged for all replicate injections. Normalization to WT and relative MRM area ratios were compared to the area and the area ratio of GroES and used to correct for a response due to cellular lysis and loading/autosampler variance. Our normalization is designed to mimic the response of Western blotting but is substantially more sensitive, multiplexed, and capable of absolute quantification. Absolute quantification of proteins was taken as the average quantitative response normalized per 1 μg of total protein as this enables comparison to other laboratories' nonstandard concentrations from culture filtrate preparations. Since equal culture volumes and growth rates were represented from each sample, this serves as a reasonable estimate of the response and quantification obtained on a per/volume/CFU basis. To determine the statistical differences between the acetylated and unacetylated forms of EsxA, we performed Student's t test using the TTEST function in Microsoft Excel. We performed a one-way analysis of variance (ANOVA) test to determine if the trends of the percent acetylation from each fraction were significantly different. Finally, we performed a one-way ANOVA test to determine if there were statistically significant differences between the levels of EsxA on the cell surface of the WT, 120A3, and complemented strains.
RESULTS
Identification of an M. marinum strain which causes decreased cytolysis of amoebae.
We hypothesized that additional genes required for Esx-1 export could be identified by screening for M. marinum strains that cannot lyse A. castellanii. We initiated a pilot genetic screen (1,100 M. marinum strains) using a near-saturating transposon (Tn) insertion library in Mycobacterium marinum (47). We spotted the Tn insertion library onto agar, diluted the colonies into fresh PAS (Page's Modified Neff's amoeba saline) medium, and infected amoebae seeded in 96-well plates with M. marinum for 2 h using variable MOIs. We visualized the amoeba monolayer every day for 5 days postinfection until the majority of M. marinum strains lysed the amoeba monolayers. We reasoned that this approach would enrich for strains specifically defective for Esx-1-mediated cytolysis. M. marinum Tn insertion strains that failed to lyse the monolayer were used to reinfect A. castellanii at a fixed MOI (MOI of 10). The ΔRD1 M. marinum strain, which bears a deletion in several genes required for Esx-1 export, and the wild-type (WT) M strain were used as controls. Cytolysis was detected by staining the infected amoebae at 24 h postinfection with ethidium-homodimer 1 (EthD-1), a nucleic acid stain that does not cross intact cellular membranes (2, 27). In cells with permeabilized membranes, the dye binds DNA and emits a red fluorescent signal. The data from this experiment, in which we identified the 120A3 M. marinum strain, are presented in Fig. 1. Infection of the amoebae with WT M. marinum resulted in cytolysis of the amoeba monolayer and a red fluorescent signal (Fig. 1A). The ΔRD1 strain caused less cytolysis and a lower fluorescent signal. Similar to infection with the ΔRD1 strain, infection of the amoebae with the 120A3 strain resulted in low levels of cytolysis and red fluorescence. We measured changes in cytolysis by counting the number of EthD-1-stained cells across 10 fields (Fig. 1B).
FIG 1.
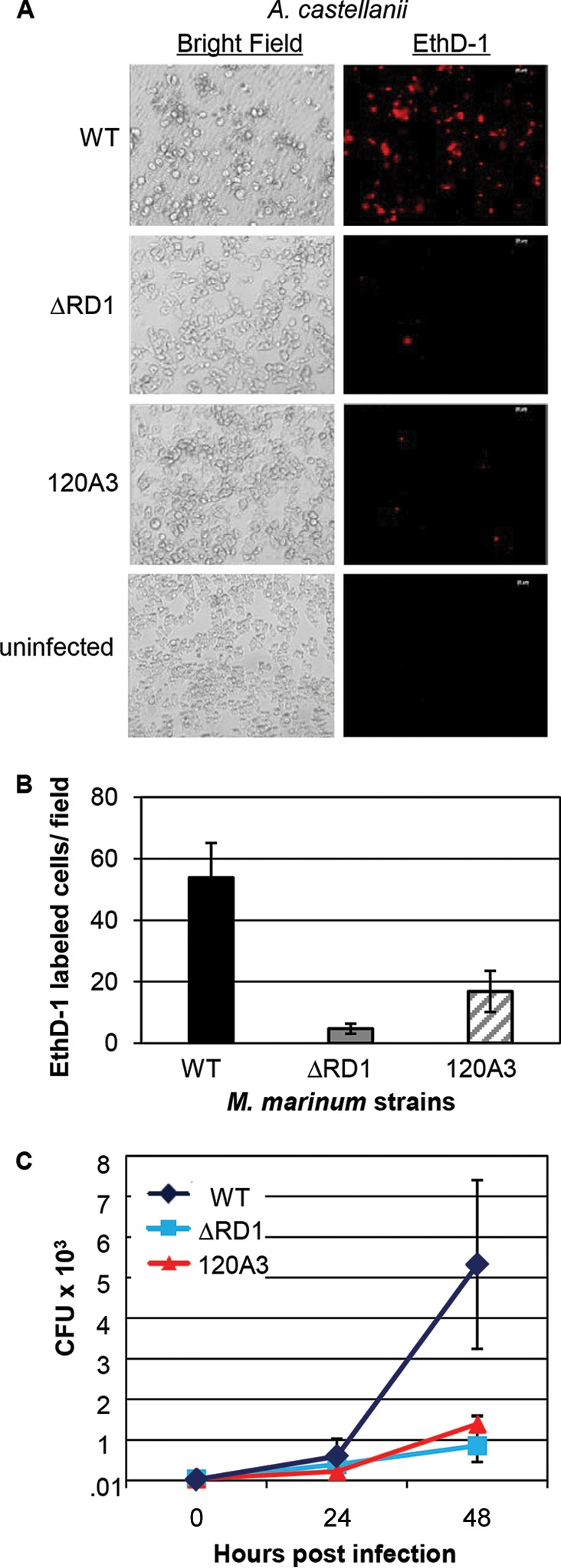
The 120A3 strain is less cytotoxic and attenuated for growth in amoebae. M. marinum infection of A. castellanii was performed at an MOI of 10. (A) Images were acquired at 24 h postinfection using a 20× objective on an AxioObserver inverted microscope (Zeiss). Scale bar, 20 μm. Ethidium homodimer (EthD-1) staining showed the permeabilization of amoeba membranes (right). (B) Quantification of the amoeba cytolysis. Red EthD-1-stained cells per field were counted; 10 independent fields from each infection were counted, and the counts were averaged. Error bars indicate the standard deviations between fields. (C) Gentamicin protection assays. Following infection, the amoeba monolayer was treated with gentamicin (100 μg/ml) for 2 h. Treated cells were washed and resuspended in fresh medium. Bacteria were harvested and plated for CFU at the indicated time points. Error bars represent the standard deviations; experiments were performed in triplicate. The differences between the CFU counts at 48 h postinfection for the WT versus the ΔRD1 strain (P = 0.02) and WT versus the 120A3 strain (P = 0.03) were statistically significant as determined by a Student's t test. The CFU counts at 0 and 24 h postinfection were not significantly different (P > 0.05).
To verify the cytolysis data, we measured the ability of each strain to replicate within the amoebae using gentamicin protection assays. Following infection, gentamicin was added to the medium to kill extracellular bacteria, while the internalized bacteria were protected by the amoebae (45). The results of this experiment are shown in Fig. 1C. The WT strain replicated within the amoebae through 48 h postinfection. The ΔRD1 and the 120A3 strains were attenuated for growth compared to the WT strain. To demonstrate that the attenuation was not due to an overall growth defect of the 120A3 strain, we measured in vitro growth of the M. marinum strains in defined medium (see Fig. S1 in the supplemental material). We observed no significant difference between the growth rates of the WT, ΔRD1, and 120A3 strains. We conclude that we have identified an M. marinum strain which caused decreased cytolysis of and was attenuated for growth within amoebae.
The 120A3 strain bears a Tn insertion in the MMAR_0039 gene.
To map the Tn insertion in the genome of the 120A3 M. marinum strain, we isolated the Tn-containing region and defined the mariner Tn/genomic DNA junction using DNA sequencing analysis (27, 47). The Tn insertion mapped between the T/A dinucleotide at position 42194/42195 bp, which is within the predicted open reading frame for the MMAR_0039 gene (annotated as bases 41992 to 42291) (Fig. 2A).
FIG 2.
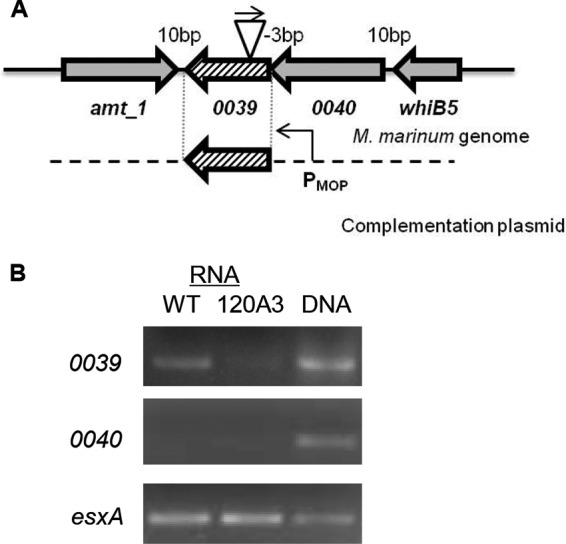
The Tn insertion in the 120A3 strain disrupts transcription from the MMAR_0039 gene. (A) Genetic organization of the MMAR_0039 locus in the genome of M. marinum. The Tn inserted within the MMAR_0039 gene in the reverse orientation (the direction of the Kan promoter on the Tn relative to the orientation of the MMAR_0039 promoter). (B) Qualitative RT-PCR analysis of MMAR_0039, MMAR_0040, and esxA in the WT and 120A3 strains. esxA was used as a positive control for transcription. DNA refers to genomic DNA.
Based on the position of the Tn insertion relative to the predicted MMAR_0039 open reading frame, we expected that transcription of the MMAR_0039 gene would be disrupted in the 120A3 strain. We measured MMAR_0039 transcription using qualitative RT-PCR with primers which flanked the Tn insertion site (Fig. 2B). We detected a band corresponding to the MMAR_0039 transcript from RNA isolated from the WT strain. A much weaker band was observed from RNA isolated from the 120A3 strain. These data indicate that transcription from the MMAR_0039 gene was reduced in the 120A3 strain compared to the WT strain.
The annotation of the region in MycoBrowser indicates that the adjacent gene, MMAR_0040, overlaps the MMAR_0039 gene by 3 bp (Fig. 2A) (50). To test if the two genes were cotranscribed, we measured transcription from the MMAR_0040 gene. We did not detect MMAR_0040 transcript from either strain under these experimental conditions (Fig. 2B). As a positive control, we measured transcription from the esxA gene, which resides at the esx-1 locus. We amplified size-appropriate products from genomic DNA for all three genes. These data indicate that despite the predicted overlap between the two genes, MMAR_0039 and MMAR_0040 are not cotranscribed in M. marinum under the conditions tested. Considering the orientation of the amt_1 gene, we conclude that the Tn insertion specifically affected transcription from the MMAR_0039 gene.
MMAR_0039 is required for virulence in M. marinum.
Because the 120A3 strain causes decreased cytolysis toward amoebae, we expected that it would also be attenuated in macrophage models of infection. Because cytolysis requires uptake, replication, and cytosolic access of pathogenic mycobacteria (2, 8, 12, 46), we used cytolysis to measure the endpoint of mycobacterial infection in macrophages. We infected RAW 246.7 murine macrophage-like cells with the WT, eccD1::Tn, and 120A3 strains. The eccD1::Tn strain bears a Tn insertion in the eccD1 gene, which is required for Esx-1 export (4, 6, 79). To measure Esx-1-dependent cytolysis, we infected RAW 246.7 cells at an MOI of 25. At 3 h postinfection, we stained the macrophage monolayers for lysis and viability using EthD-1 and calcein-acetoxymethyl (calcein-AM). Calcein-AM is a membrane-permeable dye that is cleaved by esterases in the cytosol of viable cells. Cleavage results in a product localized to the cytosol that emits green fluorescence. As with amoebae, infection of the RAW 246.7 cells with the WT strain resulted in decreased viability (less green signal) and increased lysis (increased red signal) compared to uninfected cells (Fig. 3A). The eccD1::Tn strain failed to appreciably lyse the macrophage monolayer compared to the WT strain. Infection of the RAW 246.7 cells with the 120A3 strain was phenotypically similar to that with the eccD1::Tn strain. Cytolysis was quantified by counting the number of EthD-1-labeled RAW cells per field across at least 10 independent fields (Fig. 3B). We measured the ability of each strain to replicate within the RAW cells using gentamicin protection assays (see Fig. S2 in the supplemental material). The WT strain replicated within the RAW 246.7 cells through 48 h postinfection. The ΔRD1 and the 120A3 strains were attenuated for growth compared to the WT strain. From these data we conclude that the 120A3 strain causes decreased cytolysis and is attenuated for growth within RAW 246.7 cells.
FIG 3.

The MMAR_0039 gene is required for macrophage cytolysis and IFN-β induction. (A) M. marinum infection of RAW 264.7 cell line. Macrophages were infected at an MOI of 25. Images were acquired at 3 h postinfection with a 20× objective on a Zeiss AxioObserver microscope. Scale bar, 20 μm. EthD-1 staining reflected membrane permeabilization of the macrophages. Calcein-AM labeled live macrophages. Comp, complemented strain 120A3/PMOPMMAR_0039. (B) Quantification of RAW 246.7 cytolysis. The numbers of red EthD-1-stained cells per 10 independent fields were counted, and the numbers of stained cells were averaged. Error bars indicate the standard deviation between fields. (C) The induction of the type I IFN response was assessed in RAW 246.7 cells at 4 h postinfection by measuring the expression level of IFN-β using qRT-PCR. IFN-β transcript was normalized to the levels of GAPDH transcript. Significance was determined using a Mann-Whitney U nonparametric test.
To determine if the Tn insertion in the MMAR_0039 gene was responsible for the decreased cytotoxicity, we sought to genetically complement the 120A3 M. marinum strain. We introduced the MMAR_0039 gene behind the mycobacterial optimal promoter (MOP) and integrated it into the att site in the M. marinum genome to generate the complemented strain (120A3/PMOPMMAR_0039) (Fig. 2A) (4, 51). Infection of the RAW 246.7 cells with the complemented strain restored Esx-1-mediated cytolysis (Fig. 3A and B). From these data we conclude that the MMAR_0039 gene is specifically required for Esx-1-mediated virulence and cytolysis.
The M. tuberculosis Esx-1 system elicits a type I interferon (IFN) response during the infection of macrophages (13). We hypothesized that the M. marinum Esx-1 system would also elicit a type I IFN response during macrophage infection. We measured transcription of the type I IFN, IFN-β, using quantitative RT-PCR (qRT-PCR) on RNA isolated from RAW 246.7 cells at 4 h postinfection with M. marinum (Fig. 3C). The levels of IFN-β transcript were normalized to the levels of GAPDH transcript. Compared to uninfected macrophages, WT M. marinum significantly induced transcription of IFN-β (P = 0.0286). The level of IFN-β transcript induced by infection of the RAW 246.7 cells with the eccD1::Tn strain and the 120A3 strain was significantly reduced compared to that of the WT strain (P = 0.0286). Infection of the RAW 246.7 cells with the complemented strain restored induction of IFN-β transcript. There was no significant difference between the levels of induction by the WT and complemented strains (P = 0.2000). From these data we conclude that M. marinum induces IFN-β during macrophage infection in an Esx-1-dependent, MMAR_0039-dependent manner.
MMAR_0039 is required for Esx-1 export in M. marinum.
Because host cell cytolysis and IFN-β induction are functions mediated by Esx-1 ex vivo, we tested if the MMAR_0039 gene was required for Esx-1 function in vitro. M. marinum lyses sheep red blood cells (sRBCs) in a contact-dependent, Esx-1-dependent manner (2, 55). We measured the hemolytic activity of the 120A3 strain (Fig. 4A). WT M. marinum lysed sRBCs, as indicated by the increased OD405 compared to that of the negative control (PBS). The strains bearing Tn insertions in known Esx-1 genes (eccD1 and eccCb) failed to lyse sRBCs. The eccCb::Tn strain bears a Tn insertion in the eccCb gene which encodes an FtsK-like AAA-ATPase required for Esx-1 export (4, 6). Consistent with a loss of Esx-1 function, the 120A3 strain was nonhemolytic. Hemolysis was restored to nearly WT levels in the complemented strain. These data demonstrate that the MMAR_0039 gene is required for Esx-1-mediated hemolysis.
FIG 4.
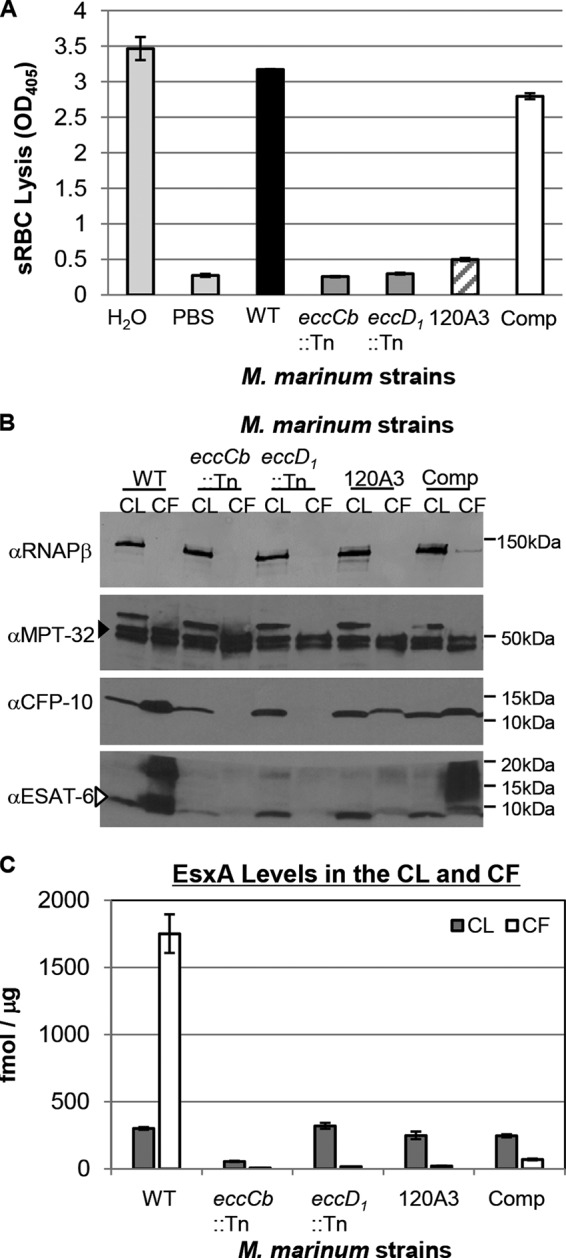
The MMAR_0039 gene is required for Esx-1 export and hemolysis in vitro. (A) Hemolysis assay to measure Esx-1 function. PBS served as a negative control, and water served as a positive control for RBC lysis. Error bars represent the standard deviations. Experiments are representative of three biological replicates. (B) M. marinum Esx-1 substrate production and secretion into the bacteriological medium as detected by Western blot analysis. The RNAP-β subunit served as a control for lysis and as a loading control for the cell lysate (CL); MPT-32 served as a loading control for the culture filtrate (CF) (filled arrowhead). The CFP-10 antibody detected the EsxB protein from M. marinum. The ESAT-6 antibody detected the EsxA protein from M. marinum (open arrowhead). (C) Absolute quantitation of EsxA from CL and CF from M. marinum WT, eccCb::Tn, eccD1::Tn, and 120A3 mutant and complemented strains. Quantification was determined by LC-MS/MS (MRM-based) analysis of tryptic digests with the addition of stable isotope dilution of peptides from each protein. EsxA values shown are normalized to those of GroES (see Fig. S4 in the supplemental material). Standard error is shown from triplicate analyses. α, anti.
Hemolysis by M. marinum has historically correlated with the Esx-1-mediated secretion of the EsxA substrate into the culture supernatant in vitro (2). To test if the 120A3 strain exported the EsxA and EsxB substrates into the culture supernatant, we prepared cell lysate (CL) and culture filtrate (CF) protein fractions. Protein production and secretion were detected by Western blot analysis (Fig. 4B). WT M. marinum produced EsxA and EsxB and exported both proteins into the culture supernatant. The eccD1::Tn and eccCb::Tn strains produced but failed to export both EsxA and EsxB into the CF. The 120A3 strain produced both EsxA and EsxB to levels comparable to those of the WT strain. The 120A3 strain exported reduced but detectable levels of EsxA and EsxB into the CF. Complementation of the MMAR_0039 gene resulted in increased EsxA and EsxB export into the culture supernatant. Notably, in both the WT and complemented strains, a higher-molecular-weight (MW) “smear” was present in the CF lanes that was absent in the CF fractions generated from the Esx-1-deficient strains. Together, these data demonstrate that MMAR_0039 specifically contributes to Esx-1 export in vitro.
To further characterize how the loss of the MMAR_0039 gene in M. marinum affected the levels and export of the Esx-1 substrates, we prepared protein tryptic digests of samples of both CL and CF fractions generated from the M. marinum strains and used an MRM-based targeted proteomics approach to specifically and accurately quantify the levels of Esx-1 substrates (see Fig. S3 in the supplemental material for MRM transitions). For EsxA, we used stable heavy-isotope dilution to absolutely quantify changes in production and export (see Fig. S4). We are unable to use stable heavy-isotope dilution to measure the levels of EsxB because, in M. marinum, the esxB gene (which encodes EsxB) is duplicated (MMAR_0187) and produces a protein identical to EsxB at the amino acid level. Levels of Esx-1 substrates were normalized to the levels of GroES, a commonly used lysis control for Western blot analysis. The measurements of EsxA levels in the CL and CF fractions are presented in Fig. 4C. Consistent with the Western blot analysis in Fig. 4B, the levels of EsxA in the CL fractions generated from the eccD1::Tn strain were comparable to those of the WT strain (319.96 ± 21.83 fmol/μg and 300.12 ± 9.48 fmol/μg, respectively). The levels of EsxA in the CLs generated from the 120A3 strain and the complemented strain were 248.89 ± 28.44 fmol/μg and 245.34 ± 11.17 fmol/μg, which corresponds to 82.93% and 81.47% of the EsxA levels in the WT strain. The level of EsxA in the CL from the eccCb::Tn strain was 55.22 ± 4.07 fmol/μg, which corresponded to 18.40% of the EsxA levels in the CL from the WT strain.
The majority of EsxA generated by the WT strain was found in the CF fraction (1,400.64 ± 115.01 fmol/μg). Relative to the WT strain, the levels of EsxA in the CF fractions generated from the eccCb::Tn, eccD1::Tn, and 120A3 strains were 5.37 ± 0.57 fmol/μg, 16.03 ± 1.33 fmol/μg, and 20.94 ± 2.14 fmol/μg, which corresponds to 0.38%, 1.14%, and 1.50%, respectively, of the EsxA found in the CF generated from the WT strain. Restoration of the MMAR_0039 gene resulted in 69.54 fmol/μg of EsxA in the CF (4.96% of WT levels).
We used targeted proteomics to quantify the relative changes in the levels of several additional Esx-1 substrates in the CL and CF generated from the Esx-1-deficient and complemented M. marinum strains compared to the WT strain (see Fig. S5 in the supplemental material). The EsxB, EspB, EspF, PPE68, EspJ, and EspK substrates were detected to various levels in the CL fractions from all strains. The levels of each substrate in the CF from the 120A3 strain were greatly reduced relative to the levels of each substrate in the CF from the WT strain (0.84% to 7.71% of WT). Restoration of the MMAR_0039 gene resulted in increased levels of each substrate in the CF (6.91% to 36.33% of the WT strain). Together, these data indicate that the MMAR_0039 gene is required for the export of Esx-1 substrates from M. marinum.
Loss of the MMAR_0039 gene affects N-α-terminal acetylation but not transport of EsxA to the cell surface.
Esx-1 substrates are also associated with the mycobacterial cell surface in vitro (27–29, 31). Because we observed low levels of EsxA and EsxB in the CF fractions generated from the 120A3 strain, we hypothesized that substrates may be present on the 120A3 cell surface. To test this hypothesis, we measured the presence of the EsxA and EsxB substrates on the M. marinum cell surface using the whole-colony MALDI-MS Esx-1 secretion assay we developed previously (47). M. marinum strains were grown on Sauton's agar to induce Esx-1 export. Surface proteins (SPs) were isolated by performing an aqueous extraction highly enriched for proteins extrinsically associated with the mycobacterial cell surface (47). We detected EsxA and EsxB in SP fractions generated from the surface of the WT strain (Fig. 5A). The EsxA protein resulted in two peaks, corresponding to the unacetylated and N-α-terminally acetylated (Ac-EsxA) forms of EsxA (47). The eccCb::Tn strain, which produced but did not export EsxA and EsxB into the CF (Fig. 4B), served as a control for lysis in the MALDI assay. Accordingly, EsxA and EsxB were not detected on the surface of cells of the eccCb::Tn strain. The peak at ∼10,500 m/z likely represents the second identical EsxB protein encoded by MMAR_0187, as we reported earlier (27, 47). Both EsxA and EsxB were present on the surface of the 120A3 strain. From these data we conclude that the 120A3 strain still functionally exported EsxA and EsxB to the mycobacterial cell surface.
FIG 5.

The N-α-terminal acetylation of EsxA, but not export to the cell surface, is altered in the 120A3 strain. (A) Whole-colony MALDI assay to measure EsxA and EsxB export to the cell surface. Two MALDI peaks represent unacetylated EsxA and N-α-terminally acetylated EsxA (Ac-EsxA). The eccCb::Tn strain served as a control for lysis. au, arbitrary units. (B) Absolute levels of EsxA from surface proteins from M. marinum WT, 120A3, and complemented strains. Quantification was determined by LC-MS/MS (MRM-based) analysis of tryptic digests with the addition of stable isotope dilution of peptides from each protein. EsxA values shown are normalized to GroES values (see Fig. S4 in the supplemental material). Standard error is shown from triplicate analyses. There is no statistically significant difference between the levels of EsxA on the surface for all three strains as determined by a one-way ANOVA (F2,6 = 2.161, P = 0.1964). (C) Model of the Salmonella RimI NAT bound to acetyl-CoA based on the structure by Vetting et al. (44) generated using PyMOL (78). The Phyre 2 model MMAR_0039 was generated using PyMOL. The alignment of RimI and MMAR_0039 was performed using FatCat (57) and modeled using PyMol. (D) MALDI peaks for unacetylated EsxA and acetylated EsxA (Ac-EsxA) (dotted lines) generated from SPs from M. marinum strains grown on Sauton's agar. The peaks are enlarged versions of the peaks boxed in panel A for emphasis. (E) Quantitation of unacetylated and N-α-terminally acetylated EsxA (Nt-EsxA) from surface proteins from M. marinum WT, 120A3, and complemented strains using MRM analysis. The average area ratio normalized to GroES is shown. Standard error is shown from triplicate analysis. The statistical significance of the differences between the WT and 120A3 strains, as well as the WT and complemented strains, were determined using Student's t test and are listed in the text. (F) Percentage of the EsxA peptides that were N-α-terminally acetylated (%Nt-Ac) in the CL, SP, and CF fractions generated from M. marinum WT, 120A3, and complemented strains. Percent acetylation was calculated as follows: (area ratio of N-α-terminally acetylated peptide)/(area ratio of N-α-terminally acetylated peptide + area ratio of unacetylated N-terminal peptide) × 100. The statistical significance of the differences in percent acetylation were determined using Student's t test; the P values are indicated in the text.
The MALDI-MS assay is not generally quantitative. To accurately measure the levels of EsxA present on the mycobacterial cell surface, we isolated SPs, digested them with trypsin, and conducted targeted MRM proteomics with stable heavy-isotope dilution for both an internal tryptic peptide of EsxA (LAAAWGGSGSEAYR) and GroES (27). We normalized the levels of EsxA to GroES to control for lysis. The levels of EsxA on the cell surface were 84.57 ± 7.30 fmol/50 μl, 88.23 ± 24.62 fmol/50 μl, and 69.38 ± 6.08 fmol/50 μl for the WT, 120A3, and complemented strains, respectively (Fig. 5B). There is no statistically significant difference between the means of the levels of EsxA on the surface for all three strains as determined by a one-way ANOVA (F2,6 = 2.161, P = 0.1964), indicating that surface export of EsxA is not disrupted in the 120A3 strain.
Our prior work indicated that the presence of EsxA on the M. marinum cell surface was a virulence determinant (27). Yet the 120A3 strain was attenuated despite the presence of EsxA on the cell surface. One explanation is that the EsxA on the surface of the 120A3 strain was nonfunctional or altered from the EsxA on the surface of the WT and complemented strains. MMAR_0039 encodes a conserved hypothetical protein with a predicted N-acetyltransferase (NAT) activity (50). NATs promote the N-α-terminal acetylation of proteins. Structural predictions using the Phyre 2 server confirmed that MMAR_0039 resembles known and predicted bacterial and eukaryotic NATs at the structural level (56). The structure of the RimI GNAT (Gcn5-related NAT) from Salmonella bound to acetyl-coenzyme A (CoA) (44) and the structural model of MMAR_0039 are shown in Fig. 5C (left and center, respectively). Structural alignment between the RimI GNAT and the model for MMAR_0039 (Fig. 5C, right) demonstrated that the two proteins are likely structurally significantly similar (P = 2.90e−04; a P value of <0.05 is significant) (57).
We sought to determine if the N-α-terminal acetylation of EsxA was altered by the loss of the MMAR_0039 gene. Figure 5D shows a magnified version of the EsxA peaks from the MALDI data presented in Fig. 5A (boxed). Relative to the EsxA peaks generated from the WT SPs, the peak corresponding to unacetylated EsxA was diminished in the SPs from the 120A3 strain (Fig. 5D). Complementation of the MMAR_0039 gene restored the mass equal to that of unacetylated EsxA from the M. marinum cell surface.
To address the magnitude of the changes we observed in the N-α-terminal acetylation of EsxA, we used targeted mass spectrometry to perform differential quantification of N-α-terminally acetylated and unacetylated EsxA associated with the surface. We measured changes in the levels of the unacetylated and N-α-terminally acetylated N-terminal tryptic peptide from EsxA (TEQQWNFAGIEAASSAIQGNVTSISHLLDEGK and Ac-TEQQWNFAGIEAASSAIQGNVTSISHLLDEGK) present in the SP fractions from the WT, 120A3, and complemented strains (Fig. 5E). We observed a statistically significant increase in the levels of acetylated EsxA in the 120A3 strain compared to the level in the WT strain (P = 0.0485). Similarly, we observed a statistically significant decrease in the levels of unacetylated EsxA on the cell surface from the 120A3 strain compared to the level of the WT strain (P = 0.0026). The levels of acetylated and unacetylated EsxA on the cell surface of the complemented strain were not significantly different from the levels on the surface of the wild-type strain (P = 0.1125 and P = 0.2982, respectively).
The area ratios of the N-terminal peptides (unacetylated N-terminal EsxA peptide plus N-α-terminally acetylated N-terminal EsxA peptide) provided a second analytical measurement of the total population of EsxA on the cell surface (Fig. 5B), and confirmed that the 120A3 strain had exported EsxA to the cell surface at WT levels (Fig. 5E, add striped and white bars).
From these data we calculated the percentage of N-α-terminally acetylated EsxA on the mycobacterial cell surface (Fig. 5F). We found that 30.01% of N-terminal tryptic peptides from EsxA in the SPs from the WT strain were N-α-terminally acetylated. In contrast, 72.21% of N-terminal tryptic peptides from EsxA in the SPs generated from the 120A3 strain were N-α-terminally acetylated. The difference in the percent acetylation values was significant between the WT and 120A3 strains (P = 0.0006). Complementation resulted in an intermediate level of N-α-terminally acetylated EsxA (51.83%) that was not statistically different from the percent acetylation of EsxA on the WT strain (P = 0.09). We conducted the same analysis on the EsxA N-terminal peptides found in the CL and CF fractions (Fig. 5F). For the WT strain 23.88% of EsxA in the CL fraction and 31.35% of EsxA in the CF fraction were N-α-terminally acetylated. For the 120A3 strain, 45.35% of EsxA the CL fraction and 36.89% of EsxA in the CF fraction were N-α-terminally acetylated. Statistical analysis indicated that the change in percent acetylation in the CL but not the CF generated from the 120A3 strain was significantly different from the percent acetylation in the wild-type fractions (CL, P = 0.0165; CF, P = 0.3625). Restoration of the MMAR_0039 gene failed to return the percent acetylation of EsxA in the CL and CF fractions to wild-type levels (percent acetylated EsxA in CL fraction, 42.66%; percent acetylated EsxA in the CF fraction, 19.52%). Both of these measurements were statistically different from the percent acetylation in the wild-type strain (CL, P = 0.0331; CF, P = 0.0291). Together, these data indicate that loss of the MMAR_0039 gene altered the homeostasis of N-α-terminal acetylation of EsxA. The only change in EsxA acetylation that was genetically linked to the loss of the MMAR_0039 gene was at the cell surface. Together these findings indicate that the loss of the MMAR_0039 gene specifically affected the levels of N-α-terminal acetylation of surface-associated EsxA.
We have observed that the Esx-1 substrates, EspB and EspF, were also N-α-terminally acetylated (see Fig. S6 in the supplemental material) (24, 58). We measured the levels N-α-terminally acetylated and unacetylated N-terminal tryptic peptides of EspF and EspB. A total of 76.65% of the N-terminal EspB peptides were N-α-terminally acetylated in the CF fraction generated from the WT strain. We were unable to detect unacetylated N-terminal EspB peptides in the CL fraction or any N-terminal EspB peptides in SP fractions. In the CL, CF, and SP fractions from the WT strain, 43.18%, 43.95%, and 53.13% of the N-terminal EspF peptides were N-α-terminally acetylated, respectively (see Fig. S7 in the supplemental material). Although there were significant differences between the percent acetylation of EspF in the 120A3 strain compared to the WT strain, an additional copy of the MMAR_0039 gene failed to restore WT acetylation. EsxN, a substrate of the Esx-5 system in M. marinum, is N-α-terminally acetylated (42, 59–61). Relative to the Esx-1 substrates measured in this study, the vast majority of the N-terminal EsxN peptides detected in the CL, SP, and CF fractions were acetylated for all three strains (88 to 93%) (see Fig. S7). The changes to the N-α-terminal acetylation of EsxN across the three strains were minimal. Yet an additional copy of the MMAR_0039 gene restored WT acetylation of EsxN in the CF fraction. Based on these findings we cannot rule out a global role for MMAR_0039 in mediating the acetylation of the EsxA paralog, EsxN.
DISCUSSION
This study has yielded several novel findings that expand our understanding of the Esx-1 export system in M. marinum. We identified MMAR_0039, a novel gene specifically required for Esx-1-mediated export and virulence in M. marinum. MMAR_0039 was discovered in a genetic screen for M. marinum strains with decreased cytolytic properties. We demonstrated that the MMAR_0039 gene, which has not previously been linked to mycobacterial pathogenesis, was required for the cytolysis of amoebae and RAW 264.7 mouse macrophage-like cells. We showed that the MMAR_0039 gene was required for Esx-1-mediated hemolysis, induction of IFN-β transcription in RAW 264.7 cells, protein export into the bacteriological medium, and virulence in M. marinum. The addition of an exogenous copy of the MMAR_0039 gene restored cytolysis and the IFN-β response and partially restored the export of Esx-1 substrates to the culture supernatant in vitro. This study demonstrates that there are additional esx-1 loci in the M. marinum genome.
Although Esx-1 substrate export to the bacteriological medium was disrupted, the loss of the MMAR_0039 gene in the 120A3 strain did not alter the export of EsxA to the surface of the mycobacterial cell. Quantitative analysis of the populations of EsxA revealed hyperacetylation of the N-α-terminal acetylation of EsxA in the 120A3 strain on the cell surface, which was restored by the addition of the MMAR_0039 gene. Although we observed changes in the acetylation of EspF in the 120A3 strain, exogenous expression of the MMAR_0039 gene failed to restore acetylation to wild-type levels, indicating that the changes we observed in the N-α-terminal acetylation of EspF did not correlate with virulence. It remains possible that MMAR_0039 is required for mediating the acetylation level of EsxN, though to a lesser extent than for EsxA. To our knowledge, this is the first report of a mycobacterial strain in which N-α-terminal acetylation is altered. Moreover, there is an inverse relationship between EsxA acetylation and Esx-1-mediated virulence.
It is generally thought that Esx-1-mediated export of substrates into the bacteriological medium is directly linked to virulence. Mycobacterial strains that export Esx-1 substrates at wild-type levels are virulent; strains which fail to export Esx-1 substrates at wild-type levels are attenuated. Indeed, the vast majority of literature supports this premise. It is assumed that the levels of Esx substrate export in vitro are a meaningful measure of virulence. Yet despite the ease of measuring Esx-1 export into the bacteriological medium, there has been no successful measurement of the levels of Esx-1 substrate export in the host.
Several recent papers directly support the idea that the relationship between the levels of in vitro secretion into the medium and virulence is not straightforward (27, 62–65). Chen et al. reported a virulent M. tuberculosis strain with single point mutations in the EspA substrate which failed to secrete Esx-1 substrates into the culture supernatant as detected by Western blot analysis (65). One interpretation of this finding is that Esx-1 substrate export into the culture supernatant is not required for virulence.
We recently reported an attenuated M. marinum strain (MMAR_1663::Tn::1664) which did not export EsxA and EsxB to the cell surface or into the CF in vitro (27). The addition of the WT locus to the mutant strain partially restored virulence and low levels of export of EsxA and EsxB to the cell surface but not the release of these proteins into the CF. We interpreted these findings to mean that the presence of EsxA and EsxB on the cell surface, rather than in CF fractions generated in vitro, correlates with M. marinum virulence ex vivo (27).
Most recently, Siegrist et al. (66) reported that in Mycobacterium smegmatis, very low levels of Esx-3 substrates in the culture supernatant (from <1% to 40% of WT levels), as measured by quantitative isotope dilution proteomics, correlates with Esx-3 function (survival under low-iron conditions). In this study, complementation was measured by Esx-3 function, not export. Esx-3 function did not correlate with WT levels of Esx-3 substrates in the culture supernatant.
Here, we showed that the 120A3 M. marinum strain was deficient for the Esx-1-mediated export of several protein substrates. Exogenous expression of the MMAR_0039 gene in the 120A3 strain resulted in a complete restoration of virulence, as measured by cytolysis, hemolysis, and IFN-β induction during macrophage infection. Yet we found that expression of the MMAR_0039 gene resulted in only partial restoration of Esx-1 substrates in the culture supernatant (∼7 to 35%, similar to the measurements for Esx-3 in Siegrist et al. [66]). Rather than conclude that complementation was incomplete, we conclude that the wild-type levels of Esx-1 substrates in the culture supernatant in vitro do not correlate with virulence. This is the first study to define the levels of each Esx-1 substrate in the supernatant that correlate with virulence. Our findings directly support the findings of Siegrist et al. and demonstrate that this phenotype extends to Esx systems with distinct functions and in different mycobacterial species. We propose that the levels of Esx-1/Esx-3 substrates generated under laboratory conditions do not reflect the levels required for function in the natural environment (i.e., host, water, or soil). This may indicate that in vitro export may not be subject to regulatory mechanisms that occur in the actual environmental niche.
Although there are few examples of N-α-terminal acetylation of bacterial proteins, the vast majority of yeast and human proteins are acetylated at their N termini (67). The functional consequences of this modification are unclear, but N-α-terminal acetylation has been linked to regulating protein stability, degradation, and protein-protein and protein-membrane interactions (68–70). EsxA is the only known bacterial virulence factor that is N-α-terminally acetylated (43, 44). This study defines the first M. marinum strain in which EsxA acetylation is altered. This strain is attenuated, but it is unclear why. We can rule out that the attenuation is due to alterations in the levels of EsxA export to the cell surface. Because we used MRM with isotope dilution with the same peptide standards to measure EsxA export in our previous and current studies, we can directly compare the levels of EsxA between separate experiments (27). The levels of extracytoplasmic EsxA (cell surface and CF) generated from the attenuated 120A3 strain were greater than those for the partially virulent strain we described previously (MMAR_1663::Tn::1664/pMMAR_1663–1668) (27). Moreover, the surface levels of EsxA in the 120A3 strain are not different from those of the wild-type strain. Therefore, the attenuation of the 120A3 strain is not simply due to low levels of extracytoplasmic EsxA.
One possibility is that the hyperacetylation of EsxA disrupts the Esx-1 system, either at the level of substrate export or function, leading to attenuation. Structural and biochemical studies have demonstrated that EsxA (ESAT-6) heterodimerizes with EsxB (CFP-10), forming a four-helix bundle (6, 71, 72). Okkels et al. showed that EsxB preferentially interacts with unacetylated EsxA in vitro using 2D gel electrophoresis of CF fractions from M. tuberculosis followed by far-Western blot analysis. The authors proposed that N-α-terminal acetylation of EsxA disrupts its interaction with EsxB, allowing EsxA to promote virulence (42, 43). Several studies have supported the idea that EsxA is a pore-forming toxin; EsxA and/or the EsxA/EsxB dimer can bind to and form pores within membranes (5, 28, 73–75). The pore-forming activity of EsxA/EsxB associated with the mycobacterial cell is reported to be more potent than EsxA alone, indicating that either EsxA/EsxB oligomerization or other cell-associated proteins contribute to pore formation (28, 75). Indeed, it has been suggested that EsxA and/or the EsxA/EsxB form higher-order oligomers (64, 76). The N-terminal charge reduction that results from N-α-terminal acetylation could affect not only the ability of EsxA to interact with EsxB but also oligomerization of the complex, membrane interaction, or pore-forming activity. EsxA and EsxB export is required for the export of all of the other M. marinum substrates measured here (79). Consistent with this observation, EsxA and EsxB may not only be substrates of the Esx-1 system but may also be part of the apparatus. For example, if EsxA/EsxB dimers oligomerize to form the channel across the MOM, disruption of oligomerization could affect the export of the other Esx-1 substrates. Alternatively, hyperacetylation of EsxA could be a phenotype associated with a nonfunctional Esx-1 exporter. Further experimentation is required to distinguish between the two possibilities.
Regardless of mechanism, our findings indicate that EsxA acetylation is important in Esx-1-mediated virulence. Brodin et al. reported that the N-α-terminal acetylation of EsxA (ESAT-6) does not affect function (64). In this study, the Thr at position 2 of EsxA, the site of N-α-terminal acetylation, was changed to a His (T2H) and expressed in M. tuberculosis H37Rv. The M. tuberculosis strain exported EsxA(T2H) into the CF and retained virulence in SCID mice (64). The acetylation status of EsxA(T2H) was not determined. The idea that substitution of the Thr at the second position of EsxA would lead to the loss of virulence relies upon the assumption that the N-α-terminal acetylated form of EsxA correlates with virulence. We propose that there is an inverse relationship between EsxA acetylation and virulence (Fig. 6). Consistent with this idea, whether EsxA(T2H) is unacetylated or N-α-terminally acetylated, like the WT protein (which is not 100%), we would predict that this strain would retain virulence, in agreement with both Lange et al. and Brodin et al. (42, 64).
FIG 6.
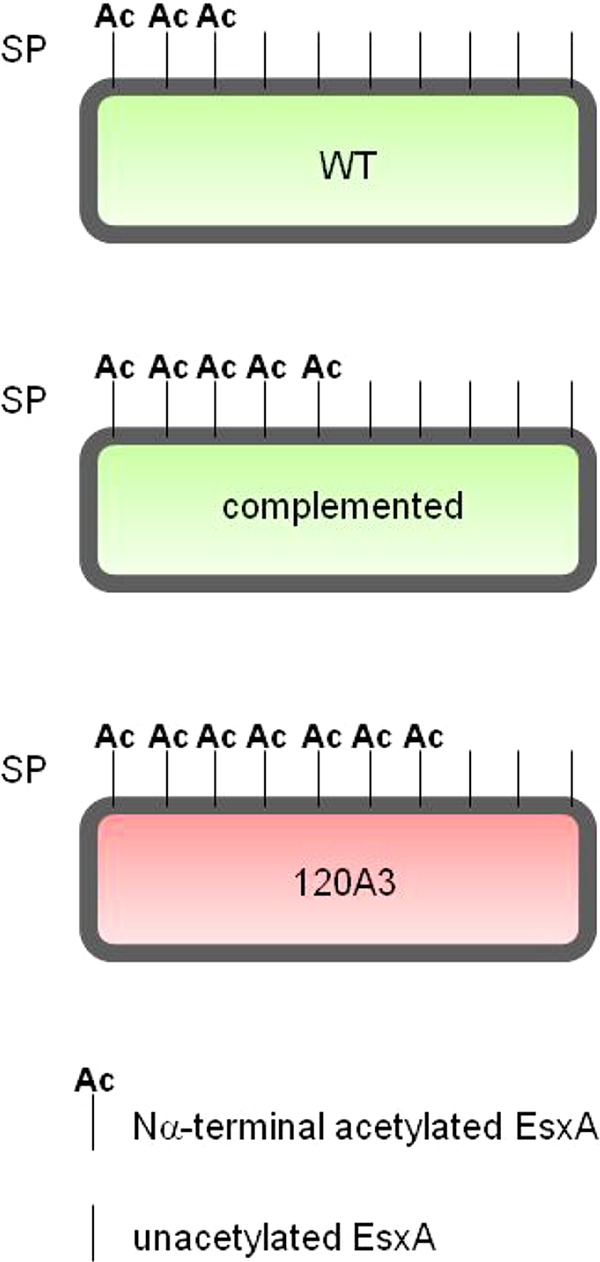
The unacetylated form of EsxA on the cell surface correlates with virulence. Model of the populations of EsxA on the M. marinum cell surface based on the quantification shown in Fig. 6. We propose that unacetylated EsxA mediates virulence. SP, surface proteins. Green, virulence; red, attenuation.
We can propose direct and indirect models to explain how MMAR_0039 promotes Esx-1 acetylation homeostasis or export. MMAR_0039 could function directly to deacetylate EsxA and other Esx-1 substrates. The loss of a deacetylase would result in increased EsxA acetylation. Structural predictions of MMAR_0039 indicate that it is an N-terminal acetyltransferase (NAT). Our efforts to demonstrate biochemically that MMAR_0039 is the EsxA acetylase have thus far been unsuccessful (data not shown). However, NATs often function in complexes, which may explain why the protein alone cannot N-α-terminally acetylate EsxA. MMAR_0039 could affect EsxA acetylation indirectly by N-α-terminally acetylating another target that affects Esx. These models will be the subject of future investigation. Because EsxN N-α-terminal acetylation was not greatly affected by the loss of MMAR_0039, we propose that the effects on EsxA acetylation are specific. Thus, there may be proteins required to mediate the acetylation state of substrates from each Esx system which could serve to regulate function.
BLAST analysis revealed that although the MMAR_0039 gene is highly conserved in several mycobacterial species, there is no clear ortholog of MMAR_0039 in M. tuberculosis. Yet EsxA from M. tuberculosis is N-α-terminally acetylated (42, 43). EsxA proteins from M. marinum and M. tuberculosis are 91.5% identical and 97.9% similar at the amino acid level. The first six amino acids which generally target substrates to NATs are identical. One of the hallmarks of NATs, in all species, is that although they all perform the same catalytic reaction, there is very low conservation at the amino acid level (44, 69).
Our findings raise the idea for the first time that surface-localized unacetylated EsxA and other Esx-1 substrates play a role in export or virulence. We defined the acetylated and unacetylated populations of EsxA in the wild-type strain for the first time in vitro. The fact that two populations exist in and on the wild-type strain may indicate that N-α-terminal acetylation of EsxA is a means to regulate Esx-1 export or function. A limitation of the current study is that we do not know the N-α-terminal acetylation state of EsxA on the mycobacterial cell surface in the host. This is difficult to measure as there are currently no antibodies available to specifically detect N-α-terminal acetylation. To fully define the role of EsxA acetylation, a mycobacterial strain in which all of the EsxA is N-α-terminally acetylated is required. Such a strain cannot currently be engineered by substituting amino acids at the second position of EsxA. However, the disruption of the MMAR_0039 gene resulted in an M. marinum strain in which the majority of surface-associated EsxA was acetylated. Clarifying the mechanism of hyperacetylation in the 120A3 strain and the role of MMAR_0039 in this process will directly improve our ability to generate such a strain.
Recent work identified several putative N-α-terminally acetylated proteins in a wide range of nonpathogenic bacterial species, suggesting that the N-α-terminal acetylation in bacteria is far more widespread than currently appreciated (77). Indeed, additional mycobacterial proteins are likely N-α-terminally acetylated. Here, we provide the first insight into how this poorly understood modification in bacteria is related to virulence. Collectively, the evidence presented here suggests that the MMAR_0039 gene is required for maintaining the appropriate balance of acetylated versus unacetylated EsxA and that maintaining this homeostasis is required for Esx-1-mediated mycobacterial virulence.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this study was supported by the National Institute of Allergies and Infectious Diseases of the National Institutes of Health under award numbers R01AI106872 to P.A.D.C. F.M.M. is supported by a postdoctoral fellowship from the Eck Institute of Global Health at the University of Notre Dame. E.A.W. is supported by a graduate fellowship from the Eck Institute of Global Health at the University of Notre Dame.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We thank the Mass Spectrometry and Proteomics Facility and the Genomics and Bioinformatics Facility at the University of Notre Dame for their assistance.
P.A.D.C., M.M.C, and F.M.M. designed the experiments. F.M.M, M.M.C, and E.A.W. conducted the experiments. M.M.C, P.A.D.C., and F.M.M. analyzed the data. All authors discussed the findings and wrote and edited the manuscript.
Footnotes
Published ahead of print 18 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02153-14.
REFERENCES
- 1.Burts ML, Williams WA, DeBord K, Missiakas DM. 2005. EsxA and EsxB are secreted by an ESAT-6-like system that is required for the pathogenesis of Staphylococcus aureus infections. Proc. Natl. Acad. Sci. U. S. A. 102:1169–1174. 10.1073/pnas.0405620102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gao LY, Guo S, McLaughlin B, Morisaki H, Engel JN, Brown EJ. 2004. A mycobacterial virulence gene cluster extending RD1 is required for cytolysis, bacterial spreading and ESAT-6 secretion. Mol. Microbiol. 53:1677–1693. 10.1111/j.1365-2958.2004.04261.x. [DOI] [PubMed] [Google Scholar]
- 3.Garufi G, Butler E, Missiakas D. 2008. ESAT-6-like protein secretion in Bacillus anthracis. J. Bacteriol. 190:7004–7011. 10.1128/JB.00458-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guinn KM, Hickey MJ, Mathur SK, Zakel KL, Grotzke JE, Lewinsohn DM, Smith S, Sherman DR. 2004. Individual RD1-region genes are required for export of ESAT-6/CFP-10 and for virulence of Mycobacterium tuberculosis. Mol. Microbiol. 51:359–370. 10.1046/j.1365-2958.2003.03844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hsu T, Hingley-Wilson SM, Chen B, Chen M, Dai AZ, Morin PM, Marks CB, Padiyar J, Goulding C, Gingery M, Eisenberg D, Russell RG, Derrick SC, Collins FM, Morris SL, King CH, Jacobs WR., Jr 2003. The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc. Natl. Acad. Sci. U. S. A. 100:12420–12425. 10.1073/pnas.1635213100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stanley SA, Raghavan S, Hwang WW, Cox JS. 2003. Acute infection and macrophage subversion by Mycobacterium tuberculosis require a specialized secretion system. Proc. Natl. Acad. Sci. U. S. A. 100:13001–13006. 10.1073/pnas.2235593100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Volkman HE, Clay H, Beery D, Chang JC, Sherman DR, Ramakrishnan L. 2004. Tuberculous granuloma formation is enhanced by a mycobacterium virulence determinant. PLoS Biol. 2:e367. 10.1371/journal.pbio.0020367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Houben D, Demangel C, van Ingen J, Perez J, Baldeon L, Abdallah AM, Caleechurn L, Bottai D, van Zon M, de Punder K, van der Laan T, Kant A, Bossers-de Vries R, Willemsen P, Bitter W, van Soolingen D, Brosch R, van der Wel N, Peters PJ. 2012. ESX-1-mediated translocation to the cytosol controls virulence of mycobacteria. Cell. Microbiol. 14:1287–1298. 10.1111/j.1462-5822.2012.01799.x. [DOI] [PubMed] [Google Scholar]
- 9.Manzanillo PS, Shiloh MU, Portnoy DA, Cox JS. 2012. Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe 11:469–480. 10.1016/j.chom.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simeone R, Bottai D, Brosch R. 2009. ESX/type VII secretion systems and their role in host-pathogen interaction. Curr. Opin. Microbiol. 12:4–10. 10.1016/j.mib.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 11.Stanley SA, Cox JS. 2013. Host-pathogen interactions during Mycobacterium tuberculosis infections. Curr. Top. Microbiol. Immunol. 374:211–241. 10.1007/82_2013_332. [DOI] [PubMed] [Google Scholar]
- 12.van der Wel N, Hava D, Houben D, Fluitsma D, van Zon M, Pierson J, Brenner M, Peters PJ. 2007. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 129:1287–1298. 10.1016/j.cell.2007.05.059. [DOI] [PubMed] [Google Scholar]
- 13.Stanley SA, Johndrow JE, Manzanillo P, Cox JS. 2007. The type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J. Immunol. 178:3143–3152. 10.4049/jimmunol.178.5.3143. [DOI] [PubMed] [Google Scholar]
- 14.Akpe San Roman S, Facey PD, Fernandez-Martinez L, Rodriguez C, Vallin C, Del Sol R, Dyson P. 2010. A heterodimer of EsxA and EsxB is involved in sporulation and is secreted by a type VII secretion system in Streptomyces coelicolor. Microbiology 156:1719–1729. 10.1099/mic.0.037069-0. [DOI] [PubMed] [Google Scholar]
- 15.Baptista C, Barreto HC, Sao-Jose C. 2013. High levels of DegU-P activate an Esat-6-like secretion system in Bacillus subtilis. PLoS One 8:e67840. 10.1371/journal.pone.0067840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coros A, Callahan B, Battaglioli E, Derbyshire KM. 2008. The specialized secretory apparatus ESX-1 is essential for DNA transfer in Mycobacterium smegmatis. Mol. Microbiol. 69:794–808. 10.1111/j.1365-2958.2008.06299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flint JL, Kowalski JC, Karnati PK, Derbyshire KM. 2004. The RD1 virulence locus of Mycobacterium tuberculosis regulates DNA transfer in Mycobacterium smegmatis. Proc. Natl. Acad. Sci. U. S. A. 101:12598–12603. 10.1073/pnas.0404892101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fyans JK, Bignell D, Loria R, Toth I, Palmer T. 2013. The ESX/type VII secretion system modulates development, but not virulence, of the plant pathogen Streptomyces scabies. Mol. Plant Pathol. 14:119–130. 10.1111/j.1364-3703.2012.00835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gray TA, Krywy JA, Harold J, Palumbo MJ, Derbyshire KM. 2013. Distributive conjugal transfer in mycobacteria generates progeny with meiotic-like genome-wide mosaicism, allowing mapping of a mating identity locus. PLoS Biol. 11:e1001602. 10.1371/journal.pbio.1001602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Converse SE, Cox JS. 2005. A protein secretion pathway critical for Mycobacterium tuberculosis virulence is conserved and functional in Mycobacterium smegmatis. J. Bacteriol. 187:1238–1245. 10.1128/JB.187.4.1238-1245.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berthet FX, Rasmussen PB, Rosenkrands I, Andersen P, Gicquel B. 1998. A Mycobacterium tuberculosis operon encoding ESAT-6 and a novel low-molecular-mass culture filtrate protein (CFP-10). Microbiology 144:3195–3203. 10.1099/00221287-144-11-3195. [DOI] [PubMed] [Google Scholar]
- 22.Brodin P, Majlessi L, Marsollier L, de Jonge MI, Bottai D, Demangel C, Hinds J, Neyrolles O, Butcher PD, Leclerc C, Cole ST, Brosch R. 2006. Dissection of ESAT-6 system 1 of Mycobacterium tuberculosis and impact on immunogenicity and virulence. Infect. Immun. 74:88–98. 10.1128/IAI.74.1.88-98.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carlsson F, Joshi SA, Rangell L, Brown EJ. 2009. Polar localization of virulence-related Esx-1 secretion in mycobacteria. PLoS Pathog. 5:e1000285. 10.1371/journal.ppat.1000285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Champion PA, Champion MM, Manzanillo P, Cox JS. 2009. ESX-1 secreted virulence factors are recognized by multiple cytosolic AAA ATPases in pathogenic mycobacteria. Mol. Microbiol. 73:950–962. 10.1111/j.1365-2958.2009.06821.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daleke MH, Ummels R, Bawono P, Heringa J, Vandenbroucke-Grauls CM, Luirink J, Bitter W. 2012. General secretion signal for the mycobacterial type VII secretion pathway. Proc. Natl. Acad. Sci. U. S. A. 109:11342–11347. 10.1073/pnas.1119453109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fortune SM, Jaeger A, Sarracino DA, Chase MR, Sassetti CM, Sherman DR, Bloom BR, Rubin EJ. 2005. Mutually dependent secretion of proteins required for mycobacterial virulence. Proc. Natl. Acad. Sci. U. S. A. 102:10676–10681. 10.1073/pnas.0504922102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kennedy GM, Hooley GC, Champion MM, Medie FM, Champion PA. 2014. A novel ESX-1 locus reveals that surface associated ESX-1 substrates mediate virulence in Mycobacterium marinum. J. Bacteriol. 196:1877–1888. 10.1128/JB.01502-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kinhikar AG, Verma I, Chandra D, Singh KK, Weldingh K, Andersen P, Hsu T, Jacobs WR, Jr, Laal S. 2010. Potential role for ESAT6 in dissemination of M. tuberculosis via human lung epithelial cells. Mol. Microbiol. 75:92–106. 10.1111/j.1365-2958.2009.06959.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Majlessi L, Brodin P, Brosch R, Rojas MJ, Khun H, Huerre M, Cole ST, Leclerc C. 2005. Influence of ESAT-6 secretion system 1 (RD1) of Mycobacterium tuberculosis on the interaction between mycobacteria and the host immune system. J. Immunol. 174:3570–3579. 10.4049/jimmunol.174.6.3570. [DOI] [PubMed] [Google Scholar]
- 30.McLaughlin B, Chon JS, MacGurn JA, Carlsson F, Cheng TL, Cox JS, Brown EJ. 2007. A mycobacterium ESX-1-secreted virulence factor with unique requirements for export. PLoS Pathog. 3:e105. 10.1371/journal.ppat.0030105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pym AS, Brodin P, Brosch R, Huerre M, Cole ST. 2002. Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol. Microbiol. 46:709–717. 10.1046/j.1365-2958.2002.03237.x. [DOI] [PubMed] [Google Scholar]
- 32.Raghavan S, Manzanillo P, Chan K, Dovey C, Cox JS. 2008. Secreted transcription factor controls Mycobacterium tuberculosis virulence. Nature 454:717–721. 10.1038/nature07219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sani M, Houben EN, Geurtsen J, Pierson J, de Punder K, van Zon M, Wever B, Piersma SR, Jimenez CR, Daffe M, Appelmelk BJ, Bitter W, van der Wel N, Peters PJ. 2010. Direct visualization by cryo-EM of the mycobacterial capsular layer: a labile structure containing ESX-1-secreted proteins. PLoS Pathog. 6:e1000794. 10.1371/journal.ppat.1000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chagnot C, Zorgani MA, Astruc T, Desvaux M. 2013. Proteinaceous determinants of surface colonization in bacteria: bacterial adhesion and biofilm formation from a protein secretion perspective. Front. Microbiol. 4:303. 10.3389/fmicb.2013.00303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van der Woude AD, Sarkar D, Bhatt A, Sparrius M, Raadsen SA, Boon L, Geurtsen J, van der Sar AM, Luirink J, Houben EN, Besra GS, Bitter W. 2012. Unexpected link between lipooligosaccharide biosynthesis and surface protein release in Mycobacterium marinum. J. Biol. Chem. 287:20417–20429. 10.1074/jbc.M111.336461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shiloh MU, Champion PA. 2010. To catch a killer. What can mycobacterial models teach us about Mycobacterium tuberculosis pathogenesis? Curr. Opin. Microbiol. 13:86–92. 10.1016/j.mib.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stinear TP, Seemann T, Harrison PF, Jenkin GA, Davies JK, Johnson PD, Abdellah Z, Arrowsmith C, Chillingworth T, Churcher C, Clarke K, Cronin A, Davis P, Goodhead I, Holroyd N, Jagels K, Lord A, Moule S, Mungall K, Norbertczak H, Quail MA, Rabbinowitsch E, Walker D, White B, Whitehead S, Small PL, Brosch R, Ramakrishnan L, Fischbach MA, Parkhill J, Cole ST. 2008. Insights from the complete genome sequence of Mycobacterium marinum on the evolution of Mycobacterium tuberculosis. Genome Res. 18:729–741. 10.1101/gr.075069.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tobin DM, Ramakrishnan L. 2008. Comparative pathogenesis of Mycobacterium marinum and Mycobacterium tuberculosis. Cell. Microbiol. 10:1027–1039. 10.1111/j.1462-5822.2008.01133.x. [DOI] [PubMed] [Google Scholar]
- 39.Joshi SA, Ball DA, Sun MG, Carlsson F, Watkins BY, Aggarwal N, McCracken JM, Huynh KK, Brown EJ. 2012. EccA1, a component of the Mycobacterium marinum ESX-1 protein virulence factor secretion pathway, regulates mycolic acid lipid synthesis. Chem. Biol. 19:372–380. 10.1016/j.chembiol.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 40.Abdallah AM, Gey van Pittius NC, Champion PA, Cox J, Luirink J, Vandenbroucke-Grauls CM, Appelmelk BJ, Bitter W. 2007. Type VII secretion—mycobacteria show the way. Nat. Rev. Microbiol. 5:883–891. 10.1038/nrmicro1773. [DOI] [PubMed] [Google Scholar]
- 41.Champion PA, Cox JS. 2007. Protein secretion systems in mycobacteria. Cell. Microbiol. 9:1376–1384. 10.1111/j.1462-5822.2007.00943.x. [DOI] [PubMed] [Google Scholar]
- 42.Lange S, Rosenkrands I, Stein R, Andersen P, Kaufmann SH, Jungblut PR. 2014. Analysis of protein species differentiation among mycobacterial low-Mr-secreted proteins by narrow pH range Immobiline gel 2-DE-MALDI-MS. J. Proteomics 97:235–244. 10.1016/j.jprot.2013.06.036. [DOI] [PubMed] [Google Scholar]
- 43.Okkels LM, Muller EC, Schmid M, Rosenkrands I, Kaufmann SH, Andersen P, Jungblut PR. 2004. CFP10 discriminates between nonacetylated and acetylated ESAT-6 of Mycobacterium tuberculosis by differential interaction. Proteomics 4:2954–2960. 10.1002/pmic.200400906. [DOI] [PubMed] [Google Scholar]
- 44.Vetting MW, Bareich DC, Yu M, Blanchard JS. 2008. Crystal structure of RimI from Salmonella typhimurium LT2, the GNAT responsible for Nα-acetylation of ribosomal protein S18. Protein Sci. 17:1781–1790. 10.1110/ps.035899.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kennedy GM, Morisaki JH, Champion PA. 2012. Conserved mechanisms of Mycobacterium marinum pathogenesis within the environmental amoeba, Acanthamoeba castellanii. Appl. Environ. Microbiol. 78:2049–2052. 10.1128/AEM.06965-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simeone R, Bobard A, Lippmann J, Bitter W, Majlessi L, Brosch R, Enninga J. 2012. Phagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. PLoS Pathog. 8:e1002507. 10.1371/journal.ppat.1002507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Champion MM, Williams EA, Kennedy GM, Champion PA. 2012. Direct detection of bacterial protein secretion using whole colony proteomics. Mol. Cell. Proteomics 11:596–604. 10.1074/mcp.M112.017533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bitter W, Houben EN, Bottai D, Brodin P, Brown EJ, Cox JS, Derbyshire K, Fortune SM, Gao LY, Liu J, Gey van Pittius NC, Pym AS, Rubin EJ, Sherman DR, Cole ST, Brosch R. 2009. Systematic genetic nomenclature for type VII secretion systems. PLoS Pathog. 5:e1000507. 10.1371/journal.ppat.1000507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao LY, Groger R, Cox JS, Beverley SM, Lawson EH, Brown EJ. 2003. Transposon mutagenesis of Mycobacterium marinum identifies a locus linking pigmentation and intracellular survival. Infect. Immun. 71:922–929. 10.1128/IAI.71.2.922-929.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kapopoulou A, Lew JM, Cole ST. 2011. The MycoBrowser portal: a comprehensive and manually annotated resource for mycobacterial genomes. Tuberculosis (Edinb.) 91:8–13. 10.1016/j.tube.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 51.George KM, Yuan Y, Sherman DR, Barry CE., III 1995. The biosynthesis of cyclopropanated mycolic acids in Mycobacterium tuberculosis. Identification and functional analysis of CMAS-2. J. Biol. Chem. 270:27292–27298. [DOI] [PubMed] [Google Scholar]
- 52.Mba Medie F, Ben Salah I, Henrissat B, Raoult D, Drancourt M. 2011. Mycobacterium tuberculosis complex mycobacteria as amoeba-resistant organisms. PLoS One 6:e20499. 10.1371/journal.pone.0020499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schubert OT, Mouritsen J, Ludwig C, Rost HL, Rosenberger G, Arthur PK, Claassen M, Campbell DS, Sun Z, Farrah T, Gengenbacher M, Maiolica A, Kaufmann SH, Moritz RL, Aebersold R. 2013. The Mtb proteome library: a resource of assays to quantify the complete proteome of Mycobacterium tuberculosis. Cell Host Microbe 13:602–612. 10.1016/j.chom.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Picotti P, Clement-Ziza M, Lam H, Campbell DS, Schmidt A, Deutsch EW, Rost H, Sun Z, Rinner O, Reiter L, Shen Q, Michaelson JJ, Frei A, Alberti S, Kusebauch U, Wollscheid B, Moritz RL, Beyer A, Aebersold R. 2013. A complete mass-spectrometric map of the yeast proteome applied to quantitative trait analysis. Nature 494:266–270. 10.1038/nature11835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.King CH, Mundayoor S, Crawford JT, Shinnick TM. 1993. Expression of contact-dependent cytolytic activity by Mycobacterium tuberculosis and isolation of the genomic locus that encodes the activity. Infect. Immun. 61:2708–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4:363–371. 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 57.Ye Y, Godzik A. 2004. FATCAT: a web server for flexible structure comparison and structure similarity searching. Nucleic Acids Res. 32:W582–W585. 10.1093/nar/gkh430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li Y, Champion MM, Sun L, Champion PA, Wojcik R, Dovichi NJ. 2012. Capillary zone electrophoresis-electrospray ionization-tandem mass spectrometry as an alternative proteomics platform to ultraperformance liquid chromatography-electrospray ionization-tandem mass spectrometry for samples of intermediate complexity. Anal. Chem. 84:1617–1622. 10.1021/ac202899p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abdallah AM, Savage ND, van Zon M, Wilson L, Vandenbroucke-Grauls CM, van der Wel NN, Ottenhoff TH, Bitter W. 2008. The ESX-5 secretion system of Mycobacterium marinum modulates the macrophage response. J. Immunol. 181:7166–7175. 10.4049/jimmunol.181.10.7166. [DOI] [PubMed] [Google Scholar]
- 60.Abdallah AM, Verboom T, Weerdenburg EM, Gey van Pittius NC, Mahasha PW, Jimenez C, Parra M, Cadieux N, Brennan MJ, Appelmelk BJ, Bitter W. 2009. PPE and PE_PGRS proteins of Mycobacterium marinum are transported via the type VII secretion system ESX-5. Mol. Microbiol. 73:329–340. 10.1111/j.1365-2958.2009.06783.x. [DOI] [PubMed] [Google Scholar]
- 61.Bottai D, Di Luca M, Majlessi L, Frigui W, Simeone R, Sayes F, Bitter W, Brennan MJ, Leclerc C, Batoni G, Campa M, Brosch R, Esin S. 2012. Disruption of the ESX-5 system of Mycobacterium tuberculosis causes loss of PPE protein secretion, reduction of cell wall integrity and strong attenuation. Mol. Microbiol. 83:1195–1209. 10.1111/j.1365-2958.2012.08001.x. [DOI] [PubMed] [Google Scholar]
- 62.Garces A, Atmakuri K, Chase MR, Woodworth JS, Krastins B, Rothchild AC, Ramsdell TL, Lopez MF, Behar SM, Sarracino DA, Fortune SM. 2010. EspA acts as a critical mediator of ESX1-dependent virulence in Mycobacterium tuberculosis by affecting bacterial cell wall integrity. PLoS Pathog. 6:e1000957. 10.1371/journal.ppat.1000957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bottai D, Majlessi L, Simeone R, Frigui W, Laurent C, Lenormand P, Chen J, Rosenkrands I, Huerre M, Leclerc C, Cole ST, Brosch R. 2011. ESAT-6 secretion-independent impact of ESX-1 genes espF and espG1 on virulence of Mycobacterium tuberculosis. J. Infect. Dis. 203:1155–1164. 10.1093/infdis/jiq089. [DOI] [PubMed] [Google Scholar]
- 64.Brodin P, de Jonge MI, Majlessi L, Leclerc C, Nilges M, Cole ST, Brosch R. 2005. Functional analysis of ESAT-6, the dominant T-cell antigen of Mycobacterium tuberculosis, reveals key residues involved in secretion, complex-formation, virulence and immunogenicity. J. Biol. Chem. 80:33953–33959. 10.1074/jbc.M503515200. [DOI] [PubMed] [Google Scholar]
- 65.Chen JM, Zhang M, Rybniker J, Basterra L, Dhar N, Tischler AD, Pojer F, Cole ST. 2013. Phenotypic Profiling of Mycobacterium tuberculosis EspA point-mutants reveals blockage of ESAT-6 and CFP-10 secretion in vitro does not always correlate with attenuation of virulence. J. Bacteriol. 195:5421–5430. 10.1128/JB.00967-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Siegrist MS, Steigedal M, Ahmad R, Mehra A, Dragset MS, Schuster BM, Phillips JA, Carr SA, Rubin EJ. 2014. Mycobacterial ESX-3 requires multiple components for iron acquisition. mBio 5(3):e1073-14. 10.1128/mBio.01073-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Arnesen T, Van Damme P, Polevoda B, Helsens K, Evjenth R, Colaert N, Varhaug JE, Vandekerckhove J, Lillehaug JR, Sherman F, Gevaert K. 2009. Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc. Natl. Acad. Sci. U. S. A. 106:8157–8162. 10.1073/pnas.0901931106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Scott DC, Monda JK, Bennett EJ, Harper JW, Schulman BA. 2011. N-terminal acetylation acts as an avidity enhancer within an interconnected multiprotein complex. Science 334:674–678. 10.1126/science.1209307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Starheim KK, Gevaert K, Arnesen T. 2012. Protein N-terminal acetyltransferases: when the start matters. Trends Biochem. Sci. 37:152–161. 10.1016/j.tibs.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 70.Arnesen T. 2011. Towards a functional understanding of protein N-terminal acetylation. PLoS Biol. 9:e1001074. 10.1371/journal.pbio.1001074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Renshaw PS, Lightbody KL, Veverka V, Muskett FW, Kelly G, Frenkiel TA, Gordon SV, Hewinson RG, Burke B, Norman J, Williamson RA, Carr MD. 2005. Structure and function of the complex formed by the tuberculosis virulence factors CFP-10 and ESAT-6. EMBO J. 24:2491–2498. 10.1038/sj.emboj.7600732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Renshaw PS, Panagiotidou P, Whelan A, Gordon SV, Hewinson RG, Williamson RA, Carr MD. 2002. Conclusive evidence that the major T-cell antigens of the Mycobacterium tuberculosis complex ESAT-6 and CFP-10 form a tight, 1:1 complex and characterization of the structural properties of ESAT-6, CFP-10, and the ESAT-6*CFP-10 complex. Implications for pathogenesis and virulence. J. Biol. Chem. 277:21598–21603. 10.1074/jbc.M201625200. [DOI] [PubMed] [Google Scholar]
- 73.De Leon J, Jiang G, Ma Y, Rubin E, Fortune S, Sun J. 2012. Mycobacterium tuberculosis ESAT-6 exhibits a unique membrane-interacting activity that is not found in its ortholog from non-pathogenic Mycobacterium smegmatis. J. Biol. Chem. 287:44184–44191. 10.1074/jbc.M112.420869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.de Jonge MI, Pehau-Arnaudet G, Fretz MM, Romain F, Bottai D, Brodin P, Honore N, Marchal G, Jiskoot W, England P, Cole ST, Brosch R. 2007. ESAT-6 from Mycobacterium tuberculosis dissociates from its putative chaperone CFP-10 under acidic conditions and exhibits membrane-lysing activity. J. Bacteriol. 189:6028–6034. 10.1128/JB.00469-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Smith J, Manoranjan J, Pan M, Bohsali A, Xu J, Liu J, McDonald KL, Szyk A, LaRonde-LeBlanc N, Gao LY. 2008. Evidence for pore formation in host cell membranes by ESX-1-secreted ESAT-6 and its role in Mycobacterium marinum escape from the vacuole. Infect. Immun. 76:5478–5487. 10.1128/IAI.00614-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Teutschbein J, Schumann G, Mollmann U, Grabley S, Cole ST, Munder T. 2009. A protein linkage map of the ESAT-6 secretion system 1 (ESX-1) of Mycobacterium tuberculosis. Microbiol. Res. 164:253–259. 10.1016/j.micres.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 77.Bonissone S, Gupta N, Romine M, Bradshaw RA, Pevzner PA. 2013. N-terminal protein processing: a comparative proteogenomic analysis. Mol. Cell. Proteomics 12:14–28. 10.1074/mcp.M112.019075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schrodinger LLC. 2010. The PyMOL molecular graphics system, version 1.3r1. Schrodinger, LLC, New York, NY. [Google Scholar]
- 79.Champion MM, Williams ES, Pinapati RS, Champion PA. 8 August 2014. Correlation of phenotypic profiles using targeted proteomics identifies mycobacterial Esx-1 substrates. J. Proteome Res. 10.1021/pr500484w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.