

Abstract
Determining the pharmacokinetics (PKs) of drug candidates is essential for understanding their biological fate. The ability to obtain human PK information early in the drug development process can help determine if future development is warranted. Microdosing was developed to assess human PKs, at ultra-low doses, early in the drug development process. Microdosing has also been used in animals to confirm PK linearity across subpharmacological and pharmacological dose ranges. The current study assessed the PKs of a novel antimicrobial preclinical drug candidate (GP-4) in rats as a step toward human microdosing studies. Dose proportionality was determined at 3 proposed therapeutic doses (3, 10, and 30 mg/kg of body weight), and PK linearity between a microdose and a pharmacological dose was assessed in Sprague-Dawley rats. Plasma PKs over the 3 pharmacological doses were proportional. Over the 10-fold dose range, the maximum concentration in plasma and area under the curve (AUC) increased 9.5- and 15.8-fold, respectively. PKs from rats dosed with a 14C-labeled microdose versus a 14C-labeled pharmacological dose displayed dose linearity. In the animals receiving a microdose and the therapeutically dosed animals, the AUCs from time zero to infinity were 2.6 ng · h/ml and 1,336 ng · h/ml, respectively, and the terminal half-lives were 5.6 h and 1.4 h, respectively. When the AUC values were normalized to a dose of 1.0 mg/kg, the AUC values were 277.5 ng · h/ml for the microdose and 418.2 ng · h/ml for the pharmacological dose. This 1.5-fold difference in AUC following a 300-fold difference in dose is considered linear across the dose range. On the basis of the results, the PKs from the microdosed animals were considered to be predictive of the PKs from the therapeutically dosed animals.
INTRODUCTION
The development of new drugs is a long and costly process, often taking more than 10 years and over $1 billion to get one drug to market (1). The huge cost of new drugs is a consequence of the high failure rate of potential candidates prior to approval. One of the factors contributing to the high attrition rate during development is poor pharmacokinetics (PKs). Poor PK parameters are responsible for up to 40% of drug candidates failing to make it past the first studies in humans (2, 3). The ability to predict complete biodistribution profiles, which include PKs, tissue distribution, and route of elimination, early in the drug development process would provide critical data for decisions about the continued development of a drug lead. These data are also useful for the optimization of dosage regimes, potentiation of therapeutic efficacy, tailoring of drug delivery systems, and evaluation of safety. Early evaluation of these factors in humans would determine if further development is warranted before the initiation of costlier clinical trials.
Microdosing is a technique used to assess a compound's in vivo biological fate through the administration of subpharmacological doses of a 14C-labeled drug candidate that produce virtually no adverse effects. Microdosing is used to downselect among multiple new chemical entities, to obtain preliminary human pharmacokinetic information for investigational new drugs early in the development process, and to confirm in vitro or in silico metabolic pathways in vivo (4–7). It should be noted that microdosing provides no data on the efficacy or safety of the drug candidate. Microdosing studies have the potential to reduce the attrition rate of drugs because inadequate PK properties can be identified earlier in the development process, saving time and money (8).
The linearity of the PKs across the range from the microdose to the therapeutic dose is one of the fundamental concerns with microdosing studies. Although it is not comprehensive, there is a growing body of evidence showing that the PK parameters of several drugs are linear over a 50- to 1,000-fold dose range (8, 9). In a seminal human study, Lappin et al. showed that the pharmacokinetics of 3 prescription drugs were essentially linear (within a factor of 2) across a range of microdoses and therapeutic doses (10). Sandhu et al. used microdosing to examine whether a preclinical drug candidate (7-deaza-2′-C-methyladenosine) displayed linear kinetics across a subpharmacological and a pharmacological dose range in an animal model before the initiation of human microdose study. The results showed that the PK properties were linear over a 50-fold dose range (11).
The success of microdosing is dependent on the availability of ultrasensitive analytical methods that are able to measure drug concentrations in the low-picogram to femtogram range (6). Accelerator mass spectrometry (AMS) is one of only a few current techniques ideally suited for this application due to its extreme sensitivity and selectivity. AMS is a technique for counting rare, long-lived isotopes independently of radioactive decay by measuring the ratio of the mass of the radioisotope of interest relative to that of a stable isotope of the element (12). AMS can be used to trace the fate of any molecule in in vitro systems or whole organisms if it is labeled with an isotope appropriate for AMS analysis. Because most biological materials contain carbon, the majority of AMS studies use 14C as the radiotracer. AMS can detect and quantify a 14C-labeled compound in a biological matrix with 1% to 3% precision at levels ranging from approximately 10 pmol 14C to 1 amol (1 × 10−18) 14C in samples containing as little as 250 μg of total carbon (12–14). More recently, methods that allow even greater sensitivity using samples as small as a few micrograms have been developed (15, 16).
The sensitivity of AMS measurement gives this technique a number of major advantages over other methods for the detection of isotopes. Importantly, because of the extreme sensitivity, PK studies have the ability to determine kinetics and metabolism using ultralow doses of the chemical and radioactivity so as not to perturb the natural biological state. In addition, the collection of detailed pharmacokinetic data requires frequent sampling, which is made possible with AMS detection, by virtue of the small sample sizes needed for analysis. AMS has been the method of choice for microdosing studies because the dose required for determining PK parameters is so low that there is little or no concern for drug efficacy or toxic effects.
The objective of the current study was to determine whether the pharmacokinetics of a novel GyrB/ParE inhibitor preclinical drug candidate (GP-4) displayed linear kinetics across a microdose and pharmacological dose range in rats. This study served as a first step toward initiating a human microdose study and as a preliminary investigation of the feasibility of using the rat as a model. The GyrB/ParE inhibitors are a novel class of antibacterial agents, the pyrimidoindoles, which inhibit two bacterial targets: the ATP-binding domains of bacterial DNA gyrase (GyrB) and topoisomerase IV (ParE). The pyrimidoindoles were designed to be highly potent against both enzymes with broad-spectrum antibacterial activity and excellent selectivity against other mammalian enzymes that utilize ATP (17).
The PK parameters for GP-4 were assessed over a 300-fold dose range by administering to rats a microdose or a pharmacological dose of 14C-labeled GP-4. Following drug administration, blood was collected at specified time points for up to 24 h. The GP-4 concentration in plasma was determined by quantifying the amount of 14C radiolabel in the samples by AMS or liquid chromatography-tandem mass spectrometry (LC/MS/MS). The values of the PK parameters obtained after administration of both the microdose and the pharmacological dose were determined and compared. The results showed that the PK properties of GP-4 for the animals given a microdose were similar to those for the animals given a pharmacological dose, indicating linearity across the 300-fold dose range. Moreover, these results demonstrate the utility of AMS in providing exceptional sensitivity for conducting microdose studies.
MATERIALS AND METHODS
Chemicals and reagents.
14C-radiolabeled GP-4 (specific activity, 50 mCi/mmol) and unlabeled GP-4 were provided by Cubist Pharmaceuticals (San Diego, CA). Chemical purity and radiopurity were assessed by high-pressure liquid chromatography (HPLC), and the unlabeled and radiolabeled compounds were determined to be >99% pure. Captisol was obtained from Ligand Pharmaceuticals, Inc. (La Jolla, CA), and verapamil and methanesulfonic acid were purchased from Sigma-Aldrich Inc. (St. Louis, MO). All other reagents were of analytical grade or better.
Animals.
Animal experiments were conducted at the Lawrence Livermore National Laboratory (LLNL) AAALAC-accredited animal care facility. The protocols for the animal experiments were reviewed and approved by the LLNL Institutional Animal Care and Use Committee prior to the initiation of any study. Six- to 8-week-old male Sprague-Dawley rats (weight, 250 to 300 g) with a surgically implanted jugular vein catheter were obtained from Harlan Laboratories (Livermore, CA). Animals were housed individually in polystyrene cages containing hardwood bedding and kept on a 12-h light/12-h dark cycle in a ventilated room maintained at 24°C. Animals were provided food (standard lab chow) and water ad libitum.
Dose proportionality.
To establish dose proportionality at the proposed therapeutic dose levels prior to initiation of the microdose study, rats (n = 3) were administered a single intravenous (i.v.) bolus dose of 3, 10, or 30 mg/kg GP-4 formulated in 20% Captisol in 0.05 M methane sulfonic acid, pH 3.5. Following dose administration, whole-blood samples (approximately 0.3 ml) were collected from each animal via the jugular vein at 0, 0.08, 0.25, 0.5, 1, 2, 4, 8, and 24 h postdose and placed into Microtainer tubes coated with lithium heparin (Becton Dickinson, Franklin Lakes, NJ), and the tubes were placed on ice. Plasma was separated from the whole blood by centrifugation (10,000 × g for 2 min) within 1 h of collection, the volume was recorded, and the plasma was stored at −20°C until analysis by LC/MS/MS.
LC/MS/MS analysis.
Each plasma sample together with an internal standard (50 ng/ml verapamil in acetonitrile-water [1:1]) was directly injected into a Shimadzu LC-20AD HPLC system (Shimadzu, Columbia, MA) coupled to an AB SCIEX API 3200Q Otrap triple-quadrupole mass spectrometer with a TurboIonSpray interface (AB SCIEX, Framingham, MA). Samples were chromatographed using a Gemini-NX 3-μm C18 column (50 by 3 mm; Phenomenex, Torrance, CA) operating at a flow rate of 0.6 ml/min initially using a solvent of 85% solvent A (12.5 mM ammonium formate, pH 4) for 1 min. This was followed by a linear gradient up to 95% solvent B (0.1% formic acid in acetonitrile) at 3.2 min and a hold at 95% solvent B until 3.8 min and then reequilibration to 85% solvent A over 1.2 min. Quantitation was by multiple reaction monitoring (transitions of m/z ratios of 435 to 418 for GP-4 and 455 to 165 for verapamil).
Microdose versus therapeutic dose pharmacokinetics.
To evaluate the pharmacokinetics of GP-4 in rodents at a microdose and a pharmacological dose, rats (n = 3) were administered a single intravenous dose of 0.01 mg/kg of body weight or 3.0 mg/kg 14C-labeled GP-4 (specific activities, 353 μCi/mmol and 1.26 μCi/mmol, respectively) formulated in 20% Captisol in 0.05 M methanesulfonic acid, pH 3.5. The use of one microdose concentration and one therapeutic concentration for this study is in accordance with the characteristics of previous microdosing studies as well as with the guidelines set forth by the U.S. Food and Drug Administration (FDA) for microdosing studies (5, 7, 18, 19). Following dose administration, whole-blood samples (approximately 0.3 ml) were collected from each animal via the jugular vein at 0, 0.08, 0.25, 0.5, 1, 2, 4, 8, and 24 h postdose and placed into Microtainer tubes coated with lithium heparin (Becton Dickinson, Franklin Lakes, NJ), and the tubes were placed on ice. Plasma was separated from the whole blood by centrifugation (10,000 × g for 2 min) within 1 h of collection, the volume was recorded, and the plasma was stored at −20°C until analysis by AMS.
AMS analysis.
Preparation of the samples for radiocarbon analysis by AMS requires conversion of the samples to graphite. This procedure has been described previously (20). All the samples and reagents were handled carefully to avoid radiocarbon cross-contamination, including using disposable materials for any item that might come into contact with the samples. A 30-μl aliquot of each plasma sample was pipetted into a 6- by 55-mm quartz tube using aerosol-resistant tips. All samples were subsequently dried under vacuum centrifugation. The dried samples were then converted to graphite by a two-step process using published methods (14). Briefly, the dried samples were oxidized to CO2 by heating at 900°C for 4 h in the presence of copper oxide. The CO2 was then cryogenically transferred to a septum-sealed vial under vacuum and reduced to filamentous graphite in the presence of cobalt, titanium hydride, and zinc powder.
The total radiocarbon content in the plasma was quantified by AMS as described previously (14, 21, 22). The 14C/12C ratios from the graphitized samples obtained by AMS were converted to ng GP-4 per ml of plasma after subtraction of the background carbon contributed from the sample and correction for the specific activity of the 14C-labeled GP-4 dosing material and the carbon content of the sample (12). The precision of the AMS measurements is based on the standard deviation of a number of measurements for equivalent samples and is a nominal 3%. Accuracy is determined by normalization to well-defined standards and with high-accuracy counting measurements and is ∼1% (12).
Pharmacokinetics and statistics.
The pharmacokinetic parameters of GP-4 were calculated by noncompartmental analysis using either WinNonlin Professional software, version 5.2.1 (Pharsight Corp. Mountain View, CA), or PK Solutions software (Summit Research Services, Montrose, CO) using a two-stage approach by taking the plasma concentration data from each rat, fitting them independently, and then estimating the means ± standard errors (SEs). The half-life (t1/2) and initial GP-4 concentration (the maximum concentration observed in plasma [Cmax]) were determined by observations from the concentration-versus-time data. The area under the curve (AUC) was calculated for the intervals from time zero to time t (AUC0–t), where t is the time of the last measurable concentration (24 h), and time zero to infinity (AUC0–∞), using the linear trapezoidal method. The volume of distribution (V) was determined on the basis of the AUC determination and reflects the V during the elimination phase. The clearance (CL) calculation is based on the AUC0–∞ and assumes 100% bioavailability. Additionally, to estimate the absorption and disposition rate constants, a two-compartment open model was fit to the plasma concentration-time profile. Estimated parameters included the rate of GP-4 absorption to the peripheral compartment from the central compartment (k12), the rate of GP-4 transfer from the peripheral compartment to the central compartment (k21), and the elimination rate constant (k10). Values are expressed as the mean ± SE (n = 3), unless otherwise noted. Dose linearity and dose proportionality were assessed using the power regression model on log-transformed data for AUC0–∞, Cmax, and t1/2 (8). Correlation coefficients (R values), coefficients of determination (R2 values), and 95% confidence limits were determined from the power regression analysis. Results were considered linear and dose proportional when R2 was ∼1, and lower confidence limits were ≥0.8 and upper confidence limits were ≤1.25 (23).
RESULTS
Dose proportionality study.
Since GP-4 is an experimental drug lead, it was important to first establish dose proportionality at the proposed therapeutic doses before initiation of the microdose study. The PK parameters of GP-4 were evaluated over a 10-fold dose range that was designed to capture the potential therapeutic doses. The mean plasma concentrations of GP-4 following single intravenous bolus administrations of 3, 10, or 30 mg/kg of GP-4 to male rats are illustrated in Fig. 1. The calculated mean pharmacokinetic parameters are presented in Table 1.
FIG 1.

Mean ± SE (n = 3) plasma concentration-time profiles of GP-4 following single intravenous administrations of 3 (●), 10 (◼), or 30 (◆) mg/kg GP-4 to male Sprague-Dawley rats.
TABLE 1.
PK parameters of GP-4 following a single i.v. administration of 3, 10, or 30 mg/kg GP-4 to male Sprague-Dawley ratsa
| Dose (mg/kg) | Cmax (ng/ml) | Terminal t1/2 (h) | AUC0–t (ng · h/ml) | AUC0–∞ (ng · h/ml) | CL (ml/min/kg) | V (ml/kg) |
|---|---|---|---|---|---|---|
| 3 | 1,430 ± 315 | 4.4 ± 1.4 | 722 ± 66.4 | 751 ± 77.4 | 68.0 ± 6.7 | 26,600 ± 9,295 |
| 10 | 7,310 ± 762 | 7.7 ± 1.2 | 3,490 ± 72.4 | 3,660 ± 99.3 | 45.6 ± 1.3 | 30,400 ± 4,768 |
| 30 | 13,600 ± 3,227 | 7.9 ± 0.7 | 11,400 ± 1,645 | 12,500 ± 1,813 | 42.1 ± 7.0 | 29,600 ± 7,736 |
Data are expressed as the means of 3 independent determinations ± standard errors.
Following intravenous administration of GP-4, plasma samples were collected at 0, 0.08, 0.25, 0.5, 1, 2, 4, 8, and 24 h. The maximum observed plasma concentrations (Cmaxs) of GP-4 were attained at the first sampling time point (i.e., 5 min postdose), with mean values ranging from 1,430 to 13,600 ng/ml across the 3 different doses. The mean apparent terminal half-life ranged from 4.4 to 7.9 h. Across the 3 doses studied, the total clearance (CL) of GP-4 from plasma ranged from 42.1 to 68.0 ml/min/kg, and the apparent volume of distribution (V) ranged from 26,600 to 30,400 ml/kg, suggesting rapid and extensive distribution beyond the plasma compartment.
The mean Cmax and AUC0–t values of GP-4 and the relative increase in parameter values between adjacent and overall doses are summarized in Table 2. Following an increase in dose from 3 to 10 mg/kg (3.33-fold), the increases in mean Cmax (5.11-fold) and AUC0–t (4.83-fold) values were slightly greater than expected for a linear dose-response. However, following an increase in dose from 10 to 30 mg/kg (3-fold), the increase in mean Cmax (1.86-fold) was slightly lower than dose proportional, although the increase in AUC0–t (3.27-fold) was approximately dose proportional. Furthermore, a test of dose proportionality using the power regression model (8) revealed R2 values for AUC0–∞ and Cmax of 0.9968 and 0.9493, respectively, indicating dose proportionality across the dose range. Overall, for an increase in dose from 3 to 30 mg/kg (10-fold), the increases in mean Cmax (9.51-fold) and AUC0–t (15.8-fold) values were within a factor of 2 and considered dose proportional.
TABLE 2.
Dose-exposure relationship of GP-4 following a single i.v. administration of 3, 10, or 30 mg/kg GP-4 to male Sprague-Dawley rats
| Dose (mg/kg) | Fold increase in dose | Cmax (ng/ml) | Fold increase in Cmax | AUC0–t (ng · h/ml) | Fold increase in AUC0–t |
|---|---|---|---|---|---|
| 3 | 1,430 | 722 | |||
| 10 | 3.33 | 7,310 | 5.11 | 3,490 | 4.83 |
| 30 | 3.00 | 13,600 | 1.86 | 11,400 | 3.27 |
| Overall | 10.0 | 9.51 | 15.8 | ||
| DPFa | 0.951 | 1.58 |
DPF, dose proportionality factor (fold increase in parameter/fold increase in dose).
Pharmacokinetic linearity study: microdose versus pharmacological dose.
A major goal of this study was to determine the effect of dose on PK parameters for GP-4 in rats prior to conducting a microdose study in humans. Following the FDA guidelines for microdosing studies (19), male Sprague-Dawley rats received a single bolus i.v. administration of 14C-labeled GP-4 at either a microdose (0.01 mg/kg) or a pharmacological dose (3 mg/kg), and plasma was collected at the time points designated above. AMS was used to quantify the plasma concentration of 14C-labeled GP-4 by quantifying the 14C equivalents at each time point. The GP-4 plasma concentration at 24 h at both doses is represented by only one animal, due to complications during sampling and loss during sample combustion for AMS analysis. Therefore, following GP-4 quantification, the pharmacokinetics at both dose concentrations were calculated using the data collected from 0 to 8 h by noncompartmental analysis and compared.
The mean plasma concentrations of GP-4 (based on total radioactivity) following administration of single intravenous doses of 0.01 or 3.0 mg/kg of 14C-labeled GP-4 to male rats are illustrated in Fig. 2. Calculated mean pharmacokinetic parameters are presented in Table 3.
FIG 2.

(A) Mean plasma concentration-time curve ± SE (n = 3) of GP-4 following single intravenous administration of 0.01 (●) or 3.0 (◼) mg/kg 14C-labeled GP-4 to male Sprague-Dawley rats. (B) Data shown in panel A normalized to a 1-mg/kg dose. *, the data point at 24 h is derived from data for one rat due to loss of a sample.
TABLE 3.
PK parameters of GP-4 following a single i.v. administration of 0.01 or 3.0 mg/kg 14C-labeled GP-4 to male Sprague-Dawley ratsa
| Dose (mg/kg) | Cmax (ng/ml) | Terminal t1/2 (h) | AUC0–t (ng · h/ml) | AUC0–∞ (ng · hr/ml) | CL (ml/min/kg) | V (ml/kg) |
|---|---|---|---|---|---|---|
| 0.01 | 2.4 ± 0.12 | 5.6 ± 3.4 | 1.9 ± 0.55 | 2.6 ± 0.54 | 69.7 ± 17.0 | 25,745 ± 13,381 |
| 3.0 | 2,125 ± 874 | 1.4 ± 0.67 | 1,297 ± 301 | 1,336 ± 336 | 42.05 ± 9.4 | 4,012 ± 973 |
Data are expressed as the means of 3 independent determinations ± standard errors.
The observed Cmaxs of GP-4 were attained at the first sampling time point (i.e., 5 min postdose). The mean distribution half-life for both compound concentrations was 0.26 h, whereas the apparent mean terminal half-lives were 5.6 h and 1.4 h for the 0.01-mg/kg dose and the 3-mg/kg dose, respectively. The total clearance (CL) of 14C-labeled GP-4 from plasma was 1.6-fold higher in the animals that received the 0.01-mg/kg dose (CL = 69.7 ml/min/kg) than those that received the 3.0-mg/kg dose (CL = 42.05 ml/min/kg). The values for the apparent volume of distribution (V) were 25,745 ml/kg and 4,012 ml/kg in the microdosed and pharmacologically dosed animals, respectively, suggesting rapid and extensive distribution beyond the plasma compartment, with a higher rate from the microdosed animals being found. Applying a 2-compartment open model, the rates of GP-4 absorption to the peripheral compartment from the central compartment (k12 values) were 1.66 h−1 and 2.08 h−1 for the microdose and therapeutic dose, respectively. The k21 rate constants (rate of GP-4 transfer from the peripheral compartment to the central compartment) for the microdose and therapeutic dose were 0.5 h−1 and 0.22 h−1, respectively, and the elimination rate constants (k10 values) were 0.70 h−1 and 0.71 h−1, respectively (data not shown). The higher k12 values than k21 and k10 values for both doses reinforce the conclusion that the majority of GP-4 is rapidly eliminated from the plasma and distributed to tissues and/or eliminated at both dose levels.
Power regression analysis of AUC0–∞ and Cmax across the entire dose range (0.01 to 30 mg/kg) revealed a linear relationship between exposure versus dose, with coefficients of determination (R2 values) of 0.989 and 0.981 for AUC and Cmax, respectively (Fig. 3; Table 4). The dose proportionality test of t1/2 determined that this parameter was dose independent.
FIG 3.
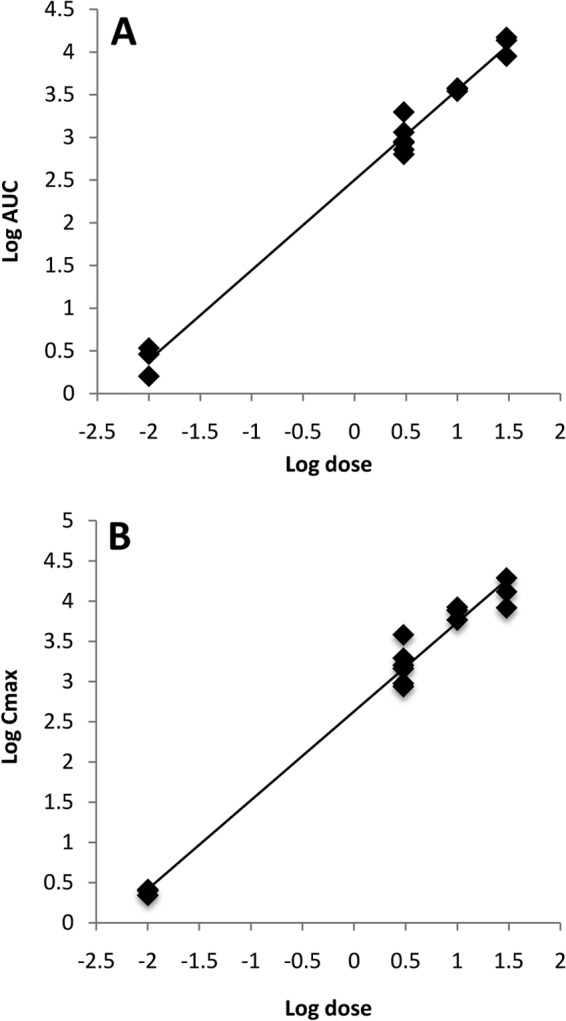
Test of dose linearity using power regression analysis for AUC0–∞ (A) and Cmax (B) of GP-4 for a dose range of 0.01 to 30 mg/kg.
TABLE 4.
Dose proportionality test using power regression model for GP-4a
| Parameter | R2 | R | Slope | 95% confidence limit |
Implication | |
|---|---|---|---|---|---|---|
| Lower | Upper | |||||
| AUC0–∞ | 0.989 | 0.994 | 1.06 | 0.99 | 1.12 | Linear |
| Cmax | 0.981 | 0.991 | 1.10 | 1.01 | 1.19 | Linear |
| t1/2 | 0.066 | 0.2 | 0.07 | −0.086 | 0.227 | Dose independent |
The dose range tested was 0.01 to 30 mg/kg.
Power regression analysis was also done on the GP-4 plasma concentration from the microdose and that from the pharmacological dose and revealed a linear relationship between the two doses with an R2 value of 0.9187 and a correlation coefficient (R) of 0.9585, indicating a strong association between the two doses (Fig. 4).
FIG 4.
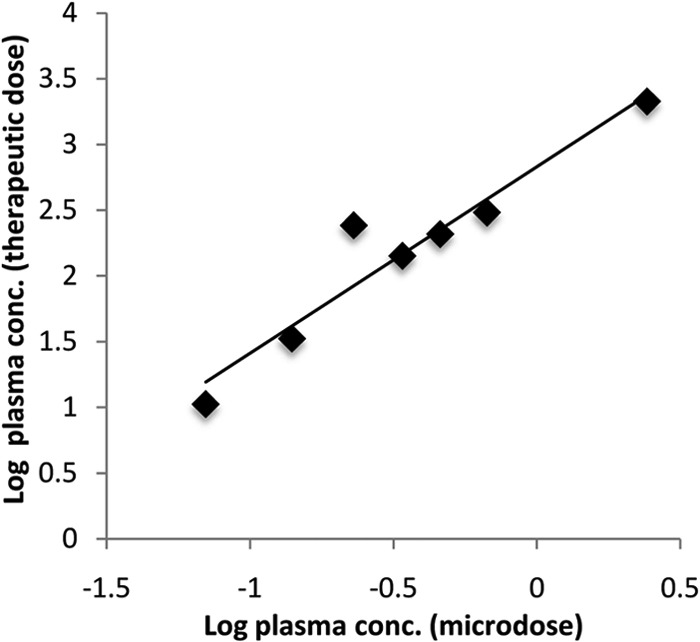
Power regression analysis of the mean (n = 3) log microdose plasma concentration of GP-4 versus the mean (n = 3) log therapeutic dose plasma concentration of GP-4 (R2 = 0.9187, R = 0.9585).
When the mean Cmaxs were dose normalized to 1.0 mg/kg, they were 243 ng/ml and 709 ng/ml for the 0.01-mg/kg and 3-mg/kg doses, respectively (Fig. 2B). This is a 2.9-fold difference across the 300-fold difference dose range. When the mean AUC0–∞ values of 14C-labeled GP-4 were dose normalized to 1.0 mg/kg, they were 277.5 ng · h/ml for the 0.01-mg/kg dose and 418.2 for the 3.0-mg/kg dose, which equates to a 1.5-fold difference between the two doses. Overall, for an increase in dose from 0.01 to 3.0 mg/kg (300-fold), the increases in mean AUC0–∞ and Cmax values were within factors of 2 and 3, respectively.
DISCUSSION
Microdosing is becoming a widely accepted way for estimating human pharmacokinetics at therapeutic dose levels for preclinical drug candidates and has been endorsed by the U.S. Food and Drug Administration and the European Medicines Agency (19–24). Microdosing studies can provide an early determination of the pharmacokinetic properties of a drug candidate, eliminating compounds with poor PK properties and allowing compounds with favorable PKs to proceed for further clinical evaluation. Estimating human PKs can greatly reduce the attrition rate of new drugs during the later stages of development, saving time and reducing costs. Key to the use of microdose studies is establishing a correlation between the PKs at the microdose and the pharmacological dose. Although the published data are limited, studies that have compared data obtained with the microdose and the therapeutic dose have shown that the microdose data are predictive of the therapeutic dose in more than 80% of the cases (6, 8, 11, 25, 26). One approach for establishing dose linearity is to validate the approach in an animal model prior to initiating a human microdosing study (8, 11).
In the present study, the rat was used as a model to determine the PKs of the tricyclic GyrB/ParE inhibitor drug candidate GP-4 at microdose and pharmacological dose levels, as a prelude to a human microdosing study. This is the first reported study to assess the PK parameters of this class of drug candidate. The results showed that the plasma PKs were proportional across 3 proposed therapeutic dose levels, indicating linear kinetics over the pharmacological dose range. A test of dose proportionality using the power regression model confirmed linearity across the dose range. It was important to establish dose proportionality across the therapeutic dose range before the initiation of the microdose study because if linearity did not exist at the higher therapeutic doses, it would be extremely difficult to establish a linear relationship between the microdose and therapeutic dose from the microdose study. Fortunately, this was not the case and the therapeutic dose range proved to be linear, as measured by AUC power regression analysis. Additionally, the levels of unchanged GP-4 after administration of a 3-mg/kg i.v. dose measured by LC/MS/MS were similar to those of the total 14C content of 14C-labeled GP-4 at the same dose measured by AMS, suggesting little or no metabolism/breakdown of GP-4 at these dose levels in vivo.
Taking advantage of the increased sensitivity of AMS compared to that of LC/MS/MS, PK parameters from animals administered a microdose (0.01 mg/kg) of GP-4 were quantified by AMS. For a direct comparison, AMS was also used to quantify PK parameters at a 3.0-mg/kg pharmacological dose over a 24-h exposure period. The lack of replicate data points at the 24-h collection time (due to complications during sampling and loss during sample combustion for AMS analysis) was unfortunate; however, because greater than 99% of the GP-4 dose was cleared from the plasma within the first 8 h after exposure, data from the 24-h collection point do not have a significant impact on the overall results and conclusions. On the basis of the results, the PKs in the rat are linear across a 300-fold dose range (R2 = 0.989). Both the microdosed and pharmacologically dosed groups exhibited a multicompartment PK profile, initially showing a rapid decline in plasma concentration postdose, followed by a slower terminal phase. Both dose groups exhibited a large apparent volume of distribution, suggesting rapid and extensive distribution beyond the plasma compartment. For an increase in dose from 0.01 to 3.0 mg/kg (300-fold), the increases in mean AUC0–∞ (1.5-fold) and Cmax (2.9-fold) values when they were dose normalized were within factors of 2 and 3, respectively. The observed variation in response could be due to the different measurement techniques (LC/MS/MS versus AMS) and/or animal-to-animal variation, given the small sample size. The possibility that the 14C label on the compound altered the PK profile also cannot be discounted; however, this is unlikely. Nonetheless, a difference in PK parameters within a factor of 2 has been regarded as an acceptable level of variation for a greater than 100-fold difference in dose to be considered dose linear (8, 10). Given the rapid distribution observed, estimates of the Cmax, which was the concentration measured at the earliest (5 min) collection time from an i.v. bolus administration, were expected to be quite variable, and the Cmax-to-dose linearity analysis likely reflects this. Therefore, a more pragmatic measure of dose linearity comes from analysis of the AUC0–∞-to-dose linearity analysis. When dose linearity was tested using power regression analysis, exposures measured by the use of both AUC and Cmax over the entire dose range of 0.01 to 30 mg/kg (by combining exposure data from both the therapeutic and microdose studies) were considered linear over the 300-fold dose range (Fig. 3; Table 4). The terminal half-life was determined to be dose independent, which is similar to what has been observed in other studies comparing PK parameters over a wide dose range (8). These results provide further confirmation of pharmacokinetic linearity over 4 orders of magnitude. In addition, the observed linearity of the GP-4 plasma concentration between the microdose and the therapeutic dose reinforces the concept of dose proportionality across a wide concentration range. This observed linearity in the rodent model provides assurance that a microdose study in humans can be predictive of the kinetics at a higher therapeutic dose level.
This study provides further evidence that the PK parameters of many drug classes, when administered at 0.01% of the intended therapeutic dose, are linear functions of the administered dose and thus can be used to estimate the PK parameters at a therapeutic dose.
ACKNOWLEDGMENTS
This work was performed under the auspices of the U.S. DOE by Lawrence Livermore National Laboratory at the Research Resource for Biomedical AMS under contract DE-AC52-07NA27344 and was supported by grants from the National Institute of General Medical Sciences (8 P41 GM103483-14) and NIAID/NIH under contract no. HHSN272200800042C, as well as by Cubist Pharmaceuticals.
At the time that the studies were conducted, Voon S. Ong and Courtney L. Ramos were employees of Cubist Pharmaceuticals.
Footnotes
Published ahead of print 18 August 2014
REFERENCES
- 1.Collier R. 2009. Drug development cost estimates hard to swallow. Can. Med. Assoc. J. 180:279–280. 10.1503/cmaj.082040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dimasi JA. 2001. Risks in new drug development: approval success rates for investigational drugs. Clin. Pharmacol. Ther. 69:297–307. 10.1067/mcp.2001.115446. [DOI] [PubMed] [Google Scholar]
- 3.Prentis RA, Lis Y, Walker SR. 1988. Pharmaceutical innovation by the seven UK-owned pharmaceutical companies (1964-1985). Br. J. Clin. Pharmacol. 25:387–396. 10.1111/j.1365-2125.1988.tb03318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaplan N, Garner C, Hafkin B. 2013. AFN-1252 in vitro absorption studies and pharmacokinetics following microdosing in healthy subjects. Eur. J. Pharm. Sci. 50:440–446. 10.1016/j.ejps.2013.08.019. [DOI] [PubMed] [Google Scholar]
- 5.Lappin G, Boyce MJ, Matzow T, Lociuro S, Seymour M, Warrington SJ. 2013. A microdose study of 14C-AR-709 in healthy men: pharmacokinetics, absolute bioavailability and concentrations in key compartments of the lung. Eur. J. Clin. Pharmacol. 69:1673–1682. 10.1007/s00228-013-1528-2. [DOI] [PubMed] [Google Scholar]
- 6.Garner RC, Lappin G. 2006. The phase 0 microdosing concept. Br. J. Clin. Pharmacol. 61:367–370. 10.1111/j.1365-2125.2006.02575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garner RC, Goris I, Laenen AA, Vanhoutte E, Meuldermans W, Gregory S, Garner JV, Leong D, Whattam M, Calam A, Snel CA. 2002. Evaluation of accelerator mass spectrometry in a human mass balance and pharmacokinetic study—experience with 14C-labeled (R)-6-[amino(4-chlorophenyl)(1-methyl-1H-imidazol-5-yl)methyl]-4-(3-chlorophenyl)1-methyl-2(1H)-quinolinone (R115777), a farnesyl transferase inhibitor. Drug Metab. Dispos. 30:823–830. 10.1124/dmd.30.7.823. [DOI] [PubMed] [Google Scholar]
- 8.Balani SK, Nagaraja NV, Qian MG, Costa AO, Daniels JS, Yang H, Shimoga PR, Wu JT, Gan LS, Lee FW, Miwa GT. 2006. Evaluation of microdosing to assess pharmacokinetic linearity in rats using liquid chromatography-tandem mass spectrometry. Drug Metab. Dispos. 34:384–388. 10.1124/dmd.105.007195. [DOI] [PubMed] [Google Scholar]
- 9.Boddy AV, Sludden J, Griffin MJ, Garner C, Kendrick J, Mistry P, Dutreix C, Newell DR, O'Brien SG. 2007. Pharmacokinetic investigation of imatinib using accelerator mass spectrometry in patients with chronic myeloid leukemia. Clin. Cancer Res. 13:4164–4169. 10.1158/1078-0432.CCR-06-2179. [DOI] [PubMed] [Google Scholar]
- 10.Lappin G, Kuhnz W, Jochemsen R, Kneer J, Chaudhary A, Oosterhuis B, Drijfhout WJ, Rowland M, Garner RC. 2006. Use of microdosing to predict pharmacokinetics at the therapeutic dose: experience with 5 drugs. Clin. Pharmacol. Ther. 80:203–215. 10.1016/j.clpt.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 11.Sandhu P, Vogel JS, Rose MJ, Ubick EA, Brunner JE, Wallace MA, Adelsberger JK, Baker MP, Henderson PT, Pearson PG, Baillie TA. 2004. Evaluation of microdosing strategies for studies in preclinical drug development: demonstration of linear pharmacokinetics in dogs of a nucleoside analog over a 50-fold dose range. Drug Metab. Dispos. 32:1254–1259. 10.1124/dmd.104.000422. [DOI] [PubMed] [Google Scholar]
- 12.Vogel JS, Turteltaub KW, Finkel RC, Nelson DE. 1995. Accelerator mass spectrometry. Anal. Chem. 67:A353–A359. 10.1021/ac00107a001. [DOI] [PubMed] [Google Scholar]
- 13.Lappin G, Garner RC. 2004. Current perspectives of 14C-isotope measurement in biomedical accelerator mass spectrometry. Anal. Bioanal. Chem. 378:356–364. 10.1007/s00216-003-2348-5. [DOI] [PubMed] [Google Scholar]
- 14.Ognibene TJ, Bench G, Vogel JS, Peaslee GF, Murov S. 2003. A high-throughput method for the conversion of CO2 obtained from biochemical samples to graphite in septa-sealed vials for quantification of 14C via accelerator mass spectrometry. Anal. Chem. 75:2192–2196. 10.1021/ac026334j. [DOI] [PubMed] [Google Scholar]
- 15.Thomas AT, Ognibene T, Daley P, Turteltaub K, Radousky H, Bench G. 2011. Ultrahigh efficiency moving wire combustion interface for online coupling of high-performance liquid chromatography (HPLC). Anal. Chem. 83:9413–9417. 10.1021/ac202013s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thomas AT, Stewart BJ, Ognibene TJ, Turteltaub KW, Bench G. 2013. Directly coupled high-performance liquid chromatography-accelerator mass spectrometry measurement of chemically modified protein and peptides. Anal. Chem. 85:3644–3650. 10.1021/ac303609n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tari LW, Li X, Trzoss M, Bensen DC, Chen Z, Lam T, Zhang J, Lee SJ, Hough G, Phillipson D, Akers-Rodriguez S, Cunningham ML, Kwan BP, Nelson KJ, Castellano A, Locke JB, Brown-Driver V, Murphy TM, Ong VS, Pillar CM, Shinabarger DL, Nix J, Lightstone FC, Wong SE, Nguyen TB, Shaw KJ, Finn J. 2013. Tricyclic GyrB/ParE (TriBE) inhibitors: a new class of broad-spectrum dual-targeting antibacterial agents. PLoS One 8:e84409. 10.1371/journal.pone.0084409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Henderson PT, Li T, He M, Zhang H, Malfatti M, Grimminger P, Dannenberg K, de Vere White R, Turteltaub KW, Pan C. 2011. A microdosing approach for characterizing formation and repair of carboplatin-DNA monoadducts and chemoresistance. Int. J. Cancer 129:1425–1434. 10.1002/ijc.25814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.U.S. Food and Drug Administration. 2005. Guidance for industry, investigators, and reviewers. Exploratory IND studies. Draft guidance. Center for Drug Evaluation and Research, U.S. Food and Drug Administration, U.S. Department of Health and Human Services, Rockville, MD. [Google Scholar]
- 20.Creek MR, Mani C, Vogel JS, Turteltaub KW. 1997. Tissue distribution and macromolecular binding of extremely low doses of [14C]-benzene in B6C3F1 mice. Carcinogenesis 18:2421–2427. 10.1093/carcin/18.12.2421. [DOI] [PubMed] [Google Scholar]
- 21.Turteltaub KW, Felton JS, Gledhill BL, Voge JS, Southon JR, Caffee MW, Finkel RC, Nelson DE, Proctor ID, Davis JC. 1990. Accelerator mass spectrometry in biomedical dosimetry: relationship between low-level exposure and covalent binding of heterocyclic amine carcinogens to DNA. Proc. Natl. Acad. Sci. U. S. A. 87:5288–5292. 10.1073/pnas.87.14.5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vogel JS. 1992. Rapid production of graphite without contamination for biomedical AMS. Radiocarbon 34:333–350. [Google Scholar]
- 23.Smith BP, Vandenhende FR, DeSante KA, Farid NA, Welch PA, Callaghan JT, Forgue ST. 2000. Confidence interval criteria for assessment of dose proportionality. Pharm. Res. 17:1278–1283. 10.1023/A:1026451721686. [DOI] [PubMed] [Google Scholar]
- 24.European Medicines Agency, Committee for Propriety Medicinal Products/Safety Working Party/2599. 2004. Position paper on non-clinical safety studies to support clinical trials with a single microdose. European Medicines Agency, London, United Kingdom. [Google Scholar]
- 25.Chen J, Flexner C, Liberman RG, Skipper PL, Louissaint NA, Tannenbaum SR, Hendrix CW, Fuchs EJ. 2012. Biphasic elimination of tenofovir diphosphate and nonlinear pharmacokinetics of zidovudine triphosphate in a microdosing study. J. Acquir. Immune Defic. Syndr. 61:593–599. 10.1097/QAI.0b013e3182717c98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seymour M. 2009. The best model for humans is human—how to accelerate early drug development safely. Altern. Lab. Anim. 37(Suppl 1):S61–S65. [DOI] [PubMed] [Google Scholar]