

Abstract
Recurrent exposure to hypoglycemia can impair the normal counterregulatory hormonal responses that guard against hypoglycemia, leading to hypoglycemia unawareness. This pathological condition known as hypoglycemia-associated autonomic failure (HAAF) is the main adverse consequence that prevents individuals with type 1 diabetes mellitus from attaining the long-term health benefits of tight glycemic control. The underlying molecular mechanisms responsible for the progressive loss of the epinephrine response to subsequent bouts of hypoglycemia, a hallmark sign of HAAF, are largely unknown. Normally, hypoglycemia triggers both the release and biosynthesis of epinephrine through activation of nicotinic acetylcholine receptors (nAChR) on the adrenal glands. We hypothesize that excessive cholinergic stimulation may contribute to impaired counterregulation. Here, we tested whether administration of the nAChR partial agonist cytisine to reduce postganglionic synaptic activity can preserve the counterregulatory hormone responses in an animal model of HAAF. Compared with nicotine, cytisine has limited efficacy to activate nAChRs and stimulate epinephrine release and synthesis. We evaluated adrenal catecholamine production and secretion in nondiabetic rats subjected to two daily episodes of hypoglycemia for 3 days, followed by a hyperinsulinemic hypoglycemic clamp on day 4. Recurrent hypoglycemia decreased epinephrine responses, and this was associated with suppressed TH mRNA induction (a measure of adrenal catecholamine synthetic capacity). Treatment with cytisine improved glucagon responses as well as epinephrine release and production in recurrently hypoglycemic animals. These data suggest that pharmacological manipulation of ganglionic nAChRs may be promising as a translational adjunctive therapy to avoid HAAF in type 1 diabetes mellitus.
Keywords: hypoglycemia-associated autonomic failure, tyrosine hydroxylase, epinephrine release, cytisine, partial nicotinic acetylcholine receptor agonists
an attenuated sympathoadrenal response following recurrent exposure to hypoglycemia results in a pathological inability to recover from low blood glucose levels, contributes to patient unawareness of hypoglycemia, and is associated with increased mortality in type 1 and possibly type 2 diabetic patients, a condition known as hypoglycemia-associated autonomic failure (HAAF) (1, 11). HAAF is recognized as a major public health problem for patients with diabetes. It can also be induced in healthy subjects, in infants and in animal models suggesting it may involve a maladaptive response to repeated stress (9, 12, 14, 20).
Despite the well-established role of hypoglycemia per se in the development of HAAF, the mechanism(s) underlying the key feature of the pathogenesis of HAAF in diabetes, the attenuated sympathoadrenal epinephrine response to falling plasma glucose levels in a background of imperfect insulin replacement and impaired glucagon responses (10), remains elusive. The reduced sympathoadrenal response in HAAF could result from adaptations/maladaptations within central or peripheral neural-humoral networks that regulate glucose homeostasis (7) or from alterations at the level of the afferent or efferent components of the sympathoadrenal system. Whereas central mechanisms contributing to HAAF have been studied intensively in humans and animal models (11), the capacities of efferent neural outputs have received less attention.
Nicotinic acetylcholine receptors play a critical role in this process since ganglionic blockers and/or surgical ablation of autonomic inputs markedly impair autonomic activation during insulin-induced hypoglycemia in animal models and in humans (reviewed in Ref. 42). Given that under HAAF conditions adrenal sympathetic nerve impulse activity remains elevated for 24 h (21, 40), the attenuated plasma epinephrine response and the accompanying reduction in adrenomedullary catecholamine content (15) likely arise from factors that alter the capacity of chromaffin cells to maintain the releasable pool of catecholamines separate from a possible failure of sustained presynaptic input or a putative deficiency in chromaffin cellular secretory mechanisms.
Since low-intensity nicotinic receptor activation induces catecholamine biosynthesis in situ for prolonged periods (48), and this result does not appear to be happening in vivo during HAAF, we hypothesized that a partial reduction in high-intensity synaptic transmission (rather than complete ganglionic blockade) may preserve the capacity to synthesize and release adrenal epinephrine during protracted stress in the intact animal. To test this hypothesis, we used the nicotinic receptor partial agonist cytisine to reduce peripheral autonomic neurotransmission in a well-characterized animal model of HAAF (39) known to have sustained transsynaptic adrenal nerve activity (21, 40). Cytisine was chosen because of its limited brain penetration, lack of reported adverse effects in vivo (45), and adequate pharmacokinetics. At an intravenous dose of 1 mg/kg, the plasma clearance of cytisine in rats is ∼35 ml·min−1·kg−1, and its half-life is 1.5 h (36). Cytisine binds with differing affinities to several subtypes of nicotinic acetylcholine receptors (nAChR) and exhibits lower efficacy (based on electrophysiological assays) compared with endogenous ligands and/or nicotine (36).
We demonstrated that alone cytisine acts as a typical partial agonist in vivo inducing modest catecholamine secretion and de novo catecholamine synthesis but is significantly less effective than an exogenous (i.e., nicotine) or endogenous (insulin-induced release of acetylcholine) full agonist. More importantly, treatment with cytisine significantly improved counterregulatory responses in recurrently hypoglycemic animals, showing both enhanced catecholamine release and greater expression of adrenal tyrosine hydroxylase (TH) mRNA, the rate-limiting enzyme in catecholamine biosynthesis.
MATERIALS AND METHODS
Animals
Adult male Sprague-Dawley rats weighing 285–320 g with carotid artery (CA) and jugular vein (JV) cannulation (Harlan Labs, Indianapolis, IN) were housed individually in temperature- (21°C), humidity- (35%), and light-controlled (12:12-h light-dark cycle) rooms. Regular rat chow (Agway Prolab 3,000; Agway, Syracuse, NY) and water ad libitum were provided to the animals unless otherwise stated. Experimental protocols were approved by the Institutional Animal Care and Use Committees at New York Medical College and Yale University.
Drugs
Cytisine and nicotine ditartrate (Sigma Chemical, St. Louis, MO) were dissolved in sterile saline and injected intraperitoneally (ip) at the doses and times indicated in the figures. Nicotine doses were calculated as that of the base, and cytisine doses are expressed as that of the salt. Regular human insulin (Humulin R; Eli Lilly, Indianapolis, IN) at a dose of 2 IU/kg was used to induce hypoglycemia during the antecedent hypoglycemia treatment (39).
Experimental Design
Effect of cytisine on adrenal epinephrine secretion and production in vivo: dose-response study.
Previously, in PC12 cells (a rat pheochromocytoma tissue culture model of adrenal chromaffin cell functions), we showed that cytisine exhibits typical partial agonist properties at native coexisting α3β4 and α7 nAChR with regard to its effects on catecholamine secretion and synthesis (44). In the present report, we aimed to compare the effects of cytisine and the full agonist nicotine on epinephrine secretion and their ability to alter TH mRNA levels in vivo. Cytisine doses ranging from 0.3 to 3 mg/kg or nicotine (1 mg/kg) were given ip to chronically catheterized animals twice daily (9 AM and 1 PM) for 3 consecutive days to mimic the HAAF model animal protocol (39). Control animals received an equal volume of saline injection under similar conditions. On day 4, the catheters were extended outside of the cages for stress-free blood sampling. Two hours was allowed for the animals to recover from handling stress before the final AM dose of cytisine, nicotine, or vehicle was given through the JV catheter. Arterial blood samples were collected before (baseline) and every 30 min after drug administration for the ensuing 2 h. After the collection of each blood sample, the erythrocytes were resuspended in an equivalent volume of artificial plasma and reinfused back into the animal to prevent volume depletion and anemia (8, 28). Animals were euthanized 5 h after the AM treatment (the time point at which maximal induction of TH mRNA occurs in response to hypoglycemia) (46), and the adrenal medullae were harvested and then immediately frozen at −80°C for TH mRNA analyses.
Effect of cytisine treatment on the epinephrine response to hypoglycemia in recurrently hypoglycemic rats.
By definition, partial agonists have dual functions. When endogenous ligand concentrations are low or absent, they can act as agonists with a smaller maximal effect at full receptor occupancy than the endogenous ligand (full agonist). However, since they are less effective than the endogenous ligand, partial agonists can also act as antagonists by suppressing the effects of high concentrations of endogenous ligand (32, 35, 36). To test whether treatment with cytisine can improve the epinephrine response in recurrently hypoglycemic animals, we randomly assigned chronically catheterized rats to one of four experimental groups: recurrent saline (RS), recurrent cytisine (RC), recurrent hypoglycemia (RH), and recurrent hypoglycemia + cytisine (CRH). Animals were administered the study drug ip twice daily at 9 AM and 1 PM with either saline, 1 mg/kg cytisine, 2 IU/kg insulin, or a combination of cytisine and insulin (CRH group), with cytisine given 30 min before the insulin (26, 36). Animal chow was removed in all groups for 3 h during each antecedent drug protocol treatment. Blood glucose was monitored from tail nick samples using hand-held glucometers (AlphaTrak; Abbott Laboratories, Chicago, IL) every 30 min throughout each hypoglycemic episode, and if needed, additional insulin was given to maintain glucose levels between 40 and 50 mg/dl (Fig. 3) in the RH and CRH groups. Animals were rescued with food or dextrose per os if blood glucose levels dropped below target values. Control groups (RS and RC) underwent the same procedure to ensure uniform exposure to handling stress for all groups. All antecedent treatments were for 3 consecutive days, as illustrated in Fig. 2. On day 3, only the morning treatment was given to allow the animals to fully recover before being fasted overnight prior to the clamp procedure on day 4. On day 4, all four groups underwent a 90-min hyperinsulinemic hypoglycemic glucose clamp (22, 23). Animal weights were monitored on a daily basis to ensure well being.
Fig. 3.
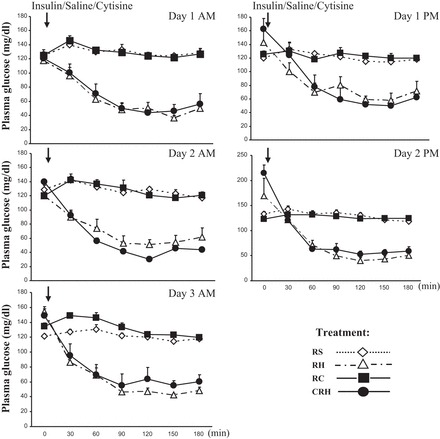
Plasma glucose levels during each antecedent treatment period. Results are presented as means ± SE; n = 9.
Fig. 2.
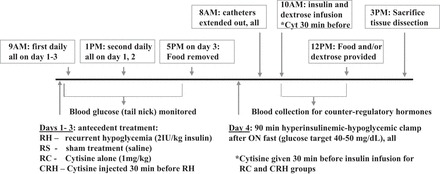
Schematic diagram depicting the main protocol used in these experiments, designed to determine the effect of cytisine given before each insulin treatment. Experimental groups are as follows: RH, recurrent hypoglycemic animals; RS, recurrent saline controls; RC, recurrent cytisine controls; CRH, animals who received cytisine before each insulin treatment. ON, overnight.
Hyperinsulinemic Hypoglycemic Clamp
Vascular catheters were extended outside of the cages for stress-free blood sampling (CA) and glucose/insulin infusion (JV), and animals were rested for ≥2 h prior to the start of the clamp. Animals treated with cytisine (RC and CRH groups) received a final dose of cytisine via the jugular vein 30 min before the clamp. A constant infusion of regular human insulin (50 mU·kg−1·min−1; Eli Lilly) and a variable 20% dextrose infusion were started, and plasma glucose levels were monitored every 5 min to guide dextrose infusion and maintain target glucose levels at ∼45 mg/dl for 90 min (Fig. 4) (22, 23). Blood samples were collected at 30-min intervals throughout the study for subsequent measurement of plasma glucagon, catecholamine, and corticosterone responses. For measuring plasma insulin levels, blood samples were collected at the beginning and at the end of the glucose clamp. At the end of the study, animals were provided with food to recover from the hypoglycemic episode and then euthanized 5 h after initiation of the hyperinsulinemic hypoglycemic clamp with an overdose of intravenous (iv) pentobarbital sodium followed by decapitation. The adrenal medullae were then dissected, frozen immediately, and stored at −80°C until further analyses.
Fig. 4.
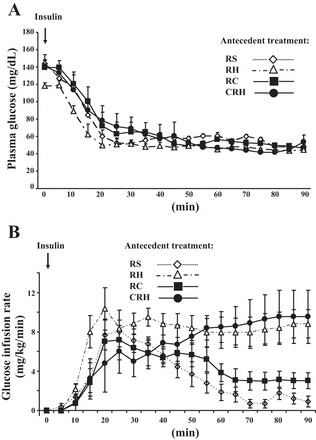
A: plasma glucose concentrations during the hyperinsulinemic hypoglycemic glucose clamp. B: glucose infusion rates during the clamp. Results are presented as means ± SE; n ≤ 9.
To determine whether there was a separate effect of refeeding on TH mRNA expression, we conducted a parallel set of studies in a smaller cohort of animals (n = 3 for each of the 4 treatment groups described above) whereby we maintained the hypoglycemic clamp for the entire 5-h period before the animals were euthanized (Fig. 6B).
Fig. 6.
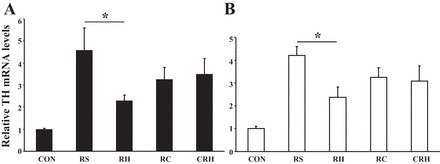
Relative adrenal TH mRNA levels correlate with the magnitude of the epinephrine response. A: after the hypoglycemic clamp the animals were provided food and euthanized 3.5 h later. The results (Northern blot) are summarized from 2 independent experiments (n = 6) and are calculated as fold induction from CON (absolute control; nontreated animals) and given as means ± SE. B: animals (n = 3 for each group) were maintained in hypoglycemic state until euthanization (5 h after the initiation of hypoglycemic clamp). *P ≤ 0.05.
Analytical Methods
Hormone analyses.
Plasma glucagon, insulin, and corticosterone were determined using commercially available radioimmunoassay kits from Linco Research (St. Charles, MO) and Diagnostic Products (Los Angeles, CA) (8). Plasma epinephrine and norepinephrine concentrations were determined using a competitive enzyme immunoassay (Rocky Mountain Diagnostics, Colorado Springs, CO) as described (44).
Isolation of RNA and Northern blot analyses.
Each left adrenal medulla was used for Northern blot analyses (31). The blots were consequently hybridized to labeled probes for TH and 18S rRNA. X-ray films were scanned and analyzed by Quantity One Software using the Bio-Rad GS 800 densitometer. The integrated optical density for each mRNA was normalized for the densities obtained for 18S rRNA levels in the same samples on the same blot.
Statistics
Statistical analysis was performed with Sigma STAT/Plot software version 12 (Sigma, San Jose, CA). All data were expressed as means ± SE. Significance was assumed at P < 0.05. Comparisons of basal and hypoglycemic responses were made using the one-way analysis of variance (ANOVA), followed by a Neuman-Keuls post hoc analysis. Sequential counterregulatory hormonal responses to hypoglycemia and glucose parameters during hypoglycemia were compared using repeated-measures ANOVA. Correlation analysis between peak plasma epinephrine levels and the observed adrenal TH mRNA levels was also performed.
RESULTS
Cytisine Modestly Stimulates Epinephrine Secretion and Elevates Adrenal TH Gene Expression In Vivo
Compared with saline, cytisine (0.3 to 3 mg/kg ip; Refs. 26 and 36) induced modest (but significant) increases in plasma epinephrine levels in a dose-dependent manner (Fig. 1A). This rise was associated with a dose-dependent increase in adrenomedullary TH mRNA levels (Fig. 1B), indicating that cytisine has the capacity to enhance both secretion and de novo synthesis of epinephrine. These effects of cytisine (3 mg/kg) were significantly less compared with nicotine (1 mg/kg) or insulin-induced release of acetylcholine during hypoglycemia (peak epinephrine values of 110 ± 10, 1,200 ± 122, and 3,310 ± 140 pg/ml, respectively), which is consistent with its limited potency to activate nicotinic receptor-mediated functions (36). Together, our data suggest that cytisine acts as a partial agonist on adrenal postganglionic nicotinic receptors in vivo serving two biologically important functions: enhancement of the secretion and de novo synthesis of epinephrine. Because the effects of cytisine appeared to plateau at a dose of 1 mg/kg, this dose was used in the remaining studies.
Fig. 1.
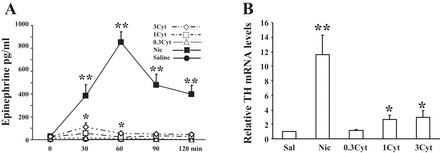
Effect of cytisine on epinephrine release and adrenal tyrosine hydroxylase (TH) mRNA levels: dose response. Experimental groups are as follows: Sal (saline-injected animals), 0.3 Cyt (animals injected ip with 0.3 mg/kg cytisine), 1 Cyt (1 mg/kg cytisine), 3 Cyt (3 mg/kg cytisine), and Nic (1 mg/kg nicotine). Each dose/drug was given twice daily, for a total of 7 episodes, to mimic the HAAF protocol. On day 4 the catheters were extended out of the cages, and arterial blood samples were collected stress free before and every 30 min after the last drug application (iv) for a total of 2 h. A: the results (plasma epinephrine values) are from 2 independent experiments (n = 6) and are presented as means ± SE. *P ≤ 0.05, Cyt vs. Sal; **P ≤ 0.002, Nic vs. Cyt. B: for RNA analyses, animals were euthanized 5 h after treatment and changes in adrenal medullary TH gene expression analyzed by Northern blots.
Can Cytisine Improve the Counterregulatory Defect Caused By Insulin-Induced Recurrent Hypoglycemia?
The outline of these experiments is shown in Fig. 2. Daily plasma glucose concentrations during the recurrent periods of treatment for all experimental groups are summarized in Fig. 3. There were no significant differences in the level of hypoglycemia that was achieved between the RH and CRH groups. Similar plasma glucose levels were also obtained for the control groups RS and RC. It should be mentioned that animals from all experimental groups displayed similar weight changes throughout treatments (data not shown).
Plasma Glucose and Insulin Levels During the Hypoglycemic Clamp
Target blood glucose levels (50 mg/dl) were achieved by 30 min and were similarly maintained at this level in all groups (Fig. 4A). Also, there was no significant difference in plasma insulin levels at baseline or after the clamp between treatment groups during the study (Table 1). These observations indicated that all animals were exposed to identical glucose and insulin stimuli during the glucose clamp portion of the protocol, differing only by their antecedent drug intervention history in the preceding 3 days. The glucose infusion rates are shown in Fig. 4B. Area under the curve (AUC) was calculated according to Tai (43). Total exogenous glucose requirements in the RH group (AUC value of 772 ± 85.79 mg/kg) were significantly higher compared with controls (calculated AUC for both, with RS and RC being 290 ± 37.24 and 357.59 ± 52.14 respectively, P < 0.05). With cytisine treatment before each bout of hypoglycemia (CRH group), the total amount of glucose administered (AUC value of 578.1 ± 30.9) was lowered compared with the RH group.
Table 1.
Average plasma insulin concentrations on day 4 before (baseline) and after hyperinsulinemic hypoglycemic clamp
| Experimental Groups | RS | RH | RC | CRH |
|---|---|---|---|---|
| Baseline | 3.2 ± 0.85 | 4.01 ± 0.98 | 4.36 ± 1.25 | 3.52 ± 1.06 |
| Hypoglycemia | 1,932 ± 120 | 2,230 ± 252 | 2,021 ± 323 | 2,032 ± 312 |
Values are means ± SE and in uIU/ml; data are summarized from 3 independent experiments (n ≥ 6/group). Experimental groups are as follows: RS, recurrent saline; RH, recurrent hypoglycemia; RC, recurrent cytisine; CRH, recurrent hypoglycemia + cytisine.
Effect of Cytisine on Counterregulatory Hormone Release Following Insulin-Induced Recurrent Hypoglycemia
There were no statistically significant differences in baseline blood levels of counterregulatory hormones between the four treatment groups (Fig. 5). In response to hypoglycemia, plasma concentrations of epinephrine (Fig. 5A) and glucagon (Fig. 5C) increased significantly from euglycemic values in all groups. Despite similar plasma glucose (Fig. 4) and insulin levels during the hypoglycemic clamp (Table 1), the rise in epinephrine (at all time points) was attenuated by nearly 50% in the RH group compared with the saline-treated (RS) group (P < 0.007; Fig. 5A). This is consistent with previously reported data for this model of HAAF (39). The epinephrine response was reduced in the cytisine-alone group (RC) compared with saline-treated animals (RS; P < 0.007 at 60 min), suggesting that cytisine may compete with acetylcholine for receptor occupancy and partially block its effects. On the other hand, cytisine treatment significantly improved the epinephrine response in animals exposed to recurrent hypoglycemia (P = 0.00173, RH vs. CRH; Fig. 5). RH animals had a significantly attenuated glucagon response, and this response was completely restored with cytisine treatment (CRH group). Cytisine treatment alone in the absence of recurrent hypoglycemia (RC group) did not affect the glucagon response (Fig. 5C).
Fig. 5.
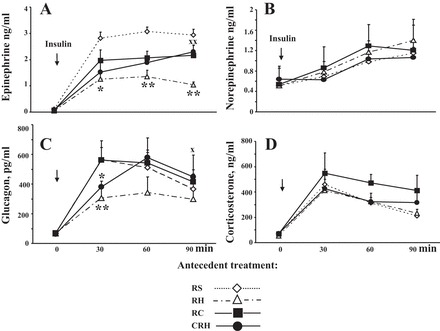
Effect of cytisine pretreatment on counterregulatory hormone release during insulin-induced recurrent hypoglycemia. Plasma epinephrine (A), norepinephrine (B), glucagon (C), and corticosterone (D) responses during hypoglycemic clamp in RS (maximal response group), RH (hypoglycemia-associated autonomic failure group), RC, and CRH. The results are summarized from 3 independent experiments (n = 9) and are presented as means ± SE. *P ≤ 0.05; **P ≤ 0.002 RH vs. RS; xP ≤ 0.05 and xxP ≤ 0.002, RH vs. CRH.
Baseline norepinephrine values and their increased increments during the hypoglycemic clamp were not significantly different between the four experimental groups (Fig. 5B) and were consistent with previously published results for RS and RH (39). These results suggest that sympathetic postganglionic outflow was similar in all experimental conditions and was not significantly affected by cytisine.
Similarly, baseline corticosterone values in all groups were not different from one another and showed the expected normal diurnal levels for that time of day (∼100 ng/ml; see Fig. 5D). During the hypoglycemic clamp, plasma corticosterone levels increased in all groups (P < 0.05 vs. corresponding baseline), but again, the responses were not different between treatment groups.
Relative Adrenal TH mRNA Levels Parallel the Magnitude of the Epinephrine Response
The release of catecholamines from the adrenal medulla in response to stress is accompanied by compensatory increases in their biosynthesis via mechanisms that result in increased TH mRNA, TH protein, and enzyme activity to maintain the releasable pool of cellular catecholamines at constant levels (24). Adrenal TH mRNA levels increased significantly in all animal groups that underwent the hypoglycemic clamp compared with nonmanipulated controls (CON). However, the rise in TH mRNA was significantly less in the group exposed to twice daily episodes of antecedent hypoglycemia (P < 0.05, RH vs. RS). The observed changes in TH mRNA levels in response to acute and recurrent hypoglycemia in the presence of cytisine (RC and CRH) were not significantly different from the RS group and correlated with peak plasma epinephrine responses (Pearson's coefficient of 0.9835). Importantly, the CRH group showed an improving trend compared with the RH group (P = 0.06; Fig. 6A).
In a control experiment, animals were euthanized at the desired time point for tissue collection (5 h), but without the recovery and refeeding interventions (Fig. 6B). A similar pattern was obtained for all experimental groups, indicating that the observed responses were in fact due to differences arising from the antecedent exposure rather than during the period of recovery following the hypoglycemic clamp.
DISCUSSION
Our data support the hypothesis that excessive adrenal nerve stimulation of nicotinic acetylcholine receptors of the adrenal chromaffin cells contributes to the development of HAAF. Consistent with all prior work (Ref. 16 and review in Ref. 24), our results confirmed that the maximal epinephrine response to hypoglycemia (Fig. 5) is associated directly with a parallel increase in TH mRNA levels (RS; Fig. 6A). However, for the first time, we also demonstrated quantitatively that steady-state levels of adrenal TH mRNA, like epinephrine blood levels, are attenuated by ∼50% during HAAF. We interpret this reduced responsiveness as arising from excessive presynaptic adrenal nerve cholinergic stimulation of adrenal chromaffin cells, since partial pharmacological inhibition of nAChRs (before each bout of hypoglycemia) significantly improved the impaired epinephrine and glucagon responses in this animal model of HAAF. Our results are intriguing and offer a proof of concept in the peripheral nervous system that parallels the previously recognized effects of nicotinic receptor partial agonists on central catecholaminergic cell responses (11, 29, 30, 36, 37).
Transsynaptic Modulation
Cytisine has been used ever since the 1960s as an aid for smoking cessation in eastern and central European countries (17), but there is only a small amount of data on its efficacy since its clinical utility is limited by its affinity for other nAChR subtypes and limited blood-brain barrier penetration (36). Its potential effects on peripheral nAChR function in vivo during whole animal responses have not been reported previously. Our data are consistent with cytisine functioning as a typical partial agonist at postsynaptic nicotinic receptors of the adrenal medulla that can induce catecholamine release (epinephrine) and de novo catecholamine synthesis (increased adrenal TH mRNA levels). In addition, as expected, the magnitude of the response is significantly lower than that of a full agonist(s) compared with either exogenously administered nicotine (Fig. 1) or insulin-induced acetylcholine release (Figs. 5A and 6).
Although binding and antagonism tests were not conducted in our studies, other reports have shown that cytisine has similar binding affinity for adrenal α3β4 receptors (27) as nicotine and no affinity for muscarinic or histamine receptors (2, 27). Furthermore, in PC12 cells (44) and in perfused adrenal glands, cytisine stimulates catecholamine release in a dose-, time-, and Ca2+-dependent manner (27, 49) and blocks the actions of acetylcholine on nicotinic receptors through a competitive mechanism (32).
Our in vivo data show that recurrent exposure to cytisine produces a modest reduction in epinephrine secretion in response to acute hypoglycemia (as observed in the RC experimental group; Fig. 5), consistent with its partial agonist properties. When animals were pretreated with cytisine before each hypoglycemic episode in an animal model of HAAF, the circulating epinephrine responses to subsequent bouts of hypoglycemia were significantly improved (CRH vs. RH group; Fig. 5). Given that ganglionic blockers and/or surgical ablation of autonomic sympathetic nerve inputs markedly impair autonomic activation of the adrenal medulla during insulin-induced hypoglycemia in both animal models and humans (42), it is reasonable to conclude that cytisine most likely acts on postganglionic neuronal nAChR to modulate their activation by the endogenously released presynaptic acetylcholine.
Insulin-induced hypoglycemia is a potent activator of the adrenomedullary hormonal system (19). This activation is mediated by indirect reflex excitation of the splanchnic nerve, which evokes large increases in catecholamine secretion (resulting in up to 70% of epinephrine content depletion) (47) accompanied by activation of TH enzyme and compensatory catecholamine biosynthesis in the adrenal medulla to maintain cellular catecholamine levels constant (41). As an index of catecholamine biosynthesis, we quantified changes in adrenal TH mRNA (25). After the hypoglycemic clamp, TH mRNA levels increased in all treatment groups compared with nonmanipulated controls. However, the rise in TH mRNA was significantly less in the RH group. This is consistent with reports showing reduced in situ staining of TH and phenylethanolamine N-methyltransferase mRNA levels in the adrenal medullas of diabetic rats that underwent the same recurrent hypoglycemia paradigm (HAAF) as our model (22, 23). Our quantitative TH mRNA data suggest that following recurrent exposure to hypoglycemia, a molecular mechanism that can limit the capacity to replenish the adrenal pool of epinephrine may exist. The possible contribution of noncholinergic receptor activation by presynaptic neuropeptides that are colocalized and coreleased with acetylcholine (24, 41) is another outstanding question that needs to be addressed in future studies. Importantly, the effect of cytisine on TH mRNA levels during acute (RC) or recurrent (CRH) hypoglycemia correlated with the corresponding rates of epinephrine secretion, and it was consistent with partial agonist properties of the drug, suggesting that future research may prove instructive as a potential therapy.
Nicotinic receptor tachyphylaxis is an unlikely mechanism to explain these results for several reasons. First, it is known that α3β4 nAchRs are only moderately susceptible to desensitization (34). Second, if desensitization had occurred, one would expect the opposite effect (or no effect) in the CRH group. Third, using intact adrenal glands in situ, reduced responsiveness was not observed; i.e., perfused rat adrenal glands had the capacity to sustain biosynthesis (increase TH mRNA, TH protein, and TH activity) and epinephrine release for prolonged periods of time via transsynaptic mechanisms (48). Taken together, sustained release in situ yet failure in vivo implicates other control signals as being operative during HAAF in an intact animal. For example, circulating factors (e.g., hormones, free fatty acids, or other substances) that are increased during recent hypoglycemia, which contribute to the inability of the adrenal medulla to sustain epinephrine biosynthesis and release, may also exist (33). Future research is necessary to ascertain whether any novel humoral factors contribute to the reduced adrenal capacity to produce catecholamines during HAAF.
Hormonal Pathways
Plasma glucagon levels were attenuated during HAAF, as reported previously (4), whereas in contrast, similar increments in glucagon were obtained in the remaining experimental groups. Notably, treatment with cytisine substantially improved the circulating glucagon responses in recurrently hypoglycemic animals to nearly the same levels as those seen in the saline group. Glucagon release from pancreatic α-cells is regulated by a number of factors, including glucose, insulin, and somatostatin secreted from neighboring β- and δ-cells, as well as by redundant autonomic inputs from sympathetic, parasympathetic, and sympathoadrenal neurotransmitters and neuropeptides (reviewed in Ref. 42). How cytisine treatment influences the relative contribution from each of these inputs remains to be elucidated.
No differences in plasma corticosterone concentrations were observed between the four treatment groups, suggesting similar levels of stress exposure and responsiveness of the HPA axis under all experimental conditions. Although several studies in nondiabetic humans (13) and normal rats (38) showed that elevated plasma glucocorticoids during antecedent hypoglycemia contribute to the development of HAAF (18), we did not observe elevated baseline corticosterone levels in our model of HAAF.
A limitation of our approach is that our current data cannot exclude a direct or indirect effect of cytisine working through central nervous system nAChRs, although it has been reported that cytisine has limited brain penetrance (36). Cholinergic receptors are crucial for acetylcholine neurotransmission in the central nervous system, and increased expression of α7 nAChR has been observed in the cerebellum of diabetic and control rats during hypoglycemia (3). We are not aware of any data supporting nAChR-mediated regulation of glucose-sensing neurons, but nAChRs in general can modulate neuronal activity at the microcircuit synaptic level by altering the cell processing of information and by influencing the velocity of action potentials or the synchrony of communication between brain areas (5). Further studies are needed to evaluate the potential peripheral vs. central effects of cytisine pertinent to glucose counterregulation. It should be noted that in some human studies a direct muscarinic cholinergic inhibition of hepatic glucose production has been observed (6), but our current data reveal that reducing cholinergic neurotransmission at nicotinic receptors improves counterregulatory responses and perhaps also hepatic glucose production, although the latter was not evaluated.
In summary, we showed that activation of nAChR during prior antecedent bouts of hypoglycemia can reduce the capacity of the adrenal medulla to replenish the releasable pool of catecholamines (compare TH mRNA in the RH vs. RS group; Fig. 6A), which in turn can lead to impairment of catecholamine secretion during subsequent bouts of hypoglycemia. This novel mechanism may represent a significant contribution to the neurogenic component of the clinical syndrome of HAAF. We speculate that modulating the nicotinic signal at the splanchnic adrenal nerve-chromaffin cell synapse may afford clinicians a new opportunity to help improve sympathoadrenal responses in compromised patients.
GRANTS
We recognize the contributions of the Juvenile Diabetes Foundation (to E. F. LaGamma and B. B. Nankova), the Yale Diabetes Research Center (DK-45735), the Empire Clinical Research Investigator Program of New York State (to E. F. LaGamma), the Children's and Women's Physicians of Westchester grant program, and the Children's Health and Research Foundation for their economic support.
DISCLOSURES
The authors declare no conflicts of interest, financial or otherwise.
AUTHOR CONTRIBUTIONS
E.F.L., O.C., and B.B.N. conception and design of research; E.F.L., O.C., and B.B.N. interpreted results of experiments; E.F.L., O.C., and B.B.N. edited and revised manuscript; E.F.L., N.K., O.C., and B.B.N. approved final version of manuscript; N.K., O.C., and B.B.N. performed experiments; N.K., O.C., and B.B.N. analyzed data; B.B.N. prepared figures; B.B.N. drafted manuscript.
ACKNOWLEDGMENTS
We appreciate the encouragement of Dr. Mladen Vranic and the helpful suggestions of Drs. Esther Sabban, Lidia Serova, and Gad Alpan during the progression of this work.
REFERENCES
- 1.No authors listed. Hypoglycemia in the Diabetes Control and Complications Trial. The Diabetes Control and Complications Trial Research Group. Diabetes 46: 271–286, 1997. [PubMed] [Google Scholar]
- 2. Anderson DJ, Arneric SP. Nicotinic receptor binding of [3H]cytisine, [3H]nicotine and [3H]methylcarbamylcholine in rat brain. Eur J Pharmacol 253: 261–267, 1994. [DOI] [PubMed] [Google Scholar]
- 3. Antony S, Peeyush Kumar T, Mathew J, Anju TR, Paulose CS. Hypoglycemia induced changes in cholinergic receptor expression in the cerebellum of diabetic rats. J Biomed Sci 17: 7, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beall C, Ashford ML, McCrimmon RJ. The physiology and pathophysiology of the neural control of the counterregulatory response. Am J Physiol Regul Integr Comp Physiol 302: R215–R223, 2012. [DOI] [PubMed] [Google Scholar]
- 5. Bertrand D. Neurocircuitry of the nicotinic cholinergic system. Dialogues Clin Neurosci 12: 463–470, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boyle PJ, Liggett SB, Shah SD, Cryer PE. Direct muscarinic cholinergic inhibition of hepatic glucose production in humans. J Clin Invest 82: 445–449, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chan O, Sherwin R. Influence of VMH fuel sensing on hypoglycemic responses. Trends Endocrinol Metab 24: 616–624, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chan O, Zhu W, Ding Y, McCrimmon RJ, Sherwin RS. Blockade of GABA(A) receptors in the ventromedial hypothalamus further stimulates glucagon and sympathoadrenal but not the hypothalamo-pituitary-adrenal response to hypoglycemia. Diabetes 55: 1080–1087, 2006. [DOI] [PubMed] [Google Scholar]
- 9. Christesen HT, Brusgaard K, Hussain K. Recurrent spontaneous hypoglycaemia causes loss of neurogenic and neuroglycopaenic signs in infants with congenital hyperinsulinism. Clin Endocrinol (Oxf) 76: 548–554, 2012. [DOI] [PubMed] [Google Scholar]
- 10. Cryer PE. Hypoglycemia-associated autonomic failure in diabetes. Am J Physiol Endocrinol Metab 281: E1115–E1121, 2001. [DOI] [PubMed] [Google Scholar]
- 11. Cryer PE. Mechanisms of hypoglycemia-associated autonomic failure in diabetes. N Engl J Med 369: 362–372, 2013. [DOI] [PubMed] [Google Scholar]
- 12. Davis SN, Mann S, Galassetti P, Neill RA, Tate D, Ertl AC, Costa F. Effects of differing durations of antecedent hypoglycemia on counterregulatory responses to subsequent hypoglycemia in normal humans. Diabetes 49: 1897–1903, 2000. [DOI] [PubMed] [Google Scholar]
- 13. Davis SN, Shavers C, Costa F, Mosqueda-Garcia R. Role of cortisol in the pathogenesis of deficient counterregulation after antecedent hypoglycemia in normal humans. J Clin Invest 98: 680–691, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Davis SN, Tate D. Effects of morning hypoglycemia on neuroendocrine and metabolic responses to subsequent afternoon hypoglycemia in normal man. J Clin Endocrinol Metab 86: 2043–2050, 2001. [DOI] [PubMed] [Google Scholar]
- 15. De Galan BE, Tack CJ, Willemsen JJ, Sweep CG, Smits P, Lenders JW. Plasma metanephrine levels are decreased in type 1 diabetic patients with a severely impaired epinephrine response to hypoglycemia, indicating reduced adrenomedullary stores of epinephrine. J Clin Endocrinol Metab 89: 2057–2061, 2004. [DOI] [PubMed] [Google Scholar]
- 16. DeCristofaro JD, LaGamma EF. Neonatal stress: effects of hypoglycemia and hypoxia on adrenal tyrosine hydroxylase gene expression. Pediatr Res 36: 719–723, 1994. [DOI] [PubMed] [Google Scholar]
- 17. Etter JF, Lukas RJ, Benowitz NL, West R, Dresler CM. Cytisine for smoking cessation: a research agenda. Drug Alcohol Depend 92: 3–8, 2008. [DOI] [PubMed] [Google Scholar]
- 18. Goldberg PA, Weiss R, McCrimmon RJ, Hintz EV, Dziura JD, Sherwin RS. Antecedent hypercortisolemia is not primarily responsible for generating hypoglycemia-associated autonomic failure. Diabetes 55: 1121–1126, 2006. [DOI] [PubMed] [Google Scholar]
- 19. Goldstein DS, Kopin IJ. Adrenomedullary, adrenocortical, and sympathoneural responses to stressors: a meta-analysis. Endocr Regul 42: 111–119, 2008. [PMC free article] [PubMed] [Google Scholar]
- 20. Heller SR, Cryer PE. Reduced neuroendocrine and symptomatic responses to subsequent hypoglycemia after 1 episode of hypoglycemia in nondiabetic humans. Diabetes 40: 223–226, 1991. [DOI] [PubMed] [Google Scholar]
- 21. Herlein JA, Morgan DA, Phillips BG, Haynes WG, Sivitz WI. Antecedent hypoglycemia, catecholamine depletion, and subsequent sympathetic neural responses. Endocrinology 147: 2781–2788, 2006. [DOI] [PubMed] [Google Scholar]
- 22. Inouye KE, Chan O, Yue JT, Matthews SG, Vranic M. Effects of diabetes and recurrent hypoglycemia on the regulation of the sympathoadrenal system and hypothalamo-pituitary-adrenal axis. Am J Physiol Endocrinol Metab 288: E422–E429, 2005. [DOI] [PubMed] [Google Scholar]
- 23. Inouye KE, Yue JT, Chan O, Kim T, Akirav EM, Park E, Riddell MC, Burdett E, Matthews SG, Vranic M. Effects of insulin treatment without and with recurrent hypoglycemia on hypoglycemic counterregulation and adrenal catecholamine-synthesizing enzymes in diabetic rats. Endocrinology 147: 1860–1870, 2006. [DOI] [PubMed] [Google Scholar]
- 24. Kvetnansky R, Sabban EL, Palkovits M. Catecholaminergic systems in stress: structural and molecular genetic approaches. Physiol Rev 89: 535–606, 2009. [DOI] [PubMed] [Google Scholar]
- 25. Lenartowski R, Goc A. Epigenetic, transcriptional and posttranscriptional regulation of the tyrosine hydroxylase gene. Int J Dev Neurosci 29: 873–883, 2011. [DOI] [PubMed] [Google Scholar]
- 26. LeSage MG, Shelley D, Ross JT, Carroll FI, Corrigall WA. Effects of the nicotinic receptor partial agonists varenicline and cytisine on the discriminative stimulus effects of nicotine in rats. Pharmacol Biochem Behav 91: 461–467, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lim DY, Jang SJ, Kim KC. Influence of cytisine on catecholamine release in isolated perfused rat adrenal glands. Arch Pharm Res 25: 932–939, 2002. [DOI] [PubMed] [Google Scholar]
- 28. McDermott JC, Hutber A, Tan MH, Bonen A. The use of a cell-free perfusate in the perfused rat hindquarter: methodological concerns. Can J Physiol Pharmacol 67: 1450–1454, 1989. [DOI] [PubMed] [Google Scholar]
- 29. Mineur YS, Eibl C, Young G, Kochevar C, Papke RL, Gundisch D, Picciotto MR. Cytisine-based nicotinic partial agonists as novel antidepressant compounds. J Pharmacol Exp Ther 329: 377–386, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mineur YS, Picciotto MR. Nicotine receptors and depression: revisiting and revising the cholinergic hypothesis. Trends Pharmacol Sci 31: 580–586, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nankova BB, Chua J, Mishra R, Kobasiuk CD, La Gamma EF. Nicotinic induction of preproenkephalin and tyrosine hydroxylase gene expression in butyrate-differentiated rat PC12 cells: a model for adaptation to gut-derived environmental signals. Pediatr Res 53: 113–118, 2003. [DOI] [PubMed] [Google Scholar]
- 32. Papke RL, Trocmé-Thibierge C, Guendisch D, Al Rubaiy SA, Bloom SA. Electrophysiological perspectives on the therapeutic use of nicotinic acetylcholine receptor partial agonists. J Pharmacol Exp Ther 337: 367–379, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Parab S, Nankova BB, La Gamma EF. Differential regulation of the tyrosine hydroxylase and enkephalin neuropeptide transmitter genes in rat PC12 cells by short chain fatty acids: concentration-dependent effects on transcription and RNA stability. Brain Res 1132: 42–50, 2007. [DOI] [PubMed] [Google Scholar]
- 34. Quick MW, Lester RA. Desensitization of neuronal nicotinic receptors. J Neurobiol 53: 457–478, 2002. [DOI] [PubMed] [Google Scholar]
- 35. Rollema H, Chambers LK, Coe JW, Glowa J, Hurst RS, Lebel LA, Lu Y, Mansbach RS, Mather RJ, Rovetti CC, Sands SB, Schaeffer E, Schulz DW, Tingley FD, 3rd, Williams KE. Pharmacological profile of the alpha4beta2 nicotinic acetylcholine receptor partial agonist varenicline, an effective smoking cessation aid. Neuropharmacology 52: 985–994, 2007. [DOI] [PubMed] [Google Scholar]
- 36. Rollema H, Shrikhande A, Ward KM, Tingley FD, 3rd, Coe JW, O'Neill BT, Tseng E, Wang EQ, Mather RJ, Hurst RS, Williams KE, de Vries M, Cremers T, Bertrand S, Bertrand D. Pre-clinical properties of the alpha4beta2 nicotinic acetylcholine receptor partial agonists varenicline, cytisine and dianicline translate to clinical efficacy for nicotine dependence. Br J Pharmacol 160: 334–345, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sajja RK, Rahman S. Lobeline and cytisine reduce voluntary ethanol drinking behavior in male C57BL/6J mice. Prog Neuropsychopharmacol Biol Psychiatry 35: 257–264, 2011. [DOI] [PubMed] [Google Scholar]
- 38. Sandoval DA, Ping L, Neill AR, Morrey S, Davis SN. Cortisol acts through central mechanisms to blunt counterregulatory responses to hypoglycemia in conscious rats. Diabetes 52: 2198–2204, 2003. [DOI] [PubMed] [Google Scholar]
- 39. Shum K, Inouye K, Chan O, Mathoo J, Bilinski D, Matthews SG, Vranic M. Effects of antecedent hypoglycemia, hyperinsulinemia, and excess corticosterone on hypoglycemic counterregulation. Am J Physiol Endocrinol Metab 281: E455–E465, 2001. [DOI] [PubMed] [Google Scholar]
- 40. Sivitz WI, Herlein JA, Morgan DA, Fink BD, Phillips BG, Haynes WG. Effect of acute and antecedent hypoglycemia on sympathetic neural activity and catecholamine responsiveness in normal rats. Diabetes 50: 1119–1125, 2001. [DOI] [PubMed] [Google Scholar]
- 41. Stroth N, Kuri BA, Mustafa T, Chan SA, Smith CB, Eiden LE. PACAP controls adrenomedullary catecholamine secretion and expression of catecholamine biosynthetic enzymes at high splanchnic nerve firing rates characteristic of stress transduction in male mice. Endocrinology 154: 330–339, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Taborsky GJ, Jr, Mundinger TO. Minireview: The role of the autonomic nervous system in mediating the glucagon response to hypoglycemia. Endocrinology 153: 1055–1062, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tai MM. A mathematical model for the determination of total area under glucose tolerance and other metabolic curves. Diabetes Care 17: 152–154, 1994. [DOI] [PubMed] [Google Scholar]
- 44. Turcanu DS, Kirtok N, Eibl C, Guendisch D, LaGamma EF, Nankova BB. Nicotinic receptor partial agonists alter catecholamine homeostasis and response to nicotine in PC12 cells. Neurosci Lett 516: 212–216, 2012. [DOI] [PubMed] [Google Scholar]
- 45. Tzankova V, Danchev N. Cytisine - from ethnomedical use to the development as a natural alternative for smoking cessation. Pharm Biotechnol 21: 151–160, 2007. [Google Scholar]
- 46. Vietor I, Rusnak M, Viskupic E, Blazicek P, Sabban EL, Kvetnansky R. Glucoprivation by insulin leads to trans-synaptic increase in rat adrenal tyrosine hydroxylase mRNA levels. Eur J Pharmacol 313: 119–127, 1996. [DOI] [PubMed] [Google Scholar]
- 47. Vollmer RR, Balcita-Pedicino JJ, Debnam AJ, Edwards DJ. Adrenal medullary catecholamine secretion patterns in rats evoked by reflex and direct neural stimulation. Clin Exp Hypertens 22: 705–715, 2000. [DOI] [PubMed] [Google Scholar]
- 48. Wakade AR. Noncholinergic transmitter(s) maintains secretion of catecholamines from rat adrenal medulla for several hours of continuous stimulation of splanchnic neurons. J Neurochem 50: 1302–1308, 1988. [DOI] [PubMed] [Google Scholar]
- 49. Yokotani K, Okada S, Nakamura K. Characterization of functional nicotinic acetylcholine receptors involved in catecholamine release from the isolated rat adrenal gland. Eur J Pharmacol 446: 83–87, 2002. [DOI] [PubMed] [Google Scholar]