

Abstract
Although the cure rate for cutaneous squamous cell carcinoma is high, the diverse spectrum of squamous cell carcinoma has made it difficult for early diagnosis, particularly the aggressive tumors that are highly associated with mortality. Therefore, molecular markers are needed as an adjunct to current staging methods for diagnosing high-risk lesions, and stratifying those patients with aggressive tumors. To identify such biomarkers, we have examined a comprehensive set of 200 histologically defined squamous cell carcinoma and normal skin samples by using a combination of microarray, QRT-PCR and immunohistochemistry analyses. A characteristic and distinguishable profile including matrix metalloproteinase (MMP) as well as other degradome components was differentially expressed in squamous cell carcinoma compared with normal skin samples. The expression levels of some of these genes including matrix metallopeptidase 1 (MMP1), matrix metallopeptidase 10 (MMP10), parathyroid hormone-like hormone (PTHLH), cyclin-dependent kinase inhibitor 2A (CDKN2A), A disintegrin and metalloproteinase with thrombospondin motifs 1 (ADAMTS1), FBJ osteosarcoma oncogene (FOS), interleukin 6 (IL6) and reversion-inducing-cysteine-rich protein with kazal motifs (RECK) were significantly differentially expressed (P≤0.02) in squamous cell carcinoma compared with normal skin. Furthermore, based on receiver operating characteristic analyses, the mRNA and protein levels of MMP1 are significantly higher in aggressive tumors compared with non-aggressive tumors. Given that MMPs represent the most prominent family of proteinases associated with tumorigenesis, we believe that they may have an important role in modulating the tumor microenvironment of squamous cell carcinoma.
Keywords: cutaneous squamous cell carcinoma, degradome, gene expression
Non-melanoma skin cancer is the most common form of human malignancy, especially among populations with lighter skin types, affecting over two million people annually in United States.1 There are over 80 different types of non-melanoma skin cancers with a wide variation in behavior and prognosis. The incidence is estimated to be increasing overall since the 1960s at a rate of 3.8% per year.2 Although the burden of non-melanoma skin cancer measured in terms of mortality and morbidity is unknown, the overall costs of non-melanoma skin cancer are thought to be quite substantial owing to its high prevalence. In the US Medicare population, it is considered a major health-care problem and among the five most costly cancers to treat.3,4 While there are many types of non-melanoma skin cancers, the most commonly seen are basal cell carcinoma and squamous cell carcinoma. Although the incidence of basal cell carcinoma is far greater, squamous cell carcinoma accounts for the majority of non-melanoma skin cancer deaths and 20% of all skin cancer-related deaths.5,6 Basal cell carcinoma very rarely metastasizes to distant sites or results in mortality. In contrast, squamous cell carcinoma found in certain areas such as in scars, sinus tracts and lip may have a > 30% risk of metastasis upon initial presentation.7,8 Moreover, squamous cell carcinoma is also associated with the development of other malignancies.9,10 For example, following squamous cell carcinoma, there appears to be an increased risk of digestive tract malignancies (relative risk 1.6, confidence interval 1.1–2.4).10 Several epidemiological studies have also evaluated the risk of squamous cell carcinoma in the general population. The causes of squamous cell carcinoma are multifactorial, including both environmental and host factors. The known environmental risk factors for squamous cell carcinoma include sun exposure (ultraviolet light exposure), ionizing radiation, cigarette smoking and certain chemical exposures such as arsenic. Induced or acquired immunosuppression as seen after solid organ transplantation11 or in patients diagnosed and treated for leukemia or lymphoma are recognized as significant risk factors for the development of squamous cell carcinoma. The incidence of squamous cell carcinoma increases with decreasing latitude, further indicating that there is an increased risk associated with more intense sun exposure.12 The known host risk factors include skin type, genetic susceptibilities, human papilloma virus infection and immunosuppression.13
The cure rate is >90% with the vast majority of tumors presenting as stage I and II. The large number of routine low-risk lesions is cured with simple excision, whereas the high-risk lesions are the most difficult subset to identify at diagnosis and to cure with simple single modality therapy. Differentiation between the high- risk and low-risk subtypes continues to be very difficult because the different skin lesions have phenotypically and microscopically similar characteristics. Thus, risk stratifications of the various subtypes required a comprehensive clinicopathologic classification system to group variants of squamous cell carcinoma based on their biologic aggressiveness or indolence. Recent changes suggested by the American Joint Committee on Cancer staging focus on incorporating clinical parameters that portend a worse prognosis to identify and stage appropriately the subset of squamous cell carcinoma that are at risk to progress to metastatic disease.14 However, these changes are a first approximation of determining those phenotypes with the worse prognosis and as such they may not capture many of the variants of squamous cell carcinoma associated with the worst biology. In addition, they are underpowered numerically for an outcome analysis given the broad spectrum of squamous cell carcinoma subtypes. With the advent of microarray techniques and other high-throughput screening, gene expression profiling can help in distinguishing the variants of different subgroups among tumors with similar morphology and may be further useful for outcome analysis and risk assessment.
The repertoire of proteases that cells and tissues coordinately regulate in order to modulate their local environment is the ‘degradome,’ which in humans is represented by several proteases including matrix metalloproteinases (MMPs), ADM (a disintegrin and metalloproteinase) and ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs).15 There is a well-established association between degradome components and cancer development, and the proteinases of every class have been linked to malignancy and invasion of tumor cells.16 Besides the invasion and metastatic activities, they also have crucial roles in early tumorigenic events such as angiogenesis, apoptosis, cell dissociation and cell migration.17 Although the signaling pathways that lead to induction of expression of MMPs are still incompletely understood, the expression patterns of some of these genes may be useful for diagnostic and therapeutic applications. Therefore, in the present study we have utilized gene and protein expression profiling for molecular markers that can be useful in the diagnostic applications, particularly to identify squamous cell carcinoma with highly aggressive behavior and metastatic potential. Identification of such tumors with the potential for aggressive behavior will set the stage for the development of optimal therapeutic regimens.
Materials and methods
Tumor Specimens
Altogether 200 skin samples were used in this study. With Institutional Review Board approval, a total of 120 skin tissues were collected from patients who underwent surgery at the Johns Hopkins Hospital (Baltimore, MD, USA) between 2004 and 2010. The first set include 69 skin specimens (32 squamous cell carcinomas and 37 normal skin samples) that were snap frozen in liquid nitrogen and stored at − 80 °C soon after surgical excision until use. A second set consisted of 51 formalin–fixed, paraffin-embedded (paraffin-preserved) tissue sections (8 non-aggressive tumors, 38 aggressive tumors and 5 normal skin samples) that were also collected after histological diagnosis at the Johns Hopkins Hospital. In addition, tissue microarrays that contained 80 skin samples (39 non-aggressive tumors, 37 aggressive tumors and 4 normal skin samples) were also procured commercially (US Biomax, Rockville, MD, USA). A number of features suggested in American Joint Committee on Cancer staging classification are associated with poor prognosis for recurrence and metastasis. Factors such as anatomic site, tumor diameter, poor differentiation, perineural invasion and a depth of invasion have been recognized as those features associated with aggressive tumor behavior.18 These criteria were identified in samples labeled as ‘aggressive’ in this study.
RNA Isolation and Quality Control
Total RNA from snap frozen tissues were isolated using Trizol reagent (Life Technologies, Grand Island, NY, USA) and purified with the RNeasy mini kit (Qiagen, Valencia, CA, USA) as per the manufacturer’s instructions. Total RNA from paraffin-preserved samples was extracted using RNeasy FFPE kit (Qiagen). The paraffin-preserved samples were briefly treated (~3 min at 56 °C) with deparaffinization solution, and subjected to a proteinase K digestion at 56 °C for 15 min to release RNA from covalently linked proteins. Finally, total RNA was purified through RNeasy MinElute® Spin Columns as per instruction. The RNA integrity was evaluated using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) and purity/concentration was determined using a Nanodrop 8000 spectrophotometer (NanoDrop Products, Wilmington, DE, USA). The RNA samples with RNA integrity number ≥7 and 260/280 ratio ≥1.9 were selected for microarray analysis.
Target Preparation and Microarray Hybridization
Microarray studies were performed on 12 RNA samples (fresh-frozen) with the Affymetrix HGU133 2.0 Plus GeneChip using standard protocols as recommended by the manufacturer (Affymetrix, Santa Clara, CA, USA). Briefly, 3.0 µg of total RNA was used to generate double-stranded cDNA using an oligo-dT primer containing the T7 RNA polymerase promoter site and the One-Cycle Target Labeling Kit (Affymetrix). cDNA was purified via column purification using the GeneChip Sample Cleanup Module, and biotinylated cRNA was synthesized by in vitro transcription using the geneChip IVT Labeling Kit. Biotin-labeled cRNA was purified with the GeneChip Sample Cleanup Module and the absorbance measured at 260 nm to determine yield. Twenty micrograms of the labeled cRNA was fragmented and quality was assessed using the Agilent 2100 Bioanalyzer and the RNA 6000 Nano Chip kit (Agilent Technologies). Labeled fragmented cRNA was hybridized to the Affymetrix GeneChip HGU 133 2.0 array for 16 h at 45°C using the recommended protocol. Washing and staining were performed on the Affymetrix 450 fluidics station using the antibody amplification protocol (Fluidics script: EukGE-WS2v5). Each GeneChip was scanned using the Affymetrix GeneChip Scanner 3000.
Bioinformatics and Statistical Analysis
Affymetrix chip image files were processed using RMA 1.0.5, the Robust Multichip Average program, using background adjustment, quantile normalization and median polishing.19 Significance analysis of microarrays was used to determine which probe sets changed significantly using two class unpaired statistics and a false discovery rate of <1% combined with a minimum fold change of 5.20 Lists of significant probe sets were annotated and analyzed in MetaMiner.21 Analysis included enrichment analysis in multiple ontologies, interactome analysis, pathway analysis and network analysis. Interactome analysis calculates the number of interactions within a data set and compares that to the whole database to determine if functional class, such as transcription factors or secreted proteins, is over- or under-represented. Network and pathway analysis examines connectivity between genes in the list to determine what metabolic or signaling pathways may be involved. Cluster analysis, heatmaps and dendrograms were constructed using Cluster, Treeview and Maple Tree analysis and visualization programs from the Eisen laboratory.22
To evaluate the significance of gene expression, Wilcoxon’s rank-sum test, χ2 or Fisher’s extract test were performed separately for testing mRNA (QRT-PCR) and protein (immunohistochemistry) expression in different tissue types (ex. normal, aggressive and non-aggressive tumors). In addition, univariate analyses via logistic regression models were also performed for exploring the association between tumor types and gene expression patterns. Receiver operating characteristics (ROC) curve was plotted at each gene, and the performances of discrimination between groups (ex. tumors vs normal, and aggressive vs non-aggressive tumors) were assessed via area under the curves. Sensitivity, specificity, positive predictive value and negative predictive value were also documented.
QRT-PCR
cDNA was first synthesized with 500 ng of total RNA in a 33 µl reverse transcription reaction mixture and then real-time PCR assays were performed on iQTM5 real-time PCR detection system (Bio-Rad Laboratories, Hercules, CA, USA). Briefly, 1 µl of cDNA was used for PCR in a 25 µl reaction mixture that contained an optimal concentration of primers (150–250 nM) and SYBR-Green Supermix according to the manufacturer’s recommendations. The thermal profile for PCR consisted of Taq polymerase activation at 95°C for 3 min, followed by 40 cycles of PCR at 95 °C for 20 s (denaturation), 55.5 °C for 30 s (annealing) and 72 °C for 60 s (extension). An average threshold cycle (Ct) from duplicate assays was used to determine the GAPDH-normalized gene expression as formulated below (relative quantification method). Results were expressed as the median of three to four independent measurements.
Relative expression levels normalized to CAPDH = 2−(gene of interest Ct − GAPDH · Ct) × 100
Immunohistochemistry
Immunohistochemical staining was performed on 4–5 µm sections of paraffin-preserved skin tissues that included 80 specimens on tissue arrays and 51 non-arrayed skin specimens. In total, these samples included 47 non-aggressive, 75 aggressive and 9 normal skins. Briefly, sections were deparaffinized in xylene and rehydrated through a series of alcohol gradients. Antigen retrieval was achieved by heating in citrate buffer at pH 6.0 for 10 min and cooled at room temperature for 30 min. Endogenous peroxidase activity was quenched in 3% hydrogen peroxide and nonspecific binding of secondary antibody blocked by incubation with normal horse serum. Individual sections were incubated with primary antibodies [rabbit polyclonal antibodies: anti-MMP10 IgG (1:100), anti-ADAMTS1 IgG (1:100); and rabbit monoclonal antibodies: anti-MMP1 IgG (1:100) from Abcam, Cambridge, MA, USA] overnight at 4°C. Conditions without primary antibody were used as technical negative controls. A streptavidin–biotin peroxidase detection system was used in accordance with the manufacturer’s instructions and developed using 3, 3′-diaminobenzidine (Vector Laboratories, Burlingame, CA, USA). Sections were counter-stained with 1% hematoxylin.
Semiquantitative immunostaining detection was used to determine the protein levels as described previously.23 Protein expression documented in immunohistochemistry was blindly scored separately by two investigators (NBP and WHW) using manual microscopic examination. Immunostaining was scored using a four-point scale system according to the intensity of staining and the percentage of positive cells: (i) high expression (intense immunostaining with >60% of tumor cells); (ii) moderate expression (intense immunostaining of 30–60% of tumor cells); (iii) low expression (intense immunostaining in <30% of cells); and (iv) negative (no expression). Scoring results were countervalidated by our study pathologist (WHW) who performed independently and in a blind manner. The intraobserver reproducibility was >95%. In statistical analysis, the scores were dichotomized as ‘weak’ (negative-to-low) and ‘strong’ (moderate-to-high) expression levels.
Results
In an effort to identify molecular signatures/markers, we have analyzed over 200 tissue samples that were surgically resected from patients who underwent treatments for skin cancer. The tissue samples including fresh-frozen and paraffin-preserved tumors were analyzed for their mRNA and protein expressions by using a combination of microarray, QRT-PCR and immunohistochemical methods.
Microarray Analysis
Microarray analysis was used to examine 12 fresh–frozen tissue samples representing 6 squamous cell carcinomas and matching normal skin. All tumors had a pathologic diagnosis of squamous cell carcinoma and the samples had both a consistent size (>1.0 cm in diameter) as well as being relatively homogeneous. By using Affymetrix arrays (GeneChip Human Genome U133 Plus 2.0), the transcriptional analysis from these samples identified 164 genes that were differentially expressed with ≥ 5-fold differences between squamous cell carcinoma and normal skin. Hierarchical cluster analysis using these differentially expressed genes further revealed distinct genetic signatures in squamous cell carcinomas compared with normal skin (Figure 1a). Of the 164 genes, 22 were upregulated (Table 1), whereas 142 were downregulated (Table 2) in squamous cell carcinomas compared with the matching normal skin. Furthermore, exploratory analysis using tree-view and maple tree tools suggest a network of genes that are highly associated in important and distinct biochemical pathways (Figure 1b).
Figure 1.

Microarray analysis of cutaneous squamous cell carcinoma. The gene expression profiles were compared between six tumors and matching normal skin samples. (a) Hierarchical cluster analysis revealing dendrogram and heatmap with distinct gene expression profiles (164 genes with ≥ 5-fold difference) in normal skin and squamous cell carcinoma. Downregulated and upregulated genes are shown in green and red blocks, respectively. (b) The network of the interaction or association between differentially expressed genes. The nodes on the network represent genes/proteins with their biological function ( , transcription factor;
, transcription factor;  , generic enzyme;
, generic enzyme;  , generic phospholipase;
, generic phospholipase;  , protein kinase;
, protein kinase;  , generic protease;
, generic protease;  , metalloprotease;
, metalloprotease;  , receptor ligand;
, receptor ligand;  , generic receptor;
, generic receptor;  , receptor with enzyme activity;
, receptor with enzyme activity;  , GPCR;
, GPCR;  , proteoglycan;
, proteoglycan;  , generic-binding protein;
, generic-binding protein;  , generic channel). Red lines are inhibitory interactions, green lines are activating and gray indicate an unspecified effect in the source literature. Red and blue circles indicate genes upregulated or downregulated, respectively, in tumors compared with normal skin. Note MMP1, MMP10 (stromelysin-2) and ADAM-TS1 are shown in ‘dashed circles’ to indicate the expression pattern and their association with other differentially expressed genes.
, generic channel). Red lines are inhibitory interactions, green lines are activating and gray indicate an unspecified effect in the source literature. Red and blue circles indicate genes upregulated or downregulated, respectively, in tumors compared with normal skin. Note MMP1, MMP10 (stromelysin-2) and ADAM-TS1 are shown in ‘dashed circles’ to indicate the expression pattern and their association with other differentially expressed genes.
Table 1.
Genes significantly overexpressed in cutaneous squamous cell carcinoma
| Gene ID | Name | Gene name | Fold change |
|---|---|---|---|
| 204475_at | MMP1a | NM_002421 | 20.68 |
| 1553973_a_at | SPINK6 | BC032003 | 14.93 |
| 239430_at | IGFL1 | AA195677 | 12.10 |
| 203691_at | PI3 | NM_002638 | 9.18 |
| 221805_at | NEFL | AL537457 | 8.58 |
| 224328_s_at | LCE3D | AB048288 | 8.37 |
| 207356_at | DEFB4P | NM_004942 | 7.22 |
| 204750_s_at | DSC2 | BF196457 | 6.71 |
| 205680_at | MMP10a | NM_002425 | 6.55 |
| 209800_at | KRT16 | AF061812 | 6.15 |
| 205916_at | S100A7 | NM_002963 | 6.08 |
| 211756_at | PTHLHa | BC005961 | 5.98 |
| 203413_at | NELL2 | NM_006159 | 5.89 |
| 227736_at | C10orf99 | AA553959 | 5.89 |
| 207039_at | CDKN2Aa | NM_000077 | 5.74 |
| 220664_at | SPRR2C | NM_006518 | 5.71 |
| 209720_s_at | SERPINB4 | BC005224 | 5.7 |
| 218755_at | KIF20A | NM_005733 | 5.68 |
| 228531_at | SAMD9 | AA741307 | 5.66 |
| 205863_at | S100A12 | NM_005621 | 5.59 |
| 209719_x_at | SERPINB3 | U19556 | 5.54 |
| 208539_x_at | SPRR2B | NM_006945 | 5.20 |
Genes also validated in separate cohorts.
Table 2.
Genes significantly underexpressed in cutaneous squamous cell carcinoma
| Gene ID | Name | Gene name | Fold change |
|---|---|---|---|
| 209613_s_at | ADH1B | M21692 | −44.69 |
| 209687_at | CXCL12 | U19495 | −37.40 |
| 227099_s_at | LOC387763 | AW276078 | −25.26 |
| 206157_at | PTX3 | NM_002852 | −22.89 |
| 223395_at | ABI3BP | AB056106 | −22.63 |
| 216248_s_at | NR4A2 | S77154 | −21.0 |
| 204719_at | ABCA8 | NM_007168 | −17.38 |
| 205382_s_at | CFDa | NM_001928 | −16.21 |
| 205030_at | FABP7 | NM_001446 | −16.10 |
| 205407_at | RECKa | NM_021111 | −15.65 |
| 202291_s_at | MGP | NM_000900 | −15.47 |
| 205200_at | CLEC3B | NM_003278 | −14.83 |
| 201525_at | APOD | NM_001647 | −13.83 |
| 229461_x_at | NEGR1 | AI123532 | −13.64 |
| 228885_at | MAMDC2 | AI862120 | −13.60 |
| 202920_at | ANK2 | BF726212 | −13.33 |
| 205399_at | DCLK1 | NM_004734 | −13.21 |
| 219908_at | DKK2 | NM_014421 | −12.45 |
| 225207_at | PDK4 | AV707102 | −12.27 |
| 213800_at | CFH | X04697 | −12.17 |
| 209101_at | CTGF | M92934 | −11.87 |
| 213764_s_at | MFAP5 | AW665892 | −11.61 |
| 210299_s_at | FHL1 | AF063002 | −11.36 |
| 201289_at | CYR61 | NM_001554 | −11.40 |
| 205207_at | IL6a | NM_000600 | −11.22 |
| 201110_s_at | THBS1a | NM_003246 | −11.21 |
| 207977_s_at | DPT | NM_001937 | −10.84 |
| 225242_s_at | CCDC80 | AW303375 | −10.66 |
| 203980_at | FABP4 | NM_001442 | −10.59 |
| 220356_at | CORIN | NM_006587 | −10.39 |
| 238018_at | FAM150B | BF449053 | −10.38 |
| 209763_at | CHRDL1 | AL049176 | −10.36 |
| 218087_s_at | SORBS1 | NM_015385 | −10.27 |
| 202995_s_at | FBLN1 | NM_006486 | −10.16 |
| 223122_s_at | SFRP2 | AF311912 | −9.55 |
| 230746_s_at | STC1 | AW003173 | −9.14 |
| 217546_at | MTM1 | B06655 | −9.09 |
| 202768_at | FOSB | NM_006732 | −9.06 |
| 205792_at | WIPS2 | NM_003881 | −9.04 |
| 225571_at | LIFR | AA701657 | −8.87 |
| 208131_s_at | PTGIS | NM_000961 | −8.86 |
| 219295_s_at | PCOLCE2 | NM_013363 | −8.64 |
| 205422_s_at | ITGBL1 | NM_004791 | −8.62 |
| 202499_s_at | SLC2A3 | NM_006931 | −8.62 |
| 227662_at | SYNPO2 | AA541622 | −8.56 |
| 202498_s_at | SCL2A3 | BE550486 | −8.52 |
| 212713_at | MFAP4 | B72286 | −8.45 |
| 212865_s_at | COL14A1 | BF449063 | −8.43 |
| 202437_s_at | CYP1B1 | NM_00104 | −8.32 |
| 209774_x_at | CXCL2 | M57731 | −8.24 |
| 239183_at | ANGPTL1 | W67461 | −8.16 |
| 204712_at | WIF1 | NM_007191 | −8.07 |
| 206101_at | ECM2 | NM_001393 | −7.99 |
| 203649_s_at | PLA2G2A | NM_000300 | −7.98 |
| 204273_at | EDNRB | NM_000115 | −7.94 |
| 202672_s_at | ATF3 | NM_001674 | −7.92 |
| 205338_s_at | DCT | NM_001922 | −7.84 |
| 223235_s_at | SMOC2 | AB014737 | −7.83 |
| 227697_at | SOCS3a | AI244908 | −7.82 |
| 222043_at | CLU | AI982754 | −7.81 |
| 1554741_s_at | FGF7 | AF523265 | −7.78 |
| 210517_s_at | AKAP12 | AB003476 | −7.69 |
| 211959_at | IGFBP5 | AW007532 | −7.69 |
| 219304_s_at | PDGFD | NM_025208 | −7.67 |
| 204955_at | SRPX | NM_006307 | −7.57 |
| 202237_at | NNMT | NM_006169 | −7.47 |
| 213397_x_at | RNASE4 | AI761728 | −7.46 |
| 212230_at | PPAP2B | AV725664 | −7.38 |
| 202149_at | NEDD9 | AL136139 | −7.36 |
| 230560_at | STXBP6 | N21096 | −7.27 |
| 225817_at | CGNL1 | AB051536 | −7.24 |
| 209189_at | FOSa | BC004490 | −7.23 |
| 209656_s_at | TMEM47 | AL136550 | −7.20 |
| 206932_at | CH25H | NM_003956 | −7.17 |
| 222453_at | CYBRD1 | AL136693 | −7.12 |
| 206432_at | HAS2 | NM_005328 | −7.12 |
| 204036_at | EDG2 | AW269335 | −7.11 |
| 209210_s_at | FERMT2 | Z24725 | −7.11 |
| 212915_at | PDZRN3 | AL569804 | −6.93 |
| 209335_at | DCN | AI281593 | −6.79 |
| 217767_at | C3 | NM_000064 | −6.78 |
| 222486_s_at | ADAMTS1a | AF060152 | −6.76 |
| 229831_at | CNTN3 | BE221817 | −6.75 |
| 203824_at | TSPAN8 | NM_004616 | −6.71 |
| 205694_at | TYRP1 | NM_000550 | −6.69 |
| 210198_s_at | PLP1 | BC002665 | −6.68 |
| 203813_s_at | SLIT3 | NM_003062 | −6.64 |
| 209094_at | DDAH1 | AL078459 | −6.50 |
| 205083_at | AOX1 | NM_001159 | −6.49 |
| 218541_s_at | C8orf4 | NM_020130 | −6.49 |
| 210139_s_at | PMP22 | L03203 | −6.47 |
| 200795_at | SPARCL1 | NM_004684 | −6.36 |
| 227399_at | VGLL3 | AI754423 | −6.30 |
| 204897_at | PTGER4 | AA897516 | −6.29 |
| 203131_at | PDGFRA | NM_006206 | −6.21 |
| 226066_at | MITF | ALII 76 5 3 | −6.20 |
| 205907_s_at | OMD | AI765819 | −6.13 |
| 221031_s_at | APOLD1 | NM_030817 | −5.99 |
| 228335_at | CLDN11 | AW264204 | −5.96 |
| 211748_x_at | PTGDS | BC005939 | −5.95 |
| 238447_at | RBMS3 | AA428240 | −5.89 |
| 209074_s_at | FAM107A | AL050264 | −5.88 |
| 201627_s_at | INSIG1 | NM_005542 | −5.86 |
| 203001_s_at | STMN2 | NM_007029 | −5.85 |
| 203305_at | F13A1 | NM_000129 | −5.82 |
| 227058_at | C13orf33 | AW084730 | −5.78 |
| 205547_s_at | TAGLN | NM_003186 | −5.77 |
| 216598_s_at | CCL2 | S69738 | −5.73 |
| 228143_at | HPS3 | AI684991 | −5.71 |
| 227613_at | ZNF331 | AW450874 | −5.65 |
| 232060_at | ROR1 | AK000776 | −5.63 |
| 207955_at | CCL27 | NM_006664 | −5.51 |
| 226625_at | TGFBR3 | AW193698 | −5.49 |
| 222722_at | OGN | AV700059 | −5.47 |
| 208782_at | FSTL1 | BC000055 | −5.47 |
| 213943_at | TWIST1 | X99268 | −5.46 |
| 218723_s_at | C13orf15 | NM_014059 | −5.42 |
| 221796_at | NTRK2 | AA707199 | −5.40 |
| 241412_at | BTC | AI620677 | −5.40 |
| 213258_at | TFPI | BF511231 | −5.38 |
| 203088_at | FBLN5 | NM_006329 | −5.38 |
| 202609_at | EPS8 | NM_004447 | −5.37 |
| 204846_at | CP | NM_000096 | −5.35 |
| 209392_at | ENPP2a | L35594 | −5.34 |
| 202766_s_at | FBN1 | NM_000138 | −5.33 |
| 228728_at | C7orf58 | BF724137 | −5.32 |
| 209047_at | AQP1 | AL518391 | −5.31 |
| 201150_s_at | TIMP3 | NM_000362 | −5.29 |
| 212977_at | CXCR7 | AI817041 | −5.28 |
| 209959_at | NR4A3 | U12767 | −5.26 |
| 223315_at | NTN4 | AF278532 | −5.25 |
| 1555097_a_at | PTGFR | BC035694 | −5.22 |
| 205404_at | HSD11B1 | NM_005525 | −5.21 |
| 227265_at | FGL2 | AW135176 | −5.18 |
| 208335_s_at | DARC | NM_002036 | −5.18 |
| 205419_at | EBI2 | NM_004951 | −5.17 |
| 202158_s_at | CUGBP2 | NM_006561 | −5.16 |
| 203543_s_at | KLF9 | NM_001206 | −5.16 |
| 225673 at | MYADM | BE908995 | −5.16 |
| 205226 at | PDGFRL | NM_006207 | −5.15 |
| 204237 at | GULP1 | NM_016315 | −5.04 |
| 213375_s_at | N4BP2L1 | N80918 | −5.02 |
Genes also validated in separate cohorts.
QRT-PCR
To validate the microarray results, QRT-PCR analysis of 12 genes (matrix metallopeptidase 1 (MMP1), matrix metallopeptidase 10 (MMP10), parathyroid hormone-like hormone (PTHLH), cyclin-dependent kinase inhibitor 2A (CDKN2A), ectonucleotide pyrophosphatase/phosphodiesterase 2 (ENPP2), A disintegrin and metalloproteinase with thrombospondin motifs 1 (ADAMTS1), FBJ osteosarcoma oncogene (FOS), suppressor of cytokine signaling 3 (SOCS3), thrombospondin 1 (THBS1), interleukin 6 (IL6), reversion-inducing-cysteine-rich protein with kazal motifs (RECK) and complement factor D (CFD)) was performed using RNA from a separate set of 27 paraffin-preserved samples that included 22 tumors and 5 normal skins (Figure 2). These genes were chosen based on the ranking order in terms of their relative expression levels and their apparent role in similar biochemical pathways. Of 12 genes tested, the expression levels of eight genes including MMP1, MMP10, PTHLH, CDKN2A, ADAMTS1, FOS, IL6 and RECK were significantly differentially expressed (P ≤ 0.02) in tumors compared with normal skin. However, the expression levels of remaining four genes including ENPP2, SOCS3, THBS1 and CFD were not statistically significant. In concordance with the microarray data, the expression levels of MMP1, MMP10, PTHLH and CDKN2A were found to be significantly high in tumors compared with normal skin. Except for the expression levels of IL6 that were found to be exceptionally high in tumors compared with normal skin, the expression levels of ADAMTS1, FOS and RECK were found to be low in tumors as expected (Figure 2).
Figure 2.

Real-time RT-PCR validation of 12 genes (matrix metallopeptidase 1 (MMP1), matrix metallopeptidase 10 (MMP10), parathyroid hormone-like hormone (PTHLH), cyclin-dependent kinase inhibitor 2A (CDKN2A), ectonucleotide pyrophosphatase/phosphodiesterase 2 (ENPP2), A disintegrin and metalloproteinase with thrombospondin motifs 1 (ADAMTS1), FBJ osteosarcoma oncogene (FOS), suppressor of cytokine signaling 3 (SOCS3), thrombospondin 1 (THBS1), interleukin 6 (IL6), reversion-inducing-cysteine-rich protein with kazal motifs (RECK) and complement factor D (CFD)) using 27 paraffin-preserved skin samples. Relative gene expression levels normalized to GAPDH in 22 squamous cell carcinoma and 5 normal skin tissues were determined using gene-specific primers as described in Materials and methods. Gray lines represent medians. Note: as expected from the microarray analysis, MMP1, MMP10, PTHLH, CDKN2A are overexpressed, and ENPP2, ADAMTS1, FOS, SOCS3, THBS1, IL-6, RECK and CFD are underexpressed in squamous cell carcinoma compared with normal skin tissues.
The QRT-PCR analysis was also performed in an additional set of fresh–frozen tissues including 32 tumors and 37 normal skin samples (Figure 3 and Table 3). However, owing to the limitation in RNA quantity, this analysis was not carried out on all genes, but performed only on three genes: MMP1, MMP10 and ADAMTS1. Similar to QRT-PCR in paraffin-preserved samples, the mRNA levels of both MMP1 and MMP10 were found to be very high in tumors compared with normal skin tissues. As expected, the expression of ADAMTS1 is significantly lower in tumors compared with normal skin tissues (Figure 3a). In addition, the mRNA expression levels were much higher in aggressive tumors compared with non-aggressive tumors (Figure 3b).
Figure 3.

Real-time reverse transcription-polymerase chain reaction (RT-PCR) validation of MMP1, MMP10 and ADAMTS1 using 69 fresh–frozen skin samples. Gene expression levels normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were used to differentiate normal skin vs tumors (a) and non-aggressive vs aggressive tumors (b) as described in Materials and methods. Gray lines represent medians.
Table 3.
Univariate analysis of the mRNA expression in 69 fresh-frozen samples
| Normal (n = 37) vs tumor (n = 32) |
Aggressive (n = 16) vs non-aggressive (n = 16) |
|||||
|---|---|---|---|---|---|---|
| MMP1 | MMP10 | ADAMTS1 | MMP1 | MMP10 | ADAMTS1 | |
| Odds ratio | 1.11 | 2.68 | 1 | 1.01 | 1 | 1 |
| 95% Confidence interval | 1.04–1.18 | 1.23–5.84 | 1.0–1.0 | 1–1.03 | 0.98–1.03 | 1–1.01 |
| P-valuea | 0.002 | 0.013 | 0.022 | 0.034 | 0.11 | 0.422 |
| Area under the curve | 0.94 | 0.9 | 0.74 | 0.72 | 0.67 | 0.58 |
| Sensitivity | 0.94 | 0.84 | 0.74 | 0.82 | 0.75 | 0.44 |
| Specificity | 0.9 | 0.94 | 0.98 | 0.62 | 0.56 | 0.75 |
| Positive predictive value | 0.91 | 0.96 | 0.74 | 0.7 | 0.63 | 0.67 |
| Negative predictive value | 0.93 | 0.76 | 0.76 | 0.77 | 0.69 | 0.55 |
P-value is based on Wilcoxon’s test method.
Immunohistochemistry
Encouraged by the mRNA validation, immunohistochemistry was also performed to further validate the protein expression of MMP1, MMP10 and ADAMTS1 in a total of 131 paraffin-preserved tissue sections (80 arrayed and 51 non-arrayed samples). As controls, 9 normal skin samples were also included in this analysis. After immunostaining, the expression intensity of each of the selected genes was assessed in all samples. The overall expression levels were found to be high in most of the tumors as compared with normal skin.
The semiquantification of protein levels documented by immunohistochemistry was further examined for differentiating aggressive tumors from non-aggressive tumors. Univariate analysis on 122 tumors (47 non-aggressive and 75 aggressive) was performed examining the association between tumor types (aggressive vs non-aggressive) and the relative expression levels (weak vs strong) for each of these three genes (Table 4). Although the expression levels of all three genes showed some association with tumor types, only MMP1 was found to be statistically significantly different (P ≤ 0.001; area under the curve ≥0.66) and was highly expressed in aggressive tumors compared with non-aggressive tumors. Representative immunostaining of MMP1 in aggressive and non-aggressive tumors is shown in Figure 4.
Table 4.
Univariate analysis of protein expression in 122 paraffin-preserved samples
| Aggressive (n = 75) vs non-aggressive (n = 47) |
|||
|---|---|---|---|
| MMP1 | MMP10 | ADAMST1 | |
| Odds ratio | 5.47 | 1.62 | 0.88 |
| 95% Confidence interval | 0.73–2.68 | −0.27 to 1.22 | −0.88 to 0.63 |
| P-valuea | < 0.001 | 0.21 | 0.74 |
| Area under the curve | 0.66 | 0.59 | 0.52 |
| Sensitivity | 0.45 | 0.51 | 0.39 |
| Specificity | 0.87 | 0.62 | 0.64 |
| Positive predictive value | 0.85 | 0.68 | 0.63 |
| Negative predictive value | 0.5 | 0.44 | 0.4 |
P-value is based on Wilcoxon’s test method.
Figure 4.
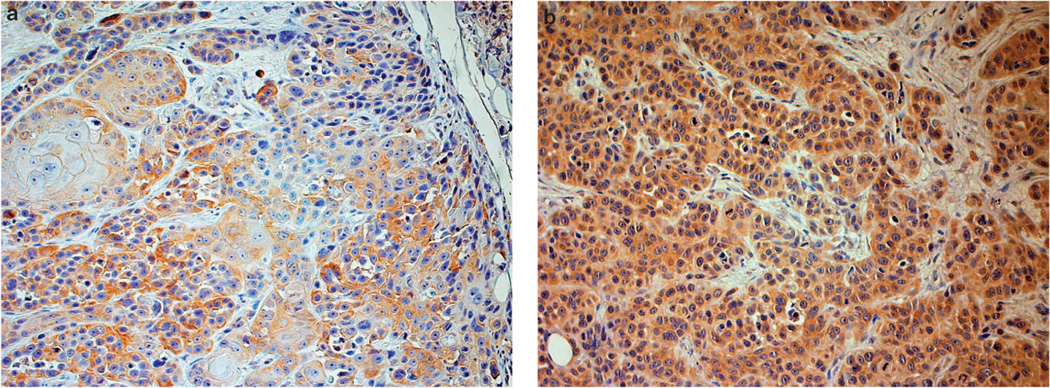
Immunohistochemistry of matrix metallopeptidase 1 (MMP1) expression in squamous cell carcinoma. High protein expression of MMP1 is detected in aggressive cancer compared with non-aggressive tumors. Note higher proportion of cells stained in aggressive tumor (b) compared with non-aggressive tumors (a); magnification × 200.
ROC Analyses
To determine the relationship between the expression patterns that associated with different tissue types, ROC curves were plotted for each gene, and ‘performance of discrimination’ was assessed via area under the curve for MMP1, MMP10 and ADAMST1. The mRNA expression levels in fresh–frozen samples (QRT-PCR) were tested for the performance of discrimination between tumors vs normal skin (Figure 5a) and aggressive vs non-aggressive tumors (Figure 5b). As there were only nine normal samples, the protein expression levels in paraffin-preserved samples were used to test between aggressive vs non-aggressive tumors (Figure 5c). On the basis of the mRNA levels, the performance of discrimination between tumors vs normal was highly significant with MMP1 (area under the curve = 0.94; P= 0.002), MMP10 (area under the curve = 0.90; P= 0.013) and ADAMTS1 (area under the curve = 0.74; P= 0.022). Although the performance of discrimination between aggressive vs non-aggressive tumors was significant with MMP1 (area under the curve = 0.72; P= 0.034), the mRNA expression of MMP10 (area under the curve = 0.67; P = 0.11) and ADAMTS1 (area under the curve = 0.58; P= 0.422) levels were not significant. Similarly, the performance of discrimination between aggressive vs non-aggressive tumors were also significant with the protein levels of MMP1 (area under the curve = 0.66; P < 0.001).
Figure 5.

Receiver operating characteristic (ROC) analysis of matrix metallopeptidase 1 (MMP1), matrix metallopeptidase 10 (MMP10), A disintegrin and metalloproteinase with thrombospondin motifs 1 (ADAMTS1) expression. The mRNA expression in 69 fresh–frozen samples was tested for the performance of discrimination between: (a) normal skin vs tumors, and (b) non-aggressive vs aggressive tumors. The protein expression in 122 paraffin-preserved samples was also tested for the performance of discrimination between non-aggressive vs aggressive tumors (c). Results are shown for the area under the curve for MMP1 (▲), MMP10 (● and ADAMTS1 (♦). The broken diagonal line denotes an area under the curve = 0.50.
Discussion
Squamous cell carcinoma is a relatively high risk of metastasis occurrence compared with basal cell carcinoma, and accounting approximately for 20% of all cutaneous malignancies observed in lighter skin population particularly in the Caucasians.24 Certain squamous cell carcinoma have a relatively high risk of metastasis and these high-risk lesions exhibit an aggressive behavior growing by lateral and vertical extension early in their natural history. Thus, the correct classification of different subtypes based on their molecular features is critically needed for both diagnosis and treatments. In the present study, we performed a comprehensive and quantitative analysis on 200 skin samples by using a combination of microarray, QRT-PCR and immunohistochemistry analyses in order to explore the subtype-specific changes that may be associated with different molecular pathways in the development of squamous cell carcinoma, especially with a particular interest in those tumors that might exhibit aggressive behavior. Also, in this study we have included a number of tissue samples identified as ‘aggressive’ as per the criteria suggested by 7th edition of American Joint Committee on Cancer staging manual.
The microarray analysis of six individually matched squamous cell carcinoma and normal skin samples identified 164 genes that were differentially expressed at least by ≥ 5-fold (Tables 1 and 2). Our microarray results are consistent with several other studies that were investigated previously under different types of skin lesions/tumors. Differentially expressed genes included those previously reported to be upregulated in non-melanoma skin cancer as compared with the unaffected skin samples (eg, MMP1, late-cornified envelope 3D (LCE3D), defensin beta 4 (DEFB4P), chromosome 10 open reading frame 99 (C10orf99), S100 calcium-binding protein A12 (S100A12), S100 calcium-binding protein A9 (S100A9), S100 calcium-binding protein A7 (S100A7), insulin growth factor-like family member 1 (IGFL1), alcohol dehydrogenase 1 beta (ADH1B), microfibrillar-associated protein 5 (MFAP5), cysteine-rich, angiogenic inducer 61 (CYR61), serine peptidase inhibitor clade B3 (SERPINB3), serine peptidase inhibitor clade B4 (SERPINB4), small proline-rich protein 2B (SPRR2B), small proline-rich protein 2B (SPRR2C), fibroblast growth factor 7 (FGF7), FBJ murine osteosarcoma viral oncogene homolog (FOS), IL6, platelet-derived growth factor alpha (PDGFRA), prostaglandin D2 synthase (PTGDS), TMP metallopeptidase inhibitor 3 (TIMP3) and collagen type IV alpha 1 (COL4A1)).25–29 Moreover, the validation studies performed in a separate set of 27 paraffin-preserved samples (22 tumors and 5 normal skin tissues) also confirmed the differential expression in squamous cell carcinoma (Figure 2). These samples were used only in QRT-PCR and not in microarray analysis. Thus, we have observed a good concordance between the microarray and QRT-PCR analyses despite different sample fixation and platforms.
Although we have primer sequence near 3′ end of the gene, the sequences are not the same as the microarray probe sequence. In some cases, the probes are on different exons entirely so the effects of alternative splicing or other mRNA expression-modifying processes are not easily determined. This may be the reason that the differential expression of some of the genes including ENPP2, SOCS3, THBS1 and CFD were not found to be statistically significant in our QRT-PCR. Nevertheless, a pattern involving a panel of genes (eg, MMP1, MMP10 and ADAMTS1) that constitute in a degradome pathway was consistently observed in both microarray and QRT-PCR analyses (Tables 1 and 2 and Figures 1b and 2). Therefore, we follow-up these three genes and further confirmed their mRNA and protein expression levels by QRT-PCR and immunohistochemistry analyses (Figure 3 and Tables 3 and 4). Indeed, these genes were chosen for additional validation mainly because of their apparent role in degradation/shredding of the extracellular matrix (degradome) that may be important in tumor formation and invasion into other tissues.
The most interesting results in this study include the differential expression of MMPs (MMP1, MMP10 and ADAMTS1), IL6 and RECK transcripts in tumor tissues. Although MMP1 is overexpressed in our microarray analysis, the expression levels of IL6 and RECK were found to be low in tumors as compared with normal skin tissues (Tables 1 and 2). Although we have observed similar expression pattern in the QRT-PCR analysis of 27 paraffin-preserved samples, the expression levels of IL6 was different in contrast to the microarray results. Indeed, IL6 was found to be high in tumor tissues compared with normal skin samples (Figure 2). This may be due to the intrinsic nature of the two different methods that have limitations in terms of transcript analysis: one known to be hybridization-based, while the other one is sequencing-based gene expression analysis. Needless to say, one comprehensive bioinformatics study emphasized the importance of independent validation of mRNA expression data even with 13–16% non-concordance between microarray and QRT-PCR analyses.30 Therefore, in our study the IL6 results from QRT-PCR analysis should be taken into the consideration as it is the most robust and highly acceptable method for transcriptome analysis. The expression levels of IL6 are high in most inflammatory processes, and are upregulated in many types of cancers including multiple myeloma, lymphoma, ovarian cancer, prostate cancer, renal cancer and carcinoma of the skin.31 Interestingly, the expression levels of IL6 is also directly linked to MMP1 expression and thereby demonstrate the regulation of a complex cytokine and protease network, particularly in the progression of cutaneous squamous cell carcinoma.32 The in vivo and in vitro analysis/studies investigated by Lederle et al.32 have clearly demonstrated an IL6-mediated upregulation of MMP1 that possibly facilitates the tumor development in rodents after they were injected with IL6-over-expressing human karatinocytes. The overexpression of IL6 and MMP1 shown in our QRT-PCR analysis (Figure 2) further support that similar mechanisms may be involved in the development of squamous cell carcinoma in humans.
On the other hand, the expression of RECK is found to be significantly low in squamous cell carcinoma compared with normal skin samples (Table 2; Figure 2). RECK is a membrane-anchored glycoprotein that contains multiple epidermal growth factor-like repeats and serine protease inhibitor-like motifs.33 It is expressed in various normal tissue types, but its expression is undetectable in tumor-derived cell lines, and downregulated in many malignancies including pancreatic, breast, lung, colorectal, prostate, gastric, cholangiocarcinoma and middle ear squamous cell cancer and osteosarcoma.34–41 Several studies have reported its antitumor activities, particularly in suppressing tumor angiogesisis, invasion and metastasis, and its high expression level often correlates with good prognosis. The increased transcriptional expression of RECK in tumor cells compared with normal tissue cells is well correlated with prolonged survival in patients with many malignant conditions including hepatocellular carcinoma, pancreatic cancer, colorectal cancer, non-small-cell lung cancer and gastric cancer.39,40,42–44 In addition, low expression of RECK indicates a shorter survival for patients with invasive breast cancer.45 Previous studies revealed that the antitumor activities of RECK is highly associated with its inhibitory effects on matrix metalloproteinases (MMPs).46,47 The decreased expression of RECK indicated the proteolytic imbalance in prostate cancer, which is associated with higher tumor aggressiveness.48 Although the underlying mechanisms are not known, the reciprocal activities of RECK and MMPs appear to be important in tumor development.33,49 In support, similar expression profiles with the downregulation of RECK and elevated levels of many MMPs including MMP1, MMP10 in tumors compared with normal skin tissues were observed in the present studies (Tables 1 and 2 and Figure 2).
MMPs are zinc-dependent endopeptidases (a.k.a. metzincin family proteinases) that act on the extracellular matrix, and they have been historically associated with tumorigenesis and metastasis by virtue of their ability to carry out ‘path-clearing’ for cancer cells. The role of another closely related families of metzincin proteinases including ADM and ADAMTS have also been implicated in development progression.50,51 Interestingly, alterations in the expression of the degradome elements including MMP1, MMP10 and ADAMTS1 were also observed in our microarray analysis (Tables 1 and 2). In addition, validation studies using QRT-PCR and immunohistochemistry analysis further confirmed the differential expression of MMP1, MMP10 and ADAMTS1 in tumors compared with normal skin tissues (Figure 3 and Table 4). The expression levels of several MMPs and other metzincin family proteinases have been previously reported to be significantly increased and directly correlate with the invasiveness of many tumors including lung, prostate, stomach, colon, breast, ovary and oral squamous cell cancers.52–57 Generally, the expression of high levels of multiple MMP family members correlates positively with tumor aggressiveness, including increased invasive capacity, metastasis and poor patient survival.24 Although we have observed similar trend in the overexpressions of MMPs in our study, only MMP1 is significantly expressed in aggressive tumors compared with non-aggressive tumors. Indeed, the expression of MMP1 is not only differentiate normal vs tumors but can also be the better discriminator/predictor of the subtypes with aggressive nature of the tumors compared with non-aggressive squamous cell carcinoma (Figures 4 and 5).
In conclusion, gene expression analysis in the present studies identified a pattern of genes with the expression of MMPs similar to that of the degradome components/pathway that may have important role in the development of skin tumors, particularly squamous cell carcinoma. The differential expression validated in the separate set of tumors suggested that there is a relationship between MMP1, MMP10, IL6, ADAMTS1 and RECK, and further substantiating their coordinated role in the development of squamous cell carcinoma. Furthermore, the increased expression of MMP1 correlates positively with tumor aggressiveness of squamous cell carcinoma, which will be useful in differentiating and identifying aggressive tumors. Thus, the genetic changes identified in this study may be useful in the development of biomarkers for differentiation of aggressive vs indolent tumors.
Acknowledgments
We thank Drs WP Andrew Lee and Gerald Bradacher for their support and encouragements.
Footnotes
Disclosure/conflict of interest
The authors declare no conflict of interest.
References
- 1.Rogers HW, Weinstock MA, Harris AR, et al. Incidence estimate of nonmelanoma skin cancer in the United States, 2006. Arch Dermatol. 2010;146:283–287. doi: 10.1001/archdermatol.2010.19. [DOI] [PubMed] [Google Scholar]
- 2.Diepgen TL, Mahler V. The epidemiology of skin cancer. Br J Dermatol. 2002;146(Suppl 61):1–6. doi: 10.1046/j.1365-2133.146.s61.2.x. [DOI] [PubMed] [Google Scholar]
- 3.Housman TS, Feldman SR, Williford PM, et al. Skin cancer is among the most costly of all cancers to treat for the Medicare population. J Am Acad Dermatol. 2003;48:425–429. doi: 10.1067/mjd.2003.186. [DOI] [PubMed] [Google Scholar]
- 4.Housman TS, Williford PM, Feldman SR, et al. Non-melanoma skin cancer: an episode of care management approach. Dermatol Surg. 2003;29:700–711. doi: 10.1046/j.1524-4725.2003.29185.x. [DOI] [PubMed] [Google Scholar]
- 5.Alam M, Ratner D. Cutaneous squamous-cell carcinoma. N Engl J Med. 2001;344:975–983. doi: 10.1056/NEJM200103293441306. [DOI] [PubMed] [Google Scholar]
- 6.Tufaro A, Prasad NB, Fischer A, et al. The clinicopathologic and molecular aspects of non-melanoma skin cancer. Skin Biopsy Perspect. 2011:197–208. http://www.intechopen.com/books/skin-biopsy-perspectives. [Google Scholar]
- 7.Ponten F, Lundeberg J. Dermatology. 2nd edn. London: Harcourt Health Publishers; 2003. Principles of tumor biology and pathogenesis of BCC’s and SCC’s; pp. 1627–1639. [Google Scholar]
- 8.Cherpelis BS, Marcusen C, Lang PG. Prognostic factors for metastasis in squamous cell carcinoma of the skin. Dermatol Surg. 2002;28:268–273. doi: 10.1046/j.1524-4725.2002.01169.x. [DOI] [PubMed] [Google Scholar]
- 9.Friedman GD, Tekawa IS. Association of basal cell skin cancers with other cancers (United States) Cancer Causes Control. 2000;11:891–897. doi: 10.1023/a:1026591016153. [DOI] [PubMed] [Google Scholar]
- 10.Efird JT, Friedman GD, Habel L, et al. Risk of subsequent cancer following invasive or in situ squamous cell skin cancer. Ann Epidemiol. 2002;12:469–475. doi: 10.1016/s1047-2797(01)00276-9. [DOI] [PubMed] [Google Scholar]
- 11.Ulrich C, Kanitakis J, Stockfleth E, et al. Skin cancer in organ transplant recipients—where do we stand today? Am J Transplant. 2008;8:2192–2198. doi: 10.1111/j.1600-6143.2008.02386.x. [DOI] [PubMed] [Google Scholar]
- 12.Arora A, Attwood J. Common skin cancers and their precursors. Surg Clin N Am. 2009;89:703–712. doi: 10.1016/j.suc.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 13.Leiter U, Garbe C. Epidemiology of melanoma and nonmelanoma skin cancer—the role of sunlight. Adv Exp Med Biol. 2008;624:89–103. doi: 10.1007/978-0-387-77574-6_8. [DOI] [PubMed] [Google Scholar]
- 14.Edge SE, Byrd DR, Compton CC, et al. AJCC Cancer Staging Manual. 7th edn. 2010. Cutaneous squamous cell carcinoma and other cutaneous carcinomas. chapter 29; pp. 301–314. [Google Scholar]
- 15.Puente XS, Sanchez LM, Overall CM, et al. Human and mouse proteases: a comparative genomic approach. Nat Rev Genet. 2003;4:544–558. doi: 10.1038/nrg1111. [DOI] [PubMed] [Google Scholar]
- 16.Skrzydlewska E, Sulkowska M, Koda M, et al. Proteolytic-antiproteolytic balance and its regulation in carcinogenesis. World J Gastroenterol. 2005;11:1251–1266. doi: 10.3748/wjg.v11.i9.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 18.Farasat S, Yu SS, Neel VA, et al. A new American Joint Committee on Cancer staging system for cutaneous squamous cell carcinoma: creation and rationale for inclusion of tumor (T) characteristics. J Am Acad Dermatol. 2011;64:1051–1059. doi: 10.1016/j.jaad.2010.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bolstad BM, Irizarry RA, Astrand M, et al. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 20.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ekins S, Nikolsky Y, Bugrim A, et al. Pathway mapping tools for analysis of high content data. Methods Mol Biol. 2007;356:319–350. doi: 10.1385/1-59745-217-3:319. [DOI] [PubMed] [Google Scholar]
- 22.Eisen MB, Spellman PT, Brown PO, et al. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prasad NB, Kowalski J, Tsai HL, et al. Three-gene molecular diagnostic model for thyroid cancer. Thyroid. 2012;22:275–284. doi: 10.1089/thy.2011.0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kerkela E, Saarialho-Kere U. Matrix metalloproteinases in tumor progression: focus on basal and squamous cell skin cancer. Exp Dermatol. 2003;12:109–125. doi: 10.1034/j.1600-0625.2003.120201.x. [DOI] [PubMed] [Google Scholar]
- 25.Hudson LG, Gale JM, Padilla RS, et al. Microarray analysis of cutaneous squamous cell carcinomas reveals enhanced expression of epidermal differentiation complex genes. Mol Carcinog. 2010;49:619–629. doi: 10.1002/mc.20636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ra SH, Li X, Binder S. Molecular discrimination of cutaneous squamous cell carcinoma from actinic keratosis and normal skin. Mod Pathol. 2011;24:963–973. doi: 10.1038/modpathol.2011.39. [DOI] [PubMed] [Google Scholar]
- 27.Dooley TP, Reddy SP, Wilborn TW, et al. Biomarkers of human cutaneous squamous cell carcinoma from tissues and cell lines identified by DNA microarrays and qRT-PCR. Biochem Biophys Res Commun. 2003;306:1026–1036. doi: 10.1016/s0006-291x(03)01099-4. [DOI] [PubMed] [Google Scholar]
- 28.Nindl I, Dang C, Forschner T, et al. Identification of differentially expressed genes in cutaneous squamous cell carcinoma by microarray expression profiling. Mol Cancer. 2006;5:30. doi: 10.1186/1476-4598-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haider AS, Peters SB, Kaporis H, et al. Genomic analysis defines a cancer-specific gene expression signature for human squamous cell carcinoma and distinguishes malignant hyperproliferation from benign hyperplasia. J Invest Dermatol. 2006;126:869–881. doi: 10.1038/sj.jid.5700157. [DOI] [PubMed] [Google Scholar]
- 30.Dallas PB, Gottardo NG, Firth MJ, et al. Gene expression levels assessed by oligonucleotide microarray analysis and quantitative real-time RT-PCR—how well do they correlate? BMC Genom. 2005;6:59. doi: 10.1186/1471-2164-6-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hong DS, Angelo LS, Kurzrock R. Interleukin-6 and its receptor in cancer: implications for translational therapeutics. Cancer. 2007;110:1911–1928. doi: 10.1002/cncr.22999. [DOI] [PubMed] [Google Scholar]
- 32.Lederle W, Depner S, Schnur S, et al. IL-6 promotes malignant growth of skin SCCs by regulating a network of autocrine and paracrine cytokines. Int J Cancer. 2011;128:2803–2814. doi: 10.1002/ijc.25621. [DOI] [PubMed] [Google Scholar]
- 33.Oh J, Takahashi R, Kondo S, et al. The membrane-anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis. Cell. 2001;107:789–800. doi: 10.1016/s0092-8674(01)00597-9. [DOI] [PubMed] [Google Scholar]
- 34.Noda M, Takahashi C. Recklessness as a hallmark of aggressive cancer. Cancer Sci. 2007;98:1659–1665. doi: 10.1111/j.1349-7006.2007.00588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clark JC, Akiyama T, Thomas DM, et al. RECK in osteosarcoma: a novel role in tumour vasculature and inhibition of tumorigenesis in an orthotopic model. Cancer. 2011;117:3517–3528. doi: 10.1002/cncr.25757. [DOI] [PubMed] [Google Scholar]
- 36.Namwat N, Puetkasichonpasutha J, Loilome W, et al. Downregulation of reversion-inducing-cysteine-rich protein with Kazal motifs (RECK) is associated with enhanced expression of matrix metalloproteinases and cholangiocarcinoma metastases. J Gastroenterol. 2011;46:664–675. doi: 10.1007/s00535-010-0345-y. [DOI] [PubMed] [Google Scholar]
- 37.Rabien A, Ergun B, Erbersdobler A, et al. RECK overexpression decreases invasive potential in prostate cancer cells. Prostate. 2012;72:948–954. doi: 10.1002/pros.21498. [DOI] [PubMed] [Google Scholar]
- 38.Shen B, Zheng MQ, Xu XY, et al. Expression of MT1-MMP and RECK protein in human gastric carcinoma. Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi. 2011;25:364–367. [PubMed] [Google Scholar]
- 39.Song SY, Son HJ, Nam E, et al. Expression of reversion-inducing-cysteine-rich protein with Kazal motifs (RECK) as a prognostic indicator in gastric cancer. Eur J Cancer. 2006;42:101–108. doi: 10.1016/j.ejca.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 40.Masui T, Doi R, Koshiba T, et al. RECK expression in pancreatic cancer: its correlation with lower invasiveness and better prognosis. Clin Cancer Res. 2003;9:1779–1784. [PubMed] [Google Scholar]
- 41.Span PN, Sweep CG, Manders P, et al. Matrix metalloproteinase inhibitor reversion-inducing cysteine-rich protein with Kazal motifs: a prognostic marker for good clinical outcome in human breast carcinoma. Cancer. 2003;97:2710–2715. doi: 10.1002/cncr.11395. [DOI] [PubMed] [Google Scholar]
- 42.Furumoto K, Arii S, Mori A, et al. RECK gene expression in hepatocellular carcinoma: correlation with invasion-related clinicopathological factors and its clinical significance. Reverse-inducing cysteine-rich protein with Kazal motifs. Hepatology. 2001;33:189–195. doi: 10.1053/jhep.2001.21048. [DOI] [PubMed] [Google Scholar]
- 43.van der Jagt MF, Sweep FC, Waas ET, et al. Correlation of reversion-inducing cysteine-rich protein with kazal motifs (RECK) and extracellular matrix metalloproteinase inducer (EMMPRIN), with MMP-2, MMP-9, and survival in colorectal cancer. Cancer Lett. 2006;237:289–297. doi: 10.1016/j.canlet.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 44.Takenaka K, Ishikawa S, Kawano Y, et al. Expression of a novel matrix metalloproteinase regulator, RECK, and its clinical significance in resected non-small cell lung cancer. Eur J Cancer. 2004;40:1617–1623. doi: 10.1016/j.ejca.2004.02.028. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, Cheng S, Zhang G, et al. Low expression of RECK indicates a shorter survival for patients with invasive breast cancer. Cancer Sci. 2012;103:1084–1089. doi: 10.1111/j.1349-7006.2012.02265.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meng N, Li Y, Zhang H, et al. A novel matrix metalloproteinase regulator. Histol Histopathol. 2008;23:1003–1010. doi: 10.14670/HH-23.1003. [DOI] [PubMed] [Google Scholar]
- 47.Noda M, Oh J, Takahashi R, et al. RECK: a novel suppressor of malignancy linking oncogenic signaling to extracellular matrix remodeling. Cancer Metas Rev. 2003;22:167–175. doi: 10.1023/a:1023043315031. [DOI] [PubMed] [Google Scholar]
- 48.Rabien A, Burkhardt M, Jung M, et al. Decreased RECK expression indicating proteolytic imbalance in prostate cancer is associated with higher tumor aggressiveness and risk of prostate-specific antigen relapse after radical prostatectomy. Eur Urol. 2007;51:1259–1266. doi: 10.1016/j.eururo.2006.05.050. [DOI] [PubMed] [Google Scholar]
- 49.Rhee JS, Coussens LM. RECKing MMP function: implications for cancer development. Trends Cell Biol. 2002;12:209–211. doi: 10.1016/s0962-8924(02)02280-8. [DOI] [PubMed] [Google Scholar]
- 50.Rocks N, Paulissen G, El Hour M, et al. Emerging roles of ADAM and ADAMTS metalloproteinases in cancer. Biochimie. 2008;90:369–379. doi: 10.1016/j.biochi.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 51.Duffy MJ, McKiernan E, O’Donovan N, et al. Role of ADAMs in cancer formation and progression. Clin Cancer Res. 2009;15:1140–1144. doi: 10.1158/1078-0432.CCR-08-1585. [DOI] [PubMed] [Google Scholar]
- 52.Baren JP, Stewart GD, Stokes A, et al. mRNA profiling of the cancer degradome in oesophago-gastric adenocarcinoma. Br J Cancer. 2012;107:143–149. doi: 10.1038/bjc.2012.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stokes A, Joutsa J, Ala-Aho R, et al. Expression profiles and clinical correlations of degradome components in the tumor microenvironment of head and neck squamous cell carcinoma. Clin Cancer Res. 2010;16:2022–2035. doi: 10.1158/1078-0432.CCR-09-2525. [DOI] [PubMed] [Google Scholar]
- 54.Nelson AR, Fingleton B, Rothenberg ML, et al. Matrix metalloproteinases: biologic activity and clinical implications. J Clin Oncol. 2000;18:1135–1149. doi: 10.1200/JCO.2000.18.5.1135. [DOI] [PubMed] [Google Scholar]
- 55.Hida Y, Hamada J. Differential expressions of matrix metalloproteinases, a disintegrin and metalloproteinases, and a disintegrin and metalloproteinases with thrombospondin motifs and their endogenous inhibitors among histologic subtypes of lung cancers. Anticancer Agents Med Chem. 2012;12:744–752. doi: 10.2174/187152012802650156. [DOI] [PubMed] [Google Scholar]
- 56.Riddick AC, Shukla CJ, Pennington CJ, et al. Identification of degradome components associated with prostate cancer progression by expression analysis of human prostatic tissues. Br J Cancer. 2005;92:2171–2180. doi: 10.1038/sj.bjc.6602630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang X, Zhu S, Luo G, et al. Expression of MMP-10 in lung cancer. Anticancer Res. 2007;27:2791–2795. [PubMed] [Google Scholar]