

Abstract
Background
Segmental genomic copy number alterations, such as loss of 11q or 3p and gain of 17q, are well established markers of poor outcome in neuroblastoma, and have been suggested to comprise tumor suppressor genes or oncogenes, respectively. The gene forkhead box P1 (FOXP1) maps to chromosome 3p14.1, a tumor suppressor locus deleted in many human cancers including neuroblastoma. FoxP1 belongs to a family of winged-helix transcription factors that are involved in processes of cellular proliferation, differentiation and neoplastic transformation.
Methods
Microarray expression profiles of 476 neuroblastoma specimens were generated and genes differentially expressed between favorable and unfavorable neuroblastoma were identified. FOXP1 expression was correlated to clinical markers and patient outcome. To determine whether hypermethylation is involved in silencing of FOXP1, methylation analysis of the 5′ region of FOXP1 in 47 neuroblastomas was performed. Furthermore, FOXP1 was re-expressed in three neuroblastoma cell lines to study the effect of FOXP1 on growth characteristics of neuroblastoma cells.
Results
Low expression of FOXP1 is associated with markers of unfavorable prognosis like stage 4, age >18 months and MYCN amplification and unfavorable gene expression-based classification (P < 0.001 each). Moreover, FOXP1 expression predicts patient outcome accurately and independently from well-established prognostic markers. Array-based CGH analysis of 159 neuroblastomas revealed that heterozygous loss of the FOXP1 locus was a rare event (n = 4), but if present, was associated with low FOXP1 expression. By contrast, DNA methylation analysis in 47 neuroblastomas indicated that hypermethylation is not regularly involved in FOXP1 gene silencing. Re-expression of FoxP1 significantly impaired cell proliferation, viability and colony formation in soft agar. Furthermore, induction of FOXP1 expression led to cell cycle arrest and apoptotic cell death of neuroblastoma cells.
Conclusions
Our results suggest that down-regulation of FOXP1 expression is a common event in high-risk neuroblastoma pathogenesis and may contribute to tumor progression and unfavorable patient outcome.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2407-14-840) contains supplementary material, which is available to authorized users.
Keywords: FoxP1, Neuroblastoma, Tumor suppressor, Cell proliferation, Disease progression
Background
Neuroblastoma is the most common extracranial solid cancer in childhood and the most common cancer in infancy [1]. The tumor is suggested to originate from immature neuroblasts giving rise to the sympathetic nervous system. The clinical hallmark of neuroblastoma is its heterogeneity, with the likelihood of cure varying widely between distinct patient subgroups [2, 3]. Like no other tumor entity, neuroblastoma provides unique features of tumor biology, including subgroups with differentiation into benign ganglioneuroma and a high incidence of complete spontaneous regression in the absence of any or with minimal therapeutic intervention [4]. By contrast, high-risk neuroblastoma patients have a particularly poor 5-year survival rate of <40% despite intensive multimodal therapy compared to an average survival rate of 80% among other pediatric malignancies [5]. The molecular mechanisms underlying these distinct neuroblastoma phenotypes are still poorly understood.
The winged helix transcription factor Forkhead box P1 (FoxP1) is one of four members of the subfamily P of the Fox transcription factor family, which is characterized by a 100-amino acid long, evolutionarily conserved DNA-binding domain called Forkhead or winged-helix domain. Members of the Fox subfamily P share several characteristics that are atypical among Fox proteins: their Forkhead domain is located near the carboxy-terminal region and they contain leucine zipper motifs that promote FoxP homo- and heterodimerization and thus highly selective, tissue- or cell-type specific activity [6]. These factors are widely but not ubiquitously expressed in human tissues and have been implicated in both embryonic development and adult tissue homeostasis by regulating cell growth, proliferation, differentiation, longevity and transformation [7, 8]. Targeted deletion of FOXP1 is lethal in embryogenesis, which is mainly due to cardiac defects [9]. Loss of FoxP1 function has been reported in several human malignancies such as endometrial cancer, lung cancer, head and neck cancer, prostate cancer, renal cell carcinoma, ovarian carcinoma, and has been shown to be associated with poor prognosis in breast cancer [8, 10, 11]. Furthermore, genomic loss at the tumor suppressor locus 3p14.1 including the FOXP1 gene has been described in many cancer types [12] including neuroblastoma [13] and has indicated the presence of a neuroblastoma-associated putative tumor suppressor gene located between 3p14.1 and 3p21.32 [14]. On the other hand, FOXP1 may also act as an oncogene as it is highly expressed in hepatocellular carcinoma and certain B cell malignancies, in which it is frequently targeted by activating chromosome translocations placing it under the transcriptional control of the IGH enhancers [15, 16].
To elucidate the role of FoxP1 in neuroblastoma pathogenesis, we examined the expression of FOXP1 in a cohort of 476 primary neuroblastomas using microarrays, and assessed its correlation with prognostic markers and patient outcome. To investigate the mechanisms of FOXP1 downregulation, we determined copy number aberrations of the FOXP1 locus in 159 neuroblastomas by array-CGH as well as DNA methylation patterns of the FOXP1 promoter in 47 tumors using mass spectrometry. Furthermore, we characterized the functional consequences of FOXP1 re-expression on cell proliferation, apoptosis, migration and colony formation in neuroblastoma cell lines.
Methods
Gene expression microarray data and patients’ characteristics
Single-color gene expression profiles were generated from 476 primary neuroblastoma samples (stage 1, n = 118; stage 2, n = 78; stage 3, n = 71; stage 4, n = 148; stage 4S, n = 61) and 3 neuroblastoma cell lines (IMR-32, CHP-212 and SK-N-BE(2)) using a 44 K oligonucleotide microarray as described elsewhere [17]. All patients were registered in respective clinical trials with written informed consent from the patient and/or a parent/legal guardian. We received ethics approval from the Ethics Commission of the Faculty of Medicine of Cologne University for the clinical trials NB97 (NCT00017225) and NB2004 (NCT00410631) including the molecular assessment of tumor material. MYCN-amplification was observed in 66 tumors, while it was absent in 405 tumors (not determined, n = 5). Patients’ age at diagnosis ranged from 0 to 296 months (median age, 13 months). Median follow-up for patients without events was 7.6 years (range, 0.4-19.0 years). Stage was classified according to the International Neuroblastoma Staging System (INSS) [18]. Total RNA of FOXP1- and GFP-expressing IMR-32, CHP-212 and SK-N-BE(2) cells was isolated at 0, 12, 24 and 72 h after transgene induction using Trizol (Life Technologies, Darmstadt, Germany). All raw and normalized microarray data are available as a subset at Gene Expression Omnibus (Accession numbers GSE45480 and GSE62419). To determine global differences in the expression profiles of FoxP1- and GFP-induced IMR-32, CHP-212 and SK-N-BE(2) cells, we compared mean expression levels of each gene between FOXP1-induced and control cells as described previously [19]. Differentially expressed genes, either up- or down-regulated after FOXP1 re-expression were determined by applying a fold-change cutoff (≥2) and performing an unpaired, two-tailed Student’s t-test. The Gene Set Enrichment Analysis (GSEA) software (Broad Institute, Cambridge, MA, USA) was used to identify coordinated changes in a priori defined sets of functionally grouped genes as described previously [20, 21]. The neuroblastoma subgroups used for methylation and array-CGH analyses consisted of 47 and 159 tumors, respectively. Risk estimation of corresponding patients was performed according to the International Neuroblastoma Risk Group (INRG) classification system (methylation analysis: high risk, n = 22; intermediate and low risk, n = 25; array-CGH analysis: high risk, n = 51; intermediate and low risk, n = 108) [22].
Analysis of genomic aberrations and promoter methylation of FOXP1
Array-CGH profiles from 159 neuroblastoma tumors were generated using 44 K or 105 K oligonucleotide microarrays as described previously [23, 24]. The raw data were analyzed by Agilent Genomics Workbench software (v. 7.0; Agilent Technologies, Santa Clara, CA, USA, 2012) [23]. Array-CGH data are available as a subset at Gene Expression Omnibus (Accession: GSE45480).
FOXP1 promoter methylation analysis was performed by Sequenom Inc. (Hamburg, Germany) as described elsewhere [19, 25]. Genomic DNA from 47 primary neuroblastoma specimens (high FOXP1 expression, n = 23; low FOXP1 expression, n = 24, as defined by the cutoff value for dichotomization of FOXP1 expression) and the neuroblastoma cell line IMR-32 was used to sequence three selected DNA regions covering 45 CpG units downstream of the FOXP1 start site (Table 1). PCR Primers were designed by using Methprimer (http://www.urogene.org/methprimer/, Additional file 1: Table S1).
Table 1.
Design statistics of genomic regions analyzed for methylation
| Number of DNA samples | 48 |
| Number of genomic regions | 1 |
| Number of amplicons | 3 |
| Number of CpG units | 45 |
| Median amplicon length | 413 bp (min = 365; max = 435) |
| Median CpG/amplicon | 14 CpG/amplicon (min = 11; max = 20) |
Data analysis and statistics
Statistical analysis was performed using SPSS software version 20.0 (IBM). Levels of FOXP1 mRNA determined by microarray analysis were compared in patient groups defined by MYCN status (normal vs. amplified), age (<18 months vs. >18 months), tumor stage (stage 1–3 vs. 4S vs. stage 4) and gene expression-based classification (favorable vs. unfavorable) according to the published PAM classifier [26]. Two-tailed nonparametric tests (Wilcoxon, Mann–Whitney U, and Kruskal–Wallis test) were used where appropriate. Kaplan-Meier estimates for OS and EFS were calculated and compared by log-rank test. Recurrence, progression and death from disease were considered as events. The cutoff value for dichotomization of FOXP1 expression was estimated by maximally selected log-rank statistics [27]. Multivariate analysis was performed for EFS and OS using Cox’s proportional hazards regression models. The factors FOXP1 (low vs. high as reference), INSS stage (4 vs. 1, 2, 3, 4S as reference), MYCN (amplified vs. normal as reference) and age at diagnosis (≥18 months vs. <18 months as reference) were fitted into a backward selection. The criterion for inclusion was a likelihood-ratio test p-value less than 0.05 and for exclusion more than 0.10. Quantitative data for functional analyses are shown as means ± SD. Unpaired two-tailed Students t-tests were used where appropriate.
Cell culture
The neuroblastoma cell lines SK-N-BE (2) and CHP-212 were obtained from American Tissue Culture Collection (ATCC, Rockville, MD, USA) and authenticated at the DSMZ-German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany). The neuroblastoma cell line IMR-32 was purchased from the DSMZ and PT67 retroviral packaging cells were obtained from Clontech (Mountain View, CA). All parental and inducible cell lines were cultured and maintained as described previously [19]. Doxycycline (Sigma-Aldrich, Deisenhofen, Germany) was used to induce transgene expression (2 μg/ml).
Plasmids and retroviruses
A human FOXP1 full open reading frame cDNA clone (pCMV6-XL5-FOXP1, clone ID SC103781) was obtained from Origene (Rockville, MD) and cloned into the pRevTRE vector (Clontech) as described elsewhere [19]. PCR primer sequences were as follows: 5′-GAGGATCCACCATGATGCAAGAATCTGGGACTGAGACAAAG-3′ (forward) and 5′-CACAAGCTTTCACTCCATGTCCTCGTTTACTGGTTC-3′ (reverse), containing restriction sites for BamH I and Hind III, respectively. The control plasmid pRevTRE-eGFP-PRE has been described previously [19].
Stable inducible neuroblastoma cell lines
Neuroblastoma cell lines stably expressing either FOXP1 or GFP under the control of the reverse tetracycline-controlled transactivator (rtTA) were generated using the RevTet™ System (Clontech) as described previously [19].
Western blot analysis
Immunoblots were prepared as described previously [28]. FoxP1 protein levels were analyzed 48 h after induction of transgene expression using primary mouse anti-FOXP1 antibody (dilution 1:1000; ab16645; Abcam, Cambridge, MA) and horseradish peroxidase-labeled secondary polyclonal goat anti-mouse antibody (dilution 1:1000; P0447, Dako, Glostrup Denmark). The antigen-antibody complex was detected with Visualizer Spray & Glow (Upstate, Schwalbach, Germany).
In vitrogrowth properties assays
The effect of FOXP1 expression on cell viability was assessed using the Trypan Blue dye exclusion test. Neuroblastoma cells were seeded in 48-well plates at a density of 1 × 104 cells/well in 200 μl RPMI-1640 (10% Tet-free FCS, 2 μg/ml doxycycline). To ensure continuous supply of nutrients throughout the measurement interval, 400 μl fresh RPMI-1640 (10% Tet-free FCS, 2 μg/ml doxycycline) per well were added on day 4. Cells were harvested at day 6 and evaluated for cell number and viability by Trypan Blue exclusion. The effect of FOXP1 expression on cell proliferation was assessed using the Alamar Blue assay (Life Technologies) according to the manufacturer’s instructions. In brief, neuroblastoma cells were seeded in 48-well plates at a density of 6 × 103 cells/well in 200 μl RPMI-1640 (10% Tet-free FCS, 2 μg/ml doxycycline). To ensure continuous supply of nutrients throughout the measurement interval, 400 μl fresh RPMI-1640 (10% Tet-free FCS, 2 μg/ml doxycycline) per well were added on day 4. At day 6, 60 μl of Alamar Blue solution were added to each well, plates were incubated for 6 h and absorbance was measured at 570 nm against a reference wavelength of 595 nm using a plate reader (Multiscan Ascent plate reader; Thermo Labsystems, Helsinki, Finland). The relative reduction of Alamar Blue was calculated as described by the manufacturer. Cell cycle distribution was assessed by FACS analysis as described previously [29].
Analysis of apoptosis
Annexin-V-binding analysis was carried out using APC-coupled Annexin-V and 7-AAD according to the manufacturer’s protocol (BD Biosciences, Heidelberg, Germany). Apoptotic cells were detected by FACS (FACS Canto; BD Biosciences) using the DIVA software (BD Biosciences). TUNEL analysis was performed 3 days after induction of FOXP1 using the In Situ Cell Death Detection Kit according to the manufacturer’s protocol (Roche Diagnostics, Mannheim, Germany) as described previously [19].
Colony formation assay
A soft agar assay was used to assess the effect of FOXP1 on colony-forming of neuroblastoma cells as described previously [19]. After 15 days of incubation at 37°C, colonies were stained with 0.005% (w/v) crystal violet (Life Technologies) and colonies larger than 100 μm diameter were counted.
Migration analysis
To assess the effect of FOXP1 on neuroblastoma cell motility, two different assays were used: a transwell (Boyden chamber) assay and a wound healing assay. First, a Boyden chamber transwell migration assay was performed according to the manufacturer’s protocol. In brief, 500 μl RPMI-1640 (20% Tet-free FCS, 2 μg/ml doxycycline) were added in each lower chamber of a 24-well transwell plate (No. 3421, 5.0 μm pore size, Costar, Corning Costar, Rochester, NY), and 300 μl cell suspension (0.5 × 106 cells/ml) of IMR-32 and SK-N-BE(2) FOXP1 or GFP transgenic cells in RPMI-1640 (FCS-free, 2 μg/ml doxycycline) were seeded in the upper chamber. Cultures were maintained for 6 days, then non-motile cells at the top of the filter were removed and the cells on the lower surface of the membrane were fixed with methanol and stained with DAPI. The number of cells that had migrated to the lower surface of the membrane was counted in four random fields at 10 × 10 magnification under a light microscope.
As a second method to determine the effect of FOXP1 re-expression on migration of neuroblastoma cells, CHP-212 and SK-N-BE(2) cells were analyzed in a wound healing assay using IBIDI culture inserts (No. 80209, IBIDI GmbH, Martinsried, Germany) according to the manufacturer’s protocol. An IBIDI culture insert consists of two reservoirs separated by a 500 mm thick wall. In brief, an IBIDI culture insert was placed into one well of the 24 well plate and slightly pressed on the top to ensure tight adhesion. Equal numbers of FOXP1 or GFP transgenic cells (70 μl per reservoir; 1.0 × 105 cells/ml) in RPMI-1640 (10% Tet-free FCS) were added into the inserts. After 24 hours, the insert was gently removed, creating a gap of 500 μm. The well was filled with RPMI-1640 (10% Tet-free FCS, 2 μg/ml doxycycline) and the migration was documented at 10 × 10 magnification under a light microscope.
All assays were conducted in triplicate and reproduced in two independent experiments.
Results
Low FOXP1transcript levels are correlated with markers of poor outcome in neuroblastoma
To investigate whether FOXP1 expression is associated with prognostic markers of poor outcome in neuroblastoma, its expression levels were evaluated in a cohort of 476 neuroblastoma microarray profiles reflecting the whole spectrum of the disease [17]. We observed similar transcript levels in localized and stage 4S tumors, while expression levels were significantly lower in stage 4 neuroblastoma (Figure 1a; P < 0.001). FOXP1 levels were substantially decreased in tumors of patients older than 18 months at diagnosis (Figure 1b; P < 0.001) as well as in tumors with MYCN amplification (Figure 1c; P < 0.001). Furthermore, low FOXP1 transcript levels significantly correlated with unfavorable gene expression-based classification (Figure 1d; P < 0.001) according to a prognostic multigene classifier that we have defined previously [26]. This predictive signature consists of 144 genes, but does not include FOXP1. Taken together, these results demonstrate that downregulation of FOXP1 is associated with unfavorable prognostic markers in neuroblastoma.
Figure 1.
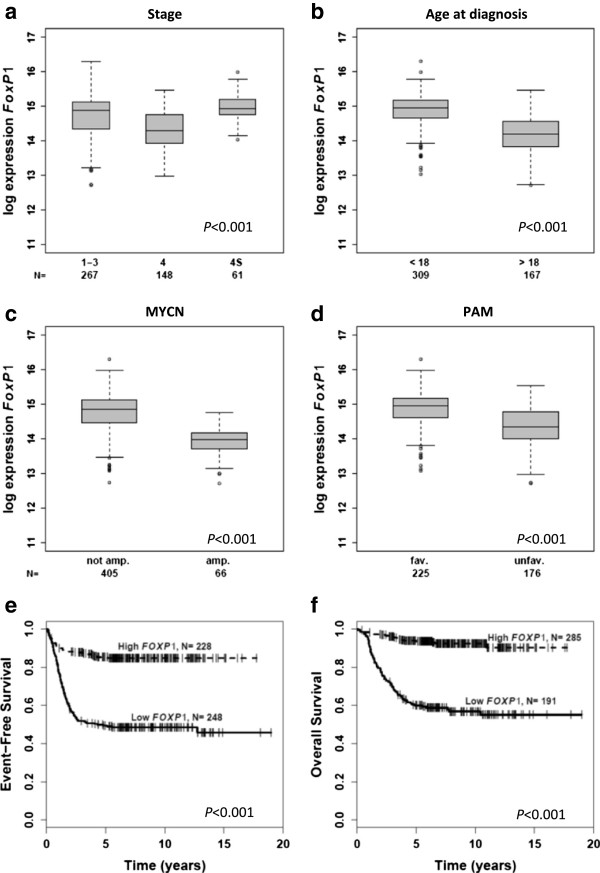
Association of FOXP1 expression levels with prognostic markers and patient outcome in 476 neuroblastomas as determined by microarray analysis. Association of (a) tumor stage, (b) age at diagnosis, (c) MYCN amplification status and (d) gene expression-based classification (PAM classifier) [26] with FOXP1 transcript levels. Boxes, median expression values (horizontal line) and 25th and 75th percentiles; whiskers, distances from the end of the box to the largest and smallest observed values that are less than 1.5 box lengths from either end of the box; open circles, outlying values. Survival curves of neuroblastoma patients for (e) EFS and (f) OS according to FOXP1 expression as determined by microarray analysis. N, patient numbers; amp, amplified; fav, favorable; unfav, unfavorable.
Low FOXP1expression is associated with adverse outcome of neuroblastoma patients
We next determined whether FOXP1 expression is associated with neuroblastoma patient event-free survival (EFS) and overall survival (OS). We noticed that low FOXP1 expression was associated with poor EFS (Figure 1e; 5-year EFS 0.49 ± 0.03 vs. 0.85 ± 0.02; P < 0.001) and OS (Figure 1f; 5-year OS 0.60 ± 0.04 vs. 0.93 ± 0.02; P < 0.001), demonstrating that FOXP1 transcript levels discriminate neuroblastoma patients with beneficial and adverse outcome. The prognostic value of FOXP1 expression was substantiated by multivariate Cox regression models based on EFS and OS considering prognostic markers that are currently used for neuroblastoma risk stratification. In these analyses, FOXP1 expression turned out to be a significant independent prognostic marker for both EFS and OS (Table 2; EFS, hazard ratio, 2.29, 95% confidence interval, 1.46 – 3.58, P < 0.001; OS, hazard ratio, 1.79, 95% confidence interval, 0.98 – 3.26, P < 0.001).
Table 2.
Multivariate Cox regression models based on EFS and OS, considering single prognostic markers and FOXP1 expression
| Marker | Patients (N) | Available cases (N) | Hazard ratio | 95% CI | P-value |
|---|---|---|---|---|---|
| Model considering single prognostic markers and FOXP1 expression based on EFS | 476 | 471 | |||
| Age (<18 months vs. >18 months) | 1.43 | 0.96-2.13 | 0.077 | ||
| Stage (stage 1-3, 4S vs. 4) | 2.31 | 1.62-3.31 | < 0.001 | ||
| MYCN status (normal vs. amplified) | 1.74 | 1.20-2.52 | 0.005 | ||
| FOXP1 expression (cutoff EFS ≥27 840 vs. <27 840) | 2.29 | 1.46-3.58 | < 0.001 | ||
| Model considering single prognostic markers and FOXP1 expression based on OS | 476 | 471 | |||
| Age (<18 months vs. >18 months) | 2.95 | 1.60-5.42 | < 0.001 | ||
| Stage (stage 1-3, 4S vs. 4) | 3.18 | 1.95-5.21 | < 0.001 | ||
| MYCN status (normal vs. amplified) | 2.89 | 1.86-4.51 | < 0.001 | ||
| FOXP1 expression (cutoff OS ≥26 428 vs. <26 428) | 1.79 | 0.98-3.26 | < 0.001 |
Abbreviations: CI confidence interval, EFS event-free survival, OS overall surviaval, vs. versus.
Genetic and epigenetic characterization of the FOXP1locus in neuroblastoma
We next aimed to evaluate whether deregulated FOXP1 expression is associated with genetic or epigenetic aberrations of the FOXP1 locus.
First, we examined the methylation status of three DNA regions covering 45 CpG units downstream of the FOXP1 start site in 47 primary neuroblastoma samples and the neuroblastoma cell line IMR-32, which expresses FOXP1 at low levels endogenously. Unsupervised one-way hierarchical clustering revealed a low degree of methylation in general, and a largely homogeneous methylation pattern in tumors of both subgroups (low expression of FOXP1 vs. high expression of FOXP1 as defined in Methods) and IMR-32 cells (Figure 2). These results suggest that aberrant DNA methylation is not regularly involved in FOXP1 gene silencing in neuroblastoma.
Figure 2.

Hierarchical clustering of FOXP1 DNA methylation ratios. A total of 45 CpG units of FOXP1 were analyzed in 47 tumor samples and the neuroblastoma cell line IMR-32 (indicated in red). FOXP1 expression levels of the tumors (blue, low; green, high; as defined by the expression cutoff value for dichotomizing patient EFS) are indicated aside. DNA methylation values are indicated by colors ranging from dark red (non-methylated) to bright yellow (65% methylated). Poor quality data are indicated in grey. A histogram is given in the inset that indicates the frequency of each color in the hierarchical clustering.
Next, we analyzed array-comparative genomic hybridization (array-CGH) profiles of 159 neuroblastoma samples and compared the results with corresponding microarray gene expression data of the same tumors. In four samples, segmental loss of chromosome 3p14.1 comprising the FOXP1 locus was detected, suggesting that deletion of FOXP1 is not a frequent genetic event in neuroblastoma. However, loss of FOXP1 was accompanied by low FOXP1 expression levels in all four cases (Figure 3), indicating that low FOXP1 expression may be result from a gene dosage effect in a subset of neuroblastoma.
Figure 3.
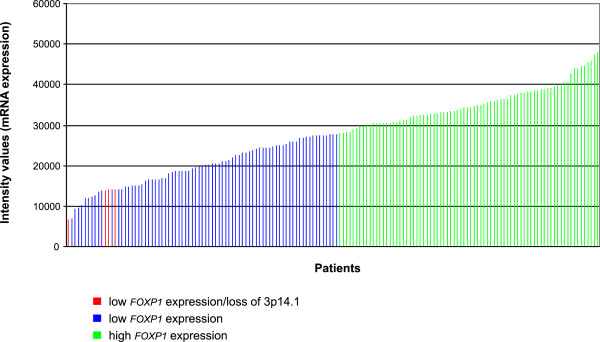
FOXP1 mRNA expression intensities of 159 tumors for which array-CGH data were available. Green and blue represent high and low FOXP1 expression as defined by the expression cutoff value for dichotomizing patient EFS. Tumors showing loss of the FOXP1 locus at 3p14.1 are indicated in red.
Doxycycline-inducible expression of FOXP1in neuroblastoma cell lines
To examine FOXP1 functions in neuroblastoma pathogenesis, we generated three stable, inducible FOXP1 transgenic cell lines (IMR-32, CHP-212 and SK-N-BE(2)) using the retroviral RevTet-On™ System (Clontech). 48 h after induction of transgene expression, polyclonal FoxP1-expressing neuroblastoma cells were compared with polyclonal GFP-expressing cells due to promoter leakage of the pRevTRE Vector System (Figure 4a). Recombinant FoxP1 protein levels were in the range of physiological levels observed in neuroblastoma patients with high FoxP1 expression (Figure 4b). Notably, Western blot hybridization analysis revealed that FOXP1 transcript levels correlate well with FoxP1 protein levels (Figure 4b), which may indicate that measurement of FOXP1 mRNA is predictive of its functional activity in neuroblastoma.
Figure 4.
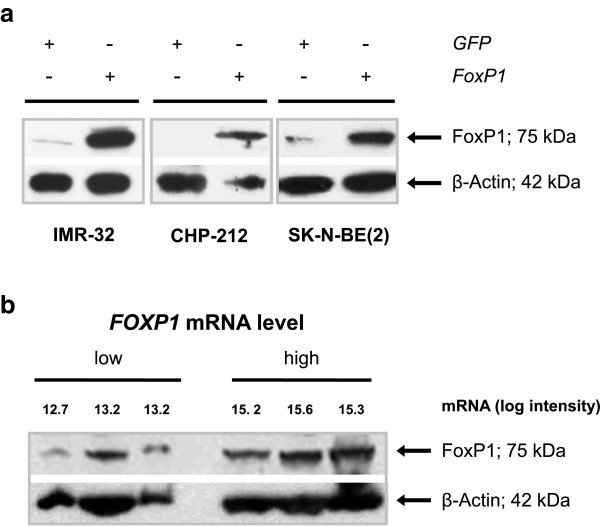
Inducible expression of FoxP1 in neuroblastoma cell lines and physiological FoxP1 levels in primary neuroblastomas. (a) Inducible FoxP1 protein expression in FOXP1- versus GFP-expressing neuroblastoma cell lines 48 h after induction of transgene expression. (b) Physiological FoxP1 protein levels in primary tumors; FOXP1 transcript levels are indicated as log intensities (microarray data).
FOXP1expression inhibits neuroblastoma cell growth
To investigate whether FOXP1 expression is involved in the regulation of growth properties in neuroblastoma cells, FOXP1-expressing IMR-32, CHP-212 and SK-N-BE(2) cells were compared with GFP-expressing controls. Cell proliferation was assayed for 6 days using Trypan Blue dye exclusion tests and Alamar Blue assays. On day 6, the number of viable cells was significantly decreased in all FOXP1-expressing cell lines as compared to GFP-expressing controls (Figure 5a and b; P < 0.01).
Figure 5.
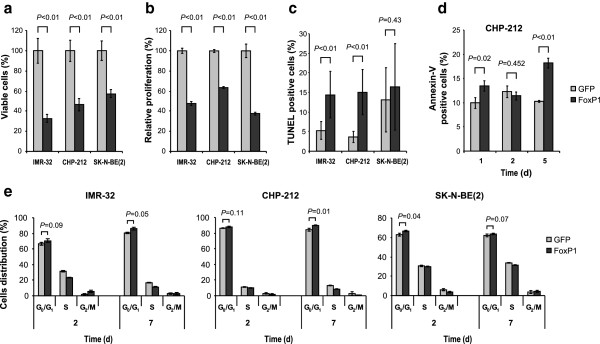
FOXP1 re-expression inhibits growth of neuroblastoma cells. FoxP1-induced changes in (a) cell viability (Trypan Blue dye exclusion analysis 6 days after induction of transgene expression), (b) cell proliferation (Alamar Blue assay 6 days after induction of transgene expression), (c) the proportion of cells showing apoptotic nuclei (72 h after induction of transgene expression), (d) the proportion of Annexin-V-positive cells (fluorescence-activated cell sorting (FACS)) and (e) cell cycle distribution (FACS). Each value represents the mean of triplicate experiments; error bars indicate S.D.
We next examined whether apoptosis may contribute to the inhibition of neuroblastoma cell growth after FOXP1 re-expression. First, DNA fragmentation was assessed using the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay in IMR-32, CHP-212 and SK-N-BE(2) cells. In IMR-32 and CHP-212, FOXP1-expressing cells showed a significantly higher fraction of TUNEL-positive cells as compared to GFP-expressing controls, while no difference in the frequency of apoptotic events was observed in SK-N-BE(2) cells (Figure 5c). Second, we analyzed the externalization of phosphatidylserine by flow cytometry using Annexin-V staining in CHP-212 cells. In accordance with the results of the TUNEL analysis, we observed a significant increase of the Annexin-V-binding fraction 5 days after FOXP1 induction in comparison to control cells (Figure 5d).
To assess whether reduced proliferation upon FOXP1 re-expression might be a consequence of impaired cell cycle progression, cell cycle distribution was determined by fluorescence-activated cell sorting (FACS) on days 2 and 7 after induction of FOXP1 expression. We observed an increased fraction of cells in the G0/G1 phase at the expense of the S-phase population in FOXP1-expressing cells in all three cell lines (Figure 5e).
Taken together, these findings indicate that FoxP1 affects growth properties in human neuroblastoma cells by both cell cycle regulation and induction of apoptosis.
FOXP1attenuates the malignant phenotype of neuroblastoma cells
To further assess the effect of FOXP1 expression on tumorigenicity, we compared anchorage-independent growth rates and migration capabilities of FOXP1-induced with GFP-induced control cells. Fifteen days after transgene induction, FOXP1-expressing IMR-32 and SK-N-BE(2) cells showed a significantly reduced colony-forming ability in soft agar to about 10 and 20% of GFP-expressing control cells, respectively (Figure 6a; P < 0.001 each). In the Boyden chamber assay, the number of migrated cells in the lower chamber was significantly smaller in the FOXP1 expressing IMR-32 and SK-N-BE(2) cells as compared to the GFP-expressing controls (Figure 6b; 30% and 50%, respectively; P < 0.001 each). Consistent with these results, gap closure was delayed in the FOXP1-induced CHP-212 and SK-N-BE(2) cells as compared to GFP-expressing controls (Figure 6c).
Figure 6.
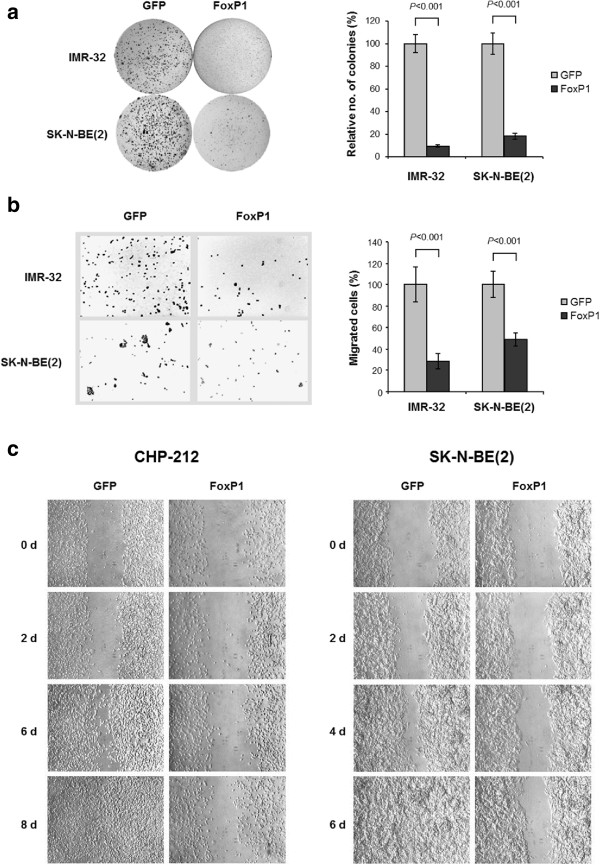
FOXP1 attenuates the malignant phenotype of neuroblastoma cells. (a) Soft agar colony formation assay of FOXP1- versus GFP-expressing control cells. Cells were cultured in soft agar for 15 days. (b) Boyden chamber migration assay of FOXP1- versus GFP-expressing control cells. Cells were seeded in Boyden chambers and incubated for 6 days. (c) Gap closure assay of FOXP1- versus GFP-expressing control cells. Images were captured at the indicated time points. Each value represents the mean of triplicate experiments; error bars indicate S.D. d, days; no., number.
Together, our observations demonstrate that reconstitution of FOXP1 expression suppresses the malignant phenotype of neuroblastoma cells.
FoxP1 modulates the expression patterns of apoptosis-, migration- and differentiation-related genes
To assess the molecular consequences of FOXP1 re-expression in neuroblastoma cells, we performed series of time-resolved expression microarray analyses of FOXP1- and GFP-expressing IMR-32, CHP-212 and SK-N-BE(2) cells. Expression profiles were generated 0, 12, 24 and 72 hours after transgene induction. Gene set enrichment analysis (GSEA) revealed a significant induction of genes involved in apoptosis in IMR-32 (Figure 7a; enrichment score =0.43; P < 0.001) and CHP-212 (Figure 7b; enrichment score =0.39; P = 0.045) neuroblastoma cells. No enrichment was found in the p53 mutant cell line SK-N-BE(2) (Figure 7c; P = 0.59). The heatmap plots in Figure 7 show a set of 39 genes involved in apoptosis. Furthermore, all three model cell lines IMR-32 (Figure 8a; enrichment score = -0.31; P < 0.001), CHP-212 (Figure 8b; enrichment score = -0.45; P < 0.001) and SK-N-BE(2) (Figure 8c; enrichment score = -0.43; P < 0.001) showed a significant down-regulation of genes involved in cell migration. The heatmap plots in Figure 8 show a set of 41 genes involved in cell migration.
Figure 7.
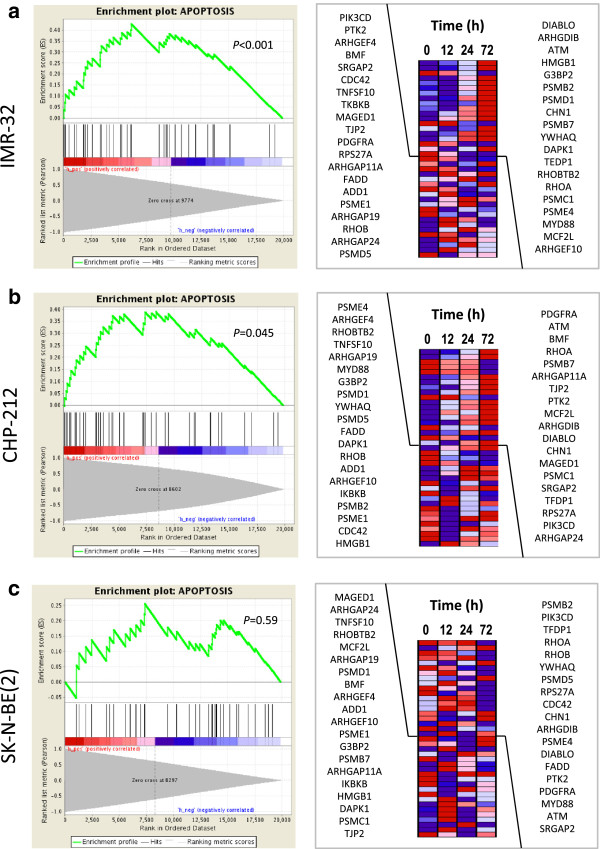
FOXP1 induces genes involved in apoptosis. Gene set enrichment analysis (GSEA) of time-resolved gene expression measurements in FoxP1-induced neuroblastoma cells. Cells were analyzed 0, 12, 24 and 72 hours after transgene induction. The enrichment score (ES) plotted as a function of the position within the ranked list of array probes is shown as a green line. The ranked list metric shown in gray illustrates the correlation of the gene expression values across the time series. Right, individual expression profiles for time leading edge probe sets contributing to the normalized enrichment score are shown. Signal intensities are illustrated by varying shades of red (up-regulation) and blue (down-regulation). FoxP1 up-regulates pro-apoptotic genes in (a) IMR-32 and (b) CHP-212 cells as compared to GFP-expressing controls. (c) Recombinant FOXP1 fails to induce pro-apoptotic genes in the p53 mutant cell line SK-N-BE(2).
Figure 8.
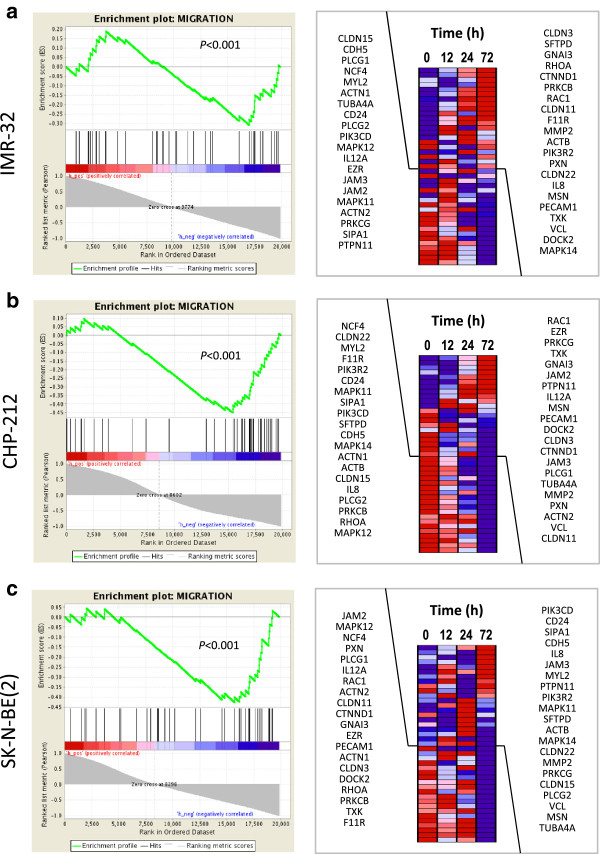
FOXP1 down-regulates genes involved in migration. Gene set enrichment analysis (GSEA) of time-resolved gene expression measurements in FoxP1-induced neuroblastoma cells. Cells were analyzed 0, 12, 24 and 72 hours after transgene induction. The enrichment score (ES) plotted as a function of the position within the ranked list of array probes is shown as a green line. The ranked list metric shown in gray illustrates the correlation of the gene expression values across the time series. Right, individual expression profiles for time leading edge probe sets contributing to the normalized enrichment score are shown. Signal intensities are illustrated by varying shades of red (up-regulation) and blue (down-regulation). FoxP1 decreases the expression of genes involved in migration processes in (a) IMR-32, (b) CHP-212 and (c) SK-N-BE (2) cells as compared to GFP-expressing controls.
To identify downstream targets and signaling pathways regulated by FOXP1, we analyzed differentially expressed genes after FOXP1 re-expression using microarray gene expression analysis (Additional file 2: Table S2). FOXP1 strongly up-regulated the expression of the homeobox C9 gene (HOXC9) and genes involved in neuronal differentiation pathways such as DBH, MAP2, RET, TH, DNER, NEFL and NPY in IMR-32 (Figure 9a) and CHP-212 cells (Figure 9b). Induction of HOXC9 or genes associated with neuronal differentiation was absent in the p53-mutated background of SK-N-BE(2) cells. By contrast, we found a strong (21.2-fold) induction of TNFSF10 (TRAIL) in SK-N-BE(2) cells immediately after FOXP1 induction (Figure 7c), indicating an alternative pathway of FOXP1-induced growth inhibition in SK-N-BE(2) cells.
Figure 9.
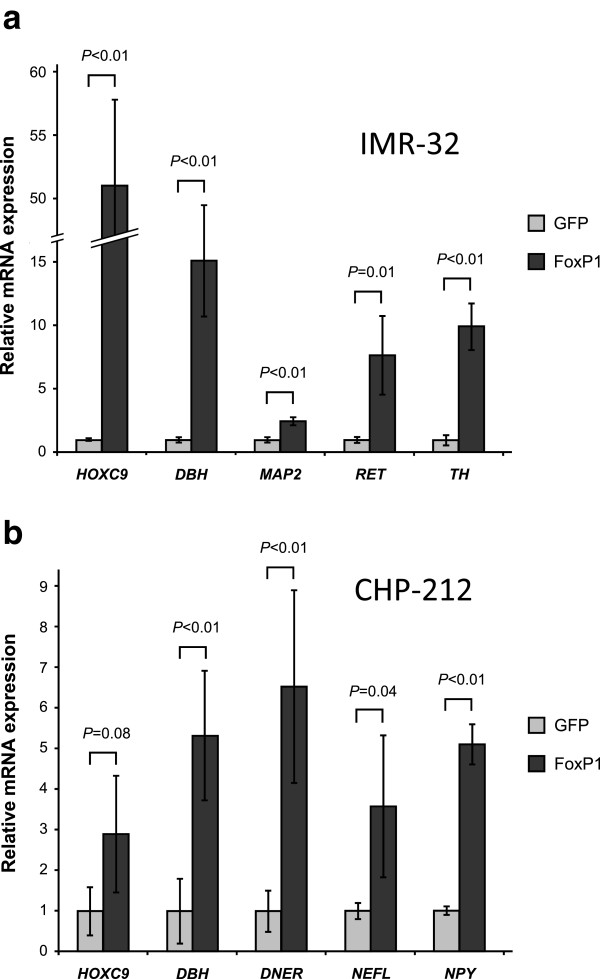
FOXP1 induces genes involved in neuronal differentiation. FOXP1 expression up-regulates neuron-related markers in (a) IMR-32 and (b) CHP-212 cells as determined by microarray gene expression analysis. Error bars indicate S.D.
Discussion
Altered patterns of gene expression arising from inappropriate regulation or function of key transcription factors cause many human diseases, including cancer. Forkhead box proteins are attracting increasing attention as tissue type dependent regulators and critical mediators of cellular processes including proliferation, migration, differentiation, cell cycle arrest or cell death. Dysregulation of Fox protein expression has been implicated in a broad spectrum of human disorders, including immunological dysfunction, infertility, speech/language disorders, and malignancies [30, 31]. FoxP1 is a transcriptional repressor that plays an important role in the development of the brain and lung in mammals and has been described to have diverse and opposing functions in different types of cancer [32]. Results of recent studies indicate that loss of FoxP1 activity promotes solid tumor proliferation, e.g. in endometrial cancer, prostate cancer, renal cell carcinoma, and breast cancer, suggesting a tumor-suppressive role in these malignancies [16, 33]. In contrast, high expression of FOXP1 was shown to correlate with poor outcome in certain types of B cell lymphomas [34–36]. While several Fox family members have been shown to play critical roles in neuroblastoma biology [37–40], the role of FoxP1 has not yet been investigated in this context.
Here, we demonstrate that low FOXP1 expression is associated with unfavorable prognostic markers and poor patient outcome in neuroblastoma. The prognostic relevance of low FOXP1 expression was validated by multivariate analysis for OS and EFS, showing that FOXP1 expression predicts neuroblastoma patient outcome independently from well-established prognostic markers. Similar results have recently been published for prostate cancer and non-small cell lung cancer, in which an inverse correlation of FOXP1 expression with increased malignancy grade and reduced patient survival was observed [33]. Together, these findings may suggest a tumor suppressive role of FoxP1 in neuroblastoma.
In line with this hypothesis, restoration of FOXP1 expression strongly decreased growth rates and tumorigenic characteristics of all three neuroblastoma cell lines analyzed in vitro. These results are consistent with recent studies of other solid tumors, in which ectopic FoxP1 exhibited strong tumor suppressor activity in prostate cancer and glioma cells [33, 41]. Although the results of the migration analyses may in part be explained by a reduction in cell number rather than a change of cell migration ability, the analysis of mRNA levels confirmed down-regulation of cell motility-associated genes such as MAPK12 [42], PLCG2 [43] and NCF4 [44] in all three FOXP1 transgenic neuroblastoma cell lines analyzed. Considering that the cell lines used in this study differ in several molecular and biological characteristics like genetic alterations, doubling time, morphology and motility, our results may suggest that diminished FOXP1 expression might act as a growth-promoting factor in neuroblastoma in general. We also found that induction of FoxP1 delayed cell cycle progression and up-regulated pro-apoptotic genes such as DIABLO [45], CDC42 [46] and DAPK1 [47], indicating the induction of the intrinsic pathway of apoptosis. In addition, we found that HOXC9 is up-regulated, a well described key regulator of neuroblastoma differentiation which links the intrinsic pathway of apoptosis, cell-cycle exit and neuronal differentiation [48]. In line with previously published analyses of HOXC9 in neuroblastoma cells, we found a significant up-regulation of genes involved in neuronal differentiation pathways such as DNER, NEFL [19], DBH, TH [48], RET [49], MAP2 [50] and NPY [51]. The combined effects of intrinsic apoptosis and induction of differentiation have been described to strongly inhibit of proliferation and reduce tumorigenicity of neuroblastoma cells previously, which is well in line with the effects FOXP1 re-expression shown here. Together, these results indicate that FoxP1 may mediate cellular growth restriction in neuroblastoma by cell cycle control, programmed cell death and induction of differentiation. Interestingly, recombinant FoxP1 failed to induce apoptosis in the p53 mutant cell line SK-N-BE(2), suggesting a p53 dependent mechanism of FoxP1 mediated cell death. In accordance with this finding, we did not observe an induction of HOXC9 or other pro-apoptotic genes in this model cell line. FoxP1 and p53 signaling have been described to be connected in B-cell lymphoma, in which the oncogenic action of FoxP1 is repressed by p53-induced miR-34a [52]. These findings emphasize the need for further studies to elucidate the interaction of these factors and their signaling partners in a tissue-specific context. Of note, SK-N-BE(2) cells showed a strong (21.2-fold) induction of TNFSF10, the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), immediately after FOXP1 induction (Figure 7c). This gene has been described to inhibit cell proliferation in several other tumor entities by regulating autophagy and extrinsic apoptosis, indicating an alternative pathway of FOXP1-induced growth inhibition in SK-N-BE(2) cells.
We also aimed to identify the molecular basis underlying the differential expression of FOXP1 observed in different neuroblastoma subgroups. Loss of chromosome 3p is a genomic aberration frequently found in unfavorable neuroblastoma, suggesting that one or multiple tumor suppressor genes are targeted by this genetic alteration [13, 53–55]. We found low FOXP1 expression in four tumor samples with segmental 3p14.1 loss containing the FOXP1 locus, pointing towards haploinsufficiency in these tumors. However, deletions of the FOXP1 locus appear to occur rarely in neuroblastoma, suggesting that other factors contribute to differential FOXP1 expression in this malignancy. Analysis of DNA methylation patterns and FOXP1 expression levels did not reveal any correlation, implicating that differential methylation is not a common cause of FOXP1 repression in neuroblastoma. In addition, somatic mutations of the coding sequence of FOXP1 have not been identified in recent reports on the mutational spectrum of neuroblastoma using next generation sequencing [56, 57]. We therefore speculate that additional molecular mechanisms may mediate FOXP1 gene silencing in neuroblastoma. Aberrant expression of FOXP1 in various pathologic conditions has been reported to be caused by a variety of molecular mechanisms, including dysregulation of upstream signaling pathways such as FoxO proteins or components of the estrogen-ER signaling pathway [58–60], differential splicing [61, 62], viral integration events [63], differential miRNA activity [64, 65] and translocations affecting upstream regulatory regions of the FOXP1 gene [66]. Further investigations are required to determine the contribution of these mechanisms to altered FOXP1 expression in neuroblastoma.
Conclusions
In summary, our study indicates that aggressive human neuroblastomas show reduced FOXP1 expression, a finding that is compatible with the previously observed loss of FoxP1 in several types of solid tumors. FOXP1 expression levels are inversely correlated with proliferative activity and tumorigenicity in vitro, supporting its role as a transcriptional regulator with critical tumor suppressor functions in solid tumor biology. Although the mechanisms that regulate FOXP1 expression in neuroblastoma remain unclear, low expression of FOXP1 may add predictive value on top of established risk stratification tools in neuroblastoma. Further analysis of this transcription factor may contribute to the understanding of the molecular processes underlying tumor regression and progression in neuroblastoma.
Electronic supplementary material
Additional file 1: Table S1: Information on oligonucleotides used as primers for DNA methylation analysis. (XLS 38 KB)
Additional file 2: Table S2: Genes up- or down-regulated after FOXP1 re-expression in IMR-32, CHP-212 and SK-N-BE(2) cells as determined by microarray gene expression analysis. (XLSX 438 KB)
Acknowledgements
We thank Anne Engesser for excellent technical assistance, Ruth Volland for statistical support and the German Neuroblastoma Tumor Bank of the German Society of Pediatric Oncology and Hematology (GPOH) for providing tumor samples. This work was supported by grants from the Cologne Center for Molecular Medicine, the Bundesministerium für Bildung und Forschung (BMBF) through the National Genome Research Network plus (NGFNplus, Grant No. 01GS0895), the Fördergesellschaft Kinderkrebs-Neuroblastom-Forschung e.V. and by the Competence Network Pediatric Oncology and Hematology (KPOH).
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SA conceived of the study, designed and carried out experimental part and wrote the manuscript. HK participated in methylation analyses and cell culture experiments and helped to draft the manuscript. BH participated in the clinical design of the study, collected clinical data and performed statistical analyses. VE performed FACS analyses. YK collected patients’ material and participated in immunoassays and microarray analyses. AO participated in the design and coordination of the neuroblastoma microarray project and provided the expression data. FR performed statistical analyses. JT performed FISH analysis. MO provided the laboratory facility and technical support necessary for the initiation of the study. FB provided the laboratory environment and financial support necessary for completion of the study. MF conceived of the study, participated in its design and coordination, participated in the design and coordination of the neuroblastoma microarray project and helped to draft the manuscript. All authors read and approved the final manuscript.
Contributor Information
Sandra Ackermann, Email: sandra.ackermann@uk-koeln.de.
Hayriye Kocak, Email: hayriye.kocak@web.de.
Barbara Hero, Email: barbara.hero@uk-koeln.de.
Volker Ehemann, Email: Volker.Ehemann@med.uni-heidelberg.de.
Yvonne Kahlert, Email: yvonne.kahlert@uk-koeln.de.
André Oberthuer, Email: andre.oberthuer@uk-koeln.de.
Frederik Roels, Email: frederik.roels@uk-koeln.de.
Jessica Theißen, Email: jessica.theissen@uk-koeln.de.
Margarete Odenthal, Email: m.odenthal@uni-koeln.de.
Frank Berthold, Email: frank.berthold@uk-koeln.de.
Matthias Fischer, Email: matthias.fischer@uk-koeln.de.
References
- 1.Schwab M, Westermann F, Hero B, Berthold F. Neuroblastoma: biology and molecular and chromosomal pathology. Lancet Oncol. 2003;4(8):472–480. doi: 10.1016/S1470-2045(03)01166-5. [DOI] [PubMed] [Google Scholar]
- 2.Coldman AJ, Fryer CJ, Elwood JM, Sonley MJ. Neuroblastoma: influence of age at diagnosis, stage, tumor site, and sex on prognosis. Cancer. 1980;46(8):1896–1901. doi: 10.1002/1097-0142(19801015)46:8<1896::AID-CNCR2820460833>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 3.Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369(9579):2106–2120. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- 4.Davidoff AM. Neuroblastoma. Semin Pediatr Surg. 2012;21(1):2–14. doi: 10.1053/j.sempedsurg.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith MA, Seibel NL, Altekruse SF, Ries LA, Melbert DL, O’Leary M, Smith FO, Reaman GH. Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol. 2010;28(15):2625–2634. doi: 10.1200/JCO.2009.27.0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lam EW, Brosens JJ, Gomes AR, Koo CY. Forkhead box proteins: tuning forks for transcriptional harmony. Nat Rev Cancer. 2013;13(7):482–495. doi: 10.1038/nrc3539. [DOI] [PubMed] [Google Scholar]
- 7.Kaufmann E, Knochel W. Five years on the wings of fork head. Mech Dev. 1996;57(1):3–20. doi: 10.1016/0925-4773(96)00539-4. [DOI] [PubMed] [Google Scholar]
- 8.Koon HB, Ippolito GC, Banham AH, Tucker PW. FOXP1: a potential therapeutic target in cancer. Expert Opin Ther Targets. 2007;11(7):955–965. doi: 10.1517/14728222.11.7.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang B, Weidenfeld J, Lu MM, Maika S, Kuziel WA, Morrisey EE, Tucker PW. Foxp1 regulates cardiac outflow tract, endocardial cushion morphogenesis and myocyte proliferation and maturation. Development. 2004;131(18):4477–4487. doi: 10.1242/dev.01287. [DOI] [PubMed] [Google Scholar]
- 10.Bates GJ, Fox SB, Han C, Launchbury R, Leek RD, Harris AL, Banham AH. Expression of the forkhead transcription factor FOXP1 is associated with that of estrogen receptor-beta in primary invasive breast carcinomas. Breast Cancer Res Treat. 2008;111(3):453–459. doi: 10.1007/s10549-007-9812-4. [DOI] [PubMed] [Google Scholar]
- 11.Kim YS, Do Hwan J, Bae S, Bae DH, Ahn Shick W. Identification of differentially expressed genes using an annealing control primer system in stage III serous ovarian carcinoma. BMC Cancer. 2010;10:576. doi: 10.1186/1471-2407-10-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bieche I, Lidereau R. Genetic alterations in breast cancer. Genes Chromosomes Cancer. 1995;14(4):227–251. doi: 10.1002/gcc.2870140402. [DOI] [PubMed] [Google Scholar]
- 13.Vandesompele J, Speleman F, Van Roy N, Laureys G, Brinskchmidt C, Christiansen H, Lampert F, Lastowska M, Bown N, Pearson A, Nicholson JC, Ross F, Combaret V, Delattre O, Feuerstein BG, Plantaz D. Multicentre analysis of patterns of DNA gains and losses in 204 neuroblastoma tumors: how many genetic subgroups are there? Med Pediatr Oncol. 2001;36(1):5–10. doi: 10.1002/1096-911X(20010101)36:1<5::AID-MPO1003>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 14.Scaruffi P, Coco S, Cifuentes F, Albino D, Nair M, Defferrari R, Mazzocco K, Tonini GP. Identification and characterization of DNA imbalances in neuroblastoma by high-resolution oligonucleotide array comparative genomic hybridization. Cancer Genet Cytogenet. 2007;177(1):20–29. doi: 10.1016/j.cancergencyto.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 15.Espinet B, Garcia-Herrera A, Gallardo F, Baro C, Salgado R, Servitje O, Estrach T, Colomo L, Romagosa V, Barranco C, Serrano S, Campo E, Pujol RM, Sole F. FOXP1 molecular cytogenetics and protein expression analyses in primary cutaneous large B cell lymphoma, leg-type. Histol Histopathol. 2012;26(2):213–221. doi: 10.14670/HH-26.213. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Zhang S, Wang X, Liu J, Yang L, He S, Chen L, Huang J. Prognostic significance of FOXP1 as an oncogene in hepatocellular carcinoma. J Clin Pathol. 2012;65(6):528–533. doi: 10.1136/jclinpath-2011-200547. [DOI] [PubMed] [Google Scholar]
- 17.Oberthuer A, Juraeva D, Li L, Kahlert Y, Westermann F, Eils R, Berthold F, Shi L, Wolfinger RD, Fischer M, Brors B. Comparison of performance of one-color and two-color gene-expression analyses in predicting clinical endpoints of neuroblastoma patients. Pharmacogenomics J. 2010;10(4):258–266. doi: 10.1038/tpj.2010.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brodeur GM, Pritchard J, Berthold F, Carlsen NL, Castel V, Castelberry RP, De Bernardi B, Evans AE, Favrot M, Hedborg F, Kaneko M, Kemshead J, Lampert F, Lee REJ, Look T, Pearson ADJ, Philip T, Roald B, Sawada T, Seeger RC, Tsuchida Y, Voute PA. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol. 1993;11(8):1466–1477. doi: 10.1200/JCO.1993.11.8.1466. [DOI] [PubMed] [Google Scholar]
- 19.Kocak H, Ackermann S, Hero B, Kahlert Y, Oberthuer A, Juraeva D, Roels F, Theissen J, Westermann F, Deubzer H, Ehemann V, Brors B, Odenthal M, Berthold F, Fischer M. Hox-C9 activates the intrinsic pathway of apoptosis and is associated with spontaneous regression in neuroblastoma. Cell Death Dis. 2013;4:e586. doi: 10.1038/cddis.2013.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prukop T, Epplen DB, Nientiedt T, Wichert SP, Fledrich R, Stassart RM, Rossner MJ, Edgar JM, Werner HB, Nave KA, Sereda MW. Progesterone antagonist therapy in a Pelizaeus-Merzbacher mouse model. Am J Hum Genet. 2014;94(4):533–546. doi: 10.1016/j.ajhg.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cohn SL, Pearson AD, London WB, Monclair T, Ambros PF, Brodeur GM, Faldum A, Hero B, Iehara T, Machin D, Mosseri V, Simon T, Garaventa A, Castel V, Matthay KK. The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol. 2009;27(2):289–297. doi: 10.1200/JCO.2008.16.6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fischer M, Bauer T, Oberthur A, Hero B, Theissen J, Ehrich M, Spitz R, Eils R, Westermann F, Brors B, Konig R, Berthold F. Integrated genomic profiling identifies two distinct molecular subtypes with divergent outcome in neuroblastoma with loss of chromosome 11q. Oncogene. 2009;29(6):865–875. doi: 10.1038/onc.2009.390. [DOI] [PubMed] [Google Scholar]
- 24.Spitz R, Oberthuer A, Zapatka M, Brors B, Hero B, Ernestus K, Oestreich J, Fischer M, Simon T, Berthold F. Oligonucleotide array-based comparative genomic hybridization (aCGH) of 90 neuroblastomas reveals aberration patterns closely associated with relapse pattern and outcome. Genes Chromosomes Cancer. 2006;45(12):1130–1142. doi: 10.1002/gcc.20376. [DOI] [PubMed] [Google Scholar]
- 25.Ehrich M, Nelson MR, Stanssens P, Zabeau M, Liloglou T, Xinarianos G, Cantor CR, Field JK, van den Boom D. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proc Natl Acad Sci U S A. 2005;102(44):15785–15790. doi: 10.1073/pnas.0507816102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oberthuer A, Berthold F, Warnat P, Hero B, Kahlert Y, Spitz R, Ernestus K, Konig R, Haas S, Eils R, Schwab M, Brors B, Westermann F, Fischer M. Customized oligonucleotide microarray gene expression-based classification of neuroblastoma patients outperforms current clinical risk stratification. J Clin Oncol. 2006;24(31):5070–5078. doi: 10.1200/JCO.2006.06.1879. [DOI] [PubMed] [Google Scholar]
- 27.Lausen B, Sauerbrei W, Schumacher M. Classification and regression trees (CART) used for the exploration of prognostic factors measured on different scales. In: Dirschedl P, Ostermann R, editors. Computational Statistics 25th Conference on Statistical Computing Edn. Heidelberg, Germany: Physica-Verlag; 1994. pp. 483–496. [Google Scholar]
- 28.Ackermann S, Goeser F, Schulte JH, Schramm A, Ehemann V, Hero B, Eggert A, Berthold F, Fischer M. Polo-like kinase 1 is a therapeutic target in high-risk neuroblastoma. Clin Cancer Res. 2011;17(4):731–741. doi: 10.1158/1078-0432.CCR-10-1129. [DOI] [PubMed] [Google Scholar]
- 29.Ehemann V, Hashemi B, Lange A, Otto HF. Flow cytometric DNA analysis and chromosomal aberrations in malignant glioblastomas. Cancer Lett. 1999;138(1–2):101–106. doi: 10.1016/S0304-3835(98)00383-8. [DOI] [PubMed] [Google Scholar]
- 30.Benayoun BA, Caburet S, Veitia RA. Forkhead transcription factors: key players in health and disease. Trends Genet. 2011;27(6):224–232. doi: 10.1016/j.tig.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 31.Hannenhalli S, Kaestner KH. The evolution of Fox genes and their role in development and disease. Nat Rev Genet. 2009;10(4):233–240. doi: 10.1038/nrg2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang B, Becanovic K, Desplats PA, Spencer B, Hill AM, Connolly C, Masliah E, Leavitt BR, Thomas EA. Forkhead box protein p1 is a transcriptional repressor of immune signaling in the CNS: implications for transcriptional dysregulation in Huntington disease. Hum Mol Genet. 2012;21(14):3097–3111. doi: 10.1093/hmg/dds132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krohn A, Seidel A, Burkhardt L, Bachmann F, Mader M, Grupp K, Eichenauer T, Becker A, Adam M, Graefen M, Huland H, Kurtz S, Steurer S, Tsourlakis MC, Minner S, Michl U, Schlomm T, Sauter G, Simon R, Sirma H. Recurrent deletion of 3p13 targets multiple tumor suppressor genes and defines a distinct subgroup of aggressive ERG fusion positive prostate cancers. J Pathol. 2013;231(1):130–141. doi: 10.1002/path.4223. [DOI] [PubMed] [Google Scholar]
- 34.Hoeller S, Schneider A, Haralambieva E, Dirnhofer S, Tzankov A. FOXP1 protein overexpression is associated with inferior outcome in nodal diffuse large B-cell lymphomas with non-germinal centre phenotype, independent of gains and structural aberrations at 3p14.1. Histopathology. 2010;57(1):73–80. doi: 10.1111/j.1365-2559.2010.03600.x. [DOI] [PubMed] [Google Scholar]
- 35.Lenz G, Wright GW, Emre NC, Kohlhammer H, Dave SS, Davis RE, Carty S, Lam LT, Shaffer AL, Xiao W, Powell J, Rosenwald A, Ott G, Muller-Hermelink HK, Gascoyne RD, Connors JM, Campo E, Jaffe ES, Delabie J, Smeland EB, Rimsza LM, Fisher RI, Weisenburger DD, Chan WC, Staudt LM. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci U S A. 2008;105(36):13520–13525. doi: 10.1073/pnas.0804295105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wlodarska I, Veyt E, De Paepe P, Vandenberghe P, Nooijen P, Theate I, Michaux L, Sagaert X, Marynen P, Hagemeijer A, De Wolf-Peeters C. FOXP1, a gene highly expressed in a subset of diffuse large B-cell lymphoma, is recurrently targeted by genomic aberrations. Leukemia. 2005;19(8):1299–1305. doi: 10.1038/sj.leu.2403813. [DOI] [PubMed] [Google Scholar]
- 37.Li D, Mei H, Qi M, Yang D, Zhao X, Xiang X, Pu J, Huang K, Zheng L, Tong Q. FOXD3 is a novel tumor suppressor that affects growth, invasion, metastasis and angiogenesis of neuroblastoma. Oncotarget. 2013;4(11):2021–2044. doi: 10.18632/oncotarget.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Santo EE, Ebus ME, Koster J, Schulte JH, Lakeman A, van Sluis P, Vermeulen J, Gisselsson D, Ora I, Lindner S, Buckley PG, Stallings RL, Vandesompele J, Eggert A, Caron HN, Versteeg R, Molenaar JJ. Oncogenic activation of FOXR1 by 11q23 intrachromosomal deletion-fusions in neuroblastoma. Oncogene. 2012;31(12):1571–1581. doi: 10.1038/onc.2011.344. [DOI] [PubMed] [Google Scholar]
- 39.Wang Z, Park HJ, Carr JR, Chen YJ, Zheng Y, Li J, Tyner AL, Costa RH, Bagchi S, Raychaudhuri P. FoxM1 in tumorigenicity of the neuroblastoma cells and renewal of the neural progenitors. Cancer Res. 2011;71(12):4292–4302. doi: 10.1158/0008-5472.CAN-10-4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Obexer P, Hagenbuchner J, Unterkircher T, Sachsenmaier N, Seifarth C, Bock G, Porto V, Geiger K, Ausserlechner M. Repression of BIRC5/survivin by FOXO3/FKHRL1 sensitizes human neuroblastoma cells to DNA damage-induced apoptosis. Mol Biol Cell. 2009;20(7):2041–2048. doi: 10.1091/mbc.E08-07-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xue L, Yue S, Zhang J. FOXP1 has a low expression in human gliomas and its overexpression inhibits proliferation, invasion and migration of human glioma U251 cells. Mol Med Rep. 2014;10(1):467–472. doi: 10.3892/mmr.2014.2197. [DOI] [PubMed] [Google Scholar]
- 42.Briet M, Schiffrin EL. Aldosterone: effects on the kidney and cardiovascular system. Nat Rev Nephrol. 2010;6(5):261–273. doi: 10.1038/nrneph.2010.30. [DOI] [PubMed] [Google Scholar]
- 43.Lamant L, de Reynies A, Duplantier MM, Rickman DS, Sabourdy F, Giuriato S, Brugieres L, Gaulard P, Espinos E, Delsol G. Gene-expression profiling of systemic anaplastic large-cell lymphoma reveals differences based on ALK status and two distinct morphologic ALK + subtypes. Blood. 2007;109(5):2156–2164. doi: 10.1182/blood-2006-06-028969. [DOI] [PubMed] [Google Scholar]
- 44.Kim YM, Cho M. Activation of NADPH oxidase subunit NCF4 induces ROS-mediated EMT signaling in HeLa cells. Cell Signal. 2014;26(4):784–796. doi: 10.1016/j.cellsig.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 45.Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102(1):43–53. doi: 10.1016/S0092-8674(00)00009-X. [DOI] [PubMed] [Google Scholar]
- 46.Bazenet CE, Mota MA, Rubin LL. The small GTP-binding protein Cdc42 is required for nerve growth factor withdrawal-induced neuronal death. Proc Natl Acad Sci U S A. 1998;95(7):3984–3989. doi: 10.1073/pnas.95.7.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Raval A, Tanner SM, Byrd JC, Angerman EB, Perko JD, Chen SS, Hackanson B, Grever MR, Lucas DM, Matkovic JJ, Lin TS, Kipps TJ, Murray F, Weisenburger D, Sanger W, Lynch J, Watson P, Jansen M, Yoshinaga Y, Rosenquist R, de Jong PJ, Coggill P, Beck S, Lynch H, de la Chapelle A, Plass C. Downregulation of death-associated protein kinase 1 (DAPK1) in chronic lymphocytic leukemia. Cell. 2007;129(5):879–890. doi: 10.1016/j.cell.2007.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mao L, Ding J, Zha Y, Yang L, McCarthy BA, King W, Cui H, Ding HF. HOXC9 links cell-cycle exit and neuronal differentiation and is a prognostic marker in neuroblastoma. Cancer Res. 2011;71(12):4314–4324. doi: 10.1158/0008-5472.CAN-11-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zha Y, Ding E, Yang L, Mao L, Wang X, McCarthy BA, Huang S, Ding HF. Functional dissection of HOXD cluster genes in regulation of neuroblastoma cell proliferation and differentiation. PLoS One. 2012;7(8):e40728. doi: 10.1371/journal.pone.0040728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Soltani MH, Pichardo R, Song Z, Sangha N, Camacho F, Satyamoorthy K, Sangueza OP, Setaluri V. Microtubule-associated protein 2, a marker of neuronal differentiation, induces mitotic defects, inhibits growth of melanoma cells, and predicts metastatic potential of cutaneous melanoma. Am J Pathol. 2005;166(6):1841–1850. doi: 10.1016/S0002-9440(10)62493-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Andersson G, Pahlman S, Parrow V, Johansson I, Hammerling U. Activation of the human NPY gene during neuroblastoma cell differentiation: induced transcriptional activities of AP-1 and AP-2. Cell Growth Differ. 1994;5(1):27–36. [PubMed] [Google Scholar]
- 52.Craig VJ, Cogliatti SB, Imig J, Renner C, Neuenschwander S, Rehrauer H, Schlapbach R, Dirnhofer S, Tzankov A, Muller A. Myc-mediated repression of microRNA-34a promotes high-grade transformation of B-cell lymphoma by dysregulation of FoxP1. Blood. 2011;117(23):6227–6236. doi: 10.1182/blood-2010-10-312231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buckley PG, Alcock L, Bryan K, Bray I, Schulte JH, Schramm A, Eggert A, Mestdagh P, De Preter K, Vandesompele J, Speleman F, Stallings RL. Chromosomal and microRNA expression patterns reveal biologically distinct subgroups of 11q- neuroblastoma. Clin Cancer Res. 2010;16(11):2971–2978. doi: 10.1158/1078-0432.CCR-09-3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scaruffi P, Stigliani S, Moretti S, Coco S, De Vecchi C, Valdora F, Garaventa A, Bonassi S, Tonini GP. Transcribed-Ultra Conserved Region expression is associated with outcome in high-risk neuroblastoma. BMC Cancer. 2009;9:441. doi: 10.1186/1471-2407-9-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Spitz R, Hero B, Ernestus K, Berthold F. Deletions in chromosome arms 3p and 11q are new prognostic markers in localized and 4 s neuroblastoma. Clin Cancer Res. 2003;9(1):52–58. [PubMed] [Google Scholar]
- 56.Molenaar JJ, Koster J, Zwijnenburg DA, van Sluis P, Valentijn LJ, van der Ploeg I, Hamdi M, van Nes J, Westerman BA, van Arkel J, Ebus ME, Haneveld F, Lakeman A, Schild L, Molenaar P, Stroeken P, van Noesel MM, Ora I, Santo EE, Caron HN, Westerhout EM, Versteeg R. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature. 2012;483(7391):589–593. doi: 10.1038/nature10910. [DOI] [PubMed] [Google Scholar]
- 57.Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M, Kiezun A, Kim J, Lawrence MS, Lichenstein L, McKenna A, Pedamallu CS, Ramos AH, Shefler E, Sivachenko A, Sougnez C, Stewart C, Ally A, Birol I, Chiu R, Corbett RD, Hirst M, Jackman SD, Kamoh B, Khodabakshi AH, Krzywinski M, Lo A, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. 2013;45(3):279–284. doi: 10.1038/ng.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Katoh M, Igarashi M, Fukuda H, Nakagama H. Cancer genetics and genomics of human FOX family genes. Cancer Lett. 2013;328(2):198–206. doi: 10.1016/j.canlet.2012.09.017. [DOI] [PubMed] [Google Scholar]
- 59.Shigekawa T, Ijichi N, Takayama S, Tsuda H, Ikeda K, Horie K, Osaki A, Saeki T, Inoue S. FOXP1 as a Potential ER Coregulator in Human Breast Cancer. Cancer Res. 2009;69(24):757S-757S. [Google Scholar]
- 60.van Boxtel R, Gomez-Puerto C, Mokry M, Eijkelenboom A, van der Vos KE, Nieuwenhuis EE, Burgering BM, Lam EW, Coffer PJ. FOXP1 acts through a negative feedback loop to suppress FOXO-induced apoptosis. Cell Death Differ. 2013;20(9):1219–1229. doi: 10.1038/cdd.2013.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gabut M, Samavarchi-Tehrani P, Wang X, Slobodeniuc V, O’Hanlon D, Sung HK, Alvarez M, Talukder S, Pan Q, Mazzoni EO, Nedelec S, Wichterle H, Woltjen K, Hughes TR, Zandstra PW, Nagy A, Wrana JL, Blencowe BJ. An alternative splicing switch regulates embryonic stem cell pluripotency and reprogramming. Cell. 2011;147(1):132–146. doi: 10.1016/j.cell.2011.08.023. [DOI] [PubMed] [Google Scholar]
- 62.Quesada V, Conde L, Villamor N, Ordonez GR, Jares P, Bassaganyas L, Ramsay AJ, Bea S, Pinyol M, Martinez-Trillos A, Lopez-Guerra M, Colomer D, Navarro A, Baumann T, Aymerich M, Rozman M, Delgado J, Gine E, Hernandez JM, Gonzalez-Diaz M, Puente DA, Velasco G, Freije JM, Tubio JM, Royo R, Gelpi JL, Orozco M, Pisano DG, Zamora J, Vazquez M, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2012;44(1):47–52. doi: 10.1038/ng.1032. [DOI] [PubMed] [Google Scholar]
- 63.Pajer P, Pecenka V, Kralova J, Karafiat V, Prukova D, Zemanova Z, Kodet R, Dvorak M. Identification of potential human oncogenes by mapping the common viral integration sites in avian nephroblastoma. Cancer Res. 2006;66(1):78–86. doi: 10.1158/0008-5472.CAN-05-1728. [DOI] [PubMed] [Google Scholar]
- 64.Datta J, Kutay H, Nasser MW, Nuovo GJ, Wang B, Majumder S, Liu CG, Volinia S, Croce CM, Schmittgen TD, Ghoshal K, Jacob ST. Methylation mediated silencing of MicroRNA-1 gene and its role in hepatocellular carcinogenesis. Cancer Res. 2008;68(13):5049–5058. doi: 10.1158/0008-5472.CAN-07-6655. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 65.Rao DS, O’Connell RM, Chaudhuri AA, Garcia-Flores Y, Geiger TL, Baltimore D. MicroRNA-34a perturbs B lymphocyte development by repressing the forkhead box transcription factor Foxp1. Immunity. 2010;33(1):48–59. doi: 10.1016/j.immuni.2010.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Streubel B, Vinatzer U, Lamprecht A, Raderer M, Chott A. T(3;14) (p14.1;q32) involving IGH and FOXP1 is a novel recurrent chromosomal aberration in MALT lymphoma. Leukemia. 2005;19(4):652–658. doi: 10.1038/sj.leu.2403644. [DOI] [PubMed] [Google Scholar]
Pre-publication history
- The pre-publication history for this paper can be accessed here: http://www.biomedcentral.com/1471-2407/14/840/prepub
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1: Information on oligonucleotides used as primers for DNA methylation analysis. (XLS 38 KB)
Additional file 2: Table S2: Genes up- or down-regulated after FOXP1 re-expression in IMR-32, CHP-212 and SK-N-BE(2) cells as determined by microarray gene expression analysis. (XLSX 438 KB)