

Abstract
The human gut microbiome is a major contributor to human metabolism and health, yet the metabolic processes that are carried out by various community members, the way these members interact with each other and with the host, and the impact of such interactions on the overall metabolic machinery of the microbiome have not yet been mapped. Here, we discuss recent efforts to study the metabolic inner workings of this complex ecosystem. We will specifically highlight two interrelated lines of work, the first aiming to deconvolve the microbiome and to characterize the metabolic capacity of various microbiome species, and the second aiming to utilize computational modeling to infer and study metabolic interactions between these species.
Introduction
The human microbiome – the collection of microorganisms that colonize the human body – plays an important role in several physiological processes and ultimately in our health (Clemente et al., 2012; White et al., 2011; Zarco et al., 2012). Of the many communities that comprise the microbiome, the one that resides in our gut is especially intertwined with our own physiology and is tightly linked to our metabolism. The metabolic capacities of this gut microbiome allows us, for example, to harvest otherwise inaccessible energy from our diet (Bäckhed et al., 2004; Gill et al., 2006; Thomas et al., 2011; Xu et al., 2003) and to biotransform various xenobiotics (Clayton et al., 2009; Maurice et al., 2013; Sousa et al., 2008; Ursell and Knight, 2013). To a large extent, the microbiome can therefore be viewed as a human organ, with specific tasks, operation modes, and capacities (Baquero and Nombela, 2012; O’Hara and Shanahan, 2006).
Considering its profound impact on our own metabolism, it is perhaps not surprising that the gut microbiome has been associated with several metabolic diseases including obesity (Ley et al., 2005, 2006; Turnbaugh et al., 2006, 2008; Upadhyay et al., 2012) and type 2 diabetes (Qin et al., 2012). The discovery of such associations and the growing appreciation for the impact that the gut microbiome has on our health have promoted extensive efforts to study the microbiome and to characterize its taxonomic and genetic composition. Much of this effort has been focused on identifying compositional shifts associated with diseases or with specific perturbations (Theriot et al., 2014). Such studies provide crucial clues into the way the microbiome affects our health and contributes to various host phenotypes. To date, however, much less is known about the inner workings of the microbiome and the various metabolic mechanisms that are at play in this ecosystem.
Two major obstacles impede progress in particular: The first concerns the inherently inextricable nature of the microbiome and of much of the data generated to study it. The gut microbiota of each individual comprises hundreds to thousands of taxa, each encoding a unique collection of enzymatic genes and carrying out a unique set of metabolic reactions. An important prerequisite to any mechanistic understanding of the microbiome is therefore a detailed characterization of the capabilities of each species. Unfortunately, however, a species-level analysis is often challenging in the context of naturally occurring communities, as many community members resist isolation and culturing efforts. In fact, it was observed over a century ago that when plating microbial samples, most species do not grow to form colonies (Winterberg, 1898), and today it is estimated that only ~1% of bacterial taxa are readily cultivable under normal conditions in vitro (Vartoukian et al., 2010). Although the fraction of culturable taxa may be higher in human-associated communities, isolation and culturing challenges may still hinder the study of key community members and of the community as a whole. These challenges, in turn, have rendered shotgun metagenomic sequencing the method of choice for studying the microbiome and for many analyses of its functional capacity. Metagenomic technologies bypass the need to isolate and culture individual species, analyzing genomic material obtained directly from a mixed sample of species and characterizing the aggregated gene content in the community (Schloss and Handelsman, 2005; Tringe et al., 2005). These technologies therefore generate convolved data, reflecting ‘community-level’ capacity rather than species-specific capabilities. Additional meta-omic technologies, such as meta-transcriptomics (Frias-Lopez et al., 2008; Turner et al., 2013; Urich et al., 2008), meta-proteomics (Erickson et al., 2012; Keiblinger et al., 2012; Klaassens et al., 2007; Kolmeder and Vos, 2014), and meta-metabolomics (Lu et al., 2014; McHardy et al., 2013; Ridaura et al., 2013; Theriot et al., 2014; Weir et al., 2013), have also been introduced, again providing community-level measures of transcripts, proteins, and metabolites, without necessarily relating these measures to specific community members (Segata et al., 2013).
The second challenge stems from the inherent complexity of the microbiome ecosystem. Complexity, especially in biology, is a product not simply of the number of parts a system is composed of, but rather of the complex and often non-linear way in which these parts interact (Weaver, 1948). Such interactions play an essential part in the microbiome clockwork and span multiple organizational scales. Within each species, the orchestrated interconnected activity of hundreds and often thousands of enzymes, both within the boundaries of a single cell and across different cells from this species population, bestows each species with a complex set of metabolic capabilities. Each species further competes for and exchanges metabolites with other taxa in its vicinity and may modulate its metabolic activity in response to environmental shifts or to the activity of other taxa in the community (Little et al., 2008; Schink, 2002). The microbiome, as a whole, in turn interacts with the host metabolism, immune response, and diet. This extremely complex web of interactions is an essential part of the microbiome’s capacity, activity, and dynamics, and it is therefore hard to imagine that a principled understanding of this multilevel and hierarchical complex system could be gained without accounting for such interactions (Borenstein, 2012). Furthermore, the composition of taxa in the microbiome can vary substantially over even short time scales (Dethlefsen and Relman, 2011), and with it the set of genes in the metagenome and the overall functional capacity of the community. Diet, for example, was shown to rapidly induce compositional shifts associated with obesity (David et al., 2014), but microbiota transplants were able to induce and subsequently rescue obesity-like symptoms (Ridaura et al., 2013). This plasticity of the microbiome and the bidirectional interaction between the composition of the microbiome and the state of the host make discerning cause and effect with respect to compositional shifts associated with a disease state an extremely challenging task (Duncan et al., 2008; Jumpertz et al., 2011; Schwiertz et al., 2010).
In this perspective, we will review and discuss recent efforts to address these challenges with an eye towards gaining an improved, systems-level, mechanistic understanding of the microbiome. We will specifically describe two complementary lines of work. The first aims to untangle the microbiome and to obtain species-specific characterization of metabolic capacities. We will discuss various approaches to isolate and sequence individual species as well as methods for deconvolving the metagenome. The second line of work aims to model the microbiome’s metabolic machinery, and to infer metabolic interactions between taxa using various in-silico metabolic modeling frameworks. Combined, this body of work provides a first peek into the inner workings of the microbiome and lays the foundation for a comprehensive, system-level understanding of its function and dynamics.
Untangling the microbiome: Inferring the metabolic capacity of community members
From cells to genomes to genes
When studying the metabolic capacity of microbial species, each species is often viewed simply as the collection of metabolic reactions it can catalyze. Attention is then focused on determining which annotated gene families or gene orthology groups (such as those defined by KEGG, Kanehisa et al., 2012) are encoded in its genome, ignoring any other sequence-level information that may be embedded in the genome. Many comparative genomic analyses, as well as metabolic modelling frameworks, use this gene-centered representation as a point of departure. In the context of metabolism-focused microbiome research, this representation provides a simple description of the various players in the ecosystem and their capabilities. It allows, for example, for studying the division of labor between different community members or the potential for competition and syntrophy.
Clearly, the convoluted nature of microbial communities makes such species-specific characterization hard to obtain, calling for the development of methods for untangling mixed communities. Importantly, however, untangling can be performed at multiple levels, ranging from a physical isolation of microbial cells, to an assembly-based sequence-level ‘isolation’, to a gene-centered metagenomic deconvolution (Figure 1). Below, we will briefly discuss such efforts, as well as the strengths and limitations of deconvolving the microbiome at the various levels.
Figure 1. A culture-independent metagenomic pipeline vs. a culture-based genomic pipeline.
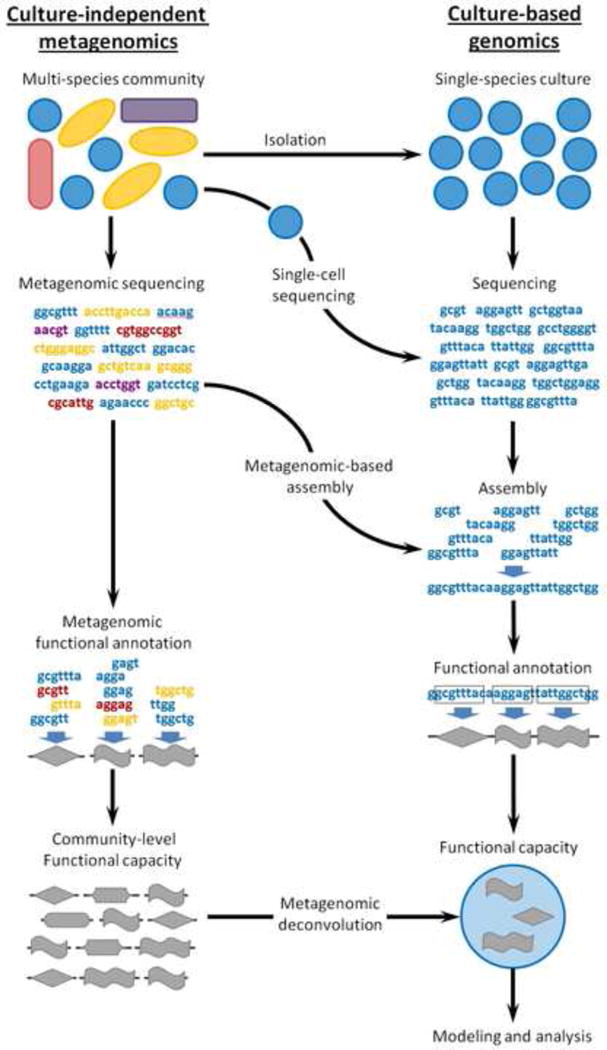
Microbiome untangling can be accomplished at several different levels, crossing over from the metagenomic pipeline into the single-species genomic pipeline at various stages as discussed in the text. Specifically, microbial cells can be physically isolated and cultured or sequenced using single-cell genomics. Shotgun metagenomic short reads can also be assembled de novo into genomes or large contigs using binning and metagenomic-based assembly. Finally, community-level functional profiles can be mathematically deconvolved into species specific functional profiles. Ultimately, the functional capacity of various community members can be characterized, facilitating species-level modeling and analysis.
Isolating and culturing ‘unculturable’ community members
Traditional, culture-based techniques have been the cornerstone of microbiology. Accordingly, the most straightforward way to map the metabolic capacities of a given community member involves the physical isolation of this species from the community and subsequent culturing. The genome of the cultured isolate can then be easily extracted and sequenced, and the various genes it encodes can be called and annotated (Delcher et al., 2007; Hyatt et al., 2010). Importantly, isolation and culturing can further facilitate extensive biochemical and experimental profiling to study the physiology of the cultured isolate and to assign functions to the many unknown enzymes encoded by its genome, augmenting any genomic-based predictions. As discussed above, however, efforts to isolate and culture individual community members under normal conditions often fail, suggesting that more sophisticated experimental techniques are required (Stewart, 2012).
Indeed, several studies have recently introduced improved culturing methods in order to grow such previously uncultivated bacteria. Using various anaerobic culturing methods on a mixture of microbes from the human gut community, one study demonstrated it was possible to recover ~56% of species-level taxa in the sample (Goodman and Kallstrom, 2011). Diluting the mixed culture further enabled obtaining 1,172 taxonomically defined isolates, which in principle could be sequenced and annotated. A different study used an in vivo cultivation method followed by plating on anaerobic medium to isolate 31 species from the human oral community, likely representing many unknown species and genera (Sizova et al., 2012).
More recently, several single-cell sequencing techniques have been introduced, bypassing the challenges involved in culturing many bacterial species. In these methods, individual cells are separated from the sample using optical trapping on a microfluidic device or fluorescent-aided cell sorting techniques (Rodrigue et al., 2009), and are subsequently lysed, amplified, and sequenced without cultivation. Such an approach was used, for example, to study hot spring sediments and to obtain two nearly complete genomes of a candidate species from a presumed novel bacterial phylum (Dodsworth et al., 2013). Examining the recent oil spill in the Gulf of Mexico, another study has constructed a draft genome of a species from the order Oceanospirillales and has examined its metabolic repertoire (Mason et al., 2012). A recent study, aimed directly at sequencing the ‘dark matter’ in the microbial phylogeny, used a similar single-cell approach to isolate and sequence 201 previously uncultivated bacterial and archaeal strains, uncovering novel metabolic features such as an archaeal-type purine synthesis proteins in bacteria (Rinke et al., 2013). Additional efforts to expand and fill-in the gaps in the microbial tree of life are currently ongoing (Wu et al., 2009).
Assembly of genomes from metagenomes
While the methods described above for isolating, culturing, and ultimately sequencing individual community members are clearly expanding the range of bacterial and archaeal genomes available, some microbial species are still likely to evade isolation efforts. Moreover, such methods are often applied on a small scale and focus on only a single, or a few microbial species at a time. A promising alternative therefore avoids isolating an individual species from the community at the physical level altogether, and instead aims to isolate its genome from the metagenome at the sequence level. This ‘isolated’ genome can then be annotated and analyzed to infer the metabolic capabilities of the species.
Various studies, for example, have used standard de novo genome assembly methods (Baker, 2012; Flicek and Birney, 2009) to assemble genomes or large genomic contigs directly from shotgun metagenomic samples (Handley et al., 2014). This approach assumes that reads originating from a given high-coverage genome in the sample will assemble successfully into contigs and thereby separate from other reads. Clearly, however, applying algorithms developed originally for assembling reads from a single-species sample without adjusting them to the complexities of metagenomic data may produce erroneous results. For example, uneven representation of species in the sample could cause genomic regions in highly abundant species to be misidentified as repeats of a single genome (Pop, 2009). Similarly, variations between closely related species in the sample could cause various assemblers to construct distinct contigs for each variant, resulting in a more fragmented assembly (Simmons et al., 2008). Recently, metagenome-specific assemblers have been developed (Beitel et al., 2014; Burton et al., 2014; Iverson et al., 2012; Lai et al., 2012; Laserson et al., 2011; Namiki et al., 2012; Peng et al., 2012; Treangen et al., 2013), more rigorously addressing these issues and accounting for the specifics of metagenomic data. Many such assemblers, for example, aim to first cluster reads or contigs into bins based on nucleotide content, abundance, graph connectivity properties, mate-pair sequences, or spatial proximity, and then use these clusters to reconstruct genome scaffolds.
Using such a binning-based approach, several studies have indeed successfully assembled various genomes of interest. In what was perhaps the first attempt to assemble a genome directly from shotgun metagenomic data, GC content was used to partition assembled scaffolds from an acid mine drainage metagenomic sample into bins (Tyson et al., 2004). These bins were then used to construct near-complete genomes of Leptospirillum group II and Ferroplasma type II. In a study of the surface seawater metagenome, mate-pairing information was used to link assembled contigs into graphs, which were then split using nucleotide composition and read-coverage statistics into scaffolds (Iverson et al., 2012). These scaffolds were eventually binned using tetra-nucleotide statistics into several near-complete genomes, including one closed genome representing a previously uncultured marine group II Euryarchaeota. In the context of the human gut microbiome, two studies of the early colonization of the infant gut (Brown et al., 2013; Sharon et al., 2012), binned contigs based on time-series abundance data rather than on nucleotide content, obtaining ~20 complete genomes and studying their predicted metabolic functions, whereas another study combined metagenomic data from the human gut and groundwater to assemble genomes from a new candidate phylum, Melainabacteria (Di Rienzi et al., 2013).
Computational inference of the genomic content of community-members
While metagenomic-based genome assembly methods are likely to expand with the development of long read and deeper coverage sequencing technologies, they may not be readily applicable in every setting. Even with increased coverage and longer reads it may be hard, for example, to assemble the genomes of low abundance species, especially in highly diverse communities or when closely related reference genomes are not available. Some computational methods have therefore been introduced, bypassing the assembly process completely and directly predicting the set of genes (or gene families) encoded by a yet-to-be-sequenced species. One such method, termed ‘metagenomic deconvolution’, specifically aims to decompose metagenomic community-level gene content data (e.g., obtained by mapping shotgun metagenomic reads to known genes families or orthology groups) into taxa-specific gene profiles (Carr et al., 2013). This method relies on the fact that the gene content in a given metagenome is a linear combination of the gene content of the member species and that this relationship can be mathematically modeled when both the taxonomic and the gene content profiles of a given sample are available. Metagenomic deconvolution then uses this model to analyze co-variation between the abundances of the various taxa and the abundances of the various gene families across a set of metagenomic samples, and identifies the most likely association between genes and their taxa of origin. This approach was shown to successfully reconstruct species-level gene content of microbiome taxa both in simulated data and in samples from the human microbiome project. For a mathematical formulation of the deconvolution framework see Carr et al., 2013 (and see also Shen-Orr and Gaujoux, 2013, for a general review of deconvolution approaches).
Other computational methods applied different approaches to predict the set of gene families encoded by various, yet-to-be-sequenced species. Several such methods, for example, rely on evolutionary gene content conservation, assuming that phylogenetically related species (e.g., those with similar 16S sequences) encode a similar set of genes (Zaneveld et al., 2010). With this assumption in mind, several studies mapped prevalent yet-to-be-sequenced community members to their nearest sequenced reference genome (using 16S similarity), estimating the functional capacity of each of these species (Morgan et al., 2012; Muegge et al., 2011). Taking this approach further, a computational framework has been recently introduced to predict the gene content of organisms that have not yet been sequenced based on the set of genes found in sequenced relatives and on evolutionary modeling (Langille et al., 2013). Specifically, this framework uses a phylogenetic tree and various ancestral state reconstruction methods (Csuros, 2010; Paradis et al., 2004) to infer the gene content of ancestral species and ultimately the gene content of all species found in a given community. Coupling this inference method with species abundance data, this framework was shown to predict the functional content of the metagenome as a whole.
Modeling microbiome metabolism and metabolic interactions
Why model metabolism?
As discussed above, characterizing the metabolic capacity of each community member is not sufficient to explain the complex inner workings of the microbiome. Rather, the web of interactions between these community members and the way they impact community dynamics need to be mapped in order to gain a fundamental understanding of the microbiome. Such interactions can be probed in various ways. Studying simple synthetic or naturally occurring communities, both in vitro (Kim et al., 2008; Kolenbrander, 2011; McDonald et al., 2013; Park et al., 2011; Petrof et al., 2013; Rakoff-Nahoum et al., 2014; Trosvik et al., 2010) and in vivo (e.g., using gnotobiotic animal models) (Faith et al., 2011; Lee et al., 2013; Mahowald et al., 2009; Roeselers et al., 2011) can provide valuable insights into the impact the activity of one species may have on the growth of another. Notably, the complexity of these experimental communities can vary from very few taxa (e.g., Mahowald et al., 2009; Trosvik et al., 2010) to representations of the entire microbiome (e.g., McDonald et al., 2013). Such assays, however, often do not provide any information about the mechanisms that are at play and may not offer a principled understanding of the processes underlying specific microbe-microbe or microbe-host interactions, particularly when investigating complex communities. Furthermore, since these approaches tend to be labor intensive and costly, they cannot be easily applied to an arbitrarily wide array of distinct communities. Similarly, analyzing the co-occurrence of species across samples (Faust et al., 2012) provides crucial clues into the presence of non-neutral structuring forces, but may fail to elucidate underlying mechanisms of interaction.
Below, we therefore discuss an alternative, modeling-based approach for studying species interaction. Importantly, we focus mainly on genome-scale metabolic modeling and on studies aiming specifically to provide a mechanistic understanding of such interactions at various scales (Levy and Borenstein, 2014). Other, more phenomenological modeling frameworks have been also introduced to quantify, for example, the strength of interactions or to predict microbiome dynamics (e.g., Marino et al., 2014; Stein et al., 2013), yet such models commonly ignore underlying mechanisms of interaction. Genome-scale metabolic models, by contrast, have been instrumental in mapping genotype to phenotype and in elucidating mechanisms of microbial behavior at the single-species level (Borenstein et al., 2008; Reed and Palsson, 2003), and developing methods to extend such frameworks and to model multi-species systems (such as the human microbiome) is therefore a promising route toward a more comprehensive understanding of species interactions (Borenstein, 2012). Multi-species metabolic models have the potential not only to infer unknown modes of interaction, but also to provide a detailed account of the underlying metabolic machinery that contributes to such interactions. In the context of the human microbiome, such models are also critical to the development of targeted intervention strategies and the cogent design of synthetic microbiota (Greenblum et al., 2013). Previous attempts at synthetic microbiome design utilized either random combinations of or relatively minor modification to existing genomes (Faith et al., 2014; Kotula et al., 2014; Petrof et al., 2013), and in turn the internal operation of these systems is often not well understood.
Selecting a modeling framework
Over the past few decades, a plethora of metabolic modeling frameworks have been introduced. These frameworks vary greatly in their scale and resolution, in the assumptions underlying the models, and in the information required to reconstruct them. Selection of an optimal modeling strategy is therefore critical and should clearly depend on the research objective and on the nature and scope of available data. In many cases, framework selection may entail a tradeoff between various modeling goals. For example, a simple modeling framework will often offer limited predictive power but can be easily applied to large communities and can utilize a wide variety of data. On the other hand, a more sophisticated framework may offer high-resolution predictions but may only be applied on a smaller scale or to simpler communities. A modeling framework can also be chosen according to the specific questions to be addressed and to the context of the study. For example, to address questions concerning community metabolism, models may be reconstructed at the metagenome-scale (Greenblum et al., 2012), whereas genome-scale models may be used to describe metabolic exchanges between community members or the specific contribution of each member to the community at large. Below, we describe recent advances in modeling metabolic interactions and discuss considerations of complexity and scale.
Network-based models of species interaction
One of the simplest, yet most powerful approaches for modeling microbial metabolism employs a naïve, network-based model to represent the set of biochemical reactions carried out by a given species and focuses on studying the connectivity and topology of such networks as a way to obtain insights into the species’ metabolism and ecology (Borenstein et al., 2008; Jeong et al., 2000; Kreimer et al., 2008; Levy and Borenstein, 2012; Stelling et al., 2002; Wunderlich and Mirny, 2006). In such network-based models, parameters such as enzyme kinetics, reaction fluxes, and stoichiometry are often disregarded in favor of a simpler, connectivity-focused representation. While these simplifications potentially reduce the predictive power of the model, network-based models are easy to reconstruct, require minimal information, and can therefore be applied on a very large scale – a critical feature for modeling complex microbiome systems.
Recently, several studies have introduced preliminary attempts to expand such network-based frameworks, moving beyond models of a single species and aiming to model multi-species microbial communities and to obtain insights into their behavior and assembly. Some of these studies, for example, developed methods for integrating several genome-scale metabolic networks and for inferring cross-species metabolic interactions (Figure 2A). This approach has been used, for example, to study host-parasite interactions (Borenstein and Feldman, 2009), bacterial ecological strategies (Freilich et al., 2009), or the global organization of bacterial co-occurrence interactions (Freilich et al., 2010). Focusing specifically on the human gut microbiome, a recent study used a similar approach to reconstruct genome-scale metabolic network models of >150 gut dwelling species and introduced a graph theory-based method to predict potential metabolic competition and syntrophy between each pair of species (Levy and Borenstein, 2013). Comparing these predicted interactions with the tendency of species to co-occur in the human gut microbiome (as measured through metagenomic sequencing), this study was able to reveal organizational forces that may govern the assembly of the gut microbiome. Specifically, it was shown that species tend to co-occur with their strongest competitors, suggesting that habitat filtering is a major determinant of community assembly. Importantly, analyzing the metagenome through the lens of genome-scale metabolic models, this study was able to address questions that metagenomic-based studies cannot. Similar models that directly link the network topologies of interacting species can further identify specific metabolic exchanges between microbial species, or between such species and the host (Cottret et al., 2008, 2010).
Figure 2. Alternative multi-species modeling frameworks.
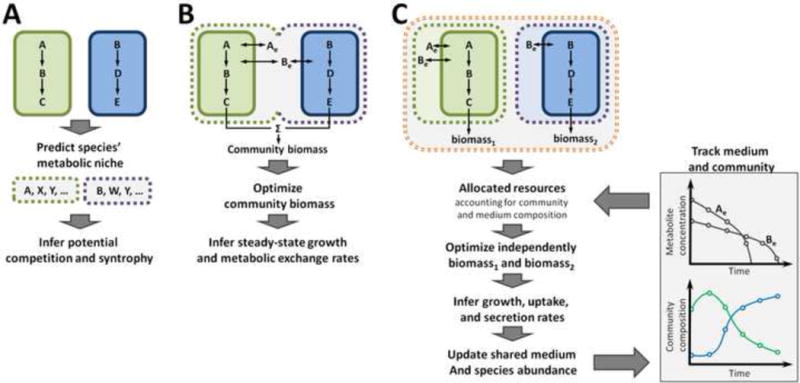
(A) Simple network-based models can be used to predict the metabolic niche of each species (e.g., using the algorithm introduced in Borenstein et al., 2008). The potential for competition and syntrophy between a pair of species can be inferred by comparing their predicted niches (Levy and Borenstein, 2013). (B) A common compartmentalization scheme in multi-species constraint-based models includes a separate compartment for each species, a shared medium compartment, and explicit shuttle reactions. Community objective is often defined as a weighted sum of the species’ biomass (see, for example, Stolyar et al., 2007). (C) A dynamics-based multi-species model, as introduced, for example, in Chiu et al., 2014. The growth of each species is independently optimized according to nutrient availability and allocated resources. This optimization step is used to infer the growth rate of each species, as well as uptake and secretion rates. These are used to update the composition of the shared medium and of the community. By iterating this process, community temporal dynamics can be tracked.
An additional benefit of this simple, network-based representation of metabolism is that it can be used to model metabolic processes at many different scales, including the scale of the microbiome as a whole. Indeed, network-based models have been applied successfully to a wide range of meta-omic analyses, revealing salient characteristics of the human microbiome. These metagenome-scale models pool all the metabolic reactions that can be catalyzed by the microbiome with no regard for organism boundaries. For example, in one of the first studies to introduce this metagenomic-systems biology framework, Greenblum et al. projected enzyme abundances from human gut microbiome metagenomic-data onto a metagenome-scale metabolic network (Greenblum et al., 2012), demonstrating that microbiome enzymes associated with obesity and with inflammatory bowel disease tend to be located at the periphery of the network. These enzymes represent ‘contact points’ between the community and the host, and conceivably would have more influence on or be more influenced by the state of the host. Applying this framework to other meta-omic data could similarly provide insight not available without metabolic modeling. One study, for example, combined network topology with meta-metabolomics to identify modules differentially abundant between preand post-small bowel transplant communities, providing a more comprehensive picture of how the human gut community responds to major ecological changes (Hartman et al., 2009). Other studies used meta-transcriptome derived networks to identify pathways not previously associated with periodontal disease (Jorth et al., 2014), or alternative pathways to biomass production (Xiong et al., 2012).
Constraint-based models of species interaction
A fundamentally different and complementary approach for modeling metabolic interactions between microbial species utilizes the well-established framework of constraint-based modeling (Reed and Palsson, 2003). Such models represent metabolic stoichiometry and known bounds on metabolic fluxes as a set of mathematical constraints and identify a distribution of fluxes that meets the various constraints and that optimizes some predefined objective (Price et al., 2003). Flux-Balance Analysis (FBA), for example, aims to infer the flux through each reaction by optimizing biomass production, ultimately predicting various organismal phenotypes such as growth rate and metabolite uptake and secretion. Often, constraint-based models are thoroughly curated and rigorously validated, thereby sacrificing broad applicability in favor of predictive and descriptive power. A detailed description of the theoretical basis and mathematical formulation of FBA can be found in Orth et al., 2010.
While such constraint-based models have proved extremely successful in predicting the behavior of single species (Edwards et al., 2001), several crucial elements in their formulation must be extended before they can be applied to model multi-species systems. One such element concerns the compartmentalization of the various reactions included in the model (Taffs et al., 2009). In the first multi-species FBA model of microbial metabolism, which aimed to study the interaction between sulfate-reducing bacteria and methanogenic archaea (commonly found in the cow rumen), the stoichiometry matrix was expanded to represent each species as a separate compartment (Stolyar et al., 2007). Metabolic exchange between the two species was then represented by external shuttle reactions between these compartments through a third compartment representing a shared environment (Figure 2B). This compartmentalization approach became standard and was later used in several other multi-species FBA models (e.g., Freilich et al., 2011; Klitgord and Segrè, 2010; Wintermute and Silver, 2010). One such study has utilized a flux balance approach to predict metabolic interaction among >100 species, demonstrating a close interplay between ecological patterns and the potential for species to compete or cooperate (Freilich et al., 2011). This study further demonstrated that cooperative interactions tend to be unidirectional but that species often form cooperative loops, benefiting all species involved.
An additional modeling element that can greatly influence the applicability and interpretation of an FBA model is the choice of objective function. In the first multi-species model described above, the community objective was defined as a weighted sum of the species’ biomass, as determined from empirically measured abundance ratios. However, since growth-rate ratios are typically unknown, more generally applicable solutions have been suggested. The OptCom framework (Zomorrodi and Maranas, 2012) utilizes a multi-level objective function, wherein individual species growth represents an inner objective and total community growth represents an outer objective. A global solution can then be found via bilinear optimization. A carefully defined objective function can also facilitate the application of FBA directly to metagenomic data. For example, a recent study of the interaction between three gut dwelling species used a multi-species FBA model coupled with two distinct objective functions, each applied to address a different prediction task (Shoaie et al., 2013): First, metabolic secretion was predicted given a defined dietary input, and was solved by constraining microbial abundances while minimizing substrate uptake rates. Subsequently, species abundances were also predicted by constraining uptake rates and optimizing total community growth. Notably, fatty-acid secretion and metabolic reprogramming predicted by the model were in accord with experimentally measured values (Mahowald et al., 2009). Another solution was introduced in the first constraint-based model of a gut microbe interacting with its eukaryotic host (Heinken et al., 2013). In this framework, the growth of one organism was constrained while that of its partner was optimized, describing the trade-off between host and microbe optimal growth. Notably, this model was able to demonstrate mutualistic growth in an idealized host-microbiome system, as well as rescue of a lethal host genotype by a symbiont. Although in this study a generic mouse model (with extension for intestinal transport and absorption) was used for the host, well-curated human metabolism models (Duarte et al., 2007) (as well as tissue specific models, Jerby et al., 2010; Wang et al., 2012) have been introduced and can be similarly integrated into a more accurate host-microbiome model.
The success of the modeling approaches described above in recovering observed patterns of microbial interaction highlights the potential of multi-species constraint-based models, but also warrants a discussion of the limitations of these frameworks and of potential solutions. First, it is unclear how these frameworks scale with community complexity and how they may be used to model more complex interactions. Multi-species constraint-based models are still in their early days and efforts are mainly focused on developing approaches for modeling very simple communities. Second, naïvely defined objectives have the potential to produce biologically implausible predictions. Specifically, the commonly used objective function that aims to maximize total community growth (e.g., Shoaie et al., 2013; Stolyar et al., 2007) inherently assumes altruistic cooperation between community members and may therefore predict that one species neglects its own growth to facilitate the faster growth of another.
Introducing temporal dynamics and expanding single-species dynamic FBA models (e.g., Collins et al., 2012) to account for multiple species with a shared environment may provide a viable alternative (Figure 2C). In such dynamics-based frameworks the growth of each species is optimized independently of its partners’ growth on a short time scale (as determined by nutrient availability) and the shared environment is iteratively updated according to predicted species’ growth, uptake, and secretion rates (Chiu et al., 2014; Tzamali et al., 2011; Zhuang et al., 2011). Competitive effects and metabolic interactions are therefore mediated through the environment as a natural byproduct of niche construction, rather than being explicitly formulated in the model. Importantly, in this way, the need to optimize a more complex and potentially artificial objective function at the community scale is circumvented. Instead, community dynamics over a long timescale are revealed through integration of discrete time-steps. Using this approach, a recent study integrated dynamic FBA with flux variability analysis to model metabolic interaction across hundreds of two-species consortia and to study the emergence of metabolic capacity in such simple communities (Chiu et al., 2014). Indeed, emergent biosynthetic capacity, wherein a multi-species community secrets into the environment metabolic products that no constituent species secretes when grown in isolation, was found to be prevalent. Using this modeling framework it was also shown that emergent biosynthetic capacity commonly occurs in two distinct phases of community growth, and that it is most probable when community members have moderately similar sets of metabolic capabilities (Chiu et al., 2014).
Integrating metabolic modeling with other modeling approaches
Metabolic models, of course, are not the only modeling frameworks capable of generating hypotheses or predictions of microbiome behavior, and other frameworks, focusing on ecological dynamics or on spatial organization, have been recently introduced. Such alternative modeling frameworks are often complementary and could be coupled in exciting ways. Stein et. al. (Stein et al., 2013), for example, introduced a generalized Lotka-Volterra model of species growth rates, interactions, and antibiotic susceptibility, capable of predicting community dynamics over a large timescale, as well as response to antibiotic perturbation (and see also Marino et al., 2014). Notably, the parameters of this model were learned directly from metagenomic data, without utilizing any prior knowledge of species physiology. Similarly, Trosvik et al. (Trosvik et al., 2010) applied a Generalized Additive Model to time-series data representing growth of a model gut community to quantify the influence of intrinsic microbe-microbe interactions. Metabolic models could complement such efforts both ‘upstream’ by informing the search for optimal parameters, and ‘downstream’ by offering a mechanistic interpretation to phenomenological model predictions. Agent-based models have been proposed to account for spatial resolution in studying species interaction (Kreft et al., 2013; Schluter and Foster, 2012), and have been successfully applied to study small cooperative communities (Momeni et al., 2013a, 2013b). In such agent-based models, the microbe’s internal complexity is mostly stripped down to allow implementation and tractability, and the rule set that determines the behavior of each agent is often somewhat arbitrary. Metabolic models can offer an attractive solution and could be used, for example, to determine the general behavior of a group of agents at specific time-steps, leading to a true multi-scale model. A simpler alternative approach for integrating metabolic modeling and spatial resolution has recently been introduced, where a constraint-based metabolic model was used to predict the behavior of various microbial species over a short time scale and a lattice-based environment and classical diffusion dynamics were used to determine how metabolites are exchanged between adjacent microhabitats (Harcombe et al., 2014).
Concluding remarks
Our ability to generate computational models of complex biological systems is rapidly improving. Yet, even as we are beginning to address the challenge of modeling whole cells (Karr et al., 2012), we are still a ways from generating equally comprehensive models of whole communities. Host-associated communities pose additional challenges, as host phenotypic response and its impact on the community further complicate any modeling framework and prohibits many typical simplifying assumptions (Heinken et al., 2013; Thiele et al., 2013). The influences of gene regulation and genotypic variation are also currently largely ignored in metabolic models, but must be taken into account to achieve the desired level of comprehensiveness, or for applications to personalized medicine. Similarly, microbe-microbe signaling and antibiotic production are not within the scope of metabolic models, but greatly impact growth. Finally, our ability to identify gene families has rapidly outpaced our ability to functionally characterize them: by some estimates as many as 75% of identified genes lack a functional annotation (Qin et al., 2010), yet enzymatic characterization is crucial to inclusion in metabolic models. Analogous to the aforementioned microbial ‘phylogenetic dark matter’, these uncharacterized genes represent a ‘functional dark matter’; it is potentially responsible for the most interesting and complex capacities of the microbial world, yet its precise function and the way such functions are carried out remain a mystery. Ironically, successfully mapping the phylogenetic dark matter, we accelerate the discovery of uncharacterized gene families and of such functional dark matter, revealing how little we know of microbial metabolism at large. As advanced as high throughput genomic technologies may be, meticulous, low throughput biochemical assays still represent the ultimate solution to this problem, highlighting the importance of constant feedback between characterizing the elements of the systems and the system as a whole.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baker M. De novo genome assembly: what every biologist should know. Nature Methods. 2012;9:333–337. [Google Scholar]
- Baquero F, Nombela C. The microbiome as a human organ. Clinical Microbiology and Infection. 2012;18:2–4. doi: 10.1111/j.1469-0691.2012.03916.x. [DOI] [PubMed] [Google Scholar]
- Beitel CW, Froenicke L, Lang JM, Korf IF, Michelmore RW, Eisen JA, Darling AE. Strain- and plasmid-level deconvolution of a synthetic metagenome by sequencing proximity ligation products. PeerJ. 2014;2:e415. doi: 10.7717/peerj.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borenstein E. Computational systems biology and in silico modeling of the human microbiome. Briefings in Bioinformatics. 2012;13:769–780. doi: 10.1093/bib/bbs022. [DOI] [PubMed] [Google Scholar]
- Borenstein E, Feldman MW. Topological signatures of species interactions in metabolic networks. Journal of Computational Biology. 2009;16:191–200. doi: 10.1089/cmb.2008.06TT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borenstein E, Kupiec M, Feldman MW, Ruppin E. Large-scale reconstruction and phylogenetic analysis of metabolic environments. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:14482–14487. doi: 10.1073/pnas.0806162105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CT, Sharon I, Thomas BC, Castelle CJ, Morowitz MJ, Banfield JF. Genome resolved analysis of a premature infant gut microbial community reveals a Varibaculum cambriense genome and a shift towards fermentation-based metabolism during the third week of life. Microbiome. 2013;1:30. doi: 10.1186/2049-2618-1-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton J, Liachko I, Dunham M, Shendure J. Species-Level Deconvolution of Metagenome Assemblies with Hi-C-Based Contact Probability Maps. G3. 2014 doi: 10.1534/g3.114.011825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäckhed F, Ding H, Wang T, Hooper L, Koh GY, Nagy A, Semenkovich CF, Gordon JI. The gut microbiota as an environmental factor that regulates fat storage. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr R, Shen-Orr SS, Borenstein E. Reconstructing the Genomic Content of Microbiome Taxa through Shotgun Metagenomic Deconvolution. PLoS Computational Biology. 2013;9:e1003292. doi: 10.1371/journal.pcbi.1003292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu HC, Levy R, Borenstein E. Emergent biosynthetic capacity in simple microbial communities. PLoS Computational Biology. 2014;10:e1003695. doi: 10.1371/journal.pcbi.1003695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton TA, Baker D, Lindon JC, Everett JR, Nicholson JK. Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism. Proceedings of the National Academy of Sciences. 2009;106:14728–14733. doi: 10.1073/pnas.0904489106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148:1258–1270. doi: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SB, Reznik E, Segrè D. Temporal expression-based analysis of metabolism. PLoS Computational Biology. 2012;8:e1002781. doi: 10.1371/journal.pcbi.1002781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottret L, Milreu PV, Acu V. Enumerating Precursor Sets of Target Metabolites in a Metabolic Network. Algorithms in Bioinformatics. 2008;5251:233–244. [Google Scholar]
- Cottret L, Milreu PV, Acuña V, Marchetti-Spaccamela A, Stougie L, Charles H, Sagot MF. Graph-Based Analysis of the Metabolic Exchanges between Two Co-Resident Intracellular Symbionts, Baumannia cicadellinicola and Sulcia muelleri, with Their Insect Host, Homalodisca coagulata. PLoS Computational Biology. 2010;6:13. doi: 10.1371/journal.pcbi.1000904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csuros M. Count: Evolutionary analysis of phylogenetic profiles with parsimony and likelihood. Bioinformatics. 2010;26:1910–1912. doi: 10.1093/bioinformatics/btq315. [DOI] [PubMed] [Google Scholar]
- David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling A, Devlin S, Varma Y, Fischbach MA, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;23:673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proceedings of the National Academy of Sciences. 2011;108:4554. doi: 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodsworth JA, Blainey PC, Murugapiran SK, Swingley WD, Ross CA, Tringe SG, Chain PSG, Scholz MB, Lo CC, Raymond J, et al. Single-cell and metagenomic analyses indicate a fermentative and saccharolytic lifestyle for members of the OP9 lineage. Nature Communications. 2013;4:1854. doi: 10.1038/ncomms2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte NC, Becker SA, Jamshidi N, Thiele I, Mo ML, Vo TD, Srivas R, Palsson BØ. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:1777–1782. doi: 10.1073/pnas.0610772104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan SH, Lobley GE, Holtrop G, Ince J, Johnstone aM, Louis P, Flint HJ. Human colonic microbiota associated with diet, obesity and weight loss. International Journal of Obesity. 2008;32:1720–1724. doi: 10.1038/ijo.2008.155. (2005) [DOI] [PubMed] [Google Scholar]
- Edwards JS, Ibarra RU, Palsson BO. In silico predictions of Escherichia coli metabolic capabilities are consistent with experimental data. Nature Biotechnology. 2001;19:125–130. doi: 10.1038/84379. [DOI] [PubMed] [Google Scholar]
- Erickson AR, Cantarel BL, Lamendella R, Darzi Y, Mongodin EF, Pan C, Shah M, Halfvarson J, Tysk C, Henrissat B, et al. Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of Crohn’s disease. PloS One. 2012;7:e49138. doi: 10.1371/journal.pone.0049138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith JJ, McNulty NP, Rey FE, Gordon JI. Predicting a human gut microbiota’s response to diet in gnotobiotic mice. Science. 2011;333:101–104. doi: 10.1126/science.1206025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith JJ, Ahern PP, Ridaura VK, Cheng J, Gordon JI. Identifying gut microbe-host phenotype relationships using combinatorial communities in gnotobiotic mice. Science Translational Medicine. 2014;6:220ra11. doi: 10.1126/scitranslmed.3008051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust K, Sathirapongsasuti JF, Izard J, Segata N, Gevers D, Raes J, Huttenhower C. Microbial Co-occurrence Relationships in the Human Microbiome. PLoS Computational Biology. 2012;8:e1002606. doi: 10.1371/journal.pcbi.1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Birney E. Sense from sequence reads: methods for alignment and assembly. Nature Methods. 2009;6:S6–S12. doi: 10.1038/nmeth.1376. [DOI] [PubMed] [Google Scholar]
- Freilich S, Kreimer A, Borenstein E, Yosef N, Sharan R, Gophna U, Ruppin E. Metabolic-network-driven analysis of bacterial ecological strategies. Genome Biology. 2009;10:R61. doi: 10.1186/gb-2009-10-6-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freilich S, Kreimer A, Meilijson I, Gophna U, Sharan R, Ruppin E. The large-scale organization of the bacterial network of ecological co-occurrence interactions. Nucleic Acids Research. 2010;38:3857–3868. doi: 10.1093/nar/gkq118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freilich S, Zarecki R, Eilam O, Segal ES, Henry CS, Kupiec M, Gophna U, Sharan R, Ruppin E. Competitive and cooperative metabolic interactions in bacterial communities. Nature Communications. 2011;2:589. doi: 10.1038/ncomms1597. [DOI] [PubMed] [Google Scholar]
- Frias-Lopez J, Shi Y, Tyson GW, Coleman ML, Schuster SC, Chisholm SW, Delong EF. Microbial community gene expression in ocean surface waters. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:3805–3810. doi: 10.1073/pnas.0708897105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, Nelson KE. Metagenomic analysis of the human distal gut microbiome. Science (New York, NY) 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman A, Kallstrom G. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proceedings of the National Academy of Sciences. 2011 doi: 10.1073/pnas.1102938108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenblum S, Turnbaugh PJ, Borenstein E. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:594–599. doi: 10.1073/pnas.1116053109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenblum S, Chiu H, Levy R, Carr R, Borenstein E. Towards a predictive systems-level model of the human microbiome: progress, challenges, and opportunities. Current Opinion in Biotechnology. 2013;24:810–820. doi: 10.1016/j.copbio.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handley KM, Bartels D, O Loughlin EJ, Williams KH, Trimble WL, Skinner K, Gilbert Ja, Desai N, Glass EM, Paczian T, et al. The complete genome sequence for putative H2 – and S-oxidizer Candidatus Sulfuricurvum sp., assembled de novo from an aquifer-derived metagenome. Environmental Microbiology. 2014:1–61. doi: 10.1111/1462-2920.12453. [DOI] [PubMed] [Google Scholar]
- Harcombe WR, Riehl WJ, Dukovski I, Granger BR, Betts A, Lang AH, Bonilla G, Kar A, Leiby N, Mehta P, et al. Metabolic Resource Allocation in Individual Microbes Determines Ecosystem Interactions and Spatial Dynamics. Cell Reports. 2014:1–12. doi: 10.1016/j.celrep.2014.03.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman AL, Lough DM, Barupal DK, Fiehn O, Fishbein T, Zasloff M, Eisen Ja. Human gut microbiome adopts an alternative state following small bowel transplantation. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:17187–17192. doi: 10.1073/pnas.0904847106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinken A, Sahoo S, Fleming RMT, Thiele I. Systems-level characterization of a host-microbe metabolic symbiosis in the mammalian gut. Gut Microbes. 2013;4:1–13. doi: 10.4161/gmic.22370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iverson V, Morris RM, Frazar CD, Berthiaume CT, Morales RL, Armbrust EV. Untangling Genomes from Metagenomes: Revealing an Uncultured Class of Marine Euryarchaeota. Science. 2012;587 doi: 10.1126/science.1212665. [DOI] [PubMed] [Google Scholar]
- Jeong H, Tombor B, Albert R, Oltvai ZN, Barabasi AL. The large-scale organization of metabolic networks. Nature. 2000;407:651–654. doi: 10.1038/35036627. [DOI] [PubMed] [Google Scholar]
- Jerby L, Shlomi T, Ruppin E. Computational reconstruction of tissue-specific metabolic models: application to human liver metabolism. Molecular Systems Biology. 2010;6:401. doi: 10.1038/msb.2010.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorth P, Turner KH, Gumus P, Nizam N, Buduneli N, Whiteley M. Metatranscriptomics of the human oral microbiome during health and disease. mBio. 2014;5:e01012–e01014. doi: 10.1128/mBio.01012-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumpertz R, Le DS, Turnbaugh PJ, Trinidad C, Bogardus C, Gordon JI, Krakoff J. Energy-balance studies reveal associations between gut microbes, caloric load, and nutrient absorption in humans. The American Journal of Clinical Nutrition. 2011;94:58–65. doi: 10.3945/ajcn.110.010132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Research. 2012;40:D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karr JR, Sanghvi JC, Macklin DN, Gutschow MV, Jacobs JM, Bolival B, Assad-Garcia N, Glass JI, Covert MW. A Whole-Cell Computational Model Predicts Phenotype from Genotype. Cell. 2012;150:389–401. doi: 10.1016/j.cell.2012.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiblinger KM, Wilhartitz IC, Schneider T, Roschitzki B, Schmid E, Eberl L, Riedel K, Zechmeister-Boltenstern S. Soil metaproteomics – Comparative evaluation of protein extraction protocols. Soil Biology & Biochemistry. 2012;54:14–24. doi: 10.1016/j.soilbio.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Boedicker JQ, Choi JW, Ismagilov RF. Defined spatial structure stabilizes a synthetic multispecies bacterial community. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:18188–18193. doi: 10.1073/pnas.0807935105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaassens ES, Vos WM, Vaughan EE. Metaproteomics approach to study the functionality of the microbiota in the human infant gastrointestinal tract. Applied and Environmental Microbiology. 2007;73:1388–1392. doi: 10.1128/AEM.01921-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klitgord N, Segrè D. Environments that induce synthetic microbial ecosystems. PLoS Computational Biology. 2010;6:e1001002. doi: 10.1371/journal.pcbi.1001002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolenbrander PE. Multispecies communities: interspecies interactions influence growth on saliva as sole nutritional source. International Journal of Oral Science. 2011;3:49–54. doi: 10.4248/IJOS11025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolmeder CA, Vos WM. Metaproteomics of our microbiome – developing insight in function and activity in man and model systems. Journal of Proteomics. 2014;97:3–16. doi: 10.1016/j.jprot.2013.05.018. [DOI] [PubMed] [Google Scholar]
- Kotula JW, Kerns SJ, Shaket La, Siraj L, Collins JJ, Way JC, Silver Pa. Programmable bacteria detect and record an environmental signal in the mammalian gut. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:4838–4843. doi: 10.1073/pnas.1321321111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreft JU, Plugge CM, Grimm V, Prats C, Leveau JHJ, Banitz T, Baines S, Clark J, Ros a, Klapper I, et al. Mighty small: Observing and modeling individual microbes becomes big science. Proceedings of the National Academy of Sciences. 2013;110:18027–18028. doi: 10.1073/pnas.1317472110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreimer A, Borenstein E, Gophna U, Ruppin E. The evolution of modularity in bacterial metabolic networks. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:6976–6981. doi: 10.1073/pnas.0712149105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai B, Ding R, Li Y, Duan L, Zhu H. A de novo metagenomic assembly program for shotgun DNA reads. Bioinformatics (Oxford, England) 2012;28:1455–1462. doi: 10.1093/bioinformatics/bts162. [DOI] [PubMed] [Google Scholar]
- Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature Biotechnology. 2013;31:814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laserson J, Jojic V, Koller D. Genovo: de novo assembly for metagenomes. Journal of Computational Biology: a Journal of Computational Molecular Cell Biology. 2011;18:429–443. doi: 10.1089/cmb.2010.0244. [DOI] [PubMed] [Google Scholar]
- Lee SM, Donaldson GP, Mikulski Z, Boyajian S, Ley K, Mazmanian SK. Bacterial colonization factors control specificity and stability of the gut microbiota. Nature. 2013;501:426–429. doi: 10.1038/nature12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy R, Borenstein E. Reverse Ecology: From Systems to Environments and Back. Advances in Experimental Medicine and Biology. 2012;751:329–345. doi: 10.1007/978-1-4614-3567-9_15. [DOI] [PubMed] [Google Scholar]
- Levy R, Borenstein E. Metabolic modeling of species interaction in the human microbiome elucidates community-level assembly rules. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:12804–12809. doi: 10.1073/pnas.1300926110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy R, Borenstein E. Metagenomic systems biology and metabolic modeling of the human microbiome: From species composition to community assembly rules. Gut Microbes. 2014;5:265–270. doi: 10.4161/gmic.28261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- Little AEF, Robinson CJ, Peterson SB, Raffa KF, Handelsman J. Rules of engagement: interspecies interactions that regulate microbial communities. Annual Review of Microbiology. 2008;62:375–401. doi: 10.1146/annurev.micro.030608.101423. [DOI] [PubMed] [Google Scholar]
- Lu K, Abo RP, Schlieper KA, Graffam ME, Levine S, Wishnok JS, Swenberg JA, Tannenbaum SR, Fox JG. Arsenic exposure perturbs the gut microbiome and its metabolic profile in mice: an integrated metagenomics and metabolomics analysis. Environmental Health Perspectives. 2014;122:284–291. doi: 10.1289/ehp.1307429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahowald Ma, Rey FE, Seedorf H, Turnbaugh PJ, Fulton RS, Wollam A, Shah N, Wang C, Magrini V, Wilson RK, et al. Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:5859–5864. doi: 10.1073/pnas.0901529106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino S, Baxter NT, Huffnagle GB, Petrosino JF, Schloss PD. Mathematical modeling of primary succession of murine intestinal microbiota. Proceedings of the National Academy of Sciences of the United States of America. 2014 doi: 10.1073/pnas.1311322111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason OU, Hazen TC, Borglin S, Chain PSG, Dubinsky EA, Fortney JL, Han J, Holman HYN, Hultman J, Lamendella R, et al. Metagenome, metatranscriptome and single-cell sequencing reveal microbial response to Deepwater Horizon oil spill. The ISME Journal. 2012;6:1715–1727. doi: 10.1038/ismej.2012.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice CF, Haiser HJ, Turnbaugh PJ. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell. 2013;152:39–50. doi: 10.1016/j.cell.2012.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JAK, Schroeter K, Fuentes S, Heikamp-Dejong I, Khursigara CM, Vos WM, Allen-Vercoe E. Evaluation of microbial community reproducibility, stability and composition in a human distal gut chemostat model. Journal of Microbiological Methods. 2013;95:167–174. doi: 10.1016/j.mimet.2013.08.008. [DOI] [PubMed] [Google Scholar]
- McHardy IH, Goudarzi M, Tong M, Ruegger PM, Schwager E, Weger JR, Graeber TG, Sonnenburg JL, Horvath S, Huttenhower C, et al. Integrative analysis of the microbiome and metabolome of the human intestinal mucosal surface reveals exquisite inter-relationships. Microbiome. 2013;1:17. doi: 10.1186/2049-2618-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momeni B, Brileya KA, Fields MW, Shou W. Strong interpopulation cooperation leads to partner intermixing in microbial communities. eLife. 2013a;2:e00230. doi: 10.7554/eLife.00230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momeni B, Waite AJ, Shou W. Spatial self-organization favors heterotypic cooperation over cheating. eLife. 2013b;2:e00960. doi: 10.7554/eLife.00960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, Leleiko N, Snapper SB, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biology. 2012;13:R79. doi: 10.1186/gb-2012-13-9-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muegge BD, Kuczynski J, Knights D, Clemente JC, González A, Fontana L, Henrissat B, Knight R, Gordon JI. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science (New York, NY) 2011;332:970–974. doi: 10.1126/science.1198719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namiki T, Hachiya T, Tanaka H, Sakakibara Y. MetaVelvet: an extension of Velvet assembler to de novo metagenome assembly from short sequence reads. Nucleic Acids Research. 2012;40:e155. doi: 10.1093/nar/gks678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth JD, Thiele I, Palsson BØ. What is flux balance analysis? Nature Biotechnology. 2010;28:245–248. doi: 10.1038/nbt.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hara AM, Shanahan F. The gut flora as a forgotten organ. EMBO Reports. 2006;7:688–693. doi: 10.1038/sj.embor.7400731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E, Claude J, Strimmer K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- Park J, Kerner A, Burns Ma, Lin XN. Microdroplet-enabled highly parallel co-cultivation of microbial communities. PloS One. 2011;6:e17019. doi: 10.1371/journal.pone.0017019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Leung HCM, Yiu SM, Chin FYL. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics (Oxford, England) 2012;28:1420–1428. doi: 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- Petrof EO, Gloor GB, Vanner SJ, Weese SJ, Carter D, Daigneault MC, Brown EM, Schroeter K, Allen-Vercoe E. Stool substitute transplant therapy for the eradication of Clostridium difficile infection: “RePOOPulating” the gut. Microbiome. 2013;1:3. doi: 10.1186/2049-2618-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop M. Genome assembly reborn: recent computational challenges. Briefings in Bioinformatics. 2009;10:354–366. doi: 10.1093/bib/bbp026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price ND, Papin JA, Schilling CH, Palsson BO. Genome-scale microbial in silico models: The constraints-based approach. Trends in Biotechnology. 2003;21:162–169. doi: 10.1016/S0167-7799(03)00030-1. [DOI] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Coyne MJ, Comstock LE. An ecological network of polysaccharide utilization among human intestinal symbionts. Current Biology. 2014;24:40–49. doi: 10.1016/j.cub.2013.10.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed J, Palsson BØ. Thirteen years of building constraint-based in silico models of Escherichia coli. Journal of Bacteriology. 2003;185 doi: 10.1128/JB.185.9.2692-2699.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science (New York, NY) 2013;341:1241214. doi: 10.1126/science.1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rienzi SC, Sharon I, Wrighton KC, Koren O, Hug LA, Thomas BC, Goodrich JK, Bell JT, Spector TD, Banfield JF, et al. The human gut and groundwater harbor non-photosynthetic bacteria belonging to a new candidate phylum sibling to Cyanobacteria. eLife. 2013;2:e01102–e01102. doi: 10.7554/eLife.01102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng JF, Darling A, Malfatti S, Swan BK, Gies EA, et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature. 2013;499:431–437. doi: 10.1038/nature12352. [DOI] [PubMed] [Google Scholar]
- Rodrigue S, Malmstrom RR, Berlin AM, Birren BW, Henn MR, Chisholm SW. Whole genome amplification and de novo assembly of single bacterial cells. PloS One. 2009;4:e6864. doi: 10.1371/journal.pone.0006864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeselers G, Mittge EK, Stephens WZ, Parichy DM, Cavanaugh CM, Guillemin K, Rawls JF. Evidence for a core gut microbiota in the zebrafish. The ISME Journal. 2011;5:1595–1608. doi: 10.1038/ismej.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schink B. Synergistic interactions in the microbial world. Antonie Van Leeuwenhoek. 2002:257–261. doi: 10.1023/a:1020579004534. [DOI] [PubMed] [Google Scholar]
- Schloss PD, Handelsman J. Metagenomics for studying unculturable microorganisms: cutting the Gordian knot. Genome Biology. 2005;6:229. doi: 10.1186/gb-2005-6-8-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluter J, Foster KR. The Evolution of Mutualism in Gut Microbiota Via Host Epithelial Selection. PLoS Biology. 2012;10:e1001424. doi: 10.1371/journal.pbio.1001424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwiertz A, Taras D, Schäfer K, Beijer S, Bos NA, Donus C, Hardt PD. Microbiota and SCFA in lean and overweight healthy subjects. Obesity (Silver Spring, Md) 2010;18:190–195. doi: 10.1038/oby.2009.167. [DOI] [PubMed] [Google Scholar]
- Segata N, Boernigen D, Tickle TL, Morgan XC, Garrett WS, Huttenhower C. Computational meta’omics for microbial community studies. Molecular Systems Biology. 2013;9:666. doi: 10.1038/msb.2013.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon I, Morowitz MJ, Thomas BC, Costello EK, Relman DA, Banfield JF. Time series community genomics analysis reveals rapid shifts in bacterial species, strains, and phage during infant gut colonization. Genome Research. 2012;23:111–120. doi: 10.1101/gr.142315.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen-Orr SS, Gaujoux R. Computational deconvolution: extracting cell type-specific information from heterogeneous samples. Current Opinion in Immunology. 2013;25:571–578. doi: 10.1016/j.coi.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoaie S, Karlsson F, Mardinoglu A, Nookaew I, Bordel S, Nielsen J. Understanding the interactions between bacteria in the human gut through metabolic modeling. Scientific Reports. 2013;3:2532. doi: 10.1038/srep02532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons SL, Dibartolo G, Denef VJ, Goltsman DSA, Thelen MP, Banfield JF. Population genomic analysis of strain variation in Leptospirillum group II bacteria involved in acid mine drainage formation. PLoS Biology. 2008;6:e177. doi: 10.1371/journal.pbio.0060177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sizova M, Hohmann T, Hazen A, Paster BJ, Halem SR, Murphy CM, Panikov NS, Epstein SS. New approaches for isolation of previously uncultivated oral bacteria. Applied and Environmental Microbiology. 2012;78:194–203. doi: 10.1128/AEM.06813-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa T, Paterson R, Moore V, Carlsson A, Abrahamsson B, Basit AW. The gastrointestinal microbiota as a site for the biotransformation of drugs. International Journal of Pharmaceutics. 2008;363:1–25. doi: 10.1016/j.ijpharm.2008.07.009. [DOI] [PubMed] [Google Scholar]
- Stein RR, Bucci V, Toussaint NC, Buffie CG, Rätsch G, Pamer EG, Sander C, Xavier JB. Ecological modeling from time-series inference: insight into dynamics and stability of intestinal microbiota. PLoS Computational Biology. 2013;9:e1003388. doi: 10.1371/journal.pcbi.1003388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelling J, Klamt S, Bettenbrock K, Schuster S, Gilles ED. Metabolic network structure determines key aspects of functionality and regulation. Nature. 2002;420:190–193. doi: 10.1038/nature01166. [DOI] [PubMed] [Google Scholar]
- Stewart EJ. Growing unculturable bacteria. Journal of Bacteriology. 2012;194:4151–4160. doi: 10.1128/JB.00345-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolyar S, Van Dien S, Hillesland KL, Pinel N, Lie TJ, Leigh Ja, Stahl Da. Metabolic modeling of a mutualistic microbial community. Molecular Systems Biology. 2007;3:92. doi: 10.1038/msb4100131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taffs R, Aston JE, Brileya K, Jay Z, Klatt CG, McGlynn S, Mallette N, Montross S, Gerlach R, Inskeep WP, et al. In silico approaches to study mass and energy flows in microbial consortia: a syntrophic case study. BMC Systems Biology. 2009;3:114. doi: 10.1186/1752-0509-3-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot CM, Koenigsknecht MJ, Carlson PE, Hatton GE, Nelson AM, Li B, Huffnagle GB, Z Li J, Young VB. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nature Communications. 2014;5:3114. doi: 10.1038/ncomms4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele I, Heinken A, Fleming RM. A systems biology approach to studying the role of microbes in human health. Current Opinion in Biotechnology. 2013;24:4–12. doi: 10.1016/j.copbio.2012.10.001. [DOI] [PubMed] [Google Scholar]
- Thomas F, Hehemann JH, Rebuffet E, Czjzek M, Michel G. Environmental and gut bacteroidetes: the food connection. Frontiers in Microbiology. 2011;2:93. doi: 10.3389/fmicb.2011.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treangen TJ, Koren S, Sommer DD, Liu B, Astrovskaya I, Ondov B, Darling AE, Phillippy AM, Pop M. MetAMOS: a modular and open source metagenomic assembly and analysis pipeline. Genome Biology. 2013;14:R2. doi: 10.1186/gb-2013-14-1-r2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tringe SG, von Mering C, Kobayashi A, Salamov Aa, Chen K, Chang HW, Podar M, Short JM, Mathur EJ, Detter JC, et al. Comparative metagenomics of microbial communities. Science (New York, NY) 2005;308:554–557. doi: 10.1126/science.1107851. [DOI] [PubMed] [Google Scholar]
- Trosvik P, Rudi K, Straetkvern KO, Jakobsen KS, Naes T, Stenseth NC. Web of ecological interactions in an experimental gut microbiota. Environmental Microbiology. 2010;12:2677–2687. doi: 10.1111/j.1462-2920.2010.02236.x. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald Ma, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Bäckhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host & Microbe. 2008;3:213–223. doi: 10.1016/j.chom.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner TR, Ramakrishnan K, Walshaw J, Heavens D, Alston M, Swarbreck D, Osbourn A, Grant A, Poole PS. Comparative metatranscriptomics reveals kingdom level changes in the rhizosphere microbiome of plants. The ISME Journal. 2013;7:2248–2258. doi: 10.1038/ismej.2013.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM, Solovyev VV, Rubin EM, Rokhsar DS, Banfield JF. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 2004;428:37–43. doi: 10.1038/nature02340. [DOI] [PubMed] [Google Scholar]
- Tzamali E, Poirazi P, Tollis IG, Reczko M. A computational exploration of bacterial metabolic diversity identifying metabolic interactions and growth-efficient strain communities. BMC Systems Biology. 2011;5:167. doi: 10.1186/1752-0509-5-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyay V, Poroyko V, Kim T-jin, Devkota S, Fu S, Liu D, Tumanov A, Koroleva EP, Deng L, Nagler C, et al. Lymphotoxin regulates commensal responses to enable diet-induced obesity. Nature Immunology. 2012;13:947–953. doi: 10.1038/ni.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urich T, Lanzén A, Qi J, Huson DH, Schleper C, Schuster SC. Simultaneous assessment of soil microbial community structure and function through analysis of the meta-transcriptome. PloS One. 2008;3:e2527. doi: 10.1371/journal.pone.0002527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursell LK, Knight R. Xenobiotics and the human gut microbiome: metatranscriptomics reveal the active players. Cell Metabolism. 2013;17:317–318. doi: 10.1016/j.cmet.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartoukian SR, Palmer RM, Wade WG. Strategies for culture of “unculturable” bacteria. FEMS Microbiology Letters. 2010;309:1–7. doi: 10.1111/j.1574-6968.2010.02000.x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Eddy JA, Price ND. Reconstruction of genome-scale metabolic models for 126 human tissues using mCADRE. BMC Systems Biology. 2012;6:153. doi: 10.1186/1752-0509-6-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver W. Science and complexity. American Scientist. 1948;36:536–544. [PubMed] [Google Scholar]
- Weir TL, Manter DK, Sheflin AM, Barnett BA, Heuberger AL, Ryan EP. Stool microbiome and metabolome differences between colorectal cancer patients and healthy adults. PloS One. 2013;8:e70803. doi: 10.1371/journal.pone.0070803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White BA, Creedon DJ, Nelson KE, Wilson BA. The vaginal microbiome in health and disease. Trends in Endocrinology and Metabolism: TEM. 2011;22:389–393. doi: 10.1016/j.tem.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterberg H. Zur Methodik der Bakterienzählung. Zeitschrift Für Hygiene Und Infectionskrankheiten. 1898;29:75–93. [Google Scholar]
- Wintermute EH, Silver PA. Emergent cooperation in microbial metabolism. Molecular Systems Biology. 2010;6:1–7. doi: 10.1038/msb.2010.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, Kunin V, Goodwin L, Wu M, Tindall BJ, et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature. 2009;462:1056–1060. doi: 10.1038/nature08656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderlich Z, Mirny LA. Using the Topology of Metabolic Networks to Predict Viability of Mutant Strains. Biophysical Journal. 2006;91:2304–2311. doi: 10.1529/biophysj.105.080572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Frank DN, Robertson CE, Hung SS, Markle J, Canty AJ, McCoy KD, Macpherson AJ, Poussier P, Danska JS, et al. Generation and analysis of a mouse intestinal metatranscriptome through Illumina based RNA-sequencing. PloS One. 2012;7:e36009. doi: 10.1371/journal.pone.0036009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, Chiang HC, Hooper L, Gordon JI. A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science (New York, NY) 2003;299:2074–2076. doi: 10.1126/science.1080029. [DOI] [PubMed] [Google Scholar]
- Zaneveld JR, Lozupone C, Gordon JI, Knight R. Ribosomal RNA diversity predicts genome diversity in gut bacteria and their relatives. Nucleic Acids Research. 2010;38:3869. doi: 10.1093/nar/gkq066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarco MF, Vess TJ, Ginsburg GS. The oral microbiome in health and disease and the potential impact on personalized dental medicine. Oral Diseases. 2012;18:109–120. doi: 10.1111/j.1601-0825.2011.01851.x. [DOI] [PubMed] [Google Scholar]
- Zhuang K, Izallalen M, Mouser P, Richter H, Risso C, Mahadevan R, Lovley DR. Genome-scale dynamic modeling of the competition between Rhodoferax and Geobacter in anoxic subsurface environments. The ISME Journal. 2011;5:305–316. doi: 10.1038/ismej.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zomorrodi AR, Maranas CD. OptCom: A multi-level optimization framework for the metabolic modeling and analysis of microbial communities. PLoS Computational Biology. 2012;8:e1002363. doi: 10.1371/journal.pcbi.1002363. [DOI] [PMC free article] [PubMed] [Google Scholar]