

Abstract
The properties of a cell are determined both genetically by the DNA sequence of its genes and epigenetically through processes that regulate the pattern, timing and magnitude of expression of its genes. While the genetic basis of disease has been a topic of intense study for decades, recent years have seen a dramatic increase in the understanding of epigenetic regulatory mechanisms and a growing appreciation that epigenetic misregulation makes a significant contribution to human disease. Several large protein families have been identified that act in different ways to control the expression of genes through epigenetic mechanisms. Many of these protein families are finally proving tractable for the development of small molecules that modulate their function and represent new target classes for drug discovery. Here, we provide an overview of some of the key epigenetic regulatory proteins and discuss progress towards the development of pharmacological tools for use in research and therapy.
Links to online information in the IUPHAR/BPS Guide to PHARMACOLOGY
| TARGETS | ||
|---|---|---|
| ASH1L | HDA8 | PRDM2 |
| ATAD2 | HDAC9 | PRMT1 |
| ATAD2B | JMJD1C | PRMT10 |
| BAZ1A | KAT2A | PRMT2 |
| BAZ1B | KAT2B | PRMT3 |
| BAZ2A | KAT5 | PRMT5 |
| BAZ2B | KAT6A | PRMT6 |
| BPTF | KAT6B | PRMT7 |
| BRD1 | KAT7 | PRMT8 |
| BRD2 | KAT8 | SETD1A |
| BRD3 | KDM1A | SETD1B |
| BRD4 | KDM1B | SETD2 |
| BRD7 | KDM2A | SETD7 |
| BRD8 | KDM2B | SETD8 |
| BRD9 | KDM3A | SETDB1 |
| BRDT | KDM3B | SETDB2 |
| BRPF1 | KDM4A | SIRT1 |
| BRPF3 | KDM4B | SIRT2 |
| BRWD1 | KDM4C | SIRT3 |
| BRWD3 | KDM4D | SIRT4 |
| CARM1 | KDM4E | SIRT5 |
| CECR2 | KDM5A | SIRT6 |
| CLOCK | KDM5B | SIRT7 |
| CREBBP | KDM5C | SMARCA2 |
| DOT1L | KDM5D | SMARCA4 |
| EHMT1 | KDM6A | SMYD2 |
| EHMT2 | KDM6B | SP100 |
| EP300 | KDM7A | SP110 |
| EZH2 | KDM8 | SP140 |
| FBXO10 | KMT2A | SP140L |
| FBXO10 | KMT2B | SUV39H1 |
| FBXO11 | KMT2C | SUV39H2 |
| GTF3C4 | KMT2D | SUV420H1 |
| HAT1 | KMT2E | SUV420H2 |
| HDAC1 | NCOA1 | TAF1 |
| HDAC10 | NCOA2 | TAF1L |
| HDAC11 | NCOA3 | TRIM24 |
| HDAC2 | NSD1 | TRIM28 |
| HDAC3 | PBRM1 | TRIM33 |
| HDAC4 | PHF2 | TRIM66 |
| HDAC5 | PHF8 | ZMYND11 |
| HDAC6 | PHIP | ZMYND8 |
| HDAC7 |
| LIGANDS |
|---|
| AMI-1 |
| anacardic acid |
| AZ505 |
| benzo[d]imidazole inhibitors of PRMT4 from BMS |
| BIX-01294 |
| BMS pyrazole inhibitor 7f |
| BRD4770 |
| bromo-deaza-SAH |
| butyric acid |
| C21 |
| C646 |
| Compound 1 (allosteric) |
| curcumin |
| daminozide |
| EI1 |
| entinostat |
| epigallocatechin-3-gallate |
| EPZ-5676 |
| EPZ-6438 |
| garcinol |
| GSK126 |
| GSK-J1 |
| H3-CoA-20 |
| I-BET-762 |
| Lys-CoA |
| mocetinostat |
| nahuoic acid A |
| NCL-1 |
| OG-L002 |
| PBIT |
| plumbagin |
| RM65 |
| rocilinostat |
| romidepsin |
| RVX 208 |
| SGC0946 |
| trichostatin A |
| UNC0638 |
| UNC0642 |
| valproic acid |
| vorinostat |
This table lists protein targets and ligands which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013a, Alexander et al., 2013b).
Introduction
The properties of a cell are determined by its specific genetic material and the pattern in which its genes and, ultimately, proteins are expressed. While the transmissibility of the genome is well recognized, the fact that cellular phenotypes can remain stable through cell division – a dividing T-cell yields two T-cells, a dividing hepatocyte yields two hepatocytes, etc. – indicates that specific gene expression patterns are also heritable in daughter cells. The latter type of inheritance is termed epigenetic (literally, ‘above genetic’) because the generation and maintenance of differentiated cell phenotypes is not due to changes in the nucleotide sequence. While heritable, epigenetic memory is also malleable, in that gene expression patterns can change in response to environmental stimuli. This allows for the development of different cell lineages during processes such as embryogenesis or haematopoiesis, and also for more subtle changes in cell function in response to physiological requirements, or importantly pathological stress that occurs during adaptive immunity.
Epigenetic control of gene expression is linked to the manner in which eukaryotic DNA associates with nuclear proteins, in a structure known as chromatin. The basic unit of chromatin is the nucleosome, which consists of approximately 147 bp of DNA wrapped around an octamer of core histones (which includes two each of histones H3, H4, H2A and H2B) (Luger et al., 1997; Davey et al., 2002). Further higher order structuring of nucleosomes through interactions with additional histone and non-histone proteins allows for tight compaction of the DNA into the limited volume of the nucleus (Talbert and Henikoff, 2010). In addition to permitting efficient DNA packing, chromatin provides an intricate scaffold for interacting with nuclear proteins such as those of the gene transcription machinery. Importantly, chromatin is subject to modifications that generate highly ordered sophisticated heterogeneity in its structure and, as a consequence, in the potential for different gene regions to be expressed. This recognition of and response to specific combinatorial patterns of modifications allows the complex epigenetic code to be translated into selective control of coordinated clusters of genes in transcriptional modules (Taverna et al., 2007; Hardison and Taylor, 2012).
Chromatin can be altered by modifications to both the DNA and the associated histone proteins. At the level of the DNA, the main modification described so far is methylation of cytosine residues, which occurs predominantly in the context of the dinucleotide sequence CpG and is mediated by members of the DNA-methyltransferase family (DNMT1, DNMT3A, DNMT3B) (Okano et al., 1999; Pradhan et al., 1999). DNA methylation leads to suppression of gene transcription through a number of mechanisms, including direct inhibition of transcription factor binding, which is necessary for recruitment of the transcription machinery, and attraction of methyl-CpG-binding domain-containing proteins that associate with transcriptionally repressive protein complexes (Reddington et al., 2013). Methyl marks can be removed from DNA in a passive manner through a failure to remethylate daughter DNA strands during cell division, or in an active process involving ten-eleven translocation (TET1-3) family proteins or DNA glycosylases and repair-mediated excision of modified bases (He et al., 2011b; Ito et al., 2011; Maiti and Drohat, 2011; Spruijt et al., 2013). Notably, TET enzymes generate intermediates in the pathway between 5-methylcytosine and cytosine, including 5-hydroxymethylcytosine, 5-formylcytosine and 5-carboxycytosine, which can be detected in cellular chromatin and may serve as epigenetic marks in their own right (Delatte and Fuks, 2013). The therapeutic potential of targeting DNA methylation including the activity of licensed pan-DNMT inhibitors to impact certain cancers has been reviewed recently (Carey et al., 2012), and will not be discussed further here.
Histone modification has been studied most extensively for the core histones of the nucleosome. Many different amino acids in these proteins are subject to post-translational modification, although the majority of these occur in the N-terminal or C-terminal ‘tails’ which extend outside of the main, globular histone domains (Kouzarides, 2007). By contrast to the relatively limited complexity of DNA methylation, histones can be modified in a wide variety of ways; by some accounts up to 60 different chemical modifications of histones have been documented, including acetylation, methylation, phosphorylation, ubiquitination, sumoylation, ADP ribosylation, crotonylation and biotinylation (Stanley et al., 2001; Kouzarides, 2007; Tan et al., 2011; Nikolov and Fischle, 2013). As discussed further below, several families of enzymes responsible for the addition (writers) or removal (erasers) of these modifications have been identified. The multiplicity of modifications and amino acid substrates can be arranged into a very high number of distinct combinations, which has prompted the hypothesis that the different patterns represent a ‘histone code’ that provides specific instructions for a given region of DNA (Strahl and Allis, 2000). Although there is presently little understanding of a detailed code, the functional relevance of histone modification to gene regulation is evident from observed correlations between the presence of certain combinations of marks and gene expression.
Histone modifications are thought to impact gene expression in two broad ways. First, certain modifications such as acetylation alter the net charge of the histone, weakening the DNA–histone interaction and yielding a chromatin structure that is more open and hence accessible to transcription factors and gene expression machinery (Hong et al., 1993). Second, and potentially more importantly, histone modifications serve as recognition marks for proteins termed epigenetic ‘readers’, which can specifically bind to these modified amino acids (Taverna et al., 2007). Thus, while writers and erasers produce the histone code, it is the epigenetic readers that decipher and translate this information (Figure 1). Reader proteins typically possess activities that lead to activation or suppression of gene expression, and/or are able to recruit other proteins that possess such functions. As is the case for the epigenetic writers and erasers, large families of readers able to recognize different modified amino acids have been identified and recently reviewed (Arrowsmith et al., 2012).
Figure 1.
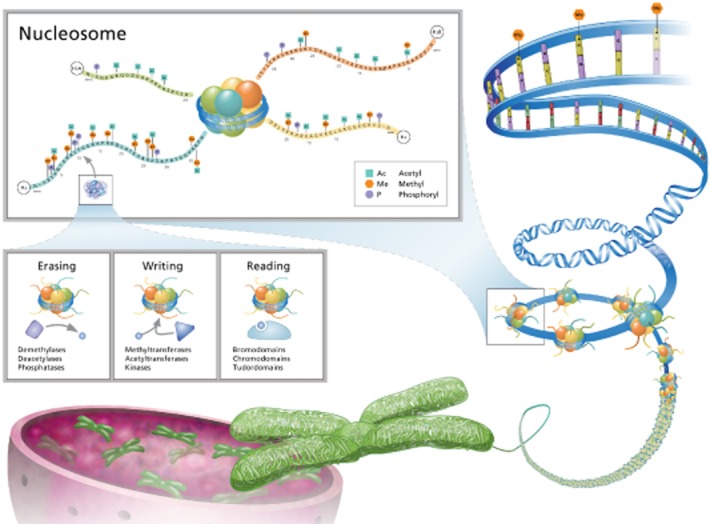
Histone modification in the regulation of chromatin structure. Nucleosomes, which represent the core subunits of chromatin, are subject to numerous modifications that influence chromatin structure and gene expression. Many different amino acids in the core histones can be altered by enzymes termed epigenetic ‘writers’ that generate various post-translational modifications, such as acetylation, methylation and phosporylation. These marks can be removed by ‘erasers’ and are recognised by ‘reader’ proteins which contain domains capable of specific recognition of the modified peptide sequences.
Together, DNA and histone modifications help control the ‘transcribability’ of genes. Thus, linked to the state of the chromatin, a gene or gene cluster may be silenced, constitutively expressed or poised for expression (or suppression) in response to a specific cell signal (Kouzarides, 2007). The dynamic nature of chromatin modifications provides a mechanism for cells to adapt their gene expression pattern in response to environmental cues. This is now well documented for fundamental biological processes such as the differentiation of activated T-cells into effector subsets, where the capacity of the cells to produce different cytokines is controlled by epigenetic modifications in cytokine gene regions that are induced by specific conditions of T-cell activation (Kanno et al., 2012). However, there is also growing evidence that aberrant epigenetic states are associated with a range of pathologies, including inflammatory, neuropsychiatric, cardiovascular, and metabolic diseases and cancer. The identification of the proteins responsible for writing, erasing and reading chromatin modifications has opened up a new area of drug discovery, with the hope of being able to reset abnormal epigenetic landscapes back to normal and thus provide lasting health benefits in these diseases. In this review, we discuss early progress in developing small molecule pharmacological modulators of epigenetic targets, focusing on a subset of histone writers, erasers and readers where understanding of biological relevance and chemical tractability is most advanced.
Lysine acetyltransferases
Multiple lysine residues in each of the core histones are subject to acetylation. Histone acetylation is mediated by a family of enzymes, the lysine acetyltransferases (KATs), which utilize acetyl CoA as a cofactor to catalyse the transfer of an acetyl group to the ε-amino group of lysine side chains (Roth et al., 2001). At least 17 mammalian KATs able to acetylate histones have been identified, although the relatively low sequence homology among protein acetyltransferases suggests that many such enzymes could yet be discovered (Yuan and Marmorstein, 2012). These enzymes have traditionally been grouped into families based on similarity in the sequence of their catalytic domains and biochemical mechanism of acetyl transfer, and are listed utilizing the simplified nomenclature proposed by Allis et al. (Sterner and Berger, 2000; Roth et al., 2001; Allis et al., 2007; Furdas et al., 2012) in Table 1. Different KATs have been shown to preferentially actetylate distinct lysine residues in histones, although considerable overlap appears to exist.
Table 1.
Lysine acetyltransferases
| Family | Proposed symbol* | Synonyms and other symbols | Uni-ProtKB/Swiss-Prot assession number |
|---|---|---|---|
| GNAT | KAT2A | GCN5, GCN5L2, HGCN5, PCAF-b | Q92830 |
| KAT2B | PCAF, P/CAF | Q92831 | |
| KAT9 | ELP3, FLJ10422 | Q9H9T3 | |
| P300/CBP | KAT3A | CBP, CREBBP, RSTS, RTS | Q92793 |
| KAT3B | P300, EP300 | Q09472 | |
| MYST | KAT5 | TIP60, PLIP, HTATIP, cPLA2, HTATIP1, ESA1, ZC2HC5 | Q92993 |
| KAT6A | MOZ, MYST3ZNF220, RUNXBP2, ZC2HC6A | Q92794 | |
| KAT6B | MORF, MYST4, querkopf, qkf, MOZ2, ZC2HC7 | Q8WYB5 | |
| KAT7 | HBO1, MYST2, HBOA, ZC2HC7 | O95251 | |
| KAT8 | hMOF, MYST1, MOF, FLJ14040, ZC2HC8 | Q9H7Z6 | |
| Transcription factor-related | KAT4 | TAF1, TAF2A, BA2R, CCG1, CCGS, DYT3, NSCL2, TAFII250, DYT3/TAF1 | P21675 |
| KAT12 | TFIIIC90, GTF3C4 | Q9UKN8 | |
| NR co-activators | KAT13A | SRC1, NCOA1, F-SRC-1, NCoA-1, RIP160, bHLHe74 | Q15788 |
| KAT13B | AIB1, ACTR, SRC3, NCOA3, RAC3, P/CIP, TRAM-1, cAGH16, TNRC16, bHLHe42, SRC-3 | Q9Y6Q9 | |
| KAT13C | P160, NCOA2, TIF2, GRIP1, NCoA-2, bHLHe75 | Q15596 | |
| KAT13D | CLOCK, KIAA0334 | O15516 | |
| Type B (cytoplasmic) | KAT1 | HAT1/HATB | O14929 |
Proposed symbols are those suggested by Allis et al. (2007). A list of the proposed lysine acetyltransferase names and symbols that have been approved by the HUGO Gene Nomenclature Committee (HGNC) can be found at http://www.genenames.org/genefamilies/kdm-kat-kmt#KAT.
Two things are worth noting with respect to the designation of these enzymes as histone acetyltransferases. First, several of these KATs are known to also acetylate non-histone proteins, which may make an important contribution to their function; this idea is supported by the detection of lysine acetylation in nearly 2000 proteins involved in many key cellular processes (Choudhary et al., 2009). Second, in some cases, enzymes have only been shown directly to acetylate histones in vitro, and their ability to do so in a cellular context remains unknown.
Acetylation of histone tails has long been associated with active gene transcription (Marushige, 1976). This is linked to the ability of acetylation to generate a more open chromatin structure (Hong et al., 1993), and to the recruitment of specific reader proteins able to bind to acetylated histones (see below). Not surprisingly, therefore, many histone acetyltransferases are known to function in transcriptional activation. However, histone acetylation is also known to influence many other processes, including cell cycle progression, chromosome dynamics, DNA recombination, DNA repair and cellular apoptosis, indicating that acetylation plays a central role in regulating chromatin-related functions (Khan and Khan, 2010).
Misregulation of histone acetyltransferase activity has been linked to many different pathogenic states, including multiple cancers, neurodegenerative disorders, plus metabolic, respiratory, inflammatory and cardiovascular diseases; as such, KATs represent attractive targets for drug development (Adcock and Lee, 2006; Avvakumov and Cote, 2007; Grabiec et al., 2008; Ghizzoni et al., 2011; Iyer et al., 2011; Pirooznia and Elefant, 2013). Several different types of KAT inhibitors have been described (Table 2). Peptide-based bisubstrate inhibitors, in which CoA is covalently linked to the ζ nitrogen of the target lysine within histone peptides, act as structural mimics and can show sub-micromolar potency and selectivity between KAT subfamilies (Lau et al., 2000). However, since these inhibitors lack cellular permeability, they have limited use for evaluation of the cellular function of the KATs. Natural products isolated from a number of different plants, as well as their synthetic derivatives, have been shown to possess KAT inhibitory activity (Balasubramanyam et al., 2003; 2004a,b; Choi et al., 2009; Ravindra et al., 2009). In addition, small molecule KAT inhibitors have been identified by high-throughput biochemical and computational (virtual) screens (Gorsuch et al., 2009; Bowers et al., 2010). One compound identified from a virtual screen, C646, was reported to be selective for KAT3A/3B over other KATs and was shown to inhibit the growth of tumour cells in vitro (Bowers et al., 2010).
Table 2.
Examples of KAT inhibitors
| Compound | Structure | Potency (µM) | Known KAT target | Source | Reference |
|---|---|---|---|---|---|
| H3-CoA-20 | 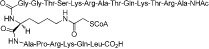 |
0.5 | KAT2B | Peptide-based bisubstrate inhibitors | (Lau et al., 2000) |
| Lys-CoA | 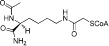 |
0.5 | KAT3B | Peptide-based bisubstrate inhibitors | (Lau et al., 2000) |
| Anacardic acid | 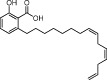 |
5–8.5 | KAT2B, KAT3B | Cashew nut shell | (Balasubramanyam et al., 2003) |
| Curcumin | 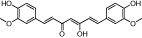 |
25 | KAT3A, KAT3B | Turmeric (spice) | (Balasubramanyam et al., 2004b) |
| Garcinol | 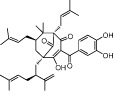 |
5–7 | KAT2B, KAT3B | Garcinia indica fruit rind | (Balasubramanyam et al., 2004a) |
| Epigallocatechin-3-gallate (EGCG) | 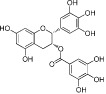 |
30–50 | Pan-KAT | Green tea | (Choi et al., 2009) |
| Plumbagin (RTK1) | 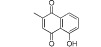 |
20–50 | KAT3A, KAT3B, KAT2B | Plumbago rosea root | (Ravindra et al., 2009) |
| C646 | 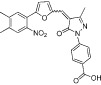 |
5 | KAT3A, KAT3B | Small molecule virtual screen | (Bowers et al., 2010) |
Overall, however, the development of therapeutic KAT inhibitors is at an early stage, and current compounds are suboptimal in a number of ways, because they generally lack potency and selectivity. Nevertheless, one natural product KAT inhibitor, curcumin, has entered into clinical trials for a number of diseases, including Alzheimer's disease, rheumatoid arthritis, cystic fibrosis and psoriasis; there are currently 85 studies at different stages in http://ClincalTrials.gov that list curcumin or related compounds. As this compound is known to affect a number of other epigenetic and non-epigenetic targets, including DNMT I and lysine deacetylases (KDACs), linking potential efficacy with effects on KATs will not be straightforward.
Lysine deacetylases
The reversal of histone acetylation is mediated by members of the KDAC family, which comprises 18 enzymes that can be divided into four classes based on their homology to the yeast orthologues Rpd3, HdaI and Sir2 (Gregoretti et al., 2004) (Table 2013b). The class I, II and IV enzymes are termed histone deacetylases (HDACs), while the class III deacetylases are named sirtuins (SIRTs). The seven sirtuins (SIRT1–7) share a conserved catalytic core domain and use NAD+ as an essential cofactor, while HDACs contain a Zn2+ ion in their active site. Notwithstanding the nomenclature, several sirtuins do function as HDACs, while at least one HDAC, HDAC6, appears to be almost exclusively localized to the cytoplasm and may not deacetylate histones in vivo (Verdel et al., 2000; Zhang et al., 2003; Martinez-Redondo and Vaquero, 2013). In addition to deacetylase activity, some SIRT proteins also possess other enzymatic activities. Thus, while SIRT1–3 and SIRT7 act primarily as KDACs, SIRT4 is an ADP-ribosyltransferase, SIRT5 is a deacetylase, demalonylase, and desuccinylase, and SIRT6 is an ADP-ribosyltransferase and a deacetylase (Roth and Chen, 2013). As for the KATs, KDACs can deacetylate many histone and non-histone proteins.
Table 3.
Lysine deacetylases
| Class | Symbol | Cellular localization | Uni-ProtKB/Swiss-Prot assession number |
|---|---|---|---|
| I (Rpd3) | HDAC1 | Nucleus | Q13547 |
| HDAC2 | Nucleus | Q92769 | |
| HDAC3 | Nucleus | O15379 | |
| HDAC8 | Nucleus/cytoplasm | Q9BY41 | |
| IIa (Hda1) | HDAC4 | Nucleus/cytoplasm | P56524 |
| HDAC5 | Nucleus/cytoplasm | Q9UQL6 | |
| HDAC7 | Nucleus/cytoplasm | Q8WUI4 | |
| HDAC9 | Nucleus/cytoplasm | Q9UKV0 | |
| IIb (Hda1) | HDAC6 | Cytoplasm | Q9UBN7 |
| HDAC10 | Cytoplasm | Q969S8 | |
| III (Sir) | SIRT1 | Nucleus/cytoplasm | Q96EB6 |
| SIRT2 | Nucleus/cytoplasm | Q8IXJ6 | |
| SIRT3 | Mitochondria | Q9NTG7 | |
| SIRT4 | Mitochondria | Q9Y6E7 | |
| SIRT5 | Mitochondria | Q9NXA8 | |
| SIRT6 | Nucleus | Q8N6T7 | |
| SIRT7 | Nucleolus | Q9NRC8 | |
| IV (Rpd3/Hda1) | HDAC11 | Nucleus/cytoplasm | Q96DB2 |
In accordance with the key role that acetylation plays in the regulation of chromatin structure, as well as the expansive function of lysine acetylation in protein networks and cellular signalling pathways (Choudhary et al., 2009), HDACs have been implicated in the regulation of gene expression and in the control of many important cellular processes such as proliferation, DNA repair and apoptosis (Lahue and Frizzell, 2012; Li et al., 2012; Matthews et al., 2012; Yao and Rahman, 2012; Joshi et al., 2013; Ververis et al., 2013). Moreover, dysregulation of HDACs has been proposed to contribute to a variety of diseases including cancer, interstitial fibrosis, autoimmune and inflammatory diseases, and metabolic disorders (Tang et al., 2013a). For this reason, considerable effort has gone into the development of HDAC inhibitors.
Most of the HDAC inhibitors generated to date show activity against multiple family members. Among these, there is a range in the breadth of activity (Table 4) – from those that inhibit essentially all HDACs [e.g. trichostatin A and suberoylanilide hydroxamic acid (SAHA)], to some that are active against class I and IIa HDACs (e.g. butyric acid, valproic acid), to others that appear more selective to class I HDACs (e.g. romidepsin, MS-275) (Khan and La Thangue, 2012) or class IIa HDACs (Lobera et al., 2013). Experiments using such non-selective HDAC compounds have provided much of the evidence that HDAC inhibition could be effective in a range of therapeutic areas (Grabiec et al., 2011; Tang et al., 2013a). More recently, inhibitors with selectivity for HDAC1, HDAC3, HDAC6 and HDAC8 have been described (Hu et al., 2003; Schlimme et al., 2011; Cantley and Haynes, 2013; Jochems et al., 2013). Although there is currently limited understanding of the biological implications of targeting individual family members, HDAC6-selective compounds have been reported to inhibit cancer cell proliferation in vitro and in xenograft models in vivo and to exert antidepressant activity in a mouse model (Schlimme et al., 2011; Santo et al., 2012; Jochems et al., 2013). The latter activity was shown to be associated with increased acetylation of the HDAC6 target α-tubulin rather than histone acetylation (Jochems et al., 2013).
Table 4.
Examples of HDAC inhibitors
| Name | Structure | Potency | HDAC specificity | Clinical trial (cancer) |
|---|---|---|---|---|
| TSA (trichostatin A) | 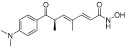 |
1.8 nM (HDAC5) | Class I, II, IV | – |
| Vorinistat (SAHA, suberoylanilide hydroxamic acid) | 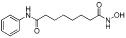 |
10 nM (HDAC1,2,3,8,9) | Class I, II, IV | FDA approved (2006) Phase II, III |
| Romidepsin (FK228) | 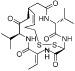 |
36–47 nM (HDAC1,2) | Class I | FDA approved (2009) Phase I, II |
| Entinostat (MS-275) | 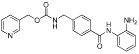 |
500 nM (HDAC1,2,3,9) | Class I | Phase II |
| Mocetinostat (MGCD0103) | 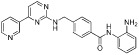 |
0.15–1.66 µM (HDAC1,2,3) | HDAC1 | Phase I, II |
| Butyric acid | 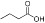 |
mM | Class I, II | Phase II |
| Valproic acid | 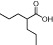 |
0.7–20 mM (HDAC1,2,3) | Class I, II | Phase I, II, III |
| ACY-1215 (rocilinostat) | 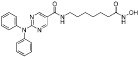 |
5 nM | HDAC6 | Phase I/II |
The ability of HDAC inhibitors to induce death, cytostasis or differentiation of tumour cells in preclinical models, combined with evidence of HDAC up-regulation in a variety of cancers, has provided a strong rationale for progressing such compounds into clinical trials for oncology (Giannini et al., 2012). Presently, there are 394 trials that include the term ‘HDAC inhibitor’ recorded in http://ClinicalTrials.gov, with the vast majority of these being in cancer. A large number of trials in multiple solid and haematological tumour types are in progress, with the most promising results obtained so far being observed when HDAC inhibitors were combined with other agents such as proteasome inhibitors (Khan and La Thangue, 2012; Qiu et al., 2013; Richardson et al., 2013; Ververis et al., 2013). Currently, there are two HDAC inhibitors that have received approval from the US FDA for the treatment of cutaneous T-cell lymphoma: vorinostat (SAHA, Zolinza®; Merck & Co., Inc., Whitehouse Station, NJ, USA) and depsipeptide (romidepsin, Istodax, Celgene Corporation, Summit, NJ, USA).
Although broadly active HDAC inhibitors are being tested in the vast majority of trials, one HDAC1-selective inhibitor (MGCD0103) has progressed into clinical trials in haematological tumours, and a HDAC6-selective inhibitor is being evaluated as a monotherapy and also in combination with other treatments in patients with relapsed or relapsed/refractory multiple myeloma (NCT01323751, NCT01583283). By selectively targeting HDAC subfamily members, it is hoped that there will be an opportunity to reduce toxic side effects associated with broadly inhibiting lysine deacetylation and hence for an improved therapeutic treatment window. Another strategy being evaluated in this respect is to design compounds that will preferentially accumulate in specific cell types. This is the basis behind the HDAC inhibitor CHR-2845, a cell-permeant ester that is metabolized to give an active acid which selectively accumulates in monocytes and macrophages. This compound has been evaluated for tolerability in patients with advanced haematological malignancies (NCT00820508), and it is hoped that this approach will be effective in haematological malignancies involving cells of the monocyte lineage.
Reflecting the broad biological effects of HDAC inhibition in preclinical studies, HDAC inhibitors have also entered into trials in other therapeutic areas, including graft versus host disease (NCT01111526), sickle cell disease (NCT01000155), Huntington's disease (NCT00212316), Rubinstein–Taybi syndrome (NCT01619644) and human immunodeficiency virus (HIV; NCT01680094). In the latter study, the aim is to evaluate the ability of the compound to reactivate HIV transcription in latently infected CD4+ T-cells, which would form part of an approach to deplete the latent pool of virus (in combination with anti-retroviral therapy). Evidence that HDAC inhibitors can induce such HIV reactivation has come from studies involving ex vivo compound treatment of cells from HIV-infected individuals, although initial small scale clinical trials have yielded conflicting results concerning the combined effects of HDAC inhibition and anti-retroviral therapy on viral load (Margolis, 2011).
Much of the interest in the SIRT family of KDACs has focused on the function of these enzymes in metabolic, oxidative/genotoxic, and oncogenic stress responses, where their deacetylation of non-histone substrates may play a predominant role. In view of the protective role of SIRTs in these processes, a major therapeutic focus has been on the development of SIRT activators for the treatment of ageing-associated pathologies, including type II diabetes, cardiovascular disease and neurodegeneration (Hall et al., 2013). SIRT1 activation has also been implicated in suppressing the immune response, leading to an interest in developing SIRT activators for treatment of autoimmune and inflammatory diseases (Kong et al., 2013). In cancer, the role of SIRT proteins is complex, with evidence for SIRTs playing roles in both promoting and suppressing tumourigenesis (Roth and Chen, 2013).
Small molecules capable of SIRT1 activation have been identified, including the weakly active polyphenol compound resveratrol, a component of red wine, and a series of compounds structurally unrelated to resveratrol with 1000-fold greater potency against SIRT1 (Dittenhafer-Reed et al., 2011). SIRT1 activators became the subject of controversy when it was shown that these compounds activated enzyme activity towards fluorescently labelled peptide substrates but not their unlabelled counterparts, suggesting a possible assay artefact (Kaeberlein et al., 2005; Pacholec et al., 2010). However, an intriguing explanation for these observations was subsequently provided when it was shown that the bulky hydrophobic fluorophore tag on the assay peptide mimicked hydrophobic amino acids present in a subset of natural SIRT1 protein substrates that are selectively subject to increased deacetylation after SIRT1 activation (Hubbard et al., 2013; Lakshminarasimhan et al., 2013). In addition, a single amino acid substitution outside of the catalytic site in SIRT1 was found to abolish activation of the enzyme as well as the cellular effects mediated by SIRT1 activators, demonstrating that SIRT1 is indeed the target of these compounds.
Trials of resveratrol in obese humans produced mild improvements in a number of different clinical parameters including systolic blood pressure, circulating cytokines, intrahepatic fat content, intramyocellular lipid content, and muscle mitochondrial oxidative phosphorylation capacity (Timmers et al., 2011). However, because resveratrol has known activity against a number of substrates besides SIRT1, including AMP-activated kinase, a fuel-sensing enzyme that is responsive to decreases in cellular energy status, the mechanism by which resveratrol mediates these effects, remains a subject of debate. Early phase clinical trials investigating higher potency SIRT1 activators (SRT2104, SRT2379) in both metabolic diseases and inflammation have been conducted (Libri et al., 2012; Hoffmann et al., 2013). Published results for SRT2104 indicate that this compound appears to be safe and well tolerated and associated with an improved lipid profile (Libri et al., 2012).
Lysine methyltransferases
A large number of enzymes capable of transferring methyl groups to lysine residues have been described. The 24 human lysine methyltransferases (KMTs) categorized by Allis et al. (2007) are listed in Table 5. All KMTs except one [KMT4/Dot1L, a unique KMT, which belongs to the class I methyltransferase family (Min et al., 2003)] contain a catalytic domain of approximately 130 amino acids, referred to as the SET domain; both SET domain-containing enzymes and KMT4 use S-adenosyl methionine (SAM) as the methyl donor. Based on a systematic screen for SET domains in the human genome, 51 putative KMTs have been identified, although the enzymatic activity of many of these is yet to be investigated (Copeland et al., 2009; Richon et al., 2011). KMTs exhibit selectivity for both the lysine residue they can modify and the degree to which that lysine residue is methylated. Thus, while lysine residues can only accept a single acetyl group, lysines can be mono-, di- or trimethylated. The site specificity of lysine methylation is determined by recognition of amino acid residues flanking the target lysine, whereas particular amino acids within the lysine-binding channel of the KMTs play an important role in dictating the methylation multiplicity of the SET domain (Qian and Zhou, 2006).
Table 5.
Lysine methyltransferases
| Proposed symbol* | Synonyms and other symbols | Histone substrate specificity | Uni-ProtKB/Swiss-Prot assession number |
|---|---|---|---|
| KMT1A | SUV39H1, SUV39H | H3K9me2/3 | O43463 |
| KMT1B | SUV39H2, FLJ23414 | H3K9me2/3 | Q9H5I1 |
| KMT1C | G9a, EHMT2, C6orf30, BAT8, Em:AF134726.3, NG36/G9a | H3K9me1/2, H1K26me1/2 | Q96KQ7 |
| KMT1D | GLP, EHMT1, Eu-HMTase1, FLJ12879, KIAA1876, bA188C12.1 | H3K9me1/2 | Q9H9B1 |
| KMT1E | ESET, SETDB1, KG1T, KIAA0067, TDRD21 | H3K9me2/3 | Q15047 |
| KMT1F | SETDB2, CLL8, C13orf4, CLLD8 | H3K9me2/3 | Q96T68 |
| KMT2A | MLL1, TRX1, HRX, ALL-1, HTRX1, CXXC7, MLL1A, MLL | H3K4me1/2/3 | Q03164 |
| KMT2B | MLL2, KIAA0304, TRX2, HRX2, WBP7, MLL1B, MLL4 | H3K4me1/2/3 | Q9UMN6 |
| KMT2C | MLL3, KIAA1506, HALR | H3K4me1/2/3 | Q8NEZ4 |
| KMT2D | MLL4, TNRC21, MLL2, ALR, CAGL114 | H3K4me1/2/3 | O14686 |
| KMT2E | MLL5, HDCMC04P | H3K4me1/2/3 | Q8IZD2 |
| KMT2F | hSET1A, SETD1A, KIAA0339, Set1 | H3K4me1/2/3 | O15047 |
| KMT2G | hSET1B, SETD1B, KIAA1076, Set1B | H3K4me1/2/3 | Q9UPS6 |
| KMT2H | ASH1, ASH1L, huASH1, ASH1L1 | H3K4me1/2/3, H3K9me1/2/3, H4K20me1/2/3 | Q9NR48 |
| KMT3A | SET2, SETD2, HYPB, HIF-1, KIAA1732, FLJ23184 | H3K36me3 | Q9BYW2 |
| KMT3B | NSD1, STO, ARA267, FLJ22263 | H3K36me1/2, H4K20me1/2 | Q96L73 |
| KMT3C | SYMD2, HSKM-B, ZMYND14 | H3K36me1/2 | Q9NRG4 |
| KMT3D | SMYD1, BOP, ZMYND22 | Unknown | Q8NB12 |
| KMT3E | SMYD3, ZNFN3A1, ZMYND1 | H3K4me2/3 | Q9H7B4 |
| KMT4 | DOT1L, KIAA1814, DOT1 | H3K79me1/2/3 | Q8TEK3 |
| KMT5A | Pr-SET7, SETD8, SET07, SET8 | H4K20me1 | Q9NQR1 |
| KMT5B | SUV420H1, CGI-85 | H4K20me2/3 | Q4FZB7 |
| KMT5C | SUV420H2, MGC2705 | H4K20me2/3 | Q86Y97 |
| KMT6A | EZH2, EZH1, ENX-1, KMT6 | H3K27me1/2/3, H1K26me2/3 | Q15910 |
| KMT6B | EZH1, KIAA0388 | H3K27me1/2/3 | Q92800 |
| KMT7 | SET7/9, KIAA1717, SET7, Set9 | H3K4me2 | Q8WTS6 |
| KMT8 | PRDM2, RIZ1, RIZ, RIZ2, MTB-ZF, HUMHOXY1 | H3K9me1/2/3 | Q13029 |
Proposed symbols are those suggested by Allis et al. (2007). A list of the proposed lysine methyltransferase names and symbols that have been approved by the HUGO Gene Nomenclature Committee (HGNC) can be found at http://www.genenames.org/genefamilies/kdm-kat-kmt#KMT.
In contrast to histone acetylation, histone methylation does not alter the charge of the histone tail, but instead influences its basicity and hydrophobicity (Migliori et al., 2010). As for acetylation, methylated lysines also serve as recognition marks for a large family of methyl reader proteins, which show selectivity for both the specific lysine residue modified and the degree of methylation. As a consequence, different types of lysine methylation are associated with divergent functions in the regulation of gene expression. For example, trimethylation of H3K4 (H3K4me3) is commonly found near the transcription start site of genes that are actively expressed or poised for expression, whereas H3K27me3 is a mark that is associated with genes for which expression is suppressed (Kouzarides, 2007). Conversely, the presence of H3K4me1 is tightly correlated with the position of gene enhancers, which are non-coding regions of DNA that promote the expression of genes that are often located a considerable distance away (Heintzman et al., 2009).
In addition to the histone tail, the residues within the core of the nucleosome can also be modified by methylation. For instance, H3K79 is subject to methylation by KMT4, and methylated H3K79 is associated with active gene transcription (Feng et al., 2002; Steger et al., 2008; Kim et al., 2013). Lysine 20 is the only well-characterized methylation site on histone H4, with methylated H4K20 being linked with transcriptional repression and a number of biological processes, including the DNA damage response, mitotic condensation and DNA replication (Jorgensen et al., 2013); the transition from H4K20me1 to me2 and me3 was recently associated with cellular quiescence (Evertts et al., 2013).
Misregulation of KMTs has been linked to a variety of human diseases, and therapeutic interest in the development of KMT inhibitors is particularly strong in cancer, where the pathological involvement of KMT overexpression, mutation and translocation has been shown (Copeland et al., 2009). One interesting example of a potential cancer target is KMT4, which is implicated in leukaemias involving chromosomal translocation of another KMT, KMT2A (MLL1). In such cancers, KMT4 is recruited into the transcriptional complex via MLL-fusion partners, and aberrant methylation of H3K79 is considered to play a causative role in disease (Steger et al., 2008; Bernt et al., 2011). Another target of major interest is KMT6, which is a key mediator of di- and trimethylation of H3K27 and is overexpressed in many types of cancers (Chang and Hung, 2012).
In recent years, small molecule KMT inhibitors have been developed that are directed at either the SAM or the substrate site of the enzymes (Wigle and Copeland, 2013). Inhibitors have been reported for various KMTs, including KMT4, KMT6, KMT1C/D, KMT3C and KMT5A (Table 6). Compounds targeting KMT4, KMT1C/D and KMT6 have shown promising efficacy in preclinical tumour models (Daigle et al., 2011; 2013; Yuan and Marmorstein, 2012; Yuan et al., 2012; Knutson et al., 2013; Liu et al., 2013), strengthening the rationale for targeting these enzymes in cancer. Building on this rationale, the KMT6 inhibitor E7438 is currently being trialled in patients with advanced solid tumours or with B-cell lymphomas (Knutson et al., 2013) (NCT01897571) while the KMT4 inhibitor EPZ-5676 has recently entered phase I studies in patients with advanced haematological malignancies (NCT01684150) (Copeland, 2013; Daigle et al., 2013).
Table 6.
Recent examples of lysine methyltransferase inhibitors
| Name | Structure | Potency | KMT specificity | Reference |
|---|---|---|---|---|
| EPZ-6438 | 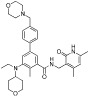 |
2.5 nM | MT6 | (Knutson et al., 2013) |
| GSK126 | 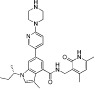 |
0.5–3 nM | KMT6 | (McCabe et al., 2012) |
| EI1 | 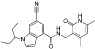 |
9.4 nM | KMT6 | (Qi et al., 2012) |
| BIX-01294 | 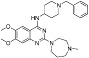 |
2.7 µM | KMT1C/D | (Kubicek et al., 2007; Lu et al., 2013) |
| UNC0638 | 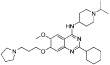 |
<15 nM | KMT1C/D | (Vedadi et al., 2011) |
| BRD4770 | 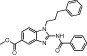 |
6.3 µM | KMT1C/D | (Yuan and Marmorstein, 2012; Yuan et al., 2012) |
| UNC0642 | 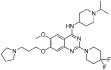 |
<2.5 nM | KMT1C/D | (Liu et al., 2013) |
| Bromo-deaza-SAH | 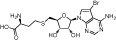 |
77 nM | KMT4 | (Yu et al., 2013) |
| EPZ-5676 | 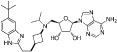 |
80 pM | KMT4 | (Daigle et al., 2011) |
| SGC0946 | 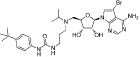 |
0.3 nM | KMT4 | (Yu et al., 2012) |
| AZ505 | 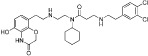 |
0.12 µM | KMT3C | (Ferguson et al., 2012) |
| Nahuoic acid | 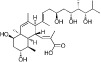 |
2 µM | KMT5A | (Williams et al., 2013) |
Lysine demethylases
Lysine-specific demethylases (KDMs) capable of removing methyl groups from histones are classified into two families (Table 7). One family is comprised of KDM1A (LSD1) and the closely related KDM1B (LSD2), demethylate lysines, utilizing a flavin adenine dinucleotide (FAD)-dependent oxidation mechanism (Shi et al., 2004; Karytinos et al., 2009). KDM1A and KDM1B are able to demethylate mono- and dimethylated lysines, but cannot remove the methyl group from trimethylated lysine because of the dependence of the demethylation mechanism on a protonated amine (Hou and Yu, 2010). The second much larger family of histone demethylases is formed by the jumonji C-domain (Jmj)-containing enzymes, which catalyse lysine demethylation through a hydroxylation pathway utilizing Fe2+ and α-ketoglutarate (α-KG) as cofactors (Tsukada et al., 2006). Unlike KDM1A and KDM1B, the Jmj family of enzymes does not depend on a protonated amine and therefore can demethylate trimethylated lysines.
Table 7.
Lysine demethylases
| Proposed symbol* | Synonyms and other symbols | Reported histone substrate specificity | Uni-ProtKB/Swiss-Prot assession number |
|---|---|---|---|
| KDM1A | LSD1, AOF2, BHC, KIAA0601, BHC110, | H3K4me1/2, H3K9me1/2 | O60341 |
| KDM1B | LSD2, AOF1, C6orf193, FLJ34109, FLJ33898, dJ298J15.2, bA204B7.3, FLJ43328 | H3K4me1/2 | Q8NB78 |
| KDM2A | JHDM1A, FBXL11, CXXC8, FBL7, KIAA1004, FBL11, LILINA, DKFZP434M1735, FLJ00115 | H3K4me3, H3K36me1/2 | Q9Y2K7 |
| KDM2B | JHDM1B, FBXL10, CXXC2, FBL10, PCCX2 | H3K36me1/2 | Q8NHM5 |
| KDM3A | JMJD1A, JHDM2A, JMJD1, KIAA0742, TSGA | H3K9me1/2 | Q9Y4C1 |
| KDM3B | JMJD1B, JHDM2B, C5orf7, KIAA1082NET22 | H3K9me1/2 | Q7LBC6 |
| KDM3C | JMJD1C, JHDM2C | H3K9me1/2 | Q15652 |
| KDM4A | JMJD2A, JHDM3A, JMJD2, KIAA0677, TDRD14A | H3K9me2/3, H3K36me2/3, H1K26me3 | O75164 |
| KDM4B | JHDM3B, JMJD2B, KIAA0876, TDRD14B | H3K9me2/3, H3K36me2/3, H1K26me3 | O94953 |
| KDM4C | JMJD2C, GASC1, JHDM3C, KIAA0780, TDRD14C | H3K9me2/3, H3K36me2/3, H1K26me3 | Q9H3R0 |
| KDM4D | JMJD2D, JHDM3D, FLJ10251 | H3K9me2/3, H1K26me2/3 | Q6B0I6 |
| KDM4E | JMJD2E, KDM4DL | H3K9me2/3 | B2RXH2 |
| KDM5A | JARID1A, RBBP2, RBP2 | H3K4me1/2/3 | P29375 |
| KDM5B | JARID1B, PLU-1, RBBP2H1, RBBP2H1A, CT31 | H3K4me1/2/3 | Q9UGL1 |
| KDM5C | JARID1C, SMCX, DXS1272E, XE169, MRX13 | H3K4me1/2/3 | P41229 |
| KDM5D | JARID1D, SMCY, HY, HYA, KIAA0234 | H3K4me2/3 | Q9BY66 |
| KDM6A | UTX | H3K27me2/3 | O15550 |
| KDM6B | JMJD3, KIAA0346 | H3K27me2/3 | O15054 |
| KDM7A | JHDM1D, KIAA1718 | H3K9me1/me2, H3K27me1/me2 | Q6ZMT4 |
| KDM7B | PHF8, JHDM1F, KIAA1111, ZNF422, DKFZp686E0868 | H3K9me2/me1, H3K27me2, H3K36me2 | Q9UPP1 |
| KDM7C | PHF2, JHDM1E, GRC5, KIAA0662, MGC176680, CENP-35 | H3K9me2 | O75151 |
| KDM8 | JMJD5, FLJ13798 | H3K36me2 | Q8N371 |
Proposed symbols are those suggested by Allis et al. (2007). A list of the proposed lysine demethylase names and symbols that have been approved by the HUGO Gene Nomenclature Committee (HGNC) can be found at http://www.genenames.org/genefamilies/kdm-kat-kmt#KDM.
As a group, the KDMs have been reported to erase a range of methyl marks on histones (Table 7). Enzymes show preferences for both the degree of lysine methylation and the histone sequence, although promiscuity with respect to the latter is exhibited by some KDMs. This promiscuity can be attributed to the presence of similar amino acids flanking the methylated lysine in distinct peptides or to a dominant role of the peptide backbone, rather than the side chains, in recognition by the enzyme (Hou and Yu, 2010). Importantly, while substrate specificity is often defined in vitro using isolated catalytic domains, the actual lysine residues targeted in vivo may be confined by other mechanisms. For example, although KDM7B can demethylate H3K9me2/me1, H3K27me2 and H3K36me2 in vitro, the intact protein contains a plant homeo domain finger that binds to H3K4me3, directing the catalytic domain towards H3K9me2 (Horton et al., 2009). In addition to regulation by other domains within the same protein, KDMs are typically found within multi-protein complexes that will greatly influence their targeting to chromatin. As for other histone-modifying enzymes, the KDMs can also act on non-histone substrates, which may play a key role in their function. For example, KDM1A has been shown to demethylate key cellular targets such as p53, DNMT 1, STAT3, E2F1 and MYPT1 (Huang et al., 2007a; Wang et al., 2009a; Kontaki and Talianidis, 2010; Yang et al., 2010; Cho et al., 2011).
Given the correlation between particular methyl marks and the transcriptional state of genes, it is expected that the activity of specific KDMs will be linked to gene activation or repression, depending on the KDM substrate. Notably, however, some KDMs such as KDM1A possess the capacity to erase marks associated with both active (H3K4me2) and silent (H3K9me2) genes. In addition, there is considerable apparent redundancy in substrate specificity, with multiple KDMs able to erase the same marks (Table 7). Thus, the functions of KDMs within cells are likely to be determined by multiple factors such as KDM expression level, enzymatic activity, and targeting to specific sites within the genome in the context of particular cells and specific environmental signals. The development of potent and selective inhibitors against members of the family combined with sophisticated epigenetic mapping of functional outcomes (mark and gene transcription level) will be essential to define these in a cellular context.
A major focus of therapeutic interest in KDMs is in oncology, as mutations and aberrant expression of KDMs have been linked to various cancers (Hojfeldt et al., 2013). For example, KDM1A is reported to be overexpressed in a number of different cancers, including neuroblastoma, breast cancer, lung cancer, prostate cancer and bladder cancer (Schulte et al., 2009; Chi et al., 2010; Lim et al., 2010; Hayami et al., 2011). The other FAD-dependent KDM, KDM2B, has also been linked to cancer, with amplification and high expression observed in urothelial carcinoma (Heidenblad et al., 2008). Likewise, Jmj KDMs including KDM2B, KDM4A, KDM4B, KDM4C, KDM5B and KDM7C have been shown to be overexpressed in breast, colorectal, lung, prostate, bladder and other tumours; the functional significance of KDM4C overexpression is further suggested by the presence of the KDM4C gene within an amplified region of a chromosome in multiple cancers (Xiang et al., 2007; Couvelard et al., 2008; Roesch et al., 2010; He et al., 2011a; Berry and Janknecht, 2013; Kogure et al., 2013; Tzatsos et al., 2013). Notably, both increased and decreased expressions of KDMs may be associated with cancer, suggesting a key role for precise control of lysine methylation in maintaining cellular homeostasis. For example, while overexpression of KDM6A is associated with breast cancer and renal cell carcinoma, inactivating mutations in KDM6A are also found in multiple cancer types (van Haaften et al., 2009; Dalgliesh et al., 2010; Gui et al., 2011; Shen et al., 2012; Paolicchi et al., 2013). Inactivating mutations in other KDMs such as KDM5A have also been linked to cancer, supporting the notion that these proteins can have a tumour suppressor function (Dalgliesh et al., 2010).
Although presently there is much less information linking KDMs to other therapeutic areas, there is suggestive genetic evidence that altered KDM activity may be relevant to a number of diseases. For instance, single nucleotide polymorphisms (SNPs) in KDM5A and KDM1A have been linked to the autoimmune diseases ankylosing spondylitis and Grave's disease respectively (Newby et al., 2010; Pointon et al., 2011). KDM6B has also been implicated in an autoimmune disease based on its overexpression in antineutrophil cytoplasmic autoantibody-associated vasculitis (Ciavatta et al., 2010). In addition, SNPs in KDM4C have been linked with autism, and SNPs in KDM3C have been linked with a number of metabolic and haematological parameters, while mutations in KDM5C cause a form of X-linked mental retardation (Jensen et al., 2005; Yuan et al., 2008; Chasman et al., 2009; Soranzo et al., 2009; Johnson et al., 2010; Kantojarvi et al., 2010). Finally, possible therapeutic applications in virus infections are suggested by the demonstrated role for KDM1A in alpha herpes virus reactivation from latency (Liang et al., 2009).
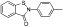
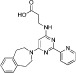
A number of small molecules have been described that inhibit the demethylase activity of KDM1A, the first histone KDM identified. Early KDM inhibitors were generated based on the homology of KDM1A/B with MAOs, which also use FAD as a cofactor. Many of these inhibitors, such as trans-2-phenylcyclopropylamine (PCPA) and paraglyne, are non-specific and broadly inhibit MAOs (Metzger et al., 2005; Lee et al., 2006; Culhane et al., 2010). Derivatives of these molecules that possess some selectivity for KDM1A over MAOs have been produced, such as OG-L002 (>30-fold selective for KDM1A) (Liang et al., 2013) (Table 8). Peptide-based inhibitors (N-propargyl lysine-containing H3 peptides) with greater potency and selectivity than the MAO inhibitors have also been developed, but these possess poor cell permeability and hence are of limited use to investigate cellular activity (Szewczuk et al., 2007; Yang et al., 2007; Culhane et al., 2010; Dancy et al., 2012). Conversely, hybrid molecules between PCPA and lysine produced cell active inhibitors with significant selectivity for KDM1A over MAOs (Ueda et al., 2009; Ogasawara et al., 2011) (Table 8). Selective KDM1 inhibitors have also been developed based on the homology of this enzyme to polyamine oxidases and on the basis of structural features of the KDM1 active site (Huang et al., 2007b; 2009b; Wang et al., 2011).
Table 8.
Examples of KDM inhibitors
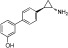
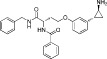
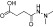
In recent years, considerable progress has also been made in the development of inhibitors targeting the more recently discovered Jmj KDMs. Based on the requirement of these enzymes for α-KG as a cofactor, α-KG analogues such as N-oxalylglycine (NOG) and α-hydroxyglutarate and their derivatives have been investigated and shown to act as low potency non-selective inhibitors of this target class (Cloos et al., 2006; Rose et al., 2008; 2010; Hamada et al., 2010; Chowdhury et al., 2011). More potent compounds that function through α-KG competition have also been identified based on small molecule screens (Rose et al., 2008; King et al., 2010; Chang et al., 2011). In addition, compounds showing selectivity for Jmj KDMs over NOG have been identified (Hamada et al., 2010).
While most of these ligand-based inhibitors are promiscuous KDM inhibitors, some compounds with more selective activities have been reported. For example, the plant growth regulator daminozide was shown to be an α-KG-competitive inhibitor that is much more potent against KDM2 and KDM7 family enzymes than other KDMs (Rose et al., 2012); other compounds with a biased activity against KDM2 and KDM7 family enzymes have been developed based on the crystal structure of KDM7B (Suzuki et al., 2013). High-throughput screening has been used to identify inhibitors of KDM4C and KDM5B (Hutchinson et al., 2012; Sayegh et al., 2013). In addition, inhibitors showing selectivity for KDM4 subfamily enzymes have been developed using a peptide-based approach, in which an α-KG analogue was linked to a small histone peptide bearing the target of these enzymes, H3(7–14)K9me3 (Woon et al., 2012). Finally, structural knowledge of mode of binding of the H3K27me3 peptide to KDM6B was used to drive the development of the compound GSK-J1, which has selectivity for KDM6A/B over other tested KDMs (Kruidenier et al., 2012) (Table 8).
Given that many of the KDM inhibitors developed so far, while improving are relatively non-selective, the therapeutic utility of targeting specific KDMs remains to be determined. However, inhibitors with some level of selectivity have shown potentially promising effects in early preclinical models. For example, certain KDM1A inhibitors have been shown to inhibit proliferation of cancer cells in vitro and to block herpes simplex virus lytic replication and reactivation from latency (Liang et al., 2009; Wang et al., 2011; Willmann et al., 2012). In addition, a cell-penetrant prodrug version of GSK-J1 (GSK-J4) was recently shown to inhibit the production of pro-inflammatory cytokines by human macrophages, supporting the notion of targeting KDM6A/B for inflammatory diseases (Kruidenier et al., 2012) consistent with initial evidence in mouse macrophages (De Santa et al., 2007; 2009).
Arginine methyltransferases
In addition to lysine residues, histone arginines are also subject to methylation (Di Lorenzo and Bedford, 2011). Such methylation is favoured by the presence of glycine-arginine rich sequences (GAR motifs), although these are neither necessary nor sufficient. Methylated guanidine nitrogen atoms on peptidyl-arginine residue confer differential packaging, structural changes and altered protein interactions. Additional levels of intricacy result from differential dimeric processing of mono-methylated arginine (MMA) into either an asymmetrical- dimethylarginine (ADMA) or symmetrical-dimethylarginine (SDMA) residue.
To date, there are 11 proteins generally accepted as being members of the family of protein arginine methyltransferases (PRMTs), identified either on the basis of demonstrable methyltransferase activity, typically using SAM as the methyl donor, or homology to other family members (Table 2004a) (Wolf, 2009). However, a systematic survey of the human genome identified 44 putative PRMTs based on sequence homology at the active site, suggesting that this number could be much higher (Richon et al., 2011). The 11 commonly accepted PRMTs have been classified into subgroups with different profiles (Yang and Bedford, 2013) (Table 2004a). All PRMTs can generate MMA, while type I PRMTs (PRMT1–4, 6 and 8) generate ADMA and type II PRMTs (PRMT5, 9) produce SDMA. PRMT7 appears to generate only MMA and has been termed as type III PRMT, although its capacity to produce SDMA remains a subject of debate (Zurita-Lopez et al., 2012). The most recent family members (PRMT10-11) await definitive characterization. PRMT1 has been proposed to be the dominant cellular PRMT enzyme on account of its driving 85% of arginine methylation in diverse cells (Tang et al., 2000).
Table 9.
Arginine methyltransferases
| Symbol | Family | Synonyms and other symbols | Reported histone substrate | Uni-ProtKB/Swiss-Prot assession number |
|---|---|---|---|---|
| PRMT1 | Type I | ANM1, HCP1, IR1B4, HRMT1L2 | H4R3 H4R3 | Q99873 |
| PRMT2 | Type I | MGC11137, HRMT1L1 | H4 | P55345 |
| PRMT3 | Type I | HRMT1L3 | O60678 | |
| PRMT4 | Type I | CARM1 | H3R17, H3R26 | Q86X55 |
| PRMT5 | Type II | JBP1, SKB1, IBP72, SKB1hs, HRMT1L5 | H3R8, H4R3 | O14744 |
| PRMT6 | Type I | FLJ10559, HRMT1L6 | H3R2, H2AR29, H4R3, H2AR3 | Q96LA8 |
| PRMT7 | Type II, III | FLJ10640, KIAA1933, [Myelin basic protein]-arginine N-methyltransferase | H4R3, H2AR3, H3R2 | Q9NVM4 |
| PRMT8 | Type I | HRMT1L4 | Unknown | Q9NR22 |
| PRMT9 | Type II | FBOX11, VIT1, UBR6, FLJ12673, MGC44383 | Unknown | Q86XK2 |
| PRMT10 | Not classified | LOC90826, FLJ46629 | Unknown | Q6P2P2 |
| PRMT11 | Not classified | FBX10, FBXO10, FLJ41992, MGC149840 | Unknown | Q9UK96 |
Most PRMTs are ubiquitously expressed, although PRMT8 is reported to be selectively expressed in the CNS (Lee et al., 2005; Kousaka et al., 2009). Lethal mouse phenotypes have been observed after knockout of at least two family members (PRMT1 and PRMT4), suggesting that these proteins have non-redundant functions (Pawlak et al., 2000; Yadav et al., 2003).
The functional consequences of protein arginine methylation are ranging wide across most physiological processes, including growing evidence for a role in epigenetic regulation. In accordance with such a function, nuclear shuttling or localization has been demonstrated for some PRMTs [e.g. for PRMT1 and PRMT6 (Frankel et al., 2002; Herrmann and Fackelmayer, 2009)]. Conversely, PRMT3 appears to be predominantly cytosolic and hence may not have a physiological role in histone methylation (Frankel and Clarke, 2000). Although understanding of histone arginine methylation has historically trailed that of lysine methylation, PRMTs have been identified as members of transcriptional complexes and can be recruited onto promoters by the action of transcription factors such as NF-κB and p53 (An et al., 2004; Covic et al., 2005). A growing number of PRMTs (including PRMT1, PRMT2, PRMT4, PRMT5, PRMT6 and PRMT7) are known to methylate different combinations of arginine residues on histones H2A, H3 and H4 (Table 2004a) with effects on chromatin accessibility.
Arginine methylation (and ADMA vs. SDMA methylation) may be stimulatory or inhibitory to transcription, depending on the histone context, the modifying enzyme and the degree of dimethylation. Co-integration with other histone code modifications has been reported. For example, PRMT1-mediated methylation of H4R3 facilitates subsequent acetylation (Wang et al., 2001) while PRMT6-mediated methylation of H3R2 prevents H3K4 methylation by the MLL complex, effectively repressing transcriptional elongation (Hyllus et al., 2007). Conversely, histone H3K18 acetylation primes the histone tail for asymmetric dimethylation at arginine 17 (H3R17me2a) by PRMT4 (Daujat et al., 2002; An et al., 2004), while H3K9ac blocks H3R8 symmetric dimethylation (H3R8me2s) by PRMT5 (Pal et al., 2004).
These subtleties further emphasize the combinatorial complexity and likely exquisite selectivity of such epigenetic changes. However, histone methylation represents only part of the regulatory transcriptional potential of PRMTs, which also includes methylation and modulation of transcription factors such as CBP (Chevillard-Briet et al., 2002) and Tat (Boulanger et al., 2005), effects on RNA stability and splicing, and genomic reorganization via methylation of AT hooks of nuclear scaffold proteins such as HMGA proteins (Sgarra et al., 2003; Edberg et al., 2004). In keeping with their broad effects, PRMT1 and PRMT4 are considered to function as general transcription factors. Although understanding of how methylarginine marks are subsequently ‘read’ to activate transcription is currently limited, a recent study reported that PRMT4-mediated asymmetric dimethylation of H3R17 (a stimulatory modification) facilitates transcription elongation through recruitment of the PAF1 complex to activate oestrogen-receptor-dependent gene transcription, suggesting a possible model that other PRMTs may also use (Wu and Xu, 2012).
Abnormal PRMT expression or activity is increasingly being associated with a growing list of diseases. At present, the major link is between PRMTs and cancer. In particular, PRMT1 is considered key for transformation by the MLL complex (Cheung et al., 2007). However, studies also support a potential role for therapeutic intervention in pulmonary and viral disorders (Boulanger et al., 2005; Sun et al., 2012; Zakrzewicz et al., 2012) as well as spinal muscular atrophy (Brahms et al., 2001).
Aided by increased understanding of catalytic mechanisms and knowledge of a number of PRMT crystal structures, several interesting tool molecules have been identified (Wigle and Copeland, 2013) (see Table 2004b for examples). Since the discovery of the first PRMT family inhibitor truly selective for methyltransferase activity [AMI-1 (Cheng et al., 2004)], screens have been run successfully and novel chemical equity has been disclosed, including the cellular inhibitor RM65 (Spannhoff et al., 2007). In a flurry of recent published activity, for example (Bissinger et al., 2011; Hart et al., 2011), the rational design of C21, a chloroacetamidine-bearing histone H4 tail analogue that acts as an irreversible PRMT1 inhibitor (Obianyo et al., 2011), has been included. Progress towards selectivity within the PRMT family, originally thought challenging due to the high sequence conservation, is also encouraging (Dillon et al., 2012; Dowden et al., 2012), because such selectivity may prove necessary to maximize therapeutic index. Additional exemplars include potent PRMT4 inhibitors from BMS (Huynh et al., 2009 and Wan et al., 2009) and a PRMT3-selective inhibitor that is also reported to be the first allosteric inhibitor of PRMTs or indeed of any reader, writer, eraser of methyl marks (Siarheyeva et al., 2012). These promising early probes raise the hope that, with appropriate lead optimization, molecules with suitable pharmacokinetic and development properties for in vivo and clinical testing may be identified.
Table 10.
Examples of PRMT inhibitors
| Inhibitor | Structure | Potency | Reported target | Reference |
|---|---|---|---|---|
| AMI-1 | 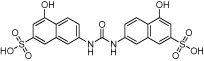 |
8.8 µM (PRMT1) | Pan-PRMT | (Cheng et al., 2004) |
| RM65 | 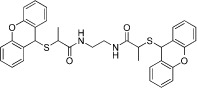 |
55 µM | PRMT1 (not tested vs. other PRMTs) | (Spannhoff et al., 2007) |
| BMS pyrazole inhibitor 7f | 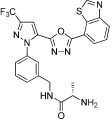 |
40 nM | PRMT4 | (Huynh et al., 2009) |
| Benzo[d]imidazole inhibitors of PRMT4 from BMS | 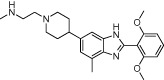 |
70 nM (best exemplar) | PRMT4 | (Wan et al., 2009) |
| C21 | 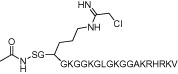 |
1.8 µM | PRMT1 (PRMT6) | (Obianyo et al., 2011) |
| Compound 1 (allosteric) | 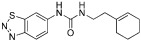 |
2.5 µM | PRMT3 | (Jones et al., 2012; Siarheyeva et al., 2012) (Siarheyeva et al., 2012) |
Arginine deiminases
Unlike the removal of lysine methyl marks, there is a scarcity of candidate enzymes with convincing demethylase activity against methylated arginines identified to date, with the exception of JmjD6 (Chang et al., 2007). Consequently, it had been suggested that enzymes of the peptidyl-arginine deiminase (PAD) family, which are able to catalyse the deimination (or citrullination) of arginine side chains into citrulline moieties, could similarly act on methylated arginines, and reverse methylation in the process (Wang et al., 2004). However, such catalysis is now thought to be unlikely under cellular conditions (reviewed by Thompson and Fast, 2006). Wider confirmation of the role of JmjD6 and the identification of additional demethylase enzymes therefore await further research.
Nevertheless, the PAD family remains a subject of interest as potential epigenetic regulators. The PADs include five members (PADs 1–4 and PAD6) with noted differences in function and substrate specificity (reviewed by Jones et al., 2009). PAD4 has been the primary focus with regard to an epigenetic function, because it is the only family member with a clear nuclear localization sequence and it has been reported to citrullinate accessible arginine residues on the tails of various histones (most notably R2, R8, R17 and R26 on H3, and R3 on H4). However, in light of recent data suggesting that PAD2 can also be found in the nucleus (Jang et al., 2011) and can effectively citrullinate histones (Zhang et al., 2012), additional PADs may cause epigenetic modulation. PAD2 represents a potential target for multiple sclerosis based on its CNS expression and ability to citrullinate, and thereby destabilize, myelin basic protein (Oguz et al., 2009). PADs 1 and 3 have roles in skin and hair follicle physiology, respectively, while PAD6 expression is limited to gametes and has not been shown to citrullinate any substrate to date.
Limited evidence for PAD4-mediated citrullination affecting transcription has come from a small number of reports, including effects on ER-responsive genes (Cuthbert et al., 2004; Wang et al., 2004; Zhang et al., 2012) and p53-regulated promoters (Li et al., 2008; 2010b). Interestingly, a reciprocal relationship between histone arginine methylation and citrullination has been demonstrated in a number of these studies [e.g. on p53 target promoters after UV treatment (Li et al., 2008)], suggesting that citrullination may indeed be an indirect barrier to methylation via depletion of naive arginine residues, and that inhibitors of histone citrullination may have widespread transcriptional effects.
Citrullinated histones are also associated with the formation of neutrophil extracellular traps (NETs). These elusive structures (Brinkmann et al., 2004; Yipp et al., 2012) offer innate immunity functions through the trapping and killing of pathogens by extruded filaments containing DNA, histones and potent granule proteins. The initial discovery of citrullinated H3 and H4 epitopes (Neeli et al., 2009; Wang et al., 2009b) followed by the reported lack of NETosis and selective interference with host defence in PAD4-deficient mice (Li et al., 2010a) emphasizes that citrullination by this enzyme is a key feature in NETosis. There is increasing evidence supportive of the rationale that a selective PAD4 inhibitor may be effective in a wide range of diseases characterized by an excessive burden of NETs. These range from thrombosis (Martinod et al., 2012) to systemic lupus erythematosus (Villanueva et al., 2011), ulcerative colitis (Savchenko et al., 2011), small-vessel vasculitis (Kessenbrock et al., 2009) and sepsis (Clark et al., 2007). NETs have also recently been linked to the pathogenesis of rheumatoid arthritis (Khandpur et al., 2012), supporting historical non-epigenetic evidence for PAD4 being important in loss of tolerance to synovial proteins in this disease.
The first notable PAD inhibitors published were peptidomimetics, rationally designed to irreversibly modify a key active site cysteine residue via covalent attachment to a haloacetaminidine moiety (Luo et al., 2006). The best studied exemplar, Cl-amidine, has subsequently been used widely as an in vitro tool. It has also demonstrated impressive efficacy in animal models of arthritis (Willis et al., 2011), colitis (Chumanevich et al., 2011) and lupus (Knight et al., 2013), despite poor pharmacokinetics in vivo and an uncertain PK–PD relationship. Cl-amidine is also a non-selective inhibitor of all PAD enzymes and subsequent efforts have focussed on the development of second-generation inhibitors with increased potency and selectivity for individual PAD enzymes (e.g. see Table 11). Molecules such as o-F-amidine and Thr-Asp-F-amidine (Causey et al., 2011; Jones et al., 2012) demonstrate signs of increased selectivity for PAD4, while PAD3-selective probes have also been described (Knuckley et al., 2010a). Encouraging signs of wider activity include the development of additional screens for PAD inhibitors and the identification of diverging and additional chemotypes (Wang et al., 2012; Bozdag et al., 2013). Ultimately, exploiting the binding determinants between the different PAD enzymes in order to identify and develop truly selective inhibitors for individual enzymes should allow definitive mechanistic understanding and guide optimal therapeutic positioning across a wider range of diseases.
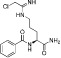
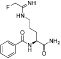
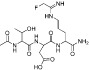
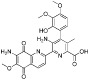
Table 11.
Examples of PAD inhibitors
Bromodomains
Acetylated lysines on histone proteins can be recognized by bromodomains (BRDs), which are small protein interaction modules of approximately 110 amino acids (Tamkun et al., 1992). There are 61 human BRDs found within 42 different proteins (Table 12), with individual proteins containing between one and six BRDs (Figure 2). The three-dimensional structure of more than half of the family of BRD containing proteins (BCPs) has been experimentally determined (Figure 2), demonstrating a conserved hydrophobic pocket that accommodates acetyl-lysine side chains (Jacobson et al., 2000; Nakamura et al., 2007; Filippakopoulos and Knapp, 2012; Filippakopoulos et al., 2012). BRDs are present in diverse nuclear proteins that possess intrinsic chromatin-modifying activity, including KATs (KAT2A, KAT2B, KAT4), KMTs (KMT2A, KMT2H), ATP-dependent chromatin-remodelling proteins (BAZ1B), helicases (SMARCA) and nuclear-scaffolding proteins (PB1) (Muller et al., 2011). In addition, BCPs are often found as components of large protein complexes controlling chromatin architecture and recruit other proteins such as epigenetic writers and readers as well as transcriptional regulatory proteins to chromatin (Dawson et al., 2011). Although the ability of BRDs to bind to acetylated lysine residues within histone proteins is linked to their gene regulatory activity, these domains have also been implicated in binding to non-histone acetylated proteins such as HIV Tat, RelA and p53 (Barlev et al., 2001; Dorr et al., 2002; Huang et al., 2009a).
Table 12.
Bromodomain-containing proteins
| Symbol | Synonyms and other symbols | Uni-ProtKB/Swiss-Prot assession number |
|---|---|---|
| ASH1L | ASH1, ASH1L1, huASH1, KMT2H, KIAA1420 | Q9NR48 |
| ATAD2 | ANCCA, CT137, DKFZp667N1320, MGC29843, MGC5254, PRO2000 | Q6PL18 |
| ATAD2B | KIAA1240 | Q9ULI0 |
| BAZ1A | ACF1, hACF1, WALp1, WCRF180 | Q9NRL2 |
| BAZ1B | WSTF, WBSCR10, WBSCR9 | Q9UIG0 |
| BAZ2A | KIAA0314, TIP5, WALp3 | Q9UIF9 |
| BAZ2B | WALp4, KIAA1476 | Q9UIF8 |
| BPTF | FALZ, FAC1, NURF301 | Q12830 |
| BRD1 | BRPF2, BRL | O95696 |
| BRD2 | KIAA9001, RING3, D6S113E, FSRG1, NAT | P25440 |
| BRD3 | KIAA0043, RING3L, ORFX | Q15059 |
| BRD4 | HUNK1, HUNKI, MCAP, CAP | O60885 |
| BRD7 | BP75, CELTIX1 | Q9NPI1 |
| BRD8 | p120, SMAP, SMAP2 | Q9H0E9 |
| BRD9 | FLJ13441 | Q9H8M2 |
| BRDT | BRD6, CT9 | Q58F21 |
| BRPF1 | BR140, PEREGRIN | P55201 |
| BRPF3 | KIAA1286 | Q9ULD4 |
| BRWD1 | C21orf107, WDR9 | Q9NSI6 |
| BRWD3 | FLJ38568, MRX93 | Q6RI45 |
| CECR2 | KIAA1740 | Q9BXF3 |
| CREBBP | CBP, KAT3A, RTS, RSTS | Q92793 |
| EP300 | KAT3B, p300 | Q09472 |
| KAT2A | GCN5, GCN5L2, HGCN5, PCAF-b | Q92830 |
| KAT2B | PCAF, GCN5, GCN5L, P/CAF | Q92831 |
| PBRM1 | BAF180, PB1 | Q86U86 |
| PHIP | WDR11, BRWD2, DCAF14, FLJ20705, ndrp | Q8WWQ0 |
| SMARCA2 | BAF190B, BRM, hBRM, hSNF2a, SNF2, SNF2LA, Sth1p, SWI2, SNF2L2 | P51531 |
| SMARCA4 | BAF190A, BRG1, FLJ39786, hSNF2b, SNF2B, SNF2L4 | P51532 |
| SP100 | – | P23497 |
| SP110 | IFI41, IFI75 | Q9HB58 |
| SP140 | LYSP100 | Q13342 |
| SP140L | – | Q9H930 |
| TAF1 | DYT3/TAF1, KAT4, NSCL2, TAFII250, BA2R, CCG1, CCGS, TAF2A | P21675 |
| TAF1L | TAFII210 | Q8IZX4 |
| TRIM24 | hTIF1, RNF82, Tif1a, TIF1A, TIF1, RNF82 | O15164 |
| TRIM28 | KAP1, RNF96, TF1B, TIF1B | Q13263 |
| TRIM33 | FLJ11429, KIAA1113, PTC7, RFG7, TF1G, TIF1G, TIF1GAMMA, TIFGAMMA, KIAA1113 | Q9UPN9 |
| TRIM66 | C11orf29, KIAA0298, TIF1D | O15016 |
| ZMYND8 | PRKCBP1, KIAA1125, RACK7 | Q9ULU4 |
| ZMYND11 | BS69 | Q15326 |
Figure 2.
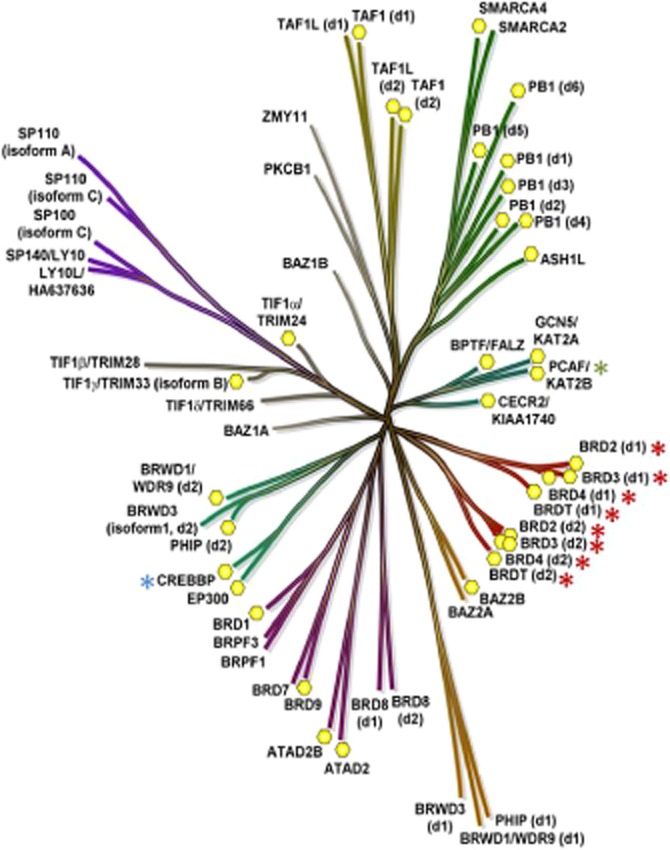
Phylogenetic tree of the human bromodomain family of proteins. The targets for which small molecule inhibitors have been identified are highlighted with asterisks. Yellow hexagons indicate X-ray structures in the public domain.
Recent studies have implicated BCPs in a wide range of human diseases, including cancer, inflammatory diseases, obesity, diabetes, infectious diseases, neurological disorders, and metabolic and cardiovascular indications (Taverna et al., 2007; Prinjha et al., 2012). Evidence for the role of BCPs includes altered expression in disease tissue, chromosomal translocations, amplifications and deletions involving BCP gene loci, genome-wide or focused gene sequence analyses linking SNPs to disease incidence, as well as phenotypes identified using knock-down or knockout studies. As an example, among the multiple BCPs reported to be overexpressed in tumours, which include ASH1L, BPTF, EP300, MLL, SMARCA2, SMARCA4, TRIM24 and TRIM28, ATAD2 has been shown to be up-regulated in various cancer types and to be significantly associated with prostate and endometrial cancer progression (Zou et al., 2009; Raeder et al., 2013), poor prognosis in breast and lung cancer (Caron et al., 2010), and occurrence of metastasis and overall survival in breast cancer (Boussouar et al., 2013). Furthermore, the identification of three sites of polymorphism in BRD2 associated with rheumatoid arthritis and the observation that Brd2-hypomorphic mice are severely obese and have reduced inflammation in fat tissue are examples of links of BCPs to inflammation (Muller et al., 2011).
An understanding of the therapeutic relevance of the regulatory function of the BRD of BCPs is beginning to emerge with the recent development of small molecule BRD inhibitors (Table 13) (Chung, 2012; Hewings et al., 2012). In some cases, these have been used to explore the interactions between BCPs and non-histone proteins. For example, compounds that bind to the BRD of PCAF with selectivity over the structurally related BRDs of CREBBP and TIF1β were identified by NMR-based small molecule screening and shown to disrupt the association of PCAF with HIV Tat-AcK50 in vitro (Zeng et al., 2005). Likewise, ischaemin (Table 13), a selective modulator of the transcriptional co-activator CREBBP, is able to block the interaction of acetylated p53 (p53K382ac) with CREBBP, leading to regulation of tumour suppressor p53-induced transcriptional activity in cells and preventing apoptosis in ischaemic cardiomyocytes (Borah et al., 2011).
Table 13.
Examples of bromodomain inhibitors
| Inhibitor | Structure | Potency | Reported target | Reference |
|---|---|---|---|---|
| Compound 2 | 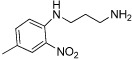 |
1.6 µM | KAT2B | (Zeng et al., 2005) |
| Ischaemin (MS120) | 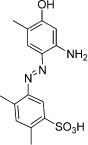 |
19 µM | CREBBP | (Borah et al., 2011) |
| I-BET-762 (GSK525762A) | 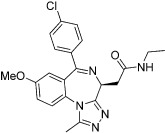 |
50–60 nM | BRD2, BRD3, BRD4, BRDT | (Nicodeme et al., 2010) |
| I-BET-151 (GSK1210151A) | 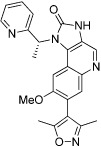 |
250–800 nM | BRD2, BRD3, BRD4, BRDT | (Seal et al., 2012) |
| JQ1 | 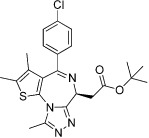 |
50–90 nM | BRD2, BRD3, BRD4, BRDT | (Filippakopoulos et al., 2010) |
| RVX-208 | 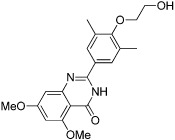 |
40 nM – 3 µM | BRD2, BRD3, BRD4, BRDT | (Khmelnitsky et al., 2013) |
The most advanced targets with respect to the development of BRD inhibitors are the members of the bromodomain and extraterminal (BET) subfamily of BCPs, which include BRD2, BRD3, BRD4 and BRDT (Table 13). Recently, a small number of potent, highly cell-permeable inhibitors with low nanomolar affinity for BET BRDs have been identified; these inhibitors appear highly selective for BET BRDs, but are active against the eight BRDs found in these four proteins due to their high degree of homology (Mirguet et al., 2013). Among the diverse chemotypes reported to date are the first inhibitors disclosed, I-BET762 (GSK525762) and JQ1, both of which originated from chemical starting points found by phenotypic screening assays aimed to identify up-regulators of apolipoprotein A1 (Apo-A1), and I-BET151 and RVX-208 (Table 13).
BET inhibitors have shown promising effects in a variety of preclinical cancer studies. One cancer of particular interest is nuclear protein in testis (NUT) midline carcinoma (NMC), a rare, aggressively lethal tumour type in which chromosomal translocations between BRD4 (and sometimes BRD3) and the NUT protein play a causative role. JQ1 has been found to induce squamous differentiation and growth arrest in patient-derived BRD4-NUT-positive NMC cell lines and to decrease tumour size and improve survival in mouse xenograft models (Filippakopoulos et al., 2010). In addition, BET inhibitors including I-BET762, I-BET151 and JQ1 have been shown to be active against myeloma (Delmore et al., 2011), lymphoma (Emadali et al., 2013), acute lymphoblastic leukaemia (Da Costa et al., 2013), prostatic cancer (Gao et al., 2013), neuroblastoma (Puissant et al., 2013; Wyce et al., 2013) and glioblastoma (Cheng et al., 2013), in vitro and in vivo, while I-BET151 has been shown to have considerable preclinical activity against acute leukaemias, including MLL-fusion protein-driven leukaemia (Dawson et al., 2011), and also against JAK2-driven myeloproliferative neoplasms (Wyspianska et al., 2013). Similarly, I-BET726 was shown to induce cytotoxicity in mouse xenograft models of human neuroblastoma (Wyce et al., 2013), and inhibition of BET has been shown to impair melanoma cell proliferation in vitro and tumour growth and metastatic behaviour in vivo (Segura et al., 2013).
Based on these promising preclinical results, BET inhibitors are now entering clinical trials (Table 14). I-BET762 (Nicodeme et al., 2010; Mirguet et al., 2013), a benzodiazepine derivative developed by GlaxoSmithKline (GSK), was recently progressed into a phase I clinical trial for treatment of NMC, as well as other cancers (Mirguet et al., 2013). In addition, CPI-0610 (Constellation Pharmaceuticals) and OTX-015 (OncoEthix) are also examples of BET inhibitors currently in phase I clinical trials for the treatment of various cancer types.
Table 14.
Clinical studies of bromodomain inhibitors
| Primary drugs | Sponsor | Trial phase | Disease type(s) | Trial ID |
|---|---|---|---|---|
| I-BET-762 | GSK | I | NMC Leukaemias, lymphomas Multiple myeloma Myeloproliferative neoplasms | NCT01587703 NCT01943851 |
| RVX-208 | Resverlogix | I/II | Dyslipidemia Atherosclerosis Acute coronary syndrome Cardiovascular disease | NCT00768274 NCT01067820 NCT01058018 NCT01423188 NCT01863225 |
| II | Type 2 diabetes | NCT01728467 | ||
| II | Alzheimer's disease | (Study planned) http://www.resverlogix.com/programs/epigenetics/neurodegenerative-diseases | ||
| CPI-0610 | Constellation Pharmaceuticals | I | Lymphomas | NCT01949883 |
| I | Leukaemias Multiple myeloma Myelodysplastic syndrome | (Study planned) http://www.constellationpharma.com/2012/09/constellation-pharmaceuticals-and-the-leukemia-lymphoma-society-partner-to-develop-novel-bet-inhibitor-for-the-treatment-of-hematologic-malignancies/ | ||
| OTX-015 | OncoEthix/ Mitsubishi Tanabe Pharma | I | Leukaemias, lymphomas Multiple myeloma Unspecified haematological cancer | NCT01713582 |
There is also great interest in the potential application of BET inhibitors in other therapeutic areas. In particular, there is considerable evidence that BET inhibitors may have utility in the treatment of autoimmune/inflammatory disease. In this regard, I-BET762 was shown to inhibit the expression of inflammatory cytokines and chemokines in activated mouse macrophages and to confer protection against LPS-induced endotoxic shock and bacteria-induced sepsis in mice (Nicodeme et al., 2010). These anti-inflammatory effects were replicated by I-BET151, which reduced pro-inflammatory cytokine production by activated human peripheral blood mononuclear cells, and was effective at suppressing LPS-induced inflammation and sepsis in mice (Seal et al., 2012), and also recently by JQ1 (Belkina et al., 2013). Moreover, I-BET762 inhibited the ability of antigen-specific T-cells, differentiated under Th1 conditions in vitro, to induce pathogenesis in an adoptive transfer model of experimental autoimmune encephalomyelitis (EAE; Bandukwala et al., 2012), and JQ1 was recently shown to suppress Th17 differentiation in vivo and to be protective in mouse models of autoimmunity (collagen-induced arthritis and EAE) (Mele et al., 2013).
One of the most clinically advanced but significantly less potent BET BRD inhibitors is RVX-208 (RVX-000222) (Picaud et al., 2013), which is currently being developed by Resverlogix Corp for the treatment of atherosclerosis and coronary artery disease based on its capacity to increase the levels of Apo-A1 and hence increase high-density lipoprotein cholesterol (McNeill, 2010) (Table 14). Toxicity and tolerability studies in animals and phase I/II clinical trials have indicated that RVX-208 is safe and well tolerated in multiple dosing regimens. It will be of great interest to see whether more potent BET inhibitors are similarly well tolerated in phase I trials mentioned above to gain an understanding of the therapeutic index of these inhibitors. RVX-208 is currently in phase 2 clinical trials for the treatment of atherosclerosis (Nicholls et al., 2011). In addition, a phase I trial indicated that RVX-208 may have potential for the removal of β-amyloid plaques in Alzheimer's disease and this will be further assessed in an ongoing phase I/II clinical trial.
Like HDAC inhibitors, BET inhibitors have been shown to reactivate HIV from latency in cell lines and primary T-cell models, indicating their possible use in clearance and cure of the latent viral pool, as described above (Zhu et al., 2012; Boehm et al., 2013a,b; Li et al., 2013). Their potential utility as therapeutics for heart failure has also been suggested recently based on the ability of JQ1 to block cardiomyocyte hypertrophy in vitro, and to prevent left ventricular hypertrophy and improve cardiac function in adult mice subjected to transverse aortic constriction (Spiltoir et al., 2013). Finally, the efficacy of BET inhibitors in blocking the pro-fibrotic responses of idiopathic pulmonary fibrosis (IPF) lung fibroblasts and attenuating bleomycin-induced lung fibrosis in mice provides a rationale to target the BET proteins for the treatment of IPF (Tang et al., 2013b).
Concluding remarks
Although the basic scientific understanding of epigenetic regulatory mechanisms is at an early stage, great progress has already been made in taking drugs targeting these pathways into the clinic. The rapid pace of these developments has been driven by the recognition that dysregulation of epigenetic processes is apparent in diverse disease states, combined with the finding that many of the proteins that generate, remove and recognize histone modifications are tractable to small molecule drug development. In turn, the tractability of epigenetic targets has led to the generation of chemical probes that are being used to accelerate investigation into the biological function of these proteins.
As work in this young field continues, it will be of great interest to understand the full potential of this target class for the treatment of human disease. There will be many important questions relating to drug development for epigenetic pathways. For example, how many proteins within the multiple target classes of writers, erasers and readers will prove tractable to the generation of potent and selective small molecule modulators? Is there redundancy among these targets, and what role will polypharmacology play? What approaches can be taken to modulate epigenetic states in disease tissue while minimizing effects on normal cellular homeostasis? As well as acutely modifying gene expression, will it be possible to reset aberrant epigenetic states to normal, allowing for short-term treatments to induce remission or even ‘cure’ disease? Answering these questions will be aided by the continuing development of pharmacological tools together with increasingly sophisticated molecular approaches for elucidating the mechanisms that regulate chromatin structure and gene expression.
Author contributions
All authors contributed to writing of sections of the manuscript.
Conflict of interest
All authors are employees and shareholders of GlaxoSmithKline, which is researching and developing epigenetic inhibitors.
Glossary
- α-KG
α-ketoglutarate
- ADMA
asymmetrical- dimethylarginine
- Apo-A1
apolipoprotein A1
- BCP
bromodomain-containing protein
- BET
bromodomain and extraterminal
- BRD
bromodomain
- DNMT
DNA-methyltransferase
- EAE
experimental autoimmune encephalomyelitis
- FAD
flavin adenine dinucleotide
- HDAC
histone deacetylase
- IPF
idiopathic pulmonary fibrosis
- Jmj
jumonji C-domain
- KAT
lysine acetyltransferase
- KDAC
lysine deacetylase
- KDM
lysine-specific demethylase
- KMT
lysine methyltransferase
- MMA
mono-methylated arginine
- NET
neutrophil extracellular trap
- NMC
NUT midline carcinoma
- NOG
N-oxalylglycine
- NUT
nuclear protein in testis
- PAD
peptidyl-arginine deiminase
- PCPA
trans-2-phenylcyclopropylamine
- PRMT
arginine methyltransferase
- SAHA
suberoylanilide hydroxamic acid
- SAM
S-adenosyl methionine
- SDMA
disymmetrical-dimethylarginine
- SIRT
sirtuin
- SNP
single nucleotide polymorphism
- TET
ten-eleven translocation
References
- Adcock IM, Lee KY. Abnormal histone acetylase and deacetylase expression and function in lung inflammation. Inflamm Res. 2006;55:311–321. doi: 10.1007/s00011-006-0081-1. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, McGrath JC, et al. The Concise Guide to PHARMACOLOGY 2013/14: overview. Br J Pharmacol. 2013a;170:1449–1458. [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The concise guide to pharmacology 2013/14: enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allis CD, Berger SL, Cote J, Dent S, Jenuwien T, Kouzarides T, et al. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131:633–636. doi: 10.1016/j.cell.2007.10.039. [DOI] [PubMed] [Google Scholar]
- An W, Kim J, Roeder RG. Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell. 2004;117:735–748. doi: 10.1016/j.cell.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012;11:384–400. doi: 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- Avvakumov N, Cote J. The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene. 2007;26:5395–5407. doi: 10.1038/sj.onc.1210608. [DOI] [PubMed] [Google Scholar]
- Balasubramanyam K, Swaminathan V, Ranganathan A, Kundu TK. Small molecule modulators of histone acetyltransferase p300. J Biol Chem. 2003;278:19134–19140. doi: 10.1074/jbc.M301580200. [DOI] [PubMed] [Google Scholar]
- Balasubramanyam K, Altaf M, Varier RA, Swaminathan V, Ravindran A, Sadhale PP, et al. Polyisoprenylated benzophenone, garcinol, a natural histone acetyltransferase inhibitor, represses chromatin transcription and alters global gene expression. J Biol Chem. 2004a;279:33716–33726. doi: 10.1074/jbc.M402839200. [DOI] [PubMed] [Google Scholar]
- Balasubramanyam K, Varier RA, Altaf M, Swaminathan V, Siddappa NB, Ranga U, et al. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J Biol Chem. 2004b;279:51163–51171. doi: 10.1074/jbc.M409024200. [DOI] [PubMed] [Google Scholar]
- Bandukwala HS, Gagnon J, Togher S, Greenbaum JA, Lamperti ED, Parr NJ, et al. Selective inhibition of CD4+ T-cell cytokine production and autoimmunity by BET protein and c-Myc inhibitors. Proc Natl Acad Sci U S A. 2012;109:14532–14537. doi: 10.1073/pnas.1212264109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlev NA, Liu L, Chehab NH, Mansfield K, Harris KG, Halazonetis TD, et al. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol Cell. 2001;8:1243–1254. doi: 10.1016/s1097-2765(01)00414-2. [DOI] [PubMed] [Google Scholar]
- Belkina AC, Nikolajczyk BS, Denis GV. BET protein function is required for inflammation: Brd2 genetic disruption and BET inhibitor JQ1 impair mouse macrophage inflammatory responses. J Immunol. 2013;190:3670–3678. doi: 10.4049/jimmunol.1202838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20:66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry WL, Janknecht R. KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res. 2013;73:2936–2942. doi: 10.1158/0008-5472.CAN-12-4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissinger E-M, Heinke R, Spannhoff A, Eberlin A, Metzger E, Cura V, et al. Acyl derivatives of p-aminosulfonamides and dapsone as new inhibitors of the arginine methyltransferase hPRMT1. Bioorg Med Chem. 2011;19:3717–3731. doi: 10.1016/j.bmc.2011.02.032. [DOI] [PubMed] [Google Scholar]
- Boehm D, Calvanese V, Dar RD, Xing S, Schroeder S, Martins L, et al. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle. 2013a;12:452–462. doi: 10.4161/cc.23309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm D, Conrad R, Ott M. Bromodomain proteins in HIV infection. Viruses. 2013b;5:1571–1586. doi: 10.3390/v5061571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borah JC, Mujtaba S, Karakikes I, Zeng L, Muller M, Patel J, et al. A small molecule binding to the coactivator CREB-binding protein blocks apoptosis in cardiomyocytes. Chem Biol. 2011;18:531–541. doi: 10.1016/j.chembiol.2010.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulanger M-C, Liang C, Russell RS, Lin R, Bedford MT, Wainberg MA, et al. Methylation of Tat by PRMT6 regulates human immunodeficiency virus type 1 gene expression. J Virol. 2005;79:124–131. doi: 10.1128/JVI.79.1.124-131.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussouar FA, Jamshidikia M, Morozumi Y, Rousseaux S, Khochbin S. Malignant genome reprogramming by ATAD2. Biochim Biophys Acta. 2013;1829:1010–1014. doi: 10.1016/j.bbagrm.2013.06.003. [DOI] [PubMed] [Google Scholar]
- Bowers EM, Yan G, Mukherjee C, Orry A, Wang L, Holbert MA, et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem Biol. 2010;17:471–482. doi: 10.1016/j.chembiol.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdag M, Dreker T, Henry C, Tosco P, Vallaro M, Fruttero R, et al. Novel small molecule protein arginine deiminase 4 (PAD4) inhibitors. Bioorg Med Chem Lett. 2013;23:715–719. doi: 10.1016/j.bmcl.2012.11.102. [DOI] [PubMed] [Google Scholar]
- Brahms H, Meheus L, De Brabandere V, Fischer U, Luhrmann R. Symmetrical dimethylation of arginine residues in spliceosomal Sm protein B/B' and the Sm-like protein LSm4, and their interaction with the SMN protein. RNA. 2001;7:1531–1542. doi: 10.1017/s135583820101442x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- Cantley M, Haynes D. Epigenetic regulation of inflammation: progressing from broad acting histone deacetylase (HDAC) inhibitors to targeting specific HDACs. Inflammopharmacology. 2013;21:301–307. doi: 10.1007/s10787-012-0166-0. [DOI] [PubMed] [Google Scholar]
- Carey N, Marques CJ, Reik W. DNA demethylases: a new epigenetic frontier in drug discovery. Drug Discov Today. 2012;16:683–690. doi: 10.1016/j.drudis.2011.05.004. [DOI] [PubMed] [Google Scholar]
- Caron C, Lestrat C, Marsal S, Escoffier E, Curtet S, Virolle V, et al. Functional characterization of ATAD2 as a new cancer/testis factor and a predictor of poor prognosis in breast and lung cancers. Oncogene. 2010;29:5171–5181. doi: 10.1038/onc.2010.259. [DOI] [PubMed] [Google Scholar]
- Causey CP, Jones JE, Slack JL, Kamei D, Jones LE, Subramanian V, et al. The development of N-alpha-(2-Carboxyl)benzoyl-N5-(2-fluoro-1-iminoethyl)-l-ornithine amide (o-F-amidine) and N-alpha-(2-Carboxyl)benzoyl-N5-(2-chloro-1-iminoethyl)-l-ornithine amide (o-Cl-amidine) as second generation protein arginine deiminase (PAD) inhibitors. J Med Chem. 2011;54:6919–6935. doi: 10.1021/jm2008985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang B, Chen Y, Zhao Y, Bruick RK. JMJD6 is a histone arginine demethylase. Science. 2007;318:444–447. doi: 10.1126/science.1145801. [DOI] [PubMed] [Google Scholar]
- Chang CJ, Hung MC. The role of EZH2 in tumour progression. Br J Cancer. 2012;106:243–247. doi: 10.1038/bjc.2011.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang K-H, King ONF, Tumber A, Woon ECY, Heightman TD, McDonough MA, et al. Inhibition of histone demethylases by 4-carboxy-2,2′-bipyridyl compounds. ChemMedChem. 2011;6:759–764. doi: 10.1002/cmdc.201100026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chasman DI, Pare G, Mora S, Hopewell JC, Peloso G, Clarke R, et al. Forty-three loci associated with plasma lipoprotein size, concentration, and cholesterol content in genome-wide analysis. PLoS Genet. 2009;5:e1000730. doi: 10.1371/journal.pgen.1000730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng D, Yadav N, King RW, Swanson MS, Weinstein EJ, Bedford MT. Small molecule regulators of protein arginine methyltransferases. J Biol Chem. 2004;279:23892–23899. doi: 10.1074/jbc.M401853200. [DOI] [PubMed] [Google Scholar]
- Cheng Z, Gong Y, Ma Y, Lu K, Lu X, Pierce LA, et al. Inhibition of BET bromodomain targets genetically diverse glioblastoma. Clin Cancer Res. 2013;19:1748–1759. doi: 10.1158/1078-0432.CCR-12-3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung N, Chan LC, Thompson A, Cleary ML, So CWE. Protein arginine-methyltransferase-dependent oncogenesis. Nat Cell Biol. 2007;9:1208–1215. doi: 10.1038/ncb1642. [DOI] [PubMed] [Google Scholar]
- Chevillard-Briet M, Trouche D, Vandel L. Control of CBP co-activating activity by arginine methylation. EMBO J. 2002;21:5457–5466. doi: 10.1093/emboj/cdf548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi P, Allis CD, Wang GG. Covalent histone modifications – miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer. 2010;10:457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H-S, Suzuki T, Dohmae N, Hayami S, Unoki M, Yoshimatsu M, et al. Demethylation of RB regulator MYPT1 by histone demethylase LSD1 promotes cell cycle progression in cancer cells. Cancer Res. 2011;71:655–660. doi: 10.1158/0008-5472.CAN-10-2446. [DOI] [PubMed] [Google Scholar]
- Choi K-C, Jung MG, Lee Y-H, Yoon JC, Kwon SH, Kang H-B, et al. Epigallocatechin-3-gallate, a histone acetyltransferase inhibitor, inhibits EBV-induced B lymphocyte transformation via suppression of RelA acetylation. Cancer Res. 2009;69:583–592. doi: 10.1158/0008-5472.CAN-08-2442. [DOI] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Chowdhury R, Yeoh KK, Tian Y-M, Hillringhaus L, Bagg EA, Rose NR, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12:463–469. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chumanevich AA, Causey CP, Knuckley BA, Jones JE, Poudyal D, Chumanevich AP, et al. Suppression of colitis in mice by Cl-amidine: a novel peptidyl arginine deiminase inhibitor. Am J Physiol Gastrointest Liver Physiol. 2011;300:G929–G938. doi: 10.1152/ajpgi.00435.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CW. Small molecule bromodomain inhibitors: extending the druggable genome. Prog Med Chem. 2012;51:1–55. doi: 10.1016/B978-0-12-396493-9.00001-7. [DOI] [PubMed] [Google Scholar]
- Ciavatta DJ, Yang J, Preston GA, Badhwar AK, Xiao H, Hewins P, et al. Epigenetic basis for aberrant upregulation of autoantigen genes in humans with ANCA vasculitis. J Clin Invest. 2010;120:3209–3219. doi: 10.1172/JCI40034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13:463–469. doi: 10.1038/nm1565. [DOI] [PubMed] [Google Scholar]
- Cloos PAC, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, et al. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature. 2006;442:307–311. doi: 10.1038/nature04837. [DOI] [PubMed] [Google Scholar]
- Copeland RA. Molecular pathways: protein methyltransferases in cancer. Clin Cancer Res. 2013;19:6344–6350. doi: 10.1158/1078-0432.CCR-13-0223. [DOI] [PubMed] [Google Scholar]
- Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery. Nat Rev Drug Discov. 2009;8:724–732. doi: 10.1038/nrd2974. [DOI] [PubMed] [Google Scholar]
- Couvelard A, Deschamps L, Rebours V, Sauvanet A, Gatter K, Pezzella F, et al. Overexpression of the oxygen sensors PHD-1, PHD-2, PHD-3, and FIH is associated with tumor aggressiveness in pancreatic endocrine tumors. Clin Cancer Res. 2008;14:6634–6639. doi: 10.1158/1078-0432.CCR-07-5258. [DOI] [PubMed] [Google Scholar]
- Covic M, Hassa PO, Saccani S, Buerki C, Meier NI, Lombardi C, et al. Arginine methyltransferase CARM1 is a promoter-specific regulator of NF-[kappa]B-dependent gene expression. EMBO J. 2005;24:85–96. doi: 10.1038/sj.emboj.7600500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culhane JC, Wang D, Yen PM, Cole PA. Comparative analysis of small molecules and histone substrate analogues as LSD1 lysine demethylase inhibitors. J Am Chem Soc. 2010;132:3164–3176. doi: 10.1021/ja909996p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbert GL, Daujat S, Snowden AW, Erdjument-Bromage H, Hagiwara T, Yamada M, et al. Histone deimination antagonizes arginine methylation. Cell. 2004;118:545–553. doi: 10.1016/j.cell.2004.08.020. [DOI] [PubMed] [Google Scholar]
- Da Costa D, Agathanggelou A, Perry T, Weston V, Petermann E, Zlatanou A, et al. BET inhibition as a single or combined therapeutic approach in primary paediatric B-precursor acute lymphoblastic leukaemia. Blood Cancer J. 2013;3:e126. doi: 10.1038/bcj.2013.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20:53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack-Sjodin PA, et al. Potent inhibition of DOT1L as treatment for MLL-fusion leukemia. Blood. 2013;122:1017–1025. doi: 10.1182/blood-2013-04-497644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463:360–363. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancy BCR, Ming SA, Papazyan R, Jelinek CA, Majumdar A, Sun Y, et al. Azalysine analogues as probes for protein lysine deacetylation and demethylation. J Am Chem Soc. 2012;134:5138–5148. doi: 10.1021/ja209574z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daujat S, Bauer U-M, Shah V, Turner B, Berger S, Kouzarides T. Crosstalk between CARM1 methylation and CBP acetylation on histone H3. Curr Biol. 2002;12:2090–2097. doi: 10.1016/s0960-9822(02)01387-8. [DOI] [PubMed] [Google Scholar]
- Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 A resolution. J Mol Biol. 2002;319:1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]
- Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan W-I, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G. The histone H3 Lysine-27 demethylase Jmjd3 links inflammation to iInhibition of polycomb-mediated gene silencing. Cell. 2007;130:1083–1094. doi: 10.1016/j.cell.2007.08.019. [DOI] [PubMed] [Google Scholar]
- De Santa F, Narang V, Yap ZH, Tusi BK, Burgold T, Austenaa L, et al. Jmjd3 contributes to the control of gene expression in LPS-activated macrophages. EMBO J. 2009;28:3341–3352. doi: 10.1038/emboj.2009.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delatte B, Fuks F. TET proteins: on the frenetic hunt for new cytosine modifications. Brief Funct Genomics. 2013;12:191–204. doi: 10.1093/bfgp/elt010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain Inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lorenzo A, Bedford MT. Histone arginine methylation. FEBS Lett. 2011;585:2024–2031. doi: 10.1016/j.febslet.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon MBC, Bachovchin DA, Brown SJ, Finn MG, Rosen H, Cravatt BF, et al. Novel inhibitors for PRMT1 discovered by high-throughput screening using activity-based fluorescence polarization. ACS Chem Biol. 2012;7:1198–1204. doi: 10.1021/cb300024c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittenhafer-Reed KE, Feldman JL, Denu JM. Catalysis and mechanistic insights into sirtuin activation. Chembiochem. 2011;12:281–289. doi: 10.1002/cbic.201000434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorr A, Kiermer V, Pedal A, Rackwitz H-R, Henklein P, Schubert U, et al. Transcriptional synergy between Tat and PCAF is dependent on the binding of acetylated Tat to the PCAF bromodomain. EMBO J. 2002;21:2715–2723. doi: 10.1093/emboj/21.11.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowden J, Pike RA, Parry RV, Hong W, Muhsen UA, Ward SG. Small molecule inhibitors that discriminate between protein arginine N-methyltransferases PRMT1 and CARM1. Org Biomol Chem. 2012;9:7814–7821. doi: 10.1039/c1ob06100c. [DOI] [PubMed] [Google Scholar]
- Edberg DD, Bruce JE, Siems WF, Reeves R. In vivo posttranslational modifications of the high mobility group A1a proteins in breast cancer cells of differing metastatic potential. Biochemistry. 2004;43:11500–11515. doi: 10.1021/bi049833i. [DOI] [PubMed] [Google Scholar]
- Emadali A, Rousseaux S, Bruder-Costa J, Rome C, Duley S, Hamaidia S, et al. Identification of a novel BET bromodomain inhibitor-sensitive, gene regulatory circuit that controls rituximab response and tumour growth in aggressive lymphoid cancers. EMBO Mol Med. 2013;5:1180–1195. doi: 10.1002/emmm.201202034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evertts AG, Manning AL, Wang X, Dyson NJ, Garcia BA, Coller HA. H4K20 methylation regulates quiescence and chromatin compaction. Mol Biol Cell. 2013;24:3025–3037. doi: 10.1091/mbc.E12-07-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Wang H, Ng HH, Erdjument-Bromage H, Tempst P, Struhl K, et al. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol. 2002;12:1052–1058. doi: 10.1016/s0960-9822(02)00901-6. [DOI] [PubMed] [Google Scholar]
- Ferguson AD, Larsen NA, Howard T, Pollard H, Green I, Grande C, et al. Structural basis of substrate methylation and inhibition of SMYD2. Structure. 2012;19:1262–1273. doi: 10.1016/j.str.2011.06.011. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P, Knapp S. The bromodomain interaction module. FEBS Lett. 2012;586:2692–2704. doi: 10.1016/j.febslet.2012.04.045. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert J-P, Barsyte-Lovejoy D, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel A, Clarke S. PRMT3 is a distinct member of the protein arginine N-methyltransferase family: conferral of substrate specificity by a zinc-finger domain. J Biol Chem. 2000;275:32974–32982. doi: 10.1074/jbc.M006445200. [DOI] [PubMed] [Google Scholar]
- Frankel A, Yadav N, Lee J, Branscombe TL, Clarke S, Bedford MT. The novel human protein arginine N-methyltransferase PRMT6 is a nuclear enzyme displaying unique substrate specificity. J Biol Chem. 2002;277:3537–3543. doi: 10.1074/jbc.M108786200. [DOI] [PubMed] [Google Scholar]
- Furdas SD, Kannan S, Sippl W, Jung M. Small molecule inhibitors of histone acetyltransferases as epigenetic tools and drug candidates. Arch Pharm. 2012;345:7–21. doi: 10.1002/ardp.201100209. [DOI] [PubMed] [Google Scholar]
- Gao L, Schwartzman J, Gibbs A, Lisac R, Kleinschmidt R, Wilmot B, et al. Androgen receptor promotes ligand-independent prostate cancer progression through c-myc upregulation. PLoS ONE. 2013;8:e63563. doi: 10.1371/journal.pone.0063563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghizzoni M, Haisma HJ, Maarsingh H, Dekker FJ. Histone acetyltransferases are crucial regulators in NF-kB mediated inflammation. Drug Discov Today. 2011;16:504–511. doi: 10.1016/j.drudis.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannini G, Cabri W, Fattorusso C, Rodriquez M. Histone deacetylase inhibitors in the treatment of cancer: overview and perspectives. Future Med Chem. 2012;4:1439–1460. doi: 10.4155/fmc.12.80. [DOI] [PubMed] [Google Scholar]
- Gorsuch S, Bavetsias V, Rowlands MG, Aherne GW, Workman P, Jarman M, et al. Synthesis of isothiazol-3-one derivatives as inhibitors of histone acetyltransferases (HATs) Bioorg Med Chem. 2009;17:467–474. doi: 10.1016/j.bmc.2008.11.079. [DOI] [PubMed] [Google Scholar]
- Grabiec A, Tak P, Reedquist K. Targeting histone deacetylase activity in rheumatoid arthritis and asthma as prototypes of inflammatory disease: should we keep our HATs on? Arthritis Res Ther. 2008;10:226. doi: 10.1186/ar2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabiec AM, Tak PP, Reedquist KA. Function of histone deacetylase inhibitors in inflammation. Crit Rev Immunol. 2011;31:233–263. doi: 10.1615/critrevimmunol.v31.i3.40. [DOI] [PubMed] [Google Scholar]
- Gregoretti I, Lee Y-M, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet. 2011;43:875–878. doi: 10.1038/ng.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Haaften G, Dalgliesh GL, Davies H, Chen L, Bignell G, Greenman C, et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet. 2009;41:521–523. doi: 10.1038/ng.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JA, Dominy JE, Lee Y, Puigserver P. The sirtuin family's role in aging and age-associated pathologies. J Clin Invest. 2013;123:973–979. doi: 10.1172/JCI64094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada S, Suzuki T, Mino K, Koseki K, Oehme F, Flamme I, et al. Design, synthesis, enzyme-inhibitory activity, and effect on human cancer cells of a novel series of jumonji domain-containing protein 2 histone demethylase inhibitors. J Med Chem. 2010;53:5629–5638. doi: 10.1021/jm1003655. [DOI] [PubMed] [Google Scholar]
- Hardison RC, Taylor J. Genomic approaches towards finding cis-regulatory modules in animals. Nat Rev Genet. 2012;13:469–483. doi: 10.1038/nrg3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart P, Lakowski TM, Thomas D, Frankel A, Martin NI. Peptidic partial bisubstrates as inhibitors of the protein arginine N-methyltransferases. Chembiochem. 2011;12:1427–1432. doi: 10.1002/cbic.201100074. [DOI] [PubMed] [Google Scholar]
- Hayami S, Kelly JD, Cho H-S, Yoshimatsu M, Unoki M, Tsunoda T, et al. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int J Cancer. 2011;128:574–586. doi: 10.1002/ijc.25349. [DOI] [PubMed] [Google Scholar]
- He J, Nguyen AT, Zhang Y. KDM2b/JHDM1b, an H3K36me2-specific demethylase, is required for initiation and maintenance of acute myeloid leukemia. Blood. 2011a;117:3869–3880. doi: 10.1182/blood-2010-10-312736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y-F, Li B-Z, Li Z, Liu P, Wang Y, Tang Q, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011b;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidenblad M, Lindgren D, Jonson T, Liedberg F, Veerla S, Chebil G, et al. Tiling resolution array CGH and high density expression profiling of urothelial carcinomas delineate genomic amplicons and candidate target genes specific for advanced tumors. BMC Med Genomics. 2008;1:3. doi: 10.1186/1755-8794-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann F, Fackelmayer FO. Nucleo-cytoplasmic shuttling of protein arginine methyltransferase 1 (PRMT1) requires enzymatic activity. Genes Cells. 2009;14:309–317. doi: 10.1111/j.1365-2443.2008.01266.x. [DOI] [PubMed] [Google Scholar]
- Hewings DS, Rooney TPC, Jennings LE, Hay DA, Schofield CJ, Brennan PE, et al. Progress in the development and application of small molecule inhibitors of bromodomain-acetyl-lysine interactions. J Med Chem. 2012;55:9393–9413. doi: 10.1021/jm300915b. [DOI] [PubMed] [Google Scholar]
- Hoffmann E, Wald J, Lavu S, Roberts J, Beaumont C, Haddad J, et al. Pharmacokinetics and tolerability of SRT2104, a first-in-class small molecule activator of SIRT1, after single and repeated oral administration in man. Br J Clin Pharmacol. 2013;75:186–196. doi: 10.1111/j.1365-2125.2012.04340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hojfeldt JW, Agger K, Helin K. Histone lysine demethylases as targets for anticancer therapy. Nat Rev Drug Discov. 2013;12:917–930. doi: 10.1038/nrd4154. [DOI] [PubMed] [Google Scholar]
- Hong L, Schroth GP, Matthews HR, Yau P, Bradbury EM. Studies of the DNA binding properties of histone H4 amino terminus. Thermal denaturation studies reveal that acetylation markedly reduces the binding constant of the H4 ‘tail’ to DNA. J Biol Chem. 1993;268:305–314. [PubMed] [Google Scholar]
- Horton JR, Upadhyay AK, Qi HH, Zhang X, Shi Y, Cheng X. Enzymatic and structural insights for substrate specificity of a family of jumonji histone lysine demethylases. Nat Struct Mol Biol. 2009;17:38–43. doi: 10.1038/nsmb.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou H, Yu H. Structural insights into histone lysine demethylation. Curr Opin Struct Biol. 2010;20:739–748. doi: 10.1016/j.sbi.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu E, Dul E, Sung C-M, Chen Z, Kirkpatrick R, Zhang G-F, et al. Identification of novel isoform-selective inhibitors within class I histone deacetylases. J Pharmacol Exp Ther. 2003;307:720–728. doi: 10.1124/jpet.103.055541. [DOI] [PubMed] [Google Scholar]
- Huang B, Yang X-D, Zhou M-M, Ozato K, Chen L-F. Brd4 coactivates transcriptional activation of NF-kB via specific binding to acetylated RelA. Mol Cell Biol. 2009a;29:1375–1387. doi: 10.1128/MCB.01365-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Sengupta R, Espejo AB, Lee MG, Dorsey JA, Richter M, et al. p53 is regulated by the lysine demethylase LSD1. Nature. 2007a;449:105–108. doi: 10.1038/nature06092. [DOI] [PubMed] [Google Scholar]
- Huang Y, Greene E, Murray ST, Goodwin AC, Baylin SB, Woster PM, et al. Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proc Natl Acad Sci U S A. 2007b;104:8023–8028. doi: 10.1073/pnas.0700720104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Stewart TM, Wu Y, Baylin SB, Marton LJ, Perkins B, et al. Novel oligoamine analogues inhibit lysine-specific demethylase 1 and induce reexpression of epigenetically silenced genes. Clin Cancer Res. 2009b;15:7217–7228. doi: 10.1158/1078-0432.CCR-09-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard BP, Gomes AP, Dai H, Li J, Case AW, Considine T, et al. Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science. 2013;339:1216–1219. doi: 10.1126/science.1231097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson SE, Leveridge MV, Heathcote ML, Francis P, Williams L, Gee M, et al. Enabling lead discovery for histone lysine demethylases by high-throughput rapid fire mass spectrometry. J Biomol Screen. 2012;17:39–48. doi: 10.1177/1087057111416660. [DOI] [PubMed] [Google Scholar]
- Huynh T, Chen Z, Pang S, Geng J, Bandiera T, Bindi S, et al. Optimization of pyrazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett. 2009;19:2924–2927. doi: 10.1016/j.bmcl.2009.04.075. [DOI] [PubMed] [Google Scholar]
- Hyllus D, Stein C, Schnabel K, Schiltz E, Imhof A, Dou Y, et al. PRMT6-mediated methylation of R2 in histone H3 antagonizes H3 K4 trimethylation. Genes Dev. 2007;21:3369–3380. doi: 10.1101/gad.447007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer A, Fairlie DP, Brown L. Lysine acetylation in obesity, diabetes and metabolic disease. Immunol Cell Biol. 2011;90:39–46. doi: 10.1038/icb.2011.99. [DOI] [PubMed] [Google Scholar]
- Jacobson RH, Ladurner AG, King DS, Tjian R. Structure and function of a human TAFII250 double bromodomain module. Science. 2000;288:1422–1425. doi: 10.1126/science.288.5470.1422. [DOI] [PubMed] [Google Scholar]
- Jang B, Shin H-Y, Choi J-K, Nguyen DPT, Jeong B-H, Ishigami A, et al. Subcellular localization of peptidyl arginine deiminase 2 and citrullinated proteins in brains of scrapie-infected mice: nuclear localization of PAD2 and membrane fraction-enriched citrullinated proteins. J Neurop Exp Neurol. 2011;70:116–124. doi: 10.1097/NEN.0b013e318207559e. [DOI] [PubMed] [Google Scholar]
- Jensen LR, Amende M, Gurok U, Moser B, Gimmel V, Tzschach A, et al. Mutations in the JARID1C gene, which Is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. Am J Hum Genet. 2005;76:227–236. doi: 10.1086/427563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jochems J, Boulden J, Lee BG, Blendy JA, Jarpe M, Mazitschek R, et al. Antidepressant-like properties of novel HDAC6 selective inhibitors with improved brain bioavailability. Neuropsychopharmacology. 2013;39:389–400. doi: 10.1038/npp.2013.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AD, Yanek LR, Chen M-H, Faraday N, Larson MG, Tofler G, et al. Genome-wide meta-analyses identifies seven loci associated with platelet aggregation in response to agonists. Nat Genet. 2010;42:608–613. doi: 10.1038/ng.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones J, Causey C, Knuckley B, Slack-Noyes J, Thompson P. Protein arginine deiminase 4 (PAD4): current understanding and future therapeutic potential. Curr Opin Drug Discov Devel. 2009;12:616–627. [PMC free article] [PubMed] [Google Scholar]
- Jones JE, Slack JL, Fang P, Zhang X, Subramanian V, Causey CP, et al. Synthesis and screening of a haloacetamidine containing library to identify PAD4 selective inhibitors. ACS Chem Biol. 2012;7:160–165. doi: 10.1021/cb200258q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen S, Schotta G, Sorensen CS. Histone H4 Lysine 20 methylation: key player in epigenetic regulation of genomic integrity. Nucleic Acids Res. 2013;41:2797–2806. doi: 10.1093/nar/gkt012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi P, Greco TM, Guise AJ, Luo Y, Yu F, Nesvizhskii AI, et al. The functional interactome landscape of the human histone deacetylase family. Mol Syst Biol. 2013;9:672. doi: 10.1038/msb.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, McDonagh T, Heltweg B, Hixon J, Westman EA, Caldwell SD, et al. Substrate-specific activation of sirtuins by resveratrol. J Biol Chem. 2005;280:17038–17045. doi: 10.1074/jbc.M500655200. [DOI] [PubMed] [Google Scholar]
- Kanno Y, Vahedi G, Hirahara K, Singleton K, O'Shea JJ. Transcriptional and epigenetic control of T helper cell specification: molecular mechanisms underlying commitment and plasticity. Annu Rev Immunol. 2012;30:707–731. doi: 10.1146/annurev-immunol-020711-075058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantojarvi K, Onkamo PI, Vanhala R, Alen R, Hedman M, Sajantila A, et al. Analysis of 9p24 and 11p12-13 regions in autism spectrum disorders: rs1340513 in the JMJD2C gene is associated with ASDs in Finnish sample. Psychiatr Genet. 2010;20:102–108. doi: 10.1097/YPG.0b013e32833a2080. [DOI] [PubMed] [Google Scholar]
- Karytinos A, Forneris F, Profumo A, Ciossani G, Battaglioli E, Binda C, et al. A novel mammalian flavin-dependent histone demethylase. J Biol Chem. 2009;284:17775–17782. doi: 10.1074/jbc.M109.003087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessenbrock K, Krumbholz M, Schonermarck U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. 2009;15:623–625. doi: 10.1038/nm.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan O, La Thangue NB. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol. 2012;90:85–94. doi: 10.1038/icb.2011.100. [DOI] [PubMed] [Google Scholar]
- Khan SN, Khan AU. Role of histone acetylation in cell physiology and diseases: an update. Clin Chim Acta. 2010;411:1401–1411. doi: 10.1016/j.cca.2010.06.020. [DOI] [PubMed] [Google Scholar]
- Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med. 2012;5:178ra40. doi: 10.1126/scitranslmed.3005580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khmelnitsky YL, Mozhaev VV, Cotterill IC, Michels PC, Boudjabi S, Khlebnikov V, et al. In vitro biosynthesis, isolation, and identification of predominant metabolites of 2-(4-(2-hydroxyethoxy)-3,5-dimethylphenyl)-5,7-dimethoxyquinazolin-4(3H)-one (RVX-208) Eur J Med Chem. 2013;64:121–128. doi: 10.1016/j.ejmech.2013.03.062. [DOI] [PubMed] [Google Scholar]
- Kim S-K, Jung I, Lee H, Kang K, Kim M, Jeong K, et al. Human histone H3K79 methyltransferase DOT1L methyltransferase binds actively transcribing RNA polymerase II to regulate gene expression. J Biol Chem. 2013;287:39698–39709. doi: 10.1074/jbc.M112.384057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King ONF, Li XS, Sakurai M, Kawamura A, Rose NR, Ng SS, et al. Quantitative high-throughput screening identifies 8-hydroxyquinolines as cell-active histone demethylase Inhibitors. PLoS ONE. 2010;5:e15535. doi: 10.1371/journal.pone.0015535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight JS, Zhao W, Luo W, Subramanian VX, O'Dell AA, Yalavarthi S, et al. Peptidyl arginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J Clin Invest. 2013;123:2981–2993. doi: 10.1172/JCI67390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuckley B, Causey CP, Jones JE, Bhatia M, Dreyton CJ, Osborne TC, et al. Substrate specificity and kinetic studies of PADs 1, 3, and 4 identify potent and selective inhibitors of protein arginine deiminase 3. Biochemistry. 2010a;49:4852–4863. doi: 10.1021/bi100363t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuckley B, Jones JE, Bachovchin DA, Slack J, Causey CP, Brown SJ, et al. A fluopol-ABPP HTS assay to identify PAD inhibitors. Chem Commun. 2010b;46:7175–7177. doi: 10.1039/c0cc02634d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013;110:7922–7927. doi: 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogure M, Takawa M, Cho H-S, Toyokawa G, Hayashi K, Tsunoda T, et al. Deregulation of the histone demethylase JMJD2A is involved in human carcinogenesis through regulation of the G1/S transition. Cancer Lett. 2013;336:76–84. doi: 10.1016/j.canlet.2013.04.009. [DOI] [PubMed] [Google Scholar]
- Kong S, Yeung P, Fang D. The class III histone deacetylase sirtuin 1 in immune suppression and its therapeutic potential in rheumatoid arthritis. J Genet Genomics. 2013;40:347–354. doi: 10.1016/j.jgg.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontaki H, Talianidis I. Lysine methylation regulates E2F1-induced cell death. Mol Cell. 2010;39:152–160. doi: 10.1016/j.molcel.2010.06.006. [DOI] [PubMed] [Google Scholar]
- Kousaka A, Mori Y, Koyama Y, Taneda T, Miyata S, Tohyama M. The distribution and characterization of endogenous protein arginine N-methyltransferase 8 in mouse CNS. Neuroscience. 2009;163:1146–1157. doi: 10.1016/j.neuroscience.2009.06.061. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin Modifications and Their Function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Kruidenier L, Chung C-W, Cheng Z, Liddle J, Che K, Joberty G, et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature. 2012;488:404–408. doi: 10.1038/nature11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubicek S, O'Sullivan RJ, August EM, Hickey ER, Zhang Q, Teodoro ML, et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell. 2007;25:473–481. doi: 10.1016/j.molcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Lahue RS, Frizzell A. Histone deacetylase complexes as caretakers of genome stability. Epigenetics. 2012;7:806–810. doi: 10.4161/epi.20922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshminarasimhan M, Rauh D, Schutkowski M, Steegborn S. Sirt1 activation by resveratrol is substrate sequence-selective. Aging. 2013;5:151–154. doi: 10.18632/aging.100542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau OD, Kundu TK, Soccio RE, Ait-si-ali S, Khalil EM, Vassilev A, et al. HATs off: selective synthetic inhibitors of the histone acetyltransferases p300 and PCAF. Mol Cell. 2000;5:589–595. doi: 10.1016/s1097-2765(00)80452-9. [DOI] [PubMed] [Google Scholar]
- Lee J, Sayegh J, Daniel J, Clarke S, Bedford MT. PRMT8, a new membrane-bound tissue-specific member of the protein arginine methyltransferase family. J Biol Chem. 2005;280:32890–32896. doi: 10.1074/jbc.M506944200. [DOI] [PubMed] [Google Scholar]
- Lee MG, Wynder C, Schmidt DM, McCafferty DG, Shiekhattar R. Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem Biol. 2006;13:563–567. doi: 10.1016/j.chembiol.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Li P, Yao H, Zhang Z, Li M, Luo Y, Thompson PR, et al. Regulation of p53 target gene expression by peptidyl arginine deiminase 4. Mol Cell Biol. 2008;28:4745–4758. doi: 10.1128/MCB.01747-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med. 2010a;207:1853–1862. doi: 10.1084/jem.20100239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Wang D, Yao H, Doret P, Hao G, Shen Q, et al. Coordination of PAD4 and HDAC2 in the regulation of p53-target gene expression. Oncogene. 2010b;29:3153–3162. doi: 10.1038/onc.2010.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Shin D, Kwon SH. Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J. 2012;280:775–793. doi: 10.1111/febs.12079. [DOI] [PubMed] [Google Scholar]
- Li Z, Guo J, Wu Y, Zhou Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res. 2013;41:277–287. doi: 10.1093/nar/gks976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Vogel JL, Narayanan A, Peng H, Kristie TM. Inhibition of the histone demethylase LSD1 blocks [alpha]-herpes virus lytic replication and reactivation from latency. Nat Med. 2009;15:1312–1317. doi: 10.1038/nm.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Quenelle D, Vogel JL, Mascaro C, Ortega A, Kristie TM. A novel selective LSD1/KDM1A inhibitor epigenetically blocks herpes simplex virus lytic replication and reactivation from latency. MBio. 2013;4:e00558–12. doi: 10.1128/mBio.00558-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libri V, Brown AP, Gambarota G, Haddad J, Shields GS, Dawes H, et al. A pilot randomized, placebo controlled, double blind phase I trial of the novel SIRT1 activator SRT2104 in elderly volunteers. PLoS ONE. 2012;7:e51395. doi: 10.1371/journal.pone.0051395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S, Janzer A, Becker A, Zimmer A, Schule R, Buettner R, et al. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis. 2010;31:512–520. doi: 10.1093/carcin/bgp324. [DOI] [PubMed] [Google Scholar]
- Liu F, Barsyte-Lovejoy D, Li F, Xiong Y, Korboukh VK, Huang X, et al. Discovery of an in vivo chemical probe of the lysine methyltransferases G9a and GLP. J Med Chem. 2013;56:8931–8942. doi: 10.1021/jm401480r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobera M, Madauss KP, Pohlhaus DT, Wright QG, Trocha M, Schmidt DR, et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat Chem Biol. 2013;9:319–325. doi: 10.1038/nchembio.1223. [DOI] [PubMed] [Google Scholar]
- Lu Z, Tian Y, Salwen HR, Chlenski A, Godley LA, Raj JU, et al. Histone-lysine methyltransferase EHMT2 is involved in proliferation, apoptosis, cell invasion, and DNA methylation of human neuroblastoma cells. Anticancer Drugs. 2013;24:484–493. doi: 10.1097/CAD.0b013e32835ffdbb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Luo Y, Arita K, Bhatia M, Knuckley B, Lee Y-H, Stallcup MR, et al. Inhibitors and inactivators of protein arginine seiminase 4: functional and structural characterization. Biochemistry. 2006;45:11727–11736. doi: 10.1021/bi061180d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti A, Drohat AC. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J Biol Chem. 2011;286:35334–35338. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis DM. Histone deacetylase inhibitors and HIV latency. Curr Opin HIV AIDS. 2011;6:25–29. doi: 10.1097/COH.0b013e328341242d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Redondo P, Vaquero A. The diversity of histone versus nonhistone sirtuin substrates. Genes Cancer. 2013;4:148–163. doi: 10.1177/1947601913483767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinod K, Demers M, Fuchs TA, Wong SL, Brill A, Gallant M, et al. Neutrophil histone modification by peptidyl arginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci U S A. 2012;110:8674–8679. doi: 10.1073/pnas.1301059110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marushige K. Activation of chromatin by acetylation of histone side chains. Proc Natl Acad Sci U S A. 1976;73:3937–3941. doi: 10.1073/pnas.73.11.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews GM, Newbold A, Johnstone RW, Steven G. Chapter five – intrinsic and extrinsic apoptotic pathway signaling as determinants of histone deacetylase inhibitor antitumor activity. Adv Cancer Res. 2012;116:165–197. doi: 10.1016/B978-0-12-394387-3.00005-7. [DOI] [PubMed] [Google Scholar]
- McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- McNeill E. RVX-208, a stimulator of apolipoprotein AI gene expression for the treatment of cardiovascular diseases. Curr Opin Investig Drugs. 2010;11:357–364. [PubMed] [Google Scholar]
- Mele DA, Salmeron A, Ghosh S, Huang H-R, Bryant BM, Lora JM. BET bromodomain inhibition suppresses TH17-mediated pathology. J Exp Med. 2013;210:2181–2190. doi: 10.1084/jem.20130376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AHFM, et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–439. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- Migliori V, Phalke S, Bezzi M, Guccione E. Arginine/lysine-methyl/methyl switches: biochemical role of histone arginine methylation in transcriptional regulation. Epigenomics. 2010;2:119–137. doi: 10.2217/epi.09.39. [DOI] [PubMed] [Google Scholar]
- Min J, Feng Q, Li Z, Zhang Y, Xu R-M. Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell. 2003;112:711–723. doi: 10.1016/s0092-8674(03)00114-4. [DOI] [PubMed] [Google Scholar]
- Mirguet O, Gosmini R, Toum J, Clement CA, Barnathan ML, Brusq J-M, et al. Discovery of epigenetic regulator I-BET762: lead optimization to afford a clinical candidate inhibitor of the BET bromodomains. J Med Chem. 2013;56:7501–7515. doi: 10.1021/jm401088k. [DOI] [PubMed] [Google Scholar]
- Muller S, Filippakopoulos P, Knapp S. Bromodomains as therapeutic targets. Expert Rev Mol Med. 2011;13:null–null. doi: 10.1017/S1462399411001992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Umehara T, Nakano K, Jang MK, Shirouzu M, Morita S, et al. Crystal structure of the human BRD2 bromodomain: insights into dimerization and recognition of acetylated histone H4. J Biol Chem. 2007;282:4193–4201. doi: 10.1074/jbc.M605971200. [DOI] [PubMed] [Google Scholar]
- Neeli I, Dwivedi N, Khan S, Radic M. Regulation of extracellular chromatin release from neutrophils. J Innate Immun. 2009;1:194–201. doi: 10.1159/000206974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newby PR, Pickles OJ, Mazumdar S, Brand OJ, Carr-Smith JD, Pearce SHS, et al. Follow-up of potential novel Graves' disease susceptibility loci, identified in the UK WTCCC genome-wide nonsynonymous SNP study. Eur J Hum Genet. 2010;18:1021–1026. doi: 10.1038/ejhg.2010.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls SJ, Gordon A, Johansson J, Wolski K, Ballantyne CM, Kastelein JJP, et al. Efficacy and safety of a novel oral inducer of apolipoprotein A-I synthesis in statin-treated patients with stable coronary artery disease: a randomized controlled trial. J Am Coll Cardiol. 2011;57:1111–1119. doi: 10.1016/j.jacc.2010.11.015. [DOI] [PubMed] [Google Scholar]
- Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung C-W, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolov M, Fischle W. Systematic analysis of histone modification readout. Mol Biosyst. 2013;9:182–194. doi: 10.1039/c2mb25328c. [DOI] [PubMed] [Google Scholar]
- Obianyo O, Causey CP, Jones JE, Thompson PR. Activity-based protein profiling of protein arginine methyltransferase 1. ACS Chem Biol. 2011;6:1127–1135. doi: 10.1021/cb2001473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogasawara D, Suzuki T, Mino K, Ueda R, Khan MNA, Matsubara T, et al. Synthesis and biological activity of optically active NCL-1, a lysine-specific demethylase 1 selective inhibitor. Bioorg Med Chem. 2011;19:3702–3708. doi: 10.1016/j.bmc.2010.12.024. [DOI] [PubMed] [Google Scholar]
- Oguz KK, Kurne A, Aksu AO, Karabulut E, Serdaroglu A, Teber S, et al. Assessment of citrullinated myelin by 1H-MR spectroscopy in early-onset multiple sclerosis. AJNR Am J Neuroradiol. 2009;30:716–721. doi: 10.3174/ajnr.A1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- Pacholec M, Bleasdale JE, Chrunyk B, Cunningham D, Flynn D, Garofalo RS, et al. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J Biol Chem. 2010;285:8340–8351. doi: 10.1074/jbc.M109.088682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S, Vishwanath SN, Erdjument-Bromage H, Tempst P, Sif S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol Cell Biol. 2004;24:9630–9645. doi: 10.1128/MCB.24.21.9630-9645.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicchi E, Crea F, Farrar WL, Green JE, Danesi R. Histone lysine demethylases in breast cancer. Crit Rev Oncol Hematol. 2013;86:97–103. doi: 10.1016/j.critrevonc.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlak MR, Scherer CA, Chen J, Roshon MJ, Ruley HE. Arginine N-methyltransferase 1 is required for early postimplantation mouse development, but cells deficient in the enzyme are viable. Mol Cell Biol. 2000;20:4859–4869. doi: 10.1128/mcb.20.13.4859-4869.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucleic Acids Res. 2014;42:D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picaud S, Wells C, Felletar I, Brotherton D, Martin S, Savitsky P, et al. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc Natl Acad Sci U S A. 2013;110:19754–19759. doi: 10.1073/pnas.1310658110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirooznia SK, Elefant F. Targeting specific HATs for neurodegenerative disease treatment: translating basic biology to therapeutic possibilities. Front Cell Neurosci. 2013;7:30. doi: 10.3389/fncel.2013.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pointon JJ, Harvey D, Karaderi T, Appleton LH, Farrar C, Wordsworth BP. The histone demethylase JARID1A is associated with susceptibility to ankylosing spondylitis. Genes Immun. 2011;12:395–398. doi: 10.1038/gene.2011.23. [DOI] [PubMed] [Google Scholar]
- Pradhan S, Bacolla A, Wells RD, Roberts RJ. Recombinant Human DNA (Cytosine-5) Methyltransferase: I. expression, purification, and comparison of de novo and maintenance methylation. J Biol Chem. 1999;274:33002–33010. doi: 10.1074/jbc.274.46.33002. [DOI] [PubMed] [Google Scholar]
- Prinjha RK, Witherington J, Lee K. Place your BETs: the therapeutic potential of bromodomains. Trends Pharmacol Sci. 2012;33:146–153. doi: 10.1016/j.tips.2011.12.002. [DOI] [PubMed] [Google Scholar]
- Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 2013;3:308–323. doi: 10.1158/2159-8290.CD-12-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci U S A. 2012;109:21360–21365. doi: 10.1073/pnas.1210371110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian C, Zhou MM. SET domain protein lysine methyltransferases: structure, specificity and catalysis. Cell Mol Life Sci. 2006;63:2755–2763. doi: 10.1007/s00018-006-6274-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu T, Zhou L, Zhu W, Wang T, Wang J, Shu Y, et al. Effects of treatment with histone deacetylase inhibitors in solid tumors: a review based on 30 clinical trials. Future Oncol. 2013;9:255–269. doi: 10.2217/fon.12.173. [DOI] [PubMed] [Google Scholar]
- Raeder MB, Birkeland E, Trovik J, Krakstad C, Shehata S, Schumacher S, et al. Integrated genomic analysis of the 8q24 amplification in endometrial cancers identifies ATAD2 as essential to MYC-dependent cancers. PLoS ONE. 2013;8:e54873. doi: 10.1371/journal.pone.0054873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindra KC, Selvi BR, Arif M, Reddy BAA, Thanuja GR, Agrawal S, et al. Inhibition of lysine acetyltransferase KAT3B/p300 activity by a naturally occurring hydroxynaphthoquinone, plumbagin. J Biol Chem. 2009;284:24453–24464. doi: 10.1074/jbc.M109.023861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddington JP, Pennings S, Meehan RR. Non-canonical functions of the DNA methylome in gene regulation. Biochem J. 2013;451:13–23. doi: 10.1042/BJ20121585. [DOI] [PubMed] [Google Scholar]
- Richardson PG, Mitsiades CS, Laubach JP, Hajek R, Spicka I, Dimopoulos MA, et al. Preclinical data and early clinical experience supporting the use of histone deacetylase inhibitors in multiple myeloma. Leuk Res. 2013;37:829–837. doi: 10.1016/j.leukres.2013.03.006. [DOI] [PubMed] [Google Scholar]
- Richon VM, Johnston D, Sneeringer CJ, Jin L, Majer CR, Elliston K, et al. Chemogenetic analysis of human protein methyltransferases. Chem Biol Drug Des. 2011;78:199–210. doi: 10.1111/j.1747-0285.2011.01135.x. [DOI] [PubMed] [Google Scholar]
- Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141:583–594. doi: 10.1016/j.cell.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose NR, Ng SS, Mecinovic J, Lienard BMR, Bello SH, Sun Z, et al. Inhibitor scaffolds for 2-oxoglutarate-dependent histone lysine demethylases. J Med Chem. 2008;51:7053–7056. doi: 10.1021/jm800936s. [DOI] [PubMed] [Google Scholar]
- Rose NR, Woon ECY, Kingham GL, King ONF, Mecinovicì J, Clifton IJ, et al. Selective inhibitors of the JMJD2 histone demethylases: combined nondenaturing mass spectrometric screening and crystallographic approaches. J Med Chem. 2010;53:1810–1818. doi: 10.1021/jm901680b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose NR, Woon ECY, Tumber A, Walport LJ, Chowdhury R, Li XS, et al. Plant growth regulator daminozide is a selective inhibitor of human KDM2/7 histone demethylases. J Med Chem. 2012;55:6639–6643. doi: 10.1021/jm300677j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth M, Chen WY. Sorting out functions of sirtuins in cancer. Oncogene. 2013;33:1609–1620. doi: 10.1038/onc.2013.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth SY, Denu JM, Allis CD. HIistone acetyl transferases. Annu Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- Santo L, Hideshima T, Kung AL, Tseng J-C, Tamang D, Yang M, et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 2012;119:2579–2589. doi: 10.1182/blood-2011-10-387365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savchenko AS, Inoue A, Ohashi R, Jiang S, Hasegawa G, Tanaka T, et al. Long pentraxin 3 (PTX3) expression and release by neutrophils in vitro and in ulcerative colitis. Pathol Int. 2011;61:290–297. doi: 10.1111/j.1440-1827.2011.02651.x. [DOI] [PubMed] [Google Scholar]
- Sayegh J, Cao J, Zou MR, Morales A, Blair LP, Norcia M, et al. Identification of small molecule inhibitors of jumonji AT-rich interactive domain 1B (JARID1B) histone demethylase by a sensitive high throughput screen. J Biol Chem. 2013;288:9408–9417. doi: 10.1074/jbc.M112.419861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlimme S, Hauser A-T, Carafa V, Heinke R, Kannan S, Stolfa DA, et al. Carbamate prodrug concept for hydroxamate HDAC inhibitors. ChemMedChem. 2011;6:1193–1198. doi: 10.1002/cmdc.201100007. [DOI] [PubMed] [Google Scholar]
- Schulte JH, Lim S, Schramm A, Friedrichs N, Koster J, Versteeg R, et al. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy. Cancer Res. 2009;69:2065–2071. doi: 10.1158/0008-5472.CAN-08-1735. [DOI] [PubMed] [Google Scholar]
- Seal J, Lamotte Y, Donche F, Bouillot A, Mirguet O, Gellibert F, et al. Identification of a novel series of BET family bromodomain inhibitors: binding mode and profile of I-BET151 (GSK1210151A) Bioorg Med Chem Lett. 2012;22:2968–2972. doi: 10.1016/j.bmcl.2012.02.041. [DOI] [PubMed] [Google Scholar]
- Segura MF, Fontanals-Cirera B, Gaziel-Sovran A, Guijarro MV, Hanniford D, Zhang G, et al. BRD4 sustains melanoma proliferation and represents a new target for epigenetic therapy. Cancer Res. 2013;73:6264–6276. doi: 10.1158/0008-5472.CAN-13-0122-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgarra R, Diana F, Bellarosa C, Dekleva V, Rustighi A, Toller M, et al. During apoptosis of tumor cells HMGA1a protein undergoes methylation: identification of the modification site by mass spectrometry. Biochemistry. 2003;42:3575–3585. doi: 10.1021/bi027338l. [DOI] [PubMed] [Google Scholar]
- Shen Y, Guo X, Wang Y, Qiu W, Chang Y, Zhang A, et al. Expression and significance of histone H3K27 demethylases in renal cell carcinoma. BMC Cancer. 2012;12:470. doi: 10.1186/1471-2407-12-470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Siarheyeva A, Senisterra G, Allali-Hassani A, Dong A, Dobrovetsky E, Wasney GA, et al. An allosteric inhibitor of protein arginine methyltransferase 3. Structure. 2012;20:1425–1435. doi: 10.1016/j.str.2012.06.001. [DOI] [PubMed] [Google Scholar]
- Soranzo N, Spector TD, Mangino M, Kuhnel B, Rendon A, Teumer A, et al. A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat Genet. 2009;41:1182–1190. doi: 10.1038/ng.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spannhoff A, Machmur R, Heinke R, Trojer P, Bauer I, Brosch G, et al. A novel arginine methyltransferase inhibitor with cellular activity. Bioorg Med Chem Lett. 2007;17:4150–4153. doi: 10.1016/j.bmcl.2007.05.088. [DOI] [PubMed] [Google Scholar]
- Spiltoir JI, Stratton MS, Cavasin MA, Demos-Davies K, Reid BG, Qi J, et al. BET acetyl-lysine binding proteins control pathological cardiac hypertrophy. J Mol Cell Cardiol. 2013;63:175–179. doi: 10.1016/j.yjmcc.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PWTC, Bauer C, et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152:1146–1159. doi: 10.1016/j.cell.2013.02.004. [DOI] [PubMed] [Google Scholar]
- Stanley JS, Griffin JB, Zempleni J. Biotinylation of histones in human cells. Eur J Biochem. 2001;268:5424–5429. doi: 10.1046/j.0014-2956.2001.02481.x. [DOI] [PubMed] [Google Scholar]
- Steger DJ, Lefterova MI, Ying L, Stonestrom AJ, Schupp M, Zhuo D, et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol Cell Biol. 2008;28:2825–2839. doi: 10.1128/MCB.02076-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner DE, Berger SL. Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev. 2000;64:435–459. doi: 10.1128/mmbr.64.2.435-459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Sun Q, Yang X, Zhong B, Jiao F, Li C, Li D, et al. Upregulated protein arginine methyltransferase 1 by IL-4 increases eotaxin-1 expression in airway epithelial cells and participates in antigen-induced pulmonary inflammation in rats. J Immunol. 2012;188:3506–3512. doi: 10.4049/jimmunol.1102635. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Ozasa H, Itoh Y, Zhan P, Sawada H, Mino K, et al. Identification of the KDM2/7 histone lysine demethylase subfamily inhibitor and its antiproliferative activity. J Med Chem. 2013;56:7222–7231. doi: 10.1021/jm400624b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szewczuk LM, Culhane JC, Yang M, Majumdar A, Yu H, Cole PA. Mechanistic analysis of a suicide inactivator of histone demethylase LSD1. Biochemistry. 2007;46:6892–6902. doi: 10.1021/bi700414b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbert PB, Henikoff S. Histone variants – ancient wrap artists of the epigenome. Nat Rev Mol Cell Biol. 2010;11:264–275. doi: 10.1038/nrm2861. [DOI] [PubMed] [Google Scholar]
- Tamkun JW, Deuring R, Scott MP, Kissinger M, Pattatucci AM, Kaufman TC, et al. Brahma: a regulator of drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2SWI2. Cell. 1992;68:561–572. doi: 10.1016/0092-8674(92)90191-e. [DOI] [PubMed] [Google Scholar]
- Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Frankel A, Cook RJ, Kim S, Paik WK, Williams KR, et al. PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. J Biol Chem. 2000;275:7723–7730. doi: 10.1074/jbc.275.11.7723. [DOI] [PubMed] [Google Scholar]
- Tang J, Yan H, Zhuang S. Histone deacetylases as targets for treatment of multiple diseases. Clin Sci. 2013a;124:651–662. doi: 10.1042/CS20120504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Peng R, Phillips JE, Deguzman J, Ren Y, Apparsundaram S, et al. Assessment of Brd4 inhibition in idiopathic pulmonary fibrosis lung fibroblasts and in vivo models of lung fibrosis. Am J Pathol. 2013b;183:470–479. doi: 10.1016/j.ajpath.2013.04.020. [DOI] [PubMed] [Google Scholar]
- Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson PR, Fast W. Histone citrullination by protein arginine deiminase: is arginine methylation a green light or a roadblock? ACS Chem Biol. 2006;1:433–441. doi: 10.1021/cb6002306. [DOI] [PubMed] [Google Scholar]
- Timmers S, Konings E, Bilet L, Houtkooper RH, Van De Weijer T, Goossens GH, et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011;14:612–622. doi: 10.1016/j.cmet.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada Y-I, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- Tzatsos A, Paskaleva P, Ferrari F, Deshpande V, Stoykova S, Contino G, et al. KDM2B promotes pancreatic cancer via polycomb-dependent and -independent transcriptional programs. J Clin Invest. 2013;123:727–739. doi: 10.1172/JCI64535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda R, Suzuki T, Mino K, Tsumoto H, Nakagawa H, Hasegawa M, et al. Identification of cell-active lysine specific demethylase 1-selective inhibitors. J Am Chem Soc. 2009;131:17536–17537. doi: 10.1021/ja907055q. [DOI] [PubMed] [Google Scholar]
- Vedadi M, Barsyte-Lovejoy D, Liu F, Rival-Gervier S, Allali-Hassani A, Labrie V, et al. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat Chem Biol. 2011;7:566–574. doi: 10.1038/nchembio.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdel A, Curtet S, Brocard M-P, Rousseaux S, Lemercier C, Yoshida M, et al. Active maintenance of mHDA2/mHDAC6 histone-deacetylase in the cytoplasm. Curr Biol. 2000;10:747–749. doi: 10.1016/s0960-9822(00)00542-x. [DOI] [PubMed] [Google Scholar]
- Ververis K, Hiong A, Karagiannis TC, Licciardi PV. Histone deacetylase inhibitors (HDACIs): multitargeted anticancer agents. Biologics. 2013;7:47–60. doi: 10.2147/BTT.S29965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol. 2011;187:538–552. doi: 10.4049/jimmunol.1100450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan H, Huynh T, Pang S, Geng J, Vaccaro W, Poss MA, et al. Benzo[d]imidazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1) – Hit to Lead studies. Bioorg Med Chem Lett. 2009;19:5063–5066. doi: 10.1016/j.bmcl.2009.07.040. [DOI] [PubMed] [Google Scholar]
- Wang H, Huang Z-Q, Xia L, Feng Q, Erdjument-Bromage H, Strahl BD, et al. Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science. 2001;293:853–857. doi: 10.1126/science.1060781. [DOI] [PubMed] [Google Scholar]
- Wang J, Hevi S, Kurash JK, Lei H, Gay F, Bajko J, et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet. 2009a;41:125–129. doi: 10.1038/ng.268. [DOI] [PubMed] [Google Scholar]
- Wang J, Lu F, Ren Q, Sun H, Xu Z, Lan R, et al. Novel histone demethylase LSD1 inhibitors selectively target cancer cells with pluripotent stem cell properties. Cancer Res. 2011;71:7238–7249. doi: 10.1158/0008-5472.CAN-11-0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wysocka J, Sayegh J, Lee Y-H, Perlin JR, Leonelli L, et al. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science. 2004;306:279–283. doi: 10.1126/science.1101400. [DOI] [PubMed] [Google Scholar]
- Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009b;184:205–213. doi: 10.1083/jcb.200806072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li P, Wang S, Hu J, Chen XA, Wu J, et al. Anticancer peptidyl arginine deiminase (PAD) inhibitors regulate the autophagy flux and the mammalian target of rapamycin complex 1 activity. J Biol Chem. 2012;287:25941–25953. doi: 10.1074/jbc.M112.375725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigle TJ, Copeland RA. Drugging the human methylome: an emerging modality for reversible control of aberrant gene transcription. Curr Opin Chem Biol. 2013;17:369–378. doi: 10.1016/j.cbpa.2013.03.035. [DOI] [PubMed] [Google Scholar]
- Williams DE, Dalisay DS, Li F, Amphlett J, Maneerat W, Chavez MAG, et al. Nahuoic acid A produced by a Streptomyces sp. isolated from a marine sediment is a selective SAM-competitive inhibitor of the histone methyltransferase SETD8. Org Lett. 2013;15:414–417. doi: 10.1021/ol303416k. [DOI] [PubMed] [Google Scholar]
- Willis VC, Gizinski AM, Banda NK, Causey CP, Knuckley B, Cordova KN, et al. N-alpha-benzoyl-N5-(2-chloro-1-iminoethyl)-l-ornithine amide, a protein arginine deiminase inhibitor, reduces the severity of murine collagen-induced arthritis. J Immunol. 2011;186:4396–4404. doi: 10.4049/jimmunol.1001620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willmann D, Lim S, Wetzel S, Metzger E, Jandausch A, Wilk W, et al. Impairment of prostate cancer cell growth by a selective and reversible lysine-specific demethylase 1 inhibitor. Int J Cancer. 2012;131:2704–2709. doi: 10.1002/ijc.27555. [DOI] [PubMed] [Google Scholar]
- Wolf SS. The protein arginine methyltransferase family: an update about function, new perspectives and the physiological role in humans. Cell Mol Life Sci. 2009;66:2109–2121. doi: 10.1007/s00018-009-0010-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woon ECY, Tumber A, Kawamura A, Hillringhaus L, Ge W, Rose NR, et al. Linking of 2-oxoglutarate and substrate binding sites enables potent and highly selective inhibition of JmjC histone demethylases. Angew Chem Int Ed Engl. 2012;51:1631–1634. doi: 10.1002/anie.201107833. [DOI] [PubMed] [Google Scholar]
- Wu J, Xu W. Histone H3R17me2a mark recruits human RNA polymerase-associated factor 1 complex to activate transcription. Proc Natl Acad Sci U S A. 2012;109:5675–5680. doi: 10.1073/pnas.1114905109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyce A, Ganji G, Smitheman KN, Chung C-W, Korenchuk S, Bai Y, et al. BET inhibition silences expression of MYCN and BCL2 and induces cytotoxicity in neuroblastoma tumor models. PLoS ONE. 2013;8:e72967. doi: 10.1371/journal.pone.0072967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyspianska BS, Bannister AJ, Barbieri I, Nangalia J, Godfrey A, Calero-Nieto FJ, et al. BET protein inhibition shows efficacy against JAK2V617F-driven neoplasms. Leukemia. 2013;28:88–97. doi: 10.1038/leu.2013.234. [DOI] [PubMed] [Google Scholar]
- Xiang Y, Zhu Z, Han G, Ye X, Xu B, Peng Z, et al. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc Natl Acad Sci U S A. 2007;104:19226–19231. doi: 10.1073/pnas.0700735104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav N, Lee J, Kim J, Shen J, Hu MCT, Aldaz CM, et al. Specific protein methylation defects and gene expression perturbations in coactivator-associated arginine methyltransferase 1-deficient mice. Proc Natl Acad Sci U S A. 2003;100:6464–6468. doi: 10.1073/pnas.1232272100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Huang J, Dasgupta M, Sears N, Miyagi M, Wang B, et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc Natl Acad Sci U S A. 2010;107:21499–21504. doi: 10.1073/pnas.1016147107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Culhane JC, Szewczuk LM, Gocke CB, Brautigam CA, Tomchick DR, et al. Structural basis of histone demethylation by LSD1 revealed by suicide inactivation. Nat Struct Mol Biol. 2007;14:535–539. doi: 10.1038/nsmb1255. [DOI] [PubMed] [Google Scholar]
- Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. 2013;13:37–50. doi: 10.1038/nrc3409. [DOI] [PubMed] [Google Scholar]
- Yao H, Rahman I. Role of histone deacetylase 2 in epigenetics and cellular senescence: implications in lung inflammaging and COPD. Am J Physiol Lung Cell Mol Physiol. 2012;303:L557–L566. doi: 10.1152/ajplung.00175.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yipp BG, Petri B, Salina D, Jenne CN, Scott BNV, Zbytnuik LD, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med. 2012;18:1386–1393. doi: 10.1038/nm.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Chory EJ, Wernimont AK, Tempel W, Scopton A, Federation A, et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat Commun. 2012;3:1288. doi: 10.1038/ncomms2304. [DOI] [PubMed] [Google Scholar]
- Yu W, Smil D, Li F, Tempel W, Fedorov O, Nguyen KT, et al. Bromo-deaza-SAH: a potent and selective DOT1L inhibitor. Bioorg Med Chem. 2013;21:1787–1794. doi: 10.1016/j.bmc.2013.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Marmorstein R. Histone acetyltransferases: rising ancient counterparts to protein kinases. Biopolymers. 2012;99:98–111. doi: 10.1002/bip.22128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Waterworth D, Perry JRB, Lim N, Song K, Chambers JC, et al. Population-based genome-wide association studies reveal six loci influencing plasma levels of liver enzymes. Am J Hum Genet. 2008;83:520–528. doi: 10.1016/j.ajhg.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Wang Q, Paulk J, Kubicek S, Kemp MM, Adams DJ, et al. A small-molecule probe of the histone methyltransferase G9a induces cellular senescence in pancreatic adenocarcinoma. ACS Chem Biol. 2012;7:1152–1157. doi: 10.1021/cb300139y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakrzewicz D, Zakrzewicz A, Preissner KT, Markart P, Wygrecka M. Protein Arginine Methyltransferases (PRMTs): promising targets for the treatment of pulmonary disorders. Int J Mol Sci. 2012;13:12383–12400. doi: 10.3390/ijms131012383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Li J, Muller M, Yan S, Mujtaba S, Pan C, et al. Selective small molecules blocking HIV-1 Tat and coactivator PCAF association. J Am Chem Soc. 2005;127:2376–2377. doi: 10.1021/ja044885g. [DOI] [PubMed] [Google Scholar]
- Zhang X, Bolt M, Guertin MJ, Chen W, Zhang S, Cherrington BD, et al. Peptidyl arginine deiminase 2-catalyzed histone H3 arginine 26 citrullination facilitates estrogen receptor target gene activation. Proc Natl Acad Sci U S A. 2012;109:13331–13336. doi: 10.1073/pnas.1203280109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Li N, Caron C, Matthias G, Hess D, Khochbin S, et al. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003;22:1168–1179. doi: 10.1093/emboj/cdg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Gaiha GD, John SP, Pertel T, Chin CR, Gao G, et al. Reactivation of latent HIV-1 by inhibition of BRD4. Cell Rep. 2012;2:807–816. doi: 10.1016/j.celrep.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou JX, Guo L, Revenko AS, Tepper CG, Gemo AT, Kung H-J, et al. Androgen-induced coactivator ANCCA mediates specific androgen receptor signaling in prostate cancer. Cancer Res. 2009;69:3339–3346. doi: 10.1158/0008-5472.CAN-08-3440. [DOI] [PubMed] [Google Scholar]
- Zurita-Lopez CI, Sandberg T, Kelly R, Clarke SG. Human protein arginine methyltransferase 7 (PRMT7) is a type III enzyme forming omega-NG-monomethylated arginine residues. J Biol Chem. 2012;287:7859–7870. doi: 10.1074/jbc.M111.336271. [DOI] [PMC free article] [PubMed] [Google Scholar]