

Abstract
Aneuploidy is a hallmark of cancer and is associated with malignancy and poor prognosis. Recent studies have revealed that aneuploidy inhibits proliferation, causes distinct alterations in the transcriptome and proteome and disturbs cellular proteostasis. However, the molecular mechanisms underlying the changes in gene expression and the impairment of proteostasis are not understood. Here, we report that human aneuploid cells are impaired in HSP90-mediated protein folding. We show that aneuploidy impairs induction of the heat shock response suggesting that the activity of the transcription factor heat shock factor 1 (HSF1) is compromised. Indeed, increased levels of HSF1 counteract the effects of aneuploidy on HSP90 expression and protein folding, identifying HSF1 overexpression as the first aneuploidy-tolerating mutation in human cells. Thus, impaired HSF1 activity emerges as a critical factor underlying the phenotypes linked to aneuploidy. Finally, we demonstrate that deficient protein folding capacity directly shapes gene expression in aneuploid cells. Our study provides mechanistic insight into the causes of the disturbed proteostasis in aneuploids and deepens our understanding of the role of HSF1 in cytoprotection and carcinogenesis.
Keywords: aneuploidy, cancer, HSF1, HSP90, protein folding
Introduction
Aneuploidy is defined by karyotypes that differ from multiples of the haploid chromosome set. Aneuploidy is not well tolerated in higher eukaryotes and represents one of the leading causes of spontaneous abortions in humans, with the rare surviving newborns suffering from multiple defects (Colnaghi et al, 2011). Moreover, aneuploidy is prevalent in cancer, where nearly 80% of solid tumors and approximately 60% of hematopoietic cancers show karyotypes differing from normal diploidy (Stankiewicz & Lupski, 2010). At the cellular level, aneuploidy is often associated with global pathway deregulation, impaired proliferation, increased energetic and metabolic demands and altered sensitivity to cytotoxic drugs (Upender et al, 2004; Niwa et al, 2006; Torres et al, 2007; Williams et al, 2008; Pavelka et al, 2010; Nawata et al, 2011; Stingele et al, 2012). The cellular response to aneuploidy is highly conserved from yeast to human and remarkably, appears to be largely independent of the exact karyotypic composition (Sheltzer et al, 2012; Stingele et al, 2012; Durrbaum et al, 2014). Experiments in budding yeast have shown that the mere presence of a transcriptionally silent additional chromosome does not lead to any apparent phenotypes; hence, taken together, it is the gene expression of the aneuploid genome that determines the phenotypic changes observed in aneuploids (Torres et al, 2007). However, in spite of these important insights, the molecular mechanisms underlying these global cellular changes are not yet fully understood.
Recent progress in understanding the cellular effects of aneuploidy was facilitated by analysis of model aneuploid cells with defined karyotypic changes such as aneuploid budding and fission yeast strains, fruit flies with segmental aneuploidy, mouse embryonic fibroblasts with unbalanced Robertsonian translocations and trisomic and tetrasomic human cell lines (Upender et al, 2004; Niwa et al, 2006; Torres et al, 2007; Williams et al, 2008; Stenberg et al, 2009; Pavelka et al, 2010; Nawata et al, 2011; Stingele et al, 2012). Analysis of disomic budding yeast revealed increased sensitivity to drugs that interfere with protein synthesis and degradation (Torres et al, 2007). Moreover, the proliferation defect of some disomies can be ameliorated by mutation of the Ubp6 protein, a deubiquitinating enzyme that was proposed to negatively regulate protein degradation (Torres et al, 2007, 2010). In line with these findings in yeast, aneuploidy in murine and human cells imposes profound changes in protein homeostasis (proteostasis). Human trisomic cells show an accumulation of cytoplasmic foci positive for both ubiquitin and SQSTM1/p62, a marker of selective autophagy (Stingele et al, 2012). Further, primary trisomic MEFs are sensitive to chemical inhibition of the chaperone HSP90 as well as to the inhibitor of autophagy chloroquine (Tang et al, 2011). In agreement with this finding, chromosomally unstable aneuploid cancer cell lines are more sensitive to HSP90 inhibition than chromosomally stable cell lines (Tang et al, 2011). A recent study demonstrates that aneuploid budding yeast harbor protein aggregates and that protein folding of HSP90 clients is compromised in these cells (Oromendia et al, 2012). These results suggest that the protein expression from supernumerary chromosomes places a burden on cellular proteostasis and that the HSP90 machinery might be particularly affected. However, the status of HSP90 function and the protein folding capacity of aneuploid cells in higher eukaryotes were so far unknown.
Here, we demonstrate for the first time that protein folding is significantly impaired by aneuploidy in human cells. Taking advantage of trisomic and tetrasomic human cells that we constructed using micronuclei-mediated chromosome transfer into the human near-diploid colorectal cancer cell line HCT116 and human immortalized retinal pigment epithelial cell line RPE-1 (Stingele et al, 2012), we show that in particular HSP90-mediated protein folding is compromised. Intriguingly, we found that HSF1-dependent activation of the heat shock response (HSR) is impaired, suggesting a mechanism by which aneuploidy impairs protein folding capacity. Importantly, endogenous or exogenous overexpression of HSF1 counteracts the effects of aneuploidy on HSP90-dependent protein folding, thereby identifying enhanced expression of HSF1 as the first aneuploidy-tolerating genetic modification in human cells. Finally, we demonstrate that the functional HSP90 and HSF1 deficiency has marked consequences for protein abundance and shapes the patterns of gene expression observed in aneuploid cells.
Results
Trisomic and tetrasomic human cell lines show defects in protein folding
Using model trisomic and tetrasomic human cell lines, we previously found an increased amount of cytoplasmic ubiquitin-positive foci in aneuploids in comparison to cognate diploids (Stingele et al, 2012). The accumulation of ubiquitinated proteins might be either due to a defect in their removal or due to their increased production in aneuploid cells. However, human aneuploid cells activate the catabolic pathway of autophagy (Stingele et al, 2012), and proteasome activity is not impaired by aneuploidy (Supplementary Fig S1A–C). This suggests that ubiquitinated proteins accumulate at higher rates, possibly due to an overwhelmed or impaired protein folding capacity, as previously proposed (Oromendia et al, 2012; Donnelly & Storchova, 2014). To directly test whether trisomic or tetrasomic human cells display protein folding defects, we employed a set of three Firefly luciferase-based sensor proteins comprising wild-type Firefly luciferase (FlucWT) and single and double mutant luciferase, FlucSM and FlucDM, that are highly sensitive to changes in the protein folding environment (Gupta et al, 2011). First, we performed luciferase refolding assays after transiently expressing FlucWT in diploid HCT116 cells and HCT116* cells, which stably express histone H2B-GFP, and their respective aneuploid derivatives. To this end, we subjected transfected cells to heat shock at 43°C for 2 h, which is sufficient to denature > 70% of luciferase, but does not result in toxicity. We then monitored refolding at 37°C over 4 h by measuring luminescence. We observed a significant impairment of FlucWT refolding in cells with trisomy and tetrasomy of chromosome 5 compared to their respective parental cell lines (Fig 1A and B). Next, we examined the effect of aneuploidy on the more sensitive mutants FlucSM and FlucDM. The mutations disrupt the stability of the Fluc protein, but do not affect its enzymatic activity. We thus hypothesized that the effect of aneuploidy on luciferase refolding should be even more pronounced in cells transfected with the destabilized mutant proteins. Indeed, the relative ability to refold both FlucDM (Fig 1C and D) and FlucSM (Supplementary Fig S1E and F) was almost threefold reduced in aneuploid cells in comparison to the parental HCT116. Thus, aneuploidy causes protein folding defects in human cell lines.
Figure 1. Trisomic and tetrasomic human cell lines show defects in protein folding.
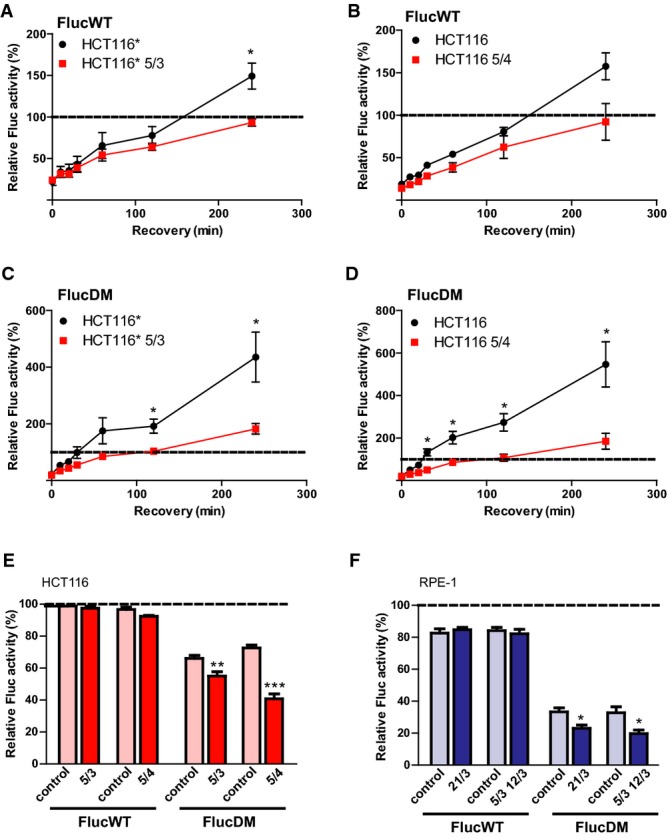
A–D Refolding of the sensor proteins upon heat shock in control cells and in respective aneuploids. HCT116 and HCT116* stably expressing histone H2B-GFP and their aneuploid derivatives were transfected either with FlucWT-mCherry (A, B) or FlucDM-mCherry (C, D) and subjected to heat stress for 2 h at 43°C. Controls were maintained at 37°C. Luminescence readings were taken immediately from heat-stressed cells (0 min) and at indicated time points after recovery at 37°C. The luminescence values of control cells maintained at 37°C were set to 100% (indicated by dotted line).
E, F Refolding of the sensor proteins upon HSP90 inhibition in control cells and in respective aneuploids. FlucWT-mCherry or FlucDM-mCherry was expressed in parental and aneuploid HCT116 or HCT116* (E) and RPE-1 or RPE-1* (F) cell lines for 36 h. Cells were then incubated with either solvent control (DMSO) or 50 nM 17-AAG for 8 h followed by measurement of luminescent activity. The depicted values show the percentage of luminescence in cells treated with 17-AAG relative to DMSO-treated cells (which were set to 100%).
Data information: All plots show the means of at least three independent experiments ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; non-parametric t-test.
The HSP90 chaperone is required for the refolding of heat-denatured luciferase, and the destabilizing mutations in FlucSM and FlucDM lead to an indispensable requirement for HSP90 for folding even in the absence of stress (Schneider et al, 1996; Gupta et al, 2011). Thus, whereas FlucWT activity is not impaired by HSP90 inhibition, the luminescent signal of FlucSM and FlucDM decreases upon treatment with 17-N-allylamino-17-demethoxygeldanamycin (17-AAG), a derivative of the antibiotic geldanamycin that binds the ATP pocket of the chaperone HSP90 (Supplementary Fig S1D). To specifically investigate HSP90-dependent protein folding capacity in aneuploid cells, we transfected diploid and aneuploid HCT116 cells with the Fluc sensors and measured luminescence in response to treatment with 17-AAG. Strikingly, we observed a consistent and significant decrease in FlucDM activity following treatment with 17-AAG in aneuploid cells compared to the control HCT116 cells (Fig 1E). These findings indicate that HSP90 function is indeed limiting in aneuploids. To exclude the possibility that the aneuploidy-induced protein folding defect is specific for aneuploid derivatives of HCT116 or for cells carrying extra copies of chromosome 5, we analyzed two additional aneuploid cell lines derived from RPE-1 (trisomy 21 and trisomy of chromosomes 5 and 12). Similarly as in HCT116-derived aneuploids, we observed consistently lower relative levels of FlucDM activity in aneuploid cells in contrast to diploid RPE-1 cells after treatment with 17-AAG (Fig 1F). These observations provide direct evidence that impaired protein folding and defective HSP90 function are common features of human aneuploid cells.
Aneuploid cells are more sensitive to inhibition of HSP90 but not to other inducers of protein folding stress
To elucidate in more detail the protein folding defect in human aneuploid cells, we analyzed their sensitivity to drugs that either directly inhibit molecular chaperones or impose a severe strain on the protein folding machinery. To this end, we measured cell viability after treatment with the HSP90 inhibitor 17-AAG, the HSC70/HSP70 inhibitor VER 155008, L-azetidine-2-carboxylic acid (AZC), a toxic L-proline analog that leads to the misfolding of newly synthesized polypeptides (Qian et al, 2010), and heat shock (45°C, 15 h). We observed a significant sensitivity of trisomic and tetrasomic HCT116 and RPE-1 cells to 17-AAG as measured by both cell viability as well as colony-forming assays (Fig 2A and B). In contrast, the response to the other compounds was less uniform: While aneuploid RPE-1 cells were more sensitive to HSC70/HSP70 inhibition by VER 155008 than diploids, aneuploid HCT116 cells were slightly more resistant to this treatment than controls (Fig 2C). Cell line-specific responses were also observed after heat shock: Only HCT116* 5/3 cells were highly sensitive to heat shock, while the other aneuploidies either showed a similar response as controls or even a marked resistance (Fig 2D). Finally, aneuploid cells were not significantly more sensitive to treatment with AZC (Supplementary Fig S2A and B). We conclude that aneuploidy exerts profound effects on cellular proteostasis, but only the increased sensitivity to HSP90 inhibition was common to all aneuploid cells that we tested. Thus, in line with previous observations from aneuploid yeast (Oromendia et al, 2012) and murine cells (Tang et al, 2011), a specific impairment of HSP90-mediated protein folding represents a general and conserved consequence of aneuploidy.
Figure 2. Sensitivity to inhibition of HSP90 but not to other inducers of protein folding stress increases in aneuploid cells.

A Wild-type, trisomic and tetrasomic cells were treated with the indicated concentrations of 17-AAG, and cell number was determined 72 h thereafter. Cell number is shown as the percentage of the DMSO-treated control.
B Colony formation efficiency of aneuploid and parental RPE-1 treated with either solvent control (DMSO) or 17-AAG at indicated concentrations. Cells were stained with crystal violet after 2 weeks.
C Wild-type, trisomic and tetrasomic cells were treated with the indicated concentration of VER 155008, and cell number was determined 72 h thereafter. Cell number is shown as the percentage of the DMSO-treated control.
D Wild-type, trisomic and tetrasomic cells were subjected to heat stress for 15 h at 45°C, and cell number was determined. Cell number is shown as the percentage of the untreated control (maintained at 37°C).
Data information: All plots show the means of at least three independent experiments ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; non-parametric t-test.
The basal and stress-induced activity of HSF1 is impaired in human aneuploid cells
Our results suggest that aneuploid cells may be compromised in their ability to mount a robust HSR when challenged with stress. Moreover, the impaired ability to refold luciferase evident already at very early time points of recovery (10, 20 min; Fig 1C and D) may point to a reduction in steady-state protein folding capacity. We hypothesized that the HSP90-specific protein folding defect might be due to changes in expression levels of factors involved in the maintenance of cellular proteostasis. Analysis of the expression changes in heat shock protein families (Kampinga et al, 2007) from our previously obtained global transcriptome and proteome data (Stingele et al, 2012) revealed a small but statistically significant reduction in both mRNA and protein abundance for the HSP90 family across a panel of six different aneuploid cell lines (Supplementary Fig S3A). Protein levels of chaperonins were also slightly decreased, although mRNA levels were not significantly changed (Supplementary Fig S3A). Analysis by immunoblotting revealed that the levels of HSP27, HSP70, and HSP90 were modestly, but consistently and significantly reduced in the majority of aneuploid cell lines analyzed (Fig 3A and B). The protein folding capacity of molecular chaperones is regulated, in part, by binding to co-chaperones and other co-factors. Phosphorylation at the C-termini of HSP70 and HSP90 enhances binding to the co-chaperone HOP, thereby increasing productive protein folding and cellular proliferation rates (Muller et al, 2013). However, the ratios of phosphorylated HSP90 and HSP70 over total levels were unchanged when comparing parental cell lines and HCT116 aneuploid derivatives (Supplementary Fig S3C and D). Our finding that the chaperone expression is impaired in aneuploid cells prompted us to analyze the expression of the heat shock response transcription factor and master regulator of chaperone expression HSF1 as well. The immunoblotting revealed a consistent reduction in protein levels in all four cell lines tested (Fig 3A and B). Notably, the transcription of the HSF1 gene is not altered in aneuploid cells, as we observed only negligible changes in HSF1 mRNA levels in qPCR experiments (Supplementary Fig S3B).
Figure 3. The basal and stress-induced activity of HSF1 is impaired in human aneuploid cells.

A, B Western blot analysis for HSP27, HSP70, HSP90 (the used antibody recognizes both constitutive and inducible forms of HSP90) and HSF1 in parental and aneuploid cell lines (A). Loading control: GAPDH; HSC70 (constitutively expressed chaperone) in RPE-1 5/3 12/3 and corresponding control (note that GAPDH is encoded on chromosome 12). Shown are representative images of at least 3 independent experiments. In panel B the quantification of the signal intensities from the Western blots shown in (A) are depicted, calculated relative to control cells (which were set to 1).
C, D HSP70-luc plasmid was expressed in parental and aneuploid HCT116 and RPE-1 cell lines for 36 h. Cells were then incubated with solvent control (DMSO), 2 μM 17-AAG or 5 μM MG132 for the indicated times. The depicted values show the fold induction in 17-AAG- or MG132-treated cells compared to DMSO-treated cells (which were set to 1).
E HCT116 (left panel) and RPE-1 (right panel) cells were transfected with siRNA targeting HSF1 or the GL2 subunit of luciferase as a control (ctrl). Cell extract was prepared 72 h after transfection and subjected to immunoblotting for HSF1 and GAPDH as a loading control. Quantification of the signal normalized to the loading control is shown above the images.
F HCT116 (left panel) and RPE-1 (right panel) cells transfected with siRNA targeting HSF1 or the GL2 subunit of luciferase as a control (ctrl). Forty-eight hours after transfection cells were incubated with the indicated concentrations of 17-AAG, and cell number was determined 72 h thereafter. Cell number is shown as the percentage of the DMSO-treated control.
Data information: All data are the mean of at least three independent experiments ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; non-parametric t-test.
Source data are available online for this figure.
In light of our observations regarding HSF1 protein levels, we asked whether the induction of HSF1 activity, that is, the ability to mount a HSR, was also impaired in aneuploid cells. To this end, we expressed a HSP70-luc construct that contains the HSP70 promoter fused to luciferase in diploid and aneuploid cells (Williams et al, 1989). We then treated the cells with 17-AAG and the proteasome inhibitor MG132, compounds that induce acute proteotoxic stress and are well-characterized activators of HSF1-dependent transcription (Mathew et al, 2001; Sharma et al, 2012). We observed that the parental cell lines induced the expression of the HSP70-luc sensor two- to threefold soon after the proteotoxic treatment and up to sevenfold in response to prolonged treatment (Fig 3C and D). In contrast, the ability of HCT116-derived and RPE-1-derived aneuploids to induce HSP70-luc was on average reduced to 50 and 60% of the control at early and later time points, respectively (Fig 3C and D). Interestingly, at the later time point, RPE-1 21/3 had recovered the ability to activate the HSP70 promoter. This is in line with the relatively mild decrease in HSF1 and chaperone levels in this cell line and with its relatively modest sensitivity to 17-AAG (Figs 2A and 3A and B). These observations might be explained by the small size of chromosome 21; hence, RPE-1 21/3 is burdened with the least amount of extra genetic material of all the aneuploid cell lines analyzed in this study. Consistent with these findings, we also observed an impaired ability to induce HSP70 expression after acute heat shock in both HCT116- and RPE-1-derived aneuploid cells (Supplementary Fig S3E). The decrease in HSF1 expression observed in aneuploid cells is relatively small, and therefore, we asked whether it is sufficient to cause the observed impairment in maintenance of proteostasis and protein folding. To address this concern, we transfected the control cell lines with siRNA to partially deplete HSF1 to 75 and 50%, respectively (Fig 3E). Indeed, consistent with previous results (Chen et al, 2013), this partial and transient depletion of HSF1 rendered cells sensitive to treatment with 17-AAG, thus suggesting a striking dosage sensitivity of the cellular response to proteotoxic stress (Fig 3F). Therefore, we conclude that the protein folding defect engendered by aneuploidy may be caused by inhibitory effects on basal and induced HSF1 activity.
Endogenous or exogenous overexpression of HSF1 ameliorates the negative effects of aneuploidy on protein folding
If the reduced protein folding capacity of aneuploid cells is due to a deficiency in HSF1 function, aneuploid cells with increased levels of HSF1 may be protected against this impairment. In fact, increased expression of HSF1 due to gene amplification is frequent in cancer, similarly as gain of chromosome 8 or its long arm where the HSF1 gene is located on 8q24.3 (Beroukhim et al, 2010). Thus, we reasoned that cells that gained chromosome 8 with the resulting increased expression of HSF1 might escape the defects in HSP90 function and protein folding caused by gain of a chromosome without HSF1. To test this possibility, we generated four clonal cell lines derived from individual HCT116 cells upon micronuclei-mediated transfer of chromosome 8. Using chromosome painting, we confirmed the presence of an extra copy of chromosome 8 in all imaged cells from all four clonal cell lines (HCT116 8/3 c1-c4; Supplementary Fig S4A). Analysis of the expression of HSF1 protein revealed an increased abundance according to expected gene copy number, that is, approximately 1.5-fold higher relative to diploid HCT116, in the clonal cell lines c1, c2, and c4. Interestingly, HSF1 levels were not substantially changed in c3 (Fig 4A and B). This was likely due to a loss of the distal region of chromosome 8 where HSF1 is located (Supplementary Fig S4B, our unpublished data). Thus, we reasoned that comparison of c1, c2, and c4 with c3 would enable us to directly test whether increased levels of HSF1 protect cells from the protein folding defects caused by the introduction of the extra copy of chromosome 8. We first analyzed the expression of HSP90. Similarly to other trisomies and tetrasomies that we analyzed (Fig 3A and B, and Supplementary Fig S3A), introduction of chromosome 8 elicited a slight decrease in HSP90 levels in c3 (Fig 4A and B). Strikingly, however, in c1, c2, and c4, we observed no decrease in HSP90 levels relative to control HCT116 (Fig 4A and B). Next, we tested the sensitivity of FlucDM to 17-AAG treatment in the four clones. The luminescent output of FlucDM was significantly lower after treatment with 17-AGG in HCT116 8/3 c3 compared to control HCT116. In contrast, the relative decreases in luminescence in response to 17-AAG treatment were comparable to the control in the trisomic cell lines c1, c2, and c4 (Fig 4C). Moreover, whereas c3 exhibited a sensitivity to 17-AAG that was comparable with the other trisomies, c1, c2, and c4 were as resistant to 17-AAG as the parental HCT116 (Fig 4D). Thus, the increased levels of HSF1 counteract the negative effect of aneuploidy on HSP90 expression and on protein folding.
Figure 4. Endogenous or exogenous overexpression of HSF1 ameliorates the negative effects of aneuploidy on protein folding.

A, B Western blot analysis of HSF1 and HSP90 expression in HCT116 8/3* c1-c4 (A). Loading control: GAPDH. Shown are representative images of at least 3 independent experiments. Quantification of the signal intensities from the Western blots (B), calculated relative to control cells (which were set to 1).
C FlucDM-mCherry was expressed in parental HCT116* and HCT116* 8/3 c1-c4 for 36 h. Cells were then incubated with either solvent control (DMSO) or 50 nM 17-AAG for 8 h followed by measurement of luminescent activity. The depicted values show the percentage of luminescence in cells treated with 17-AAG relative to DMSO-treated cells (which were set to 100%).
D Parental HCT116* and HCT116* cells trisomic for chromosome 8 (HCT116 8/3* c1-c4) were treated with the indicated concentrations of 17-AAG, and cell number was determined 72 h thereafter. Cell number is shown as the percentage of the DMSO-treated control (which was set to 100%).
E Western blot analysis of HSF1 expression and its downstream targets in the indicated aneuploid cells transfected with ca-HSF1. Loading control: GAPDH. Shown are representative images of at least 3 independent experiments. Quantification of the signal intensities normalized to the loading control is shown above the images.
F RPE-1* 21/3 cells were transiently transfected with either pCDNA or ca-HSF1 by electroporation. Forty-eight hours post-transfection cells were incubated with the indicated concentrations of 17-AAG, and cell number was determined 72 h thereafter. Cell number is shown as the percentage of the DMSO-treated control.
G FlucDM-mCherry was co-expressed with either pCDNA or ca-HSF1 in the indicated cell lines for 36 h. Cells were then incubated with either solvent control (DMSO), 50 nM 17-AAG (HCT116), or 5 nM 17-AAG (RPE-1) for 8 h followed by measurement of luminescent activity. The depicted values show the percentage of luminescence in cells treated with 17-AAG relative to DMSO-treated cells (which were set to 100%).
H Western blot analysis of HSF1 expression and its downstream targets in the indicated aneuploid cells transfected with ca-HSF1 using electroporation. Loading control: GAPDH. Shown are representative images of at least 3 independent experiments. Quantification of the signal intensities normalized to the loading control is shown above the images.
I Control HCT116* cells were transiently transfected with either pCDNA or ca-HSF1 by electroporation. Fourty-eight hours post-transfection cells were incubated with the indicated concentrations of 17-AAG and cell number was determined 72 h thereafter. Cell number is shown as the percentage of the DMSO-treated control.
J Control RPE-1* cells were transiently transfected with either pCDNA or ca-HSF1 by electroporation. Fourty-eight hours post-transfection cells were incubated with the indicated concentrations of 17-AAG and cell number was determined 72 h thereafter. Cell number is shown as the percentage of the DMSO-treated control.
Data information: The data are the mean of at least three independent experiments ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; non-parametric t-test.
To directly determine a role for increased HSF1 activity in mitigating the effects of aneuploidy on HSP90 and protein folding, we performed transient transfections with a constitutively active truncated HSF1 allele (ca-HSF1) (Zuo et al, 1995). Immunoblotting confirmed efficient expression of ca-HSF1 upon transient transfection in HCT116 8/3 c3 aneuploids as well as in HCT116 5/4 and RPE-1 21/3 and increased the expression levels of its downstream targets (Fig 4E). Importantly, the transient ca-HSF1 expression significantly improved the survival of aneuploid cells in the presence of 17-AAG as well as protected the folding of the FlucDM sensor against its effects (Fig 4F and G). Our observations suggest that cellular sensitivity to 17-AAG is finely tuned to the levels of HSF1. To test the generality of this conclusion, we transfected the control HCT116 and RPE-1 cells with ca-HSF1 (Fig 4H). In agreement with our hypothesis, the transient overexpression of ca-HSF1 also significantly protected the control cell lines against 17-AAG-associated toxicity (Fig 4I and J). Taken together, these results show that increasing the levels of the HSR master regulator HSF1 is sufficient to counteract the impaired HSP90 function of human aneuploid cells.
Since trisomy in human cells leads to proliferation defects, we asked whether the difference in HSF1 expression is reflected by changes in proliferative capacity. In line with our hypothesis that HSF1 mitigates some of the phenotypes caused by aneuploidy, we observed that HCT116 clones with high HSF1 expression proliferated markedly faster than HCT116 8/3 c3 (Supplementary Fig S4E). This also suggests that the decreased protein folding capacity contributes to the proliferation defects observed in trisomic cells.
Chromosome 8 carries approximately 4,170 open reading frames, among them the CCNE2 gene encoding cyclin E2 that plays a critical role in the G1 and in the G1-S transition and is often overexpressed in cancers, and MYC encoding the c-Myc transcription factor, a critical oncogene which upregulates a large number of genes involved in cell proliferation (Dang, 1999; Hwang & Clurman, 2005). We thus asked whether increased levels of c-Myc or cyclin E2 may be involved in improved proliferation rates in c1, c2 or c4. Western blotting revealed that cyclin E2 levels were increased on average 1.23-fold, and c-Myc levels were either unchanged or slightly reduced in all four HCT116 8/3 cell lines (Supplementary Fig S4C and D), in line with our previous finding that the abundance of some proteins is lower than expected based on the corresponding copy number changes in aneuploid cells (Stingele et al, 2012). As expected, based on these observations, Spearman correlation analysis revealed that only HSF1 levels highly correlated with the sensitivity of trisomic cells to HSP90 inhibition, their protein folding capacity as well as with proliferation rate (Supplementary Fig S4F). Taken together, both endogenous and exogenous overexpression of HSF1 ameliorates the adverse effects of aneuploidy on HSP90-dependent protein folding and proliferation in human cells.
Impaired HSP90 function in aneuploid cells affects the abundance of HSP90 client proteins
HSP90 plays a critical role in the folding of a wide variety of client proteins, in particular protein kinases as well as steroid hormone receptors and subunits of macromolecular complexes (Rohl et al, 2013). Thus, we asked whether the defect in HSP90 activity in aneuploid cells leads to a decreased abundance of client proteins that rely on HSP90. To this end, we compared recently reported data that elucidate the global HSP90 interactome and that classify interactors based on the strength of their interaction with HSP90 (Taipale et al, 2012) to the transcriptome and proteome changes that we observed in human aneuploid cell lines (Stingele et al, 2012). Our analysis revealed that the abundance of proteins that strongly interact with HSP90 was significantly lower in two out of the four aneuploid cell lines tested (HCT116* 5/4 and HCT116 5/4). In contrast, the abundance of non-interacting proteins was not affected in any of the analyzed cell lines (Fig 5A, Supplementary Datasets S1 and S2). Additionally, mRNA levels of strong interactors were unchanged, indicating that only the protein levels are affected (Fig 5A). Because this dataset of interactors may not represent a comprehensive list of all HSP90 clients, we also compared protein expression data from aneuploids with respect to another database of HSP90-interacting proteins generated by the Picard laboratory (http://www.picard.ch/Hsp90Int/index.php). Again, we observed a significant reduction in the expression of HSP90 interactors in two out of four aneuploid cells lines (HCT116 5/4 and RPE-1* 21/3; Fig 5B, Supplementary Dataset S3). Taken together, the reduced abundance of HSP90-interacting proteins in three out of four aneuploid cell lines supports the hypothesis that the HSP90 machinery is impaired in aneuploid cells and suggests that this impairment directly contributes to the altered protein composition of aneuploid cells.
Figure 5. The proteome and transcriptome changes in aneuploid cells resemble the cellular response to protein folding deficiency.

A Relative abundance (calculated aneuploid/diploid ratio) of proteins that were identified as non-interactors and strong interactors of HSP90 in human cells (left panel). Relative abundance (calculated aneuploid/diploid ratio) of corresponding mRNAs (right panel). *P < 0.05; **P < 0.01; ***P < 0.001; Mann–Whitney U-test.
B Relative abundance (calculated aneuploid/diploid ratio) of factors that were identified to interact with HSP90. *P < 0.05; **P < 0.01; ***P < 0.001; Mann–Whitney U-test.
C Changes in pathways identified in the proteome of cells upon inhibition of HSP90 compared to the proteome changes in trisomic cells using the 2D-annotation enrichment analysis.
D Changes in pathway regulation identified in the transcriptome of HCC cells upon HSF1 knockdown compared to transcriptome changes in trisomic cells using the 2D-annotation enrichment analysis.
The global expression changes in aneuploids resemble the cellular responses to HSP90 and HSF1 deficiency
HSP90 represents a critical hub in cell signaling through its chaperoning of a wide array of kinases and other proteins. Indeed, pharmacological inhibition of HSP90 results in significant alterations in the activity of multiple signaling pathways (Sharma et al, 2012). Our previous analysis of the changes in pathway regulation in human aneuploid cells identified a specific set of pathways that are up- or downregulated in response to aneuploidy, and these pathways appear to be conserved (Sheltzer et al, 2012; Stingele et al, 2012; Durrbaum et al, 2014). However, it remains unclear which molecular processes are responsible for these protein expression changes observed in aneuploid cells. Thus, we first asked whether the impairment in HSP90 function contributes to the changes of protein abundance in aneuploids. We compared the quantitative proteome changes in aneuploids cells with the proteome changes occurring upon pharmacological inhibition of HSP90 for 24 h (Sharma et al, 2012). The analysis of proteome changes upon HSP90 inhibition was performed in HeLa cells that are extensively aneuploid. Therefore, the proteome of treated cells was normalized to the proteome of untreated HeLa cells. Using 2-dimensional annotation enrichment analysis that enables direct comparison of relative pathway enrichments (Cox & Mann, 2012), we found an overlap between the proteome changes due to aneuploidy and proteome changes due to HSP90 inhibition (Supplementary Dataset S4), in particular among the downregulated pathways, which includes pathways of DNA and RNA metabolism, such as DNA repair and replication and RNA splicing, as well as cell cycle pathways (Fig 5C, Supplementary Fig S5A and B).
HSF1 predominantly regulates the expression of genes involved in proteostasis as part of the heat shock response, but has recently been shown to control the transcription of multiple additional target genes (Mendillo et al, 2012). We therefore compared the transcriptional profile of aneuploid cells with the transcriptional profile of a human hepatocellular carcinoma cell line (HCC) in which HSF1 was depleted by RNAi (Chuma et al, 2014). Comparison of the pathway enrichment in aneuploid cells and in cells depleted of HSF1 revealed a striking similarity in both downregulated and upregulated pathways (Fig 5D, Supplementary Fig S5C, Supplementary Dataset S4). Similar comparison with cells depleted of c-Myc showed no similarities between the pathways changes. This analysis suggests that the transcriptional activity of c-Myc does not affect the pathways that are deregulated by aneuploidy and supports the notion that the observed effect is specific for HSF1 (Supplementary Fig S5D). These results suggest that functional deficiency in HSF1 is a major determinant of the previously identified transcriptional aneuploidy response pattern (Stingele et al, 2012; Durrbaum et al, 2014). Taken together, our analyses suggest that the functional HSF1 and HSP90 deficiency caused by aneuploidy determines the global transcriptome and proteome changes in these cells.
Discussion
Aneuploid cells often suffer from low proliferation rates and exhibit hallmarks of cells undergoing proteotoxic stress as evidenced by their sensitivity to conditions that compromise or overburden protein folding (Torres et al, 2007; Tang et al, 2011; Oromendia et al, 2012; Stingele et al, 2012). Here, we directly demonstrate for the first time that human aneuploid cells suffer from a protein folding defect and show that in particular HSP90-dependent protein folding is affected. Additionally, we identify a pronounced impairment in the ability of aneuploids to trigger a full heat shock response, suggesting that the functionality of heat shock-associated factors, in particular, the responsible transcription factor HSF1, is compromised. Importantly, we demonstrate that increasing the gene copy number of HSF1 counters the effects of aneuploidy on HSP90 expression and protein folding. Finally, our analysis suggests that the observed functional deficiency in HSP90 and HSF1 contributes to the transcriptome and proteome changes observed in aneuploid cells. Thus, we propose that the cellular defects associated with aneuploidy may be direct consequences of impaired protein folding capacity.
Aneuploidy impairs protein folding
Both disomic budding yeast and tri- and tetrasomic human cells accumulate cytoplasmic protein deposits (Oromendia et al, 2012; Stingele et al, 2012). Previously, it has been proposed that the increased protein expression in aneuploid cells leads to a saturation of protein folding capacity and to low-level but chronic protein misfolding (Oromendia et al, 2012; Donnelly & Storchova, 2014). The misfolding, in turn, leads either to the aggregation, or destabilization and degradation of proteins with high or specific demands on the chaperone machinery. Indeed, we show that the presence of even one extra chromosome significantly impairs cellular protein folding in human aneuploid cells (Fig 1). This is mainly due to a defect in HSP90-dependent protein folding, whereas targeting the early steps in protein folding immediately after release from the ribosome, through AZC or HSP70 inhibition, does not preferentially impair the viability of human aneuploid cells (Figs 1 and 2, and Supplementary Fig S2). We hypothesize that the toxicity associated with impairment of proper protein folding at an early stage is determined by how efficiently and quickly cells can dispose of terminally misfolded proteins. Since both proteasome activity and autophagic degradation are elevated in mammalian aneuploid cells (Supplementary Fig S1B and C and Tang et al, 2011; Stingele et al, 2013), this may explain why they are not more sensitive or even slightly more resistant to such impairment. In contrast, we propose that the sensitivity to HSP90 inhibition observed in all the aneuploids analyzed in this study, regardless of the identity of the supernumerary chromosome(s) or the cell line rather reflects the loss-of-function of HSP90 clients and of HSP90-dependent processes. Therefore, our data together with previous observations in trisomic MEFs and disomic budding yeast (Tang et al, 2011; Oromendia et al, 2012) make a compelling argument that aneuploidy leads to a specific functional deficiency in HSP90-mediated protein folding.
In seeking to determine an explanation for why the HSP90 chaperone machinery is particularly affected by aneuploidy, we discovered that HSP90 family proteins were downregulated at both the mRNA and protein levels across a panel of six aneuploid cell lines. Intriguingly, this downregulation correlated with a decrease in total HSF1 levels in all four aneuploid cells lines tested and an impaired ability to induce HSF1 activity in response to acute proteotoxic stress (Fig 3). We emphasize, however, that the response to acute proteotoxic stress was not completely abolished, but rather delayed and diminished. Notably, while a delay and decrease can be detected in aneuploid budding yeast strains, this effect appears to be more modest (Oromendia et al, 2012). Interestingly, elevated levels of HSP72, but not HSP90 or other heat shock factors, were identified in aneuploid murine fibroblasts compared to diploid controls (Tang et al, 2011). The differential regulation of HSP72 and HSP90 suggests that the activation of HSP72 in murine aneuploids is not due to elevated HSF1-dependent transcription, but rather modulated by other means that are specific to HSP72. Despite this difference, aneuploidy renders murine fibroblasts sensitive to the HSP90 inhibitor 17-AAG similarly as human aneuploids, thus further strengthening the notion that HSP90-mediated protein folding is specifically limiting for aneuploid cells. In future, it will be important to address by what mechanism aneuploidy impairs HSF1 function and why HSP90-dependent protein folding is particularly affected. We propose two hypothetical mechanisms for how HSF1 function might be impaired by aneuploidy. First, the metastable protein HSF1 may be incorporated into the ubiquitin-positive cytoplasmic deposits in aneuploid cells, and thereby rendered inactive. Alternatively, the HSF1 protein may be subject to post-translational inhibitory regulation that is elevated in aneuploids. Interestingly, overexpression of model β-sheet proteins in human cells also impairs the induction of cellular stress responses (Olzscha et al, 2011). Uncovering the similarities and differences in these two models of chronic proteotoxic stress will improve our understanding of the mechanisms involved in the maintenance of protein homeostasis.
We found that increased copy numbers of HSF1 can alleviate the protein folding defect and the impaired response to proteotoxic stress in aneuploid cells. This was confirmed in two different scenarios, by transfer of chromosome 8 and by transient overexpression of transgenic HSF1 (Fig 4). We employed overexpression of the upstream regulator of the heat shock response to ensure integrated and balanced expression of HSP90 and its co-chaperones. This is essential because the concentration of essential co-chaperones is limiting for HSP90 activity (Li et al, 2012), and the chaperone-dependent processes often require all components of a given chaperone system (Rampelt et al, 2012). It should be noted that the transformed cell line HCT116 contains segmental aneuploidies, specifically copy gain on the long arms of chromosomes 8, 10, 16, and 17 and a loss of the Y-chromosome. These copy number changes are preserved in all created trisomic and tetrasomic cell lines (Stingele et al, 2012). However, our results suggest that an additional increase in copy numbers of HSF1 is necessary to rescue the defects arising in response to another whole chromosomal aneuploidy in de novo created trisomic cells so that the activity of HSF1 is sufficient to override the negative effects of aneuploidy on the protein folding machinery. More generally, our results suggest that in the context of the cellular response to severe proteotoxic stress (e.g. HSP90 inhibition), augmentation of HSF1 levels and/or activity fulfills a powerful cytoprotective function.
Previously, it was shown that a loss-of-function mutation in the gene encoding the deubiquitinating enzyme Ubp6 also markedly alleviated the negative effects of aneuploidy including impaired proliferation and accumulation of cytoplasmic protein deposits in budding yeast (Torres et al, 2010; Oromendia et al, 2012). Here, we have identified the first aneuploidy-tolerating genetic modification in human cells. These results support the interesting possibility that the adverse effects of aneuploidy can be suppressed either by enhancing protein degradation or by increasing cellular protein folding capacity. It is generally accepted that the sensitivity of cancer cells to HSP90 inhibitors stems from their reliance on heat shock proteins to chaperone the high number of overexpressed or mutated oncoproteins, and from the role of chaperones in protecting against general cellular stress associated with tumorigenesis (Dai et al, 2007). Our findings together with previous work (Tang et al, 2011) may provide an additional rationale for why inhibitors of protein degradation and protein folding emerge as a potentially effective cancer therapy and suggest that levels of HSF1 protein and/or activity may be important determinants of sensitivity to 17-AAG.
Consequences of the protein folding defects for aneuploids
Chemical or genetic impairment of HSP90 leads to the destabilization of multiple protein kinases and other proteins with critical roles in diverse cellular processes (Taipale et al, 2012). We found that the proteome of human aneuploid cells resembles the proteome of HeLa cells treated with the HSP90 inhibitor, 17-DMAG. In particular, pathways involved in DNA metabolic processes, chromatin modification, and transcription were downregulated in both conditions, whereas the overlap among upregulated pathways was rather modest. Even more similarities in both upregulated and downregulated pathways were revealed by comparison between transcriptional aneuploidy response patterns and the transcriptome changes in response to HSF1 depletion in the human HCC cell line (Chuma et al, 2014). Although HSF1, the major heat shock transcription factor, is most known for its role in regulating the expression of chaperones and proteins involved in the maintenance of proteostasis, recent discoveries have revealed its role in the regulation of a plethora of cellular processes (Mendillo et al, 2012). Based on our observations, we propose that the reduced HSP90 activity and the resulting decrease in stability of HSP90 client proteins partially underlies the pathway downregulation observed in aneuploid cells. Simultaneously, the impaired HSF1 activity affects, both directly, through the reduced expression of HSF1 target genes and indirectly, through the reduced protein folding capacity of aneuploid cells, the transcription of many targets. Taken together, we propose that the proteotoxic stress imposed by the presence of extra genetic material is a major determinant of the changes in gene expression in aneuploid cells.
We hypothesize that the HSP90 defect may have additional consequences for aneuploid cells. Two phenomena are worthy of particular mention. First, there is now a large body of evidence to suggest that HSP90 acts as a buffer against phenotypic variation by masking the effects of genetic polymorphisms (Jarosz et al, 2010). Second, HSP90 inhibition itself leads to chromosomal instability in budding yeast (Chen et al, 2012). Taken together with our results, we propose that aneuploidy is likely to further accelerate both the rate and manifestation of genetic change and our data suggest a general mechanism whereby changes in DNA copy number can lead to further genetic alterations.
Relevance for cancer and other pathologies
Aneuploidy is a hallmark of cancer, where it correlates with malignancy, drug resistance, and poor prognosis. However, trisomy and tetrasomy markedly impair cellular functions including proliferation, suggesting that aneuploidy-tolerating changes might be necessary to facilitate the growth of aneuploid cancer cells. Further, whether a similar impairment of protein folding capacity also occurs in cells of trisomy syndromes is currently not known and should be addressed in future. Intriguingly, somatic trisomy of chromosome 8 is frequently found in myeloid lineage disorders, some lymphomas and solid tumors such as breast and ovarian cancer. Interestingly, 8q24, where the HSF1 gene is located, is one of the most commonly amplified regions in cancer cells (Beroukhim et al, 2010; Davoli et al, 2013), and chromosome 8 is the largest somatic chromosome whose trisomy is compatible, although extremely rarely, with post-natal survival (Ganmore et al, 2009; Beroukhim et al, 2010). Indeed, we show that the presence of HSF1 on chromosome 8 protects against some of the adverse effects of aneuploidy. HSF1 is a critical facilitator of malignant proliferation, a role which it performs by supporting many important cellular processes (Dai et al, 2007). An additional role of HSF1 in promoting carcinogenesis may be to protect cancer cells from the proteotoxic stress induced by aneuploidy. Our results lend strong support to this notion and suggest a causal link between two recurring features of cancer cells: aneuploidy and altered HSF1 activity.
Materials and Methods
Cell lines and culturing conditions
The HCT116- and RPE-1-derived tri- and tetrasomic cell lines have been constructed by micronuclei-mediated chromosome transfer as described previously (Stingele et al, 2012). Parental cell line HCT116 (human colon carcinoma cell line): HCT116 3/3 (trisomy 3), HCT116 5/4 (tetrasomy 5) (Haugen et al, 2008); parental cell line HCT116* stably expressing histone H2B-GFP: HCT116* 5/3 (trisomy 5), HCT116* 5/4 (tetrasomy 5), (Stingele et al, 2013), HCT116 8/3 c1-c4; parental cell line RPE-1 (human retinal pigment epithelial cell line, hTERT immortalized): RPE-1 5/3 12/3 (trisomy 5, 12); parental cell line RPE-1* stably expressing histone H2B-GFP: RPE-1* 21/3 (trisomy 21). Cells were grown in DMEM GlutaMax (Gibco) supplemented with 10% FBS and 5% Pen/Strep under standard conditions.
Transfections and luciferase assays
Cells were transfected with a total of 1 or 1.5 μg of the indicated plasmids in 12-well plates using X-tremeGENE HP DNA transfection reagent (Roche) according to the manufacturer’s protocol. Cells were trypsinized, counted, and seeded into 96-well plates 24 h after transfection and then allowed to recover for 24 h. Then, cells were treated with either solvent control (DMSO), 5 or 50 nM 17-AAG (8 h), 2 μM 17-AAG (4 h), or 5 μM MG132 (8 or 20 h). 17-AAG and MG132 were purchased from Enzo Life Sciences and Tocris Bioscience, respectively. To measure luminescence, 30 μl of SteadyGlo reagent (Promega) was added directly to the wells of the 96-well plates and the plates were shaken for 10 s to ensure mixing and cell lysis. Luminescence was measured on a Tecan plate reader after 15-min incubation in the dark.
Proteasome activity assay
Cells were seeded at 2 × 104 per well in triplicates in 96-well plates. Forty minutes later, Proteasome-Glo™ Chymotrypsin-Like Cell-Based Assay (Promega) was added according to the manufacturer’s protocol. Luminescence was detected using a Fluoroskan Ascent FL plate reader operated by the Ascent software. For evaluation the mean with SEM of biological triplicates was calculated.
Western blotting
Exponentially growing cells were harvested and lyzed in RIPA buffer supplemented with protease inhibitors (Roche). 20 μg of protein were then resolved on 10% polyacrylamide gels and transferred to nitrocellulose membranes using the semi-dry technique. After blocking in low fat 5% milk in TBS-T, membranes were incubated with the following primary antibodies: HSP90 (1:1,000; Cell Signaling #4877), HSC70 (1:1,000; Enzo Life Sciences ADI-SPA-815), HSP70 (1:1,000; Enzo Life Sciences ADI-SPA-810), HSP27 (1:1,000; Enzo Life Sciences ADI-SPA-800), HSF1 (1:1,000; Enzo Life Sciences ADI-SPA-901), cyclin E2 (1:1,000; Cell Signaling #4132), c-Myc (1:200, Santa Cruz Biotechnology sc-40), GAPDH (1:2,000; Cell Signaling #2,118). The antibodies against phospho-HSP70 and phospho-HSP90 were a kind gift from Petr Müller, Masaryk University, Brno. After incubation with HRP-conjugated secondary antibodies, HRP substrate was added and luminescent signals were quantified using a LAS 3000 instrument (FujiFilm). Protein bands were quantified using ImageJ software.
Colony formation assays
Cells were seeded at 1,000 per well in 6-well plates 24 h before the treatment. Subsequently, cells were continuously treated with 17-AAG (5 or 25 nM) or DMSO for 10–12 days. Colonies were fixed with methanol:glacial acetic acid (1:1) and stained with 0.02% crystal violet for 15 min before washing with tap water and air-drying.
Cell viability assays
Cells were seeded at 2,000 per well in 96-well plates 24 h before the treatment. Subsequently, cells were treated with the indicated drugs at the indicated concentrations and cell viability was determined after 72 h, unless otherwise stated. Viability was determined using the CellTiterGlo® luminescent cell viability assay (Promega) according to the manufacturer’s instructions.
Chromosome transfer and preparation of chromosome spreads and paints
Chromosome transfer and the preparation of spreads and paints were performed as previously (Stingele et al, 2012).
RNA isolation and qPCR
Total RNA was isolated using the RNeasy kit (Qiagen) and reverse-transcribed into cDNA using the First Strand cDNA synthesis kit (Roche). qPCR was performed with a HSF1 assay from Qiagen (Cat. No. 330001 PPH00164F) on a LightCycler 480 (Roche) instrument using the KAPA SYBR FAST master mix. Absolute quantification was performed with an external standard, and the specificity of the amplicons was confirmed by melting curve analysis. HSF1 mRNA expression was normalized to ribosomal protein L27 (RPL27) as a housekeeping gene (de Jonge et al, 2007).
siRNA transfections
Cells were transfected at 50% confluency using 800 pmol siRNA and Oligofectamine according to the manufacturer’s instructions. Transfections were conducted in OptiMEM for 4 h. The siRNA sequences used were acquired from Eurofins Genomics and are as follows: HSF1 (5′ CGGAUUCAGGGAAGCAGCUGGUGCA 3′; (Jacobs & Marnett, 2009); GL2 (5′ CGUACGCGGAAUACUUCGATT 3′).
Electroporation
Cells were electroporated using the Amaxa Nucleofector II apparatus and following the manufacturer’s instructions and protocols for HCT116 cells and RPE-1 cells, respectively. Briefly, 1 million cells were resuspended in Cell Line Nucleofector Solution V containing 2 μg of either pCDNA or ca-HSF1 plasmid and transferred to cuvettes. HCT116 cells were electroporated using the D-032 Program and for RPE-1 cells the Program was U-017.
Transcriptome and proteome data analysis
The analyses of quantitative transcriptome and proteome data were performed as previously described (Durrbaum et al, 2014). The data were retrieved from the Gene Expression Omnibus database with the following accession numbers: knockdown of HSF1 in HCC: GSE47639; knockdown of c-myc in HeLa, BT-474, MCF-7, MDA-MB-231: GSE5823; HCT116- and RPE-1-derived trisomies and tetrasomies: GSE47830 and GSE39768.
Statistical analyses
All data related to viability, Fluc folding and protein expression were analyzed using Student’s t-test; *P < 0.05; **P < 0.01; ***P < 0.001. All statistically analyzed experiments were performed at least three times.
Acknowledgments
We are thankful to Ulrich Hartl, Richard Voellmy, and Petr Müller for generous gifts of reagents and acknowledge the excellent technical support of Aline Sewo-Pires de Campos. We thank Mary-Jeanette Andrews for help with analysis of the heat shock response and Abrar Zawed for assistance during the revision of the manuscript. We are grateful to Ulrich Hartl, Stefan Jentsch, Boris Pfander, Mark S Hipp and Lisa Vincenz as well as the members of the Storchova group for critical reading of our manuscript and for fruitful discussions. This work was supported by the Max Planck Society, the Center for Integrated Protein Science, Munich, and by a grant from Deutsche Forschungsgemeinschaft to ZS.
Author contributions
ND and VP performed experiments, SS performed initial experiments and analyzed the proteasome activity; MD contributed the bioinformatics analysis; ZS and ND conceived the study and wrote the manuscript, all authors analyzed the data and commented on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, McHenry KT, Pinchback RM, Ligon AH, Cho YJ, Haery L, Greulich H, Reich M, Winckler W, Lawrence MS, Weir BA, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Bradford WD, Seidel CW, Li R. Hsp90 stress potentiates rapid cellular adaptation through induction of aneuploidy. Nature. 2012;482:246–250. doi: 10.1038/nature10795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Chen J, Loo A, Jaeger S, Bagdasarian L, Yu J, Chung F, Korn J, Ruddy D, Guo R, McLaughlin ME, Feng F, Zhu P, Stegmeier F, Pagliarini R, Porter D, Zhou W. Targeting HSF1 sensitizes cancer cells to HSP90 inhibition. Oncotarget. 2013;4:816–829. doi: 10.18632/oncotarget.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuma M, Sakamoto N, Nakai A, Hige S, Nakanishi M, Natsuizaka M, Suda G, Sho T, Hatanaka K, Matsuno Y, Yokoo H, Kamiyama T, Taketomi A, Fujii G, Tashiro K, Hikiba Y, Fujimoto M, Asaka M, Maeda S. Heat shock factor 1 accelerates hepatocellular carcinoma development by activating nuclear factor-kappaB/mitogen-activated protein kinase. Carcinogenesis. 2014;35:272–281. doi: 10.1093/carcin/bgt343. [DOI] [PubMed] [Google Scholar]
- Colnaghi R, Carpenter G, Volker M, O’Driscoll M. The consequences of structural genomic alterations in humans: genomic disorders, genomic instability and cancer. Semin Cell Dev Biol. 2011;22:875–885. doi: 10.1016/j.semcdb.2011.07.010. [DOI] [PubMed] [Google Scholar]
- Cox J, Mann M. 1D and 2D annotation enrichment: a statistical method integrating quantitative proteomics with complementary high-throughput data. BMC Bioinformatics. 2012;13(Suppl. 16):S12. doi: 10.1186/1471-2105-13-S16-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C, Whitesell L, Rogers AB, Lindquist S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell. 2007;130:1005–1018. doi: 10.1016/j.cell.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19:1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli T, Xu AW, Mengwasser KE, Sack LM, Yoon JC, Park PJ, Elledge SJ. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell. 2013;155:948–962. doi: 10.1016/j.cell.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly N, Storchova Z. Dynamic karyotype, dynamic proteome: buffering the effects of aneuploidy. Biochim Biophys Acta. 2014;1843:473–481. doi: 10.1016/j.bbamcr.2013.11.017. [DOI] [PubMed] [Google Scholar]
- Durrbaum M, Kuznetsova AY, Passerini V, Stingele S, Stoehr G, Storchova Z. Unique features of the transcriptional response to model aneuploidy in human cells. BMC Genomics. 2014;15:139. doi: 10.1186/1471-2164-15-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganmore I, Smooha G, Izraeli S. Constitutional aneuploidy and cancer predisposition. Hum Mol Genet. 2009;18:R84–R93. doi: 10.1093/hmg/ddp084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Kasturi P, Bracher A, Loew C, Zheng M, Villella A, Garza D, Hartl FU, Raychaudhuri S. Firefly luciferase mutants as sensors of proteome stress. Nat Methods. 2011;8:879–884. doi: 10.1038/nmeth.1697. [DOI] [PubMed] [Google Scholar]
- Haugen AC, Goel A, Yamada K, Marra G, Nguyen TP, Nagasaka T, Kanazawa S, Koike J, Kikuchi Y, Zhong X, Arita M, Shibuya K, Oshimura M, Hemmi H, Boland CR, Koi M. Genetic instability caused by loss of MutS homologue 3 in human colorectal cancer. Cancer Res. 2008;68:8465–8472. doi: 10.1158/0008-5472.CAN-08-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang HC, Clurman BE. Cyclin E in normal and neoplastic cell cycles. Oncogene. 2005;24:2776–2786. doi: 10.1038/sj.onc.1208613. [DOI] [PubMed] [Google Scholar]
- Jacobs AT, Marnett LJ. HSF1-mediated BAG3 expression attenuates apoptosis in 4-hydroxynonenal-treated colon cancer cells via stabilization of anti-apoptotic Bcl-2 proteins. J Biol Chem. 2009;284:9176–9183. doi: 10.1074/jbc.M808656200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarosz DF, Taipale M, Lindquist S. Protein homeostasis and the phenotypic manifestation of genetic diversity: principles and mechanisms. Annu Rev Genet. 2010;44:189–216. doi: 10.1146/annurev.genet.40.110405.090412. [DOI] [PubMed] [Google Scholar]
- de Jonge HJ, Fehrmann RS, de Bont ES, Hofstra RM, Gerbens F, Kamps WA, de Vries EG, van der Zee AG, te Meerman GJ, ter Elst A. Evidence based selection of housekeeping genes. PLoS ONE. 2007;2:e898. doi: 10.1371/journal.pone.0000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampinga HH, Henning RH, van Gelder IC, Brundel BJ. Beat shock proteins and atrial fibrillation. Cell Stress Chaperones. 2007;12:97–100. doi: 10.1379/CSC-285.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Soroka J, Buchner J. The Hsp90 chaperone machinery: conformational dynamics and regulation by co-chaperones. Biochim Biophys Acta. 2012;1823:624–635. doi: 10.1016/j.bbamcr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- Mathew A, Mathur SK, Jolly C, Fox SG, Kim S, Morimoto RI. Stress-specific activation and repression of heat shock factors 1 and 2. Mol Cell Biol. 2001;21:7163–7171. doi: 10.1128/MCB.21.21.7163-7171.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendillo ML, Santagata S, Koeva M, Bell GW, Hu R, Tamimi RM, Fraenkel E, Ince TA, Whitesell L, Lindquist S. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell. 2012;150:549–562. doi: 10.1016/j.cell.2012.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller P, Ruckova E, Halada P, Coates PJ, Hrstka R, Lane DP, Vojtesek B. C-terminal phosphorylation of Hsp70 and Hsp90 regulates alternate binding to co-chaperones CHIP and HOP to determine cellular protein folding/degradation balances. Oncogene. 2013;32:3101–3110. doi: 10.1038/onc.2012.314. [DOI] [PubMed] [Google Scholar]
- Nawata H, Kashino G, Tano K, Daino K, Shimada Y, Kugoh H, Oshimura M, Watanabe M. Dysregulation of gene expression in the artificial human trisomy cells of chromosome 8 associated with transformed cell phenotypes. PLoS ONE. 2011;6:e25319. doi: 10.1371/journal.pone.0025319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa O, Tange Y, Kurabayashi A. Growth arrest and chromosome instability in aneuploid yeast. Yeast. 2006;23:937–950. doi: 10.1002/yea.1411. [DOI] [PubMed] [Google Scholar]
- Olzscha H, Schermann SM, Woerner AC, Pinkert S, Hecht MH, Tartaglia GG, Vendruscolo M, Hayer-Hartl M, Hartl FU, Vabulas RM. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell. 2011;144:67–78. doi: 10.1016/j.cell.2010.11.050. [DOI] [PubMed] [Google Scholar]
- Oromendia AB, Dodgson SE, Amon A. Aneuploidy causes proteotoxic stress in yeast. Genes Dev. 2012;26:2696–2708. doi: 10.1101/gad.207407.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavelka N, Rancati G, Zhu J, Bradford WD, Saraf A, Florens L, Sanderson BW, Hattem GL, Li R. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature. 2010;468:321–325. doi: 10.1038/nature09529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian SB, Zhang X, Sun J, Bennink JR, Yewdell JW, Patterson C. mTORC1 links protein quality and quantity control by sensing chaperone availability. J Biol Chem. 2010;285:27385–27395. doi: 10.1074/jbc.M110.120295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampelt H, Kirstein-Miles J, Nillegoda NB, Chi K, Scholz SR, Morimoto RI, Bukau B. Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. EMBO J. 2012;31:4221–4235. doi: 10.1038/emboj.2012.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohl A, Rohrberg J, Buchner J. The chaperone Hsp90: changing partners for demanding clients. Trends Biochem Sci. 2013;38:253–262. doi: 10.1016/j.tibs.2013.02.003. [DOI] [PubMed] [Google Scholar]
- Schneider C, Sepp-Lorenzino L, Nimmesgern E, Ouerfelli O, Danishefsky S, Rosen N, Hartl FU. Pharmacologic shifting of a balance between protein refolding and degradation mediated by Hsp90. Proc Natl Acad Sci USA. 1996;93:14536–14541. doi: 10.1073/pnas.93.25.14536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K, Vabulas RM, Macek B, Pinkert S, Cox J, Mann M, Hartl FU. Quantitative proteomics reveals that Hsp90 inhibition preferentially targets kinases and the DNA damage response. Mol Cell Proteomics. 2012;11:M111–M014654. doi: 10.1074/mcp.M111.014654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheltzer JM, Torres EM, Dunham MJ, Amon A. Transcriptional consequences of aneuploidy. Proc Natl Acad Sci USA. 2012;109:12644–12649. doi: 10.1073/pnas.1209227109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61:437–455. doi: 10.1146/annurev-med-100708-204735. [DOI] [PubMed] [Google Scholar]
- Stenberg P, Lundberg LE, Johansson AM, Ryden P, Svensson MJ, Larsson J. Buffering of segmental and chromosomal aneuploidies in Drosophila melanogaster. PLoS Genet. 2009;5:e1000465. doi: 10.1371/journal.pgen.1000465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele S, Stoehr G, Peplowska K, Cox J, Mann M, Storchova Z. Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol Syst Biol. 2012;8:608. doi: 10.1038/msb.2012.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele S, Stoehr G, Storchova Z. Activation of autophagy in cells with abnormal karyotype. Autophagy. 2013;9:246–248. doi: 10.4161/auto.22558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover Kenneth D, Karras Georgios I, Lindquist S. Quantitative analysis of Hsp90-client interactions reveals principles of substrate recognition. Cell. 2012;150:987–1001. doi: 10.1016/j.cell.2012.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang YC, Williams BR, Siegel JJ, Amon A. Identification of aneuploidy-selective antiproliferation compounds. Cell. 2011;144:499–512. doi: 10.1016/j.cell.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, Amon A. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science. 2007;317:916–924. doi: 10.1126/science.1142210. [DOI] [PubMed] [Google Scholar]
- Torres EM, Dephoure N, Panneerselvam A, Tucker CM, Whittaker CA, Gygi SP, Dunham MJ, Amon A. Identification of aneuploidy-tolerating mutations. Cell. 2010;143:71–83. doi: 10.1016/j.cell.2010.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upender MB, Habermann JK, McShane LM, Korn EL, Barrett JC, Difilippantonio MJ, Ried T. Chromosome transfer induced aneuploidy results in complex dysregulation of the cellular transcriptome in immortalized and cancer cells. Cancer Res. 2004;64:6941–6949. doi: 10.1158/0008-5472.CAN-04-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GT, McClanahan TK, Morimoto RI. E1a transactivation of the human HSP70 promoter is mediated through the basal transcriptional complex. Mol Cell Biol. 1989;9:2574–2587. doi: 10.1128/mcb.9.6.2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE, Amon A. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science. 2008;322:703–709. doi: 10.1126/science.1160058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo J, Rungger D, Voellmy R. Multiple layers of regulation of human heat shock transcription factor 1. Mol Cell Biol. 1995;15:4319–4330. doi: 10.1128/mcb.15.8.4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.