

Abstract
Mitochondrial fusion and fission is a dynamic process critical for the maintenance of mitochondrial function and cell viability. During excitotoxicity neuronal mitochondria are fragmented, but the mechanism underlying this process is poorly understood. Here, we show that Mfn2 is the only member of the mitochondrial fusion/fission machinery whose expression is reduced in in vitro and in vivo models of excitotoxicity. Whereas in cortical primary cultures, Drp1 recruitment to mitochondria plays a primordial role in mitochondrial fragmentation in an early phase that can be reversed once the insult has ceased, Mfn2 downregulation intervenes in a delayed mitochondrial fragmentation phase that progresses even when the insult has ceased. Downregulation of Mfn2 causes mitochondrial dysfunction, altered calcium homeostasis, and enhanced Bax translocation to mitochondria, resulting in delayed neuronal death. We found that transcription factor MEF2 regulates basal Mfn2 expression in neurons and that excitotoxicity-dependent degradation of MEF2 causes Mfn2 downregulation. Thus, Mfn2 reduction is a late event in excitotoxicity and its targeting may help to reduce excitotoxic damage and increase the currently short therapeutic window in stroke.
Keywords: excitotoxicity, mitochondrial dynamics, neuron, transcriptional regulation
Introduction
Glutamate is the main excitatory neurotransmitter in the central nervous system. It plays an essential role in development, synaptic plasticity, and neuronal survival but sustained elevated levels of extracellular glutamate kill neurons in a process called excitotoxicity (Arundine & Tymianski, 2003). Excitotoxicity takes place in both chronic neurological diseases, such as Huntington’s and Alzheimer’s disease, and in acute episodes such as traumatic brain injury and ischemic stroke. The NMDA receptor (NMDAR) is the main ionotropic glutamate receptor in the CNS and the excessive flux of Ca2+ that passes through it is a major cause of excitotoxicity. Despite evidence indicating a crucial role of NMDAR activation in brain damage during stroke, clinical trials with NMDAR blockers have failed because of poor tolerance and efficacy (Ikonomidou & Turski, 2002; Muir, 2006). In contrast to excessive NMDAR activity that causes cell death, its physiological activity triggers pro-survival signals that may play a role in promoting recovery and preventing delayed neuronal loss in the penumbra (Ikonomidou & Turski, 2002; Lo, 2008). Thus, future therapies to reduce excitotoxicity must target pro-death events downstream of NMDAR without affecting the pro-survival signals. Several mechanisms are implicated in cell death triggered by Ca2+ influx through NMDAR. Mitochondrial dysfunction caused by excessive Ca2+ uptake acts as a signaling hub for many pro-death events (Reynolds & Hastings, 1995; Stout et al, 1998; Almeida et al, 1999; Yu et al, 2002; Soriano et al, 2008).
Mitochondria are dynamic organelles that constantly fuse and divide, changing shape and localization. The equilibrium between fission and fusion is important for mitochondrial function, which is not only limited to supplying energy to the cell, but also intervenes in anabolic and catabolic biochemical pathways and the regulation of Ca2+ homeostasis, and is a key regulator of cell death progression. The core mitochondrial fusion and fission machineries are formed by a group of dynamin-related large GTPases (Liesa et al, 2009; Westermann, 2010).
Inner mitochondrial membrane fusion is mediated by Opa1. Two mitofusins (Mfn), Mfn1 and 2, mediate mitochondrial outer membrane fusion. Mfn1 and Mfn2 display high homology (81%) and around 60% identity, but nonetheless they have non-redundant roles (de Brito & Scorrano, 2008b; Liesa et al, 2009). In addition to its fusion role, Mfn2 localization in the ER is necessary to maintain the reticular morphology of the ER and control the ER–mitochondria interaction (de Brito & Scorrano, 2008a). Mfn2 may also play a role in neuronal mitochondrial trafficking, and disruption of this function can lead to axon degeneration (Baloh et al, 2007; Misko et al, 2012). Mitochondrial fission is mediated by Drp1. Drp1 is mainly cytoplasmic and its translocation to mitochondria, recruited by Fis1 and/or Mff, is essential for mitochondrial fission. Drp1 is subjected to several posttranscriptional modifications, including phosphorylation, ubiquitination, SUMOylation and nitrosylation, which can either activate or repress fission activity (Cho et al, 2010; Oettinghaus et al, 2012).
Although mitochondrial fission per se does not cause cell death, fragmentation of mitochondria has been shown to play a key role in cell death progression. Mitochondrial fragmentation occurs early in apoptosis and can be delayed by expressing a dominant-negative Drp1 (Frank et al, 2001; Breckenridge et al, 2003). Recently, Drp1 has also been implicated in the induction of necrosis (Wang et al, 2012). Mutations in Mfn2 are the most commonly identified cause of Charcot-Marie-Tooth type 2 (CMT2), a dominantly inherited disease characterized by degeneration of peripheral sensory and motor axons (Zuchner et al, 2004). Purkinje cells require Mfn2 but not Mfn1 for dendritic outgrowth, spine formation, and cell survival (Chen et al, 2007). Mitochondrial fragmentation is an early event that occurs before the release of mitochondrial proteins and neurite degeneration in an in vivo animal model of stroke (Barsoum et al, 2006). Despite the importance of mitochondrial dynamics in cell death progression, the exact mechanism that underlies the mitochondrial fragmentation in excitotoxicity is incompletely understood. In this study, we assessed how the proteins of the core mitochondrial fusion/fission machinery are regulated in excitotoxicity. We found that Mfn2 levels are reduced in both in vitro and in vivo models of excitotoxicity, via MEF2 degradation that, by acting on the Mfn2 promoter, regulates basal levels of Mfn2. Downregulation of Mfn2 causes mitochondrial dysfunction and altered Ca2+ homeostasis and facilitate Bax recruitment to mitochondria during excitotoxicity.
Results
Mfn2 protein expression is reduced in excitotoxicity
Mitochondrial dynamics plays a pivotal role in cell death. Changes in mitochondrial morphology have been observed in excitotoxicity, but the precise mechanism has not been fully defined. For a better understanding of the mechanism by which mitochondria are fragmented during excitotoxicity, we exposed primary cortical cultures to moderate doses (30 μM) of the glutamate receptor agonist NMDA over a time course of 1, 2, 4, and 8 h and analyzed the expression of the proteins of the mitochondrial fission/fusion machinery. During the first 2 h after NMDA application, there were no significant changes in either fusion or fission proteins, but after 4 h of NMDA treatment, we observed a 40% reduction in the fusion protein Mfn2 with no changes in the other fusion proteins, Mfn1 and Opa1. Surprisingly, Drp1 showed a tendency to reduce its protein levels (Fig 1A and B). To rule out the possibility that changes in the expression of mitochondrial fission/fusion protein were due to changes in mitochondrial mass, volume normalization was also performed with the mitochondrial protein porin, achieving similar results (Supplementary Fig S1A). Using oxygen and glucose deprivation (OGD) as another in vitro model of excitotoxicity, we also found that the level of Mfn2 was reduced 4 h after reoxigenation with no changes in the other proteins of the fission/fusion machinery (Supplementary Fig S1B and C).
Figure 1. Mfn2 expression is reduced in excitotoxicity in vitro and in vivo.
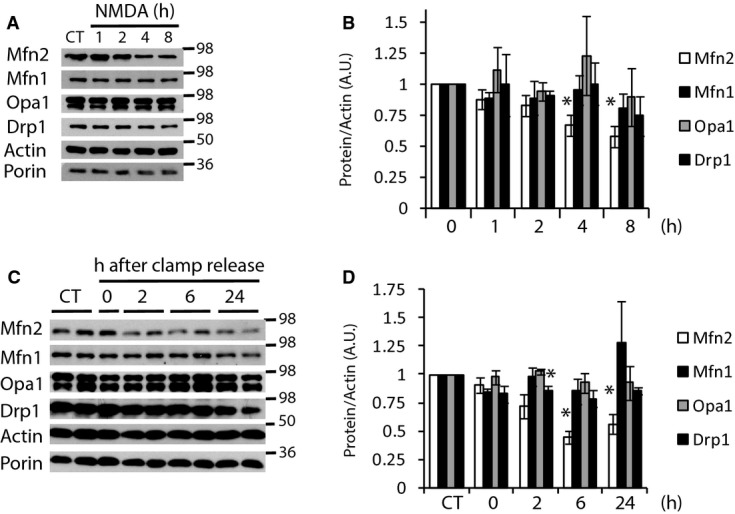
A, B Western analysis of mitochondrial fusion/fission proteins from in vitro primary cortical neurons exposed to NMDA (30 μM) for the indicated times and densitometric analysis normalized to actin (n = 3–6).
C, D Western analysis of mitochondrial fusion/fission proteins from in vivo brain extracts of rats subjected to MCAO plus 90 min of ipsilateral common carotid artery clamp followed by clamp release for the indicated times and densitometric analysis normalized to actin (n = 3–7).
Data information: Results are presented as mean ± SEM. *P < 0.05 compared to control, two-tailed t-test.
Source data are available online for this figure.
To check the pathophysiological relevance of these in vitro findings, we then used an in vivo model of cerebral ischemia in 12-day-old rats consisting in applying a permanent middle cerebral artery occlusion combined with 90 min of transient occlusion of the ipsilateral common carotid artery (Vaslin et al, 2009). During an ischemic episode, glutamate levels build up as a result of synaptic release and impaired and/or reversed uptake mechanisms (Camacho & Massieu, 2006), which induces excessive activation of NMDA glutamate receptors (NMDARs) and Ca2+-mediated cell death (Arundine & Tymianski, 2003). We analyzed the expression of the mitochondrial fusion/fission machinery at several time points after the ischemic insult. The results were in the same trend to those found in vitro. Two hours after restoration of the common carotid artery blood flow, that is 3.5 h after the ischemia onset, Mfn2 protein levels started to decline and reached a significant 50% reduction 6 h after the restoration of the common carotid artery blood flow (Fig 1C and D, and Supplementary Fig S1D). Drp1 also showed a slight but significant reduction (20%) 2 h after the restoration of blood flow. Neither of the other mitochondrial fusion proteins (Mfn1 and Opa1) showed changes in levels during the 24 h after the onset of ischemia. Thus, our in vitro neuronal cultures recapitulate well the excitotoxic in vivo model.
Activation of Drp1 induces mitochondrial fragmentation
We observed the downregulation of Mfn2 4 h after initiating the insult, but nonetheless the kinetics of excitotoxicity-mediated mitochondrial fragmentation has been reported to be fast (Rintoul et al, 2003; Barsoum et al, 2006; Young et al, 2010). To clarify this, we analyzed the mitochondrial morphology of those neurons expressing RFP targeted to the mitochondrial matrix (mtRFP) that were still alive (as shown by nuclear DAPI staining; Supplementary Fig S2A) after NMDA treatment of different durations. Untreated neurons contained mainly well-connected tubular mitochondria (60%). After only 30–60 min of NMDA application most of the neurons contained globular mitochondria that remained intact for the following 2 h with a small amount of additional fragmentation 4 h after NMDA treatment (Fig 2A and B). Although the secondary fragmentation correlates with the diminished Mfn2 expression, it is unlikely that Mfn2 is responsible for the first and early phase of mitochondrial fragmentation.
Figure 2. Activation of Drp1 induces mitochondrial fragmentation.
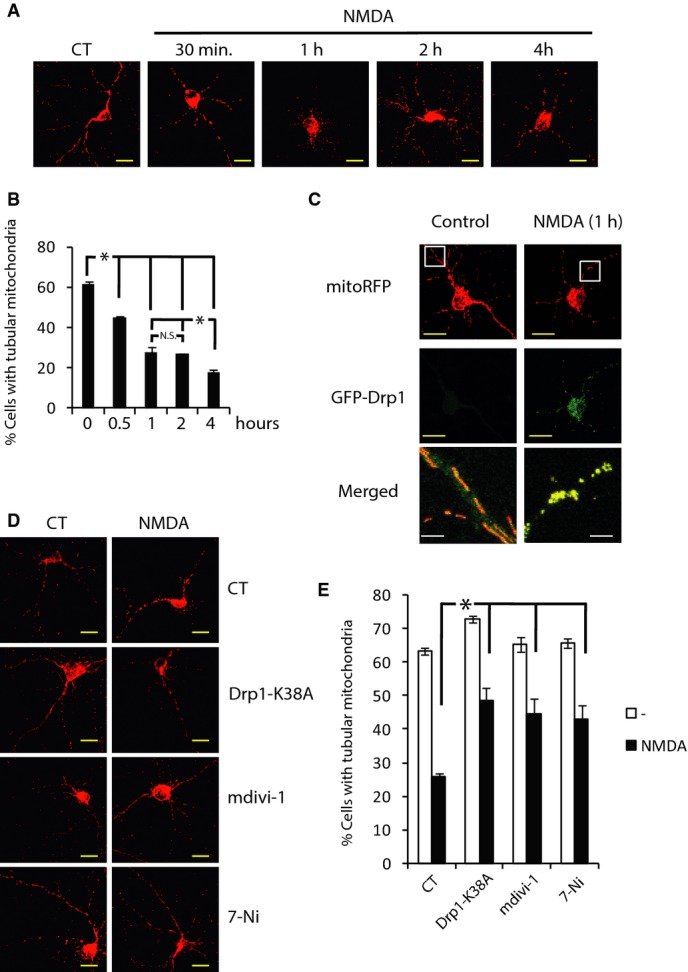
A Representative images of NMDA-induced mitochondrial fragmentation. Neurons transfected with mitochondria-targeted RFP (mtRFP) were treated with NMDA (30 μM) for the indicated times. Scale bar, 20 μm.
B Mitochondrial morphology analysis from (A) (n = 3–8). *P < 0.05 compared to control, one-way ANOVA followed by Bonferroni’s post hoc test. Results are presented as mean ± SEM.
C NMDA induces increased Drp1 translocation to mitochondria. Neurons were transfected with plasmids encoding GFP-Drp1 and mtRFP. After 48 h, neurons were stimulated with NMDA (30 μM) for 1 h or left unstimulated, fixed, and visualized under a confocal microscope. Brightness and contrast has been adjusted in the merged image to visualize GFP signal in the control condition. Scale bar, yellow = 20 μm, white = 4 μm.
D, E Representative images showing that genetic and pharmacological inhibitors of Drp1 block mitochondrial fragmentation (D). Neurons were transfected with plasmids encoding mtRFP and Drp1-K38A or control (globin). After 48 h, neurons were stimulated with NMDA (30 μM) for 1 h or they were pre-treated for 1 h before NMDA stimulation with mdiv-1 (25 μM) or 7-nitroindazole (7-Ni at 5 μM in Arg-free medium) and mitochondrial morphology was analyzed (E). (n = 3). Scale bar, 20 μm. Results are presented as mean ± SEM. *P < 0.05, one-way ANOVA followed by Bonferroni’s post hoc test.
We observed a tendency for a decline in the level of Drp1 in excitotoxicity (Fig 1). Nonetheless, what determines Drp1 activity is its subcellular localization. Drp1 is mainly cytosolic, and by posttranslational modifications, it is recruited to the mitochondria by Mff and/or Fis1 where it promotes fission (James et al, 2003; Yoon et al, 2003; Gandre-Babbe & van der Bliek, 2008; Otera et al, 2010). In basal conditions, GFP-Drp1 transfected neurons showed a weak diffuse signal within the neuron. Application of NMDA for 1 h produced a strong punctuate signal that co-localized with mitochondria (Fig 2C). Overexpression of a mutant form of Drp1 (Drp1-K38A), which acts as a dominant negative (Smirnova et al, 1998), or using the Drp1 inhibitor mdivi-1, significantly attenuated the NMDA-mediated mitochondrial fragmentation during the first hour of NMDA treatment (Fig 2D and E). Excessive calcium uptake by mitochondria in excitotoxicity causes mitochondrial depolarization (Soriano et al, 2006b; Nicholls, 2009), this produces an increase in cytosolic calcium that can activate calcineurin which has been shown to de-phosphorylate Drp1 to promote its recruitment to mitochondria and fission (Cereghetti et al, 2008). We analyzed whether mitochondrial fission preceded or was posterior to mitochondrial depolarization. We found that 1 min before fission mitochondria have lost 60% of the mitochondrial membrane potential, which continues to dissipate progressively after fission (Supplementary Fig S2B and C). We next analyzed the possible involvement of calcineurin in the NMDA-mediated mitochondrial fission. Inhibition of calcineurin with cyclosporine A (CsA) did not prevent mitochondrial fission (Supplementary Fig S2D) as neither did the inhibition of CaMK, a calcium-dependent kinase that activates Drp1 (Han et al, 2008), with KN-62. Drp1 is also activated by nitrosylation (Cho et al, 2009). As it had been reported previously (Barsoum et al, 2006; Cho et al, 2009), here, we also showed that inhibition of the nitric oxide synthase with 7-nitroindazole (7-Ni) blocked the mitochondrial fragmentation to the same extent as the mutant Drp1-K38A or mdivi-1 (Fig 2E). These experiments indicate that NO-mediated Drp1 activation plays a key role in mitochondrial fragmentation during excitotoxicity, although other mechanisms may also be involved.
Mfn2 intervenes in an irreversible late phase of mitochondrial fragmentation
Previous studies have shown that glutamate and nitric oxide mediated mitochondrial fragmentation are amenable to restoration once the stimulus has ceased (Rintoul et al, 2003; Barsoum et al, 2006). Thereby, we decided to test the reversibility of mitochondrial morphology in our system. We applied NMDA for 1 h and then washed out the NMDA and returned to fresh media. We observed that the mitochondrial tubular morphology was recovered 90 min after replacing the media but that after 180 min the mitochondria fragmented once again (Fig 3A). The events that triggered the decline in Mfn2 levels began during the first hour of NMDA exposure since subsequent washout of the agonist did not block the reduction in Mfn2 (Fig 3B). Because the delayed phase of mitochondrial fragmentation correlates with the reduction in Mfn2 protein levels (Figs 1A, 2B and 3A and B), we reasoned that the reduction in Mfn2 levels in excitotoxicity intervenes in late mitochondrial fission. We knocked down Mfn2 using shRNA, which targets its sequence (shMfn2). Importantly, shMfn2 reduced Mfn2 protein levels by around 50%, resembling the reduction observed in excitotoxicity (Fig 3C). As expected, knockdown of Mfn2 was sufficient to cause mitochondrial fragmentation (Fig 3D). Another two shRNAs targeting Mfn2 at different sequences also showed mitochondrial fragmentation (Supplementary Fig S3A and B). Conversely, exogenous expression of Mfn2 significantly blocked the late phase of mitochondrial fragmentation after NMDA washout (Fig 3E and F).
Figure 3. Mfn2 intervenes in an irreversible delayed phase of mitochondrial fragmentation.
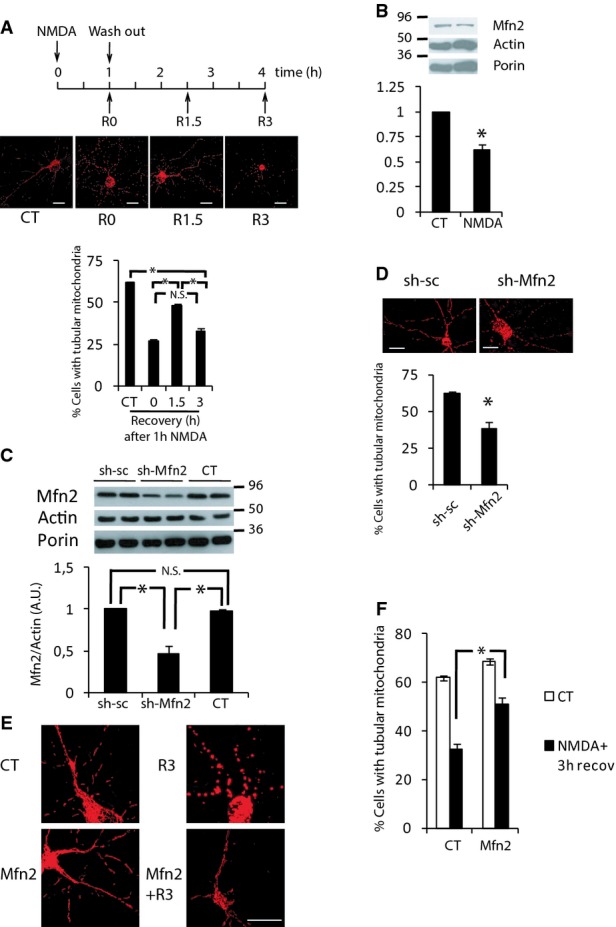
A Representative images and analysis of secondary mitochondrial fragmentation after NMDA washout. Neurons were transfected with mtRFP. After 48 h, neurons were stimulated with NMDA (30 μM) for 1 h and washed out for the indicated times and mitochondrial morphology was analyzed (n = 4–5). *P < 0.05, one-way ANOVA followed by Bonferroni’s post hoc test. Scale bar, 20 μm.
B Mfn2 is reduced after NMDA washout. Western blot and densitometric analysis of neurons treated with NMDA (30 μM) for 1 h and washed out for three additional hours (n = 4). *P < 0.05, two-tailed t-test.
C Representative Western blot and densitometric analysis of neurons transduced with AAV producing shRNA against Mfn2 or scrambled (n = 4). *P < 0.05, one-way ANOVA followed by Bonferroni’s post hoc test.
D Mfn2 knockdown causes mitochondrial fragmentation. Representative images and analysis of mitochondrial morphology of neurons transfected with plasmids shMfn2 or sh-sc and mtRFP (n = 4). *P < 0.05, two-tailed t-test. Scale bar, 20 μm.
E, F Mfn2 blocks the delayed phase of NMDA-induced mitochondrial fragmentation. Representative images of neurons transfected with plasmids encoding mtRFP and Mfn2 or globin (CT) were treated with NMDA (30 μM) for 1 h and washed out for three additional hours (E) and mitochondrial morphology was analyzed (F) (n = 3). *P < 0.05, one-way ANOVA followed by Bonferroni’s post hoc test. Scale bar, 20 μm.
Data information: Results are presented as mean ± SEM.
Source data are available online for this figure.
All of this indicates that excitotoxicity promotes mitochondrial fragmentation by at least two mechanisms, a fast mechanism relying on Drp1 activation that lasts while the insult is present, and a late mechanism dependent on the reduction in Mfn2 expression that takes place hours after the insult is generated and persists independently of the removal of the insult.
Reduced Mfn2 expression causes mitochondrial dysfunction and altered Ca2+ homeostasis
In addition to its mitochondrial fusion activity, Mfn2 has unique properties, which are not shared with Mfn1, such as activating mitochondrial oxidative metabolism or tethering of mitochondria with the ER regulating Ca2+ homeostasis (Bach et al, 2003; Pich et al, 2005; Baloh et al, 2007; de Brito & Scorrano, 2008a). Since mitochondrial dysfunction and altered Ca2+ homeostasis are hallmarks of excitotoxicity, we wondered how decreased levels of Mfn2 could affect mitochondrial function. Respirometric assays showed decreased respiratory control ratio (RCR) and spare respiratory capacity (SRC) in Mfn2 KD neurons (Fig 4A and B). Determination of mitochondrial membrane potential showed that mitochondria in Mfn2 KD neurons had 15% lower mitochondrial membrane potential than control transfected cells (Fig 4C), in concordance with what has been observed in other cell types (Bach et al, 2003; Soriano et al, 2006a). In agreement with lower SRC, when neurons were challenged with subtoxic doses of NMDA (15 μM) mitochondrial membrane potential loss was greatly enhanced in Mfn2 KD neurons (Fig 4C). Concomitantly, when the culture medium was changed to a medium with pyruvate but without glucose just before the NMDA application (to rely solely on mitochondrial metabolism), ATP levels decreased significantly in Mfn2 KD neurons (Fig 4D). All together, these results indicate an impaired mitochondrial function in the Mfn2 KD neurons.
Figure 4. Mfn2 downregulation causes mitochondrial dysfunction and altered Ca2+ homeostasis.
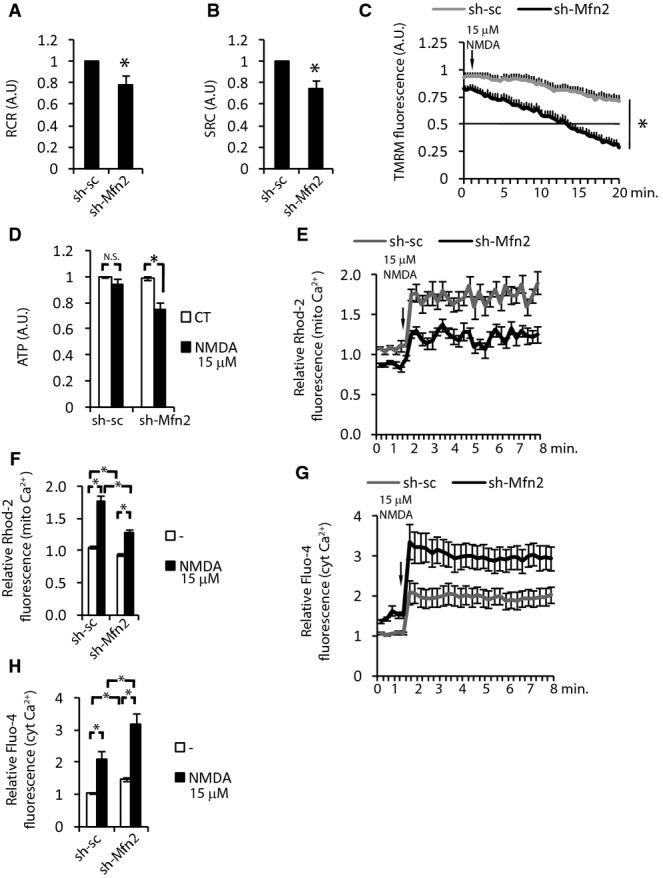
A, B Respiratory control ratio (A) and spare respiratory capacity (B) of neurons transduced with AAV expressing shRNA targeting Mfn2 (sh-Mfn2) or scrambled (sh-sc) (n = 6). *P < 0.05, two-tailed t-test.
C Loss of mitochondrial membrane potential in neurons transfected with sh-Mfn2 or sh-sc and treated with NMDA (15 μM; arrow) was determined by measuring TMRM fluorescence. The values were normalized to surrounding untransfected neurons (n = 19–21 neurons analyzed in five independent experiments). *P < 0.05, two-tailed t-test.
D ATP levels were analyzed following 30-min NMDA (15 μM) treatment to neurons transduced with AAV producing sh-Mfn2 or control sh-sc (in glucose-free medium) (n = 4). *P < 0.05, two-tailed t-test.
E Determination of mitochondrial Ca2+ with Rhodamine-2 in cortical neurons transfected with plasmids producing sh-Mfn2 or sh-sc and treated with NMDA (15 μM), the values were normalized to the signal of surrounding untransfected neurons (n = 20 neurons from five independent experiments).
F Histograms showing the average mitochondrial Ca2+ levels before and after NMDA application (15 μM). *P < 0.05, one-way ANOVA followed by Bonferroni’s post hoc test.
G Determination of intracellular Ca2+ with Fluo-4 in cortical neurons transfected with plasmids producing sh-Mfn2 or sh-sc and treated with NMDA (15 μM), the values were normalized to the signal of surrounding untransfected neurons (n = 16 neurons from 4 independent experiments).
H Histograms show the average intracellular Ca2+ levels before and after NMDA application (15 μM). *P < 0.05, one-way ANOVA followed by Bonferroni’s post hoc test.
Data information: Results are presented as mean ± SEM.
Because mitochondria play a key role in buffering the increase in cytosolic Ca2+ produced during excitotoxicity, which depends on proper mitochondrial membrane potential (Nicholls, 2009), we reasoned that Ca2+ homeostasis may be impaired in Mfn2 KD neurons. We measured mitochondrial Ca2+ with Rhodamine-2 (Rhod-2). Under basal conditions, Mfn2 KD neurons have a slightly lower (9.9 ± 3.52%) mitochondrial Ca2+ concentration than control neurons (Fig 4E and F). The application of NMDA produced a lower increase of the mitochondrial Ca2+ in the Mfn2 KD neurons than in the control neurons (Fig 4E and F). Because Rhod-2 localization to mitochondria is voltage sensitive and lower mitochondrial membrane potential could lead to reduced Rhod-2 fluorescence, we also analyzed cytoplasmic Ca2+ and we found that, oppositely to mitochondrial Ca2+, cytoplasmic Ca2+ was moderately higher in Mfn2 KD neurons and NMDA application produced bigger increase in Mfn2 KD neurons (Fig 4G and H, and Supplementary Fig S4A). Increase in cytoplasmic Ca2+ was due to reduced mitochondrial Ca2+ uptake since depolarization of mitochondria with CCCP, hence abolishment of mitochondrial Ca2+ uptake, produced the same increase in NMDA-mediated rise in cytoplasmic Ca2+ in control or Mfn2 KD neurons (Supplementary Fig S4B).
Together, these data support the notion that a reduction in Mfn2 causes mitochondrial malfunction, which causes disturbances in Ca2+ homeostasis.
Mfn2 reduction intervenes in delayed excitotoxic death
Given that reduced levels of Mfn2 had a great impact on mitochondrial membrane potential and Ca2+ handling when neurons were treated with low doses of NMDA, next we investigated whether the reduction in Mfn2 could have consequences for cell viability. We showed that this was indeed the case, as knockdown of Mfn2 sensitized neurons to subtoxic doses of NMDA (Fig 5A). Another two shRNAs targeting Mfn2 at different sequences also showed sensitization to subtoxic doses of NMDA, ruling out the possibility of an off-target effect (Supplementary Fig S5A). Next, we overexpressed Mfn2 and observed an almost twofold protection to the NMDA-induced neuronal death (Fig 5B).The protection was not due to the restoration of mitochondrial morphology since overexpression of Mfn1 did not show significant protection to NMDA (Supplementary Fig S5B). Shortly after exposure to glutamate, a subpopulation of neurons died by necrosis. Surviving neurons can recover or transiently and undergo delayed cell death, depending on mitochondrial function (Ankarcrona et al, 1995; Luetjens et al, 2000). We studied how exogenous expression of Mfn2 could affect the different phases of excitotoxic cell death. We determined cell death after an hour treatment with NMDA (30 μM) followed by the washout of NMDA and allowed the neurons to recover for different length of time. After 1.5 h recovery (i.e. 2.5 h from the onset of the insult), nearly 40% of the neurons died with no protection by Mfn2 expression. From then on, and correlating with endogenous Mfn2 reduction, cell death in the neurons that were transfected with the control vector increased progressively, but Mfn2 expression completely blocked the delayed cell death (Fig 5C).
Figure 5. Mfn2 reduction intervenes in delayed excitotoxic death.
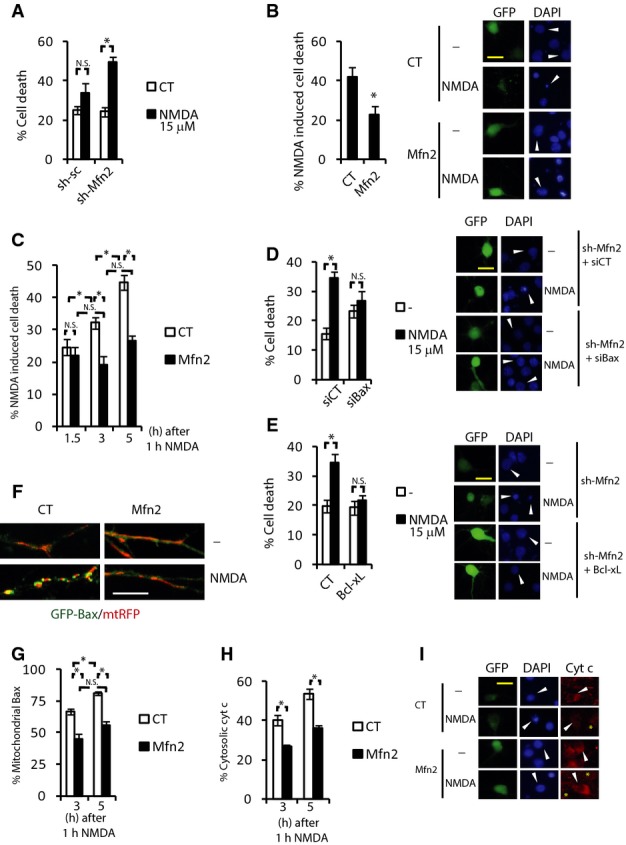
A Neurons transfected with plasmids expressing sh-Mfn2 or sh-sc were treated with subtoxic doses of NMDA (15 μM) for 6 h. Death was analyzed by fixing cells, DAPI staining and counting pyknotic nuclei of the transfected neurons (n = 5). *P < 0.05, two-tailed t-test.
B Analysis and representative images of cortical neurons transfected with plasmids expressing Mfn2 or control (globin). After 48 h, neurons were exposed to NMDA (30 μM) for 6 h. NMDA-dependent neuronal death was analyzed (n = 8). *P < 0.05, two-tailed t-test. Arrowhead points to the transfected neuron. Scale bar, 20 μm.
C Cortical neurons expressing Mfn2 or control plasmid (globin) were treated with 30 μM NMDA for 1 h followed by extensive washout and were allowed to recover for the indicated times. Cell death was analyzed by fixing cells, DAPI staining, and counting pyknotic nuclei of the transfected neurons (n = 5). *P < 0.05, one-way ANOVA followed by Bonferroni’s post hoc test.
D, E Representative images and cell death analysis of neurons transfected with a plasmid producing sh-Mfn2 plus (D) siRNA CT or targeting Bax, or (E) plasmid control or expressing Bcl-xL and treated with NMDA (15 μM) for 6 h (n = 8). *P < 0.05, two-tailed t-test. Arrowhead points to the transfected neuron. Scale bar, 20 μm.
F Representative images of neurons expressing GFP-Bax and mtRFP plus control or Mfn2 expression vector and treated with NMDA (30 μM) for 1 h followed by extensive washout and were allowed to recover for the 3 h. Scale bar, 10 μm.
G Analysis of mitochondrial Bax localization of neurons transfected as in (F) and allowed to recover after NMDA treatment (30 μM) for the indicated times (n = 3–5). *P < 0.05, one-way ANOVA followed by Bonferroni’s post hoc test.
H, I The cellular localization of cytochrome c was analyzed in neurons transfected with GFP plus control or Mfn2-expressing plasmid and treated as in (G) (n = 5). *P < 0.05, one-way ANOVA followed by Bonferroni’s post hoc test. Representative images are shown in (I). Arrowhead points to the transfected neuron, asterisk highlights cells with diffuse or lost cytochrome c. Scale bar, 20 μm.
Data information: Results are presented as mean ± SEM.
Given that several mechanism are responsible for the NMDA-mediated cell death, to better characterize the mechanism by which reduction of Mfn2 causes delayed excitotoxicity we used NMDA at 15 μM, which only causes death when Mfn2 is knocked down (Fig 5A). Analysis of the NAD+/NADH ratio as a readout of the metabolic status of the neuron did not indicate that an energetic collapse was responsible for the neuronal death when Mfn2 expression was reduced (Supplementary Fig S5C). Evidences indicate that opening of the mitochondrial permeability transition pore (mPTP) and necrotic death occurs during the early phase of excitotoxicity as opposed to apoptosis that occurs in delayed excitotoxicity (Dirnagl et al, 1999). Consequently, inhibition of the mPTP with CsA failed to protect Mfn2 KD neurons to low doses of NMDA (15 μM; Supplementary Fig S5D). During delayed excitotoxic cell death Bax translocates to mitochondria to produce the mitochondrial outer membrane permeabilization (MOMP) (Xiang et al, 1998; D’Orsi et al, 2012; Shelat et al, 2013). Bax is known to interact with Mfn2 (Karbowski et al, 2006; Brooks et al, 2007), and Mfn2 interferes with Bax activation (Sugioka et al, 2004; Neuspiel et al, 2005). Bax siRNA blocked the NMDA-dependent neuronal death in Mfn2 KD neurons (Fig 5D). In addition, overexpression of the anti-apoptotic Bcl-xL protein also blocked the neuronal death (Fig 5E). In agreement with the reported activation of calpain downstream of Bax activation (D’Orsi et al, 2012), by measuring cleavage of spectrin (calpain substrate), we observed an enhanced calpain activation in Mfn2 KD neurons (Supplementary Fig S5E). According to the role of Blc-2 family proteins regulating mitochondrial dynamics by using siBax and Bcl-xL, overexpression partially prevented the delayed mitochondrial fragmentation after NMDA application (Supplementary Fig S5F and G).
Next, we studied whether exogenous expression of Mfn2 affected Bax translocation to mitochondria in excitotoxicity. We observed that in GFP-Bax expressing neurons Bax was mainly cytoplasmic and that NMDA (30 μM) application resulted in Bax translocation to mitochondria, forming the typical foci on the tips of the mitochondria (Karbowski et al, 2002; Yuan et al, 2006; Fig 5F). Bax translocation to mitochondria was progressive after NMDA treatment and Mfn2 expressing neurons showed less mitochondrial Bax than globin (control) expressing neurons (Fig 5F and G). As expected, NMDA-mediated delayed cytochrome c release was blocked by Mfn2 expression in a similar pattern to mitochondrial Bax localization (Fig 5H and I).
All these data indicate that the late reduction of Mfn2 levels impairs mitochondrial function and enhances delayed excitotoxic death by promoting Bax recruitment to mitochondria.
Mfn2 is regulated at the transcriptional level in excitotoxicity
Next, we investigated the mechanism by which Mfn2 expression is reduced in excitotoxicity. It is well known that Mfn2 is degraded by the proteasome in a Parkin-dependent manner (Tanaka et al, 2010; Chan et al, 2011). We wondered whether the decrease in Mfn2 levels in excitotoxicity was due to its proteasomal degradation. Surprisingly, we found that pretreatment of neurons with the proteasome inhibitor MG-132 did not abolish the NMDA-dependent reduction in Mfn2 (Fig 6A and B) even when the concentration of MG-132 used (10 μM) promoted the accumulation of ubiquitinated proteins and effectively blocked the CCCP-mediated degradation of Mfn2 (Supplementary Fig S6A and B). Therefore, we can conclude that excitotoxic-dependent downregulation of Mfn2 is not dependent on proteasomal degradation.
Figure 6. Mfn2 is regulated at transcriptional level in excitotoxicity.
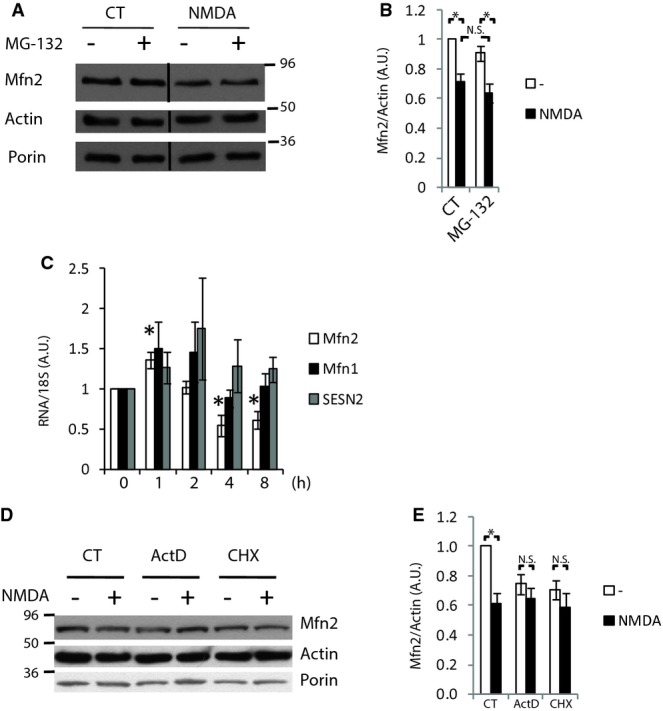
A, B Cortical neurons with or without pre-incubation with proteasome inhibitor MG-132 (10 μM) were treated for 4 h with NMDA (30 μM), and Mfn2 levels were analyzed by Western blot. Densitometric analysis is shown in (B) (n = 7). Results are presented as mean ± SEM. *P < 0.05, one-way ANOVA followed by Bonferroni’s post hoc test.
C–E Cortical neurons were treated with NMDA (30 μM) for the indicated times, and mRNA expression was determined by real-time qPCR (n = 4). *P < 0.05, two-tailed t-test. Cortical neurons were treated with transcriptional inhibitor actinomycin D, translational inhibitor cycloheximide, and NMDA (30 μM) for 4 h as indicated, and Mfn2 protein expression was analyzed by Western blot. Representative Western blot and densitometric analysis are show in (D) and (E), respectively (n = 4). Results are presented as mean ± SEM. *P < 0.05, two-tailed t-test.
Source data are available online for this figure.
Excitotoxicity causes changes in transcriptional programs (Zhang et al, 2007; Cook et al, 2012), which could be consistent with the slow effect of NMDA on Mfn2 expression. We investigated whether Mfn2 reduction was due to changes in its gene expression. Mfn2 gene expression showed an initial increase during the first hour after NMDA application, probably a defensive response to malfunctioning mitochondria. Mfn2 gene expression started to decrease during the second hour, reaching a plateau after 4 h that remained steady for up to 8 h of treatment and correlated with the time course of protein expression (Fig 6C). Neurons subjected to OGD also showed downregulation of Mfn2 mRNA expression (Supplementary Fig S6C). The repression of Mfn2 expression is specific since mitochondrial Mfn1 and cytoplasmic SESN2 expression were not affected by NMDA treatment (Fig 6C). These data indicate that Mfn2 gene expression is downregulated during excitotoxicity, but do not rule out the possibility that other proteases could participate in the excitotoxicity-mediated reduction in Mfn2. To verify that a transcriptional change is the main mechanism by which Mfn2 expression is reduced in excitotoxicity, we used the transcriptional inhibitor actinomycin D or the translational inhibitor cycloheximide. As expected, the use of these inhibitors reduced Mfn2 protein levels but additional application of NMDA did not produce a further reduction in Mfn2, as would be expected if a proteolytic process was responsible for NMDA-mediated Mfn2 downregulation (Fig 6D and E). Thus, all these sets of experiments clearly indicate that the main mechanism by which NMDA mediates Mfn2 downregulation is at the transcriptional level rather than being a proteolytic process.
Excitotoxicity-mediated Mfn2 downregulation depends on MEF2
ROS is an important regulator of cell signaling (Holmstrom & Finkel, 2014). We first examined whether mitochondrial ROS produced in excitotoxicity could mediate Mfn2 downregulation. The use of general scavenger trolox, or apocynin, an inhibitor of NADPH oxidase, which is the major source of ROS in excitotoxicity (Brennan et al, 2009), failed to block NMDA-mediated Mfn2 downregulation (Supplementary Fig S7A).
MEF2 (A–D) are transcription factors that play an important role in neuronal development and viability (Mao et al, 1999; Flavell et al, 2008). During excitotoxicity, MEF2s are cleaved by caspases generating DNA binding domains without the transactivation domain, which acts as a dominant negative interfering form (Li et al, 2001; Okamoto et al, 2002). Time-course analysis of protein extracts of cortical neurons exposed to NMDA, subjected to OGD or brains from rats that have been subjected to ischemia, showed a calcium-dependent reduction of MEF2A in a pattern that correlated well with Mfn2 expression (Fig 7A and B, and Supplementary Fig S7B and C). This raises the possibility that NMDA-dependent cleavage of MEF2s may be responsible for Mfn2 downregulation in excitotoxicity. Neurons expressing the DNA binding domain of MEF2 without the transactivation domain (MEF2-DN) showed reduced expression of Mfn2 protein and mRNA (Fig 7C). Consistent with the reduced expression of Mfn2, MEF2-DN transduced neurons contained fragmented mitochondria whose morphology was restored by expressing exogenous Mfn2 (Fig 7D and E). Mitochondria of MEF2-DN expressing neurons showed reduced mitochondrial membrane potential that could be raised by overexpression of Mfn2 (Fig 7F). MEF2-DN expression caused neuronal death when doses of NMDA at the threshold of toxicity (15 μM) were applied, but co-expression of Mfn2 completely blocked neuronal death (Fig 7G and H). All these results demonstrate that Mfn2 expression is regulated by MEF2 but do not answer the question of whether MEF2 degradation is the main factor mediating Mfn2 downregulation in excitotoxicity. Thus, we analyzed the levels of Mfn2 mRNA in neurons transduced with AAV coding MEF2-DN and observed that Mfn2 downregulation with respect to its control (GFP transduced) neurons was not additively enhanced when NMDA was applied, suggesting that they act in a common pathway (Fig 7C; note that the AAV transduction efficiency was around 75–90%, thus the results underestimate the actual MEF2-DN-dependent repression). Together, these data demonstrate a crucial role of MEF2 in the excitotoxicity-dependent downregulation of Mfn2.
Figure 7. Excitotoxicity-mediated Mfn2 downregulation depends on MEF2.
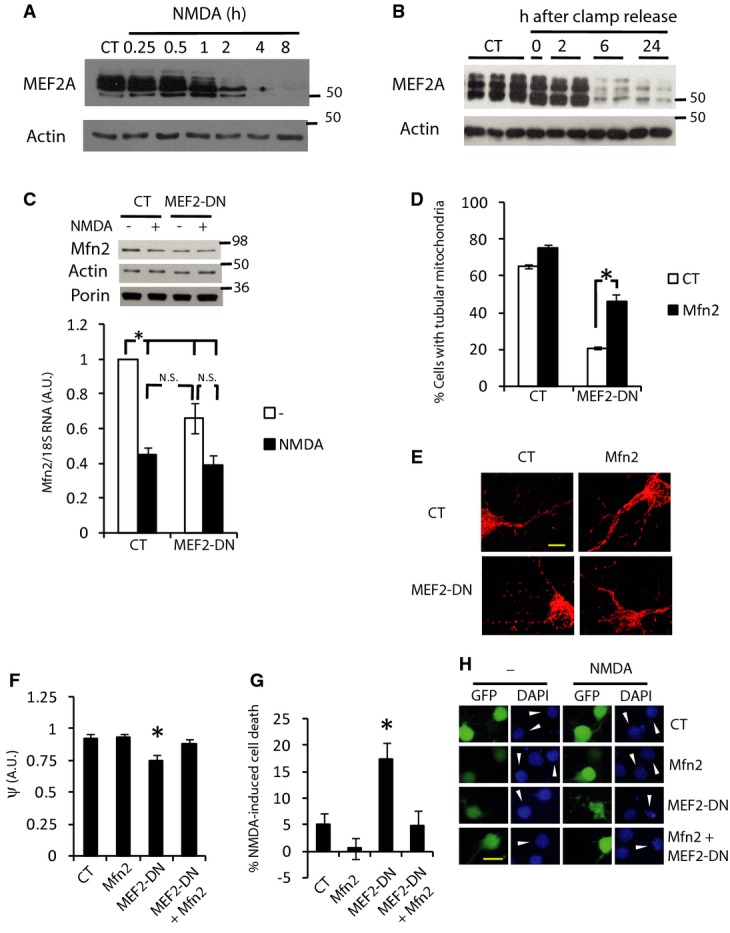
A, B Time course of excitotoxicity-dependent MEF2A degradation in (A) cortical neurons in vitro treated with NMDA (30 μM) for 4 h and (B) brain extracts of rats subjected to MCAO plus 90 min of ipsilateral common carotid artery clamp followed by clamp release for the indicated times.
C Cortical neurons transduced with AAV expressing MEF2-DN or GFP (control) were treated with NMDA (30 μM) for 4 h and Mfn2 mRNA and protein expression was determined (n = 5). *P < 0.05, one-way ANOVA followed by Bonferroni’s post hoc test.
D, E Mitochondrial morphology in cortical neurons transfected with the indicated expression vectors (D) (n = 4–5). *P < 0.05, one-way ANOVA followed by Bonferroni’s post hoc test. Representative images are shown in (E).
F Mitochondrial membrane potential of neurons expressing the indicated vectors relative to surrounding untransfected neurons (n = 9–10 cells from three independent experiments). *P < 0.05, two-tailed t-test.
G, H Cortical neurons were transfected with the indicated expression vectors. After 48 h, cells were treated with subtoxic doses of NMDA (15 μM) for 6 h and cell death was analyzed (n = 8). *P < 0.05, one-way ANOVA followed by Bonferroni’s post hoc test. Representative images are shown in (H). Arrowhead points to the transfected neuron. Scale bar, 20 μm.
Data information: Results are presented as mean ± SEM.
Source data are available online for this figure.
MEF2A directly regulates basal Mfn2 expression in neurons
Next, we investigated whether MEF2 could directly regulate Mfn2 expression. In promoter reporter assays, MEF2-DN repressed the activity of human Mfn2 promoter in neurons but did not repress the activity of SESN2 promoter, consistent with the unchanged expression of SESN2 in excitotoxicity (Fig 8A). The effect of MEF2-DN on the Mfn2 promoter was neuron specific since it did not affect the promoter activity in 10T1/2 fibroblasts, a cell line with far less expression of MEF2A than neurons (Fig 8A and Supplementary Fig S8A). These results are consistent with a prevalent role of MEF2 in the regulation of basal Mfn2 expression in neurons.
Figure 8. MEF2A directly regulates basal Mfn2 expression in neurons.
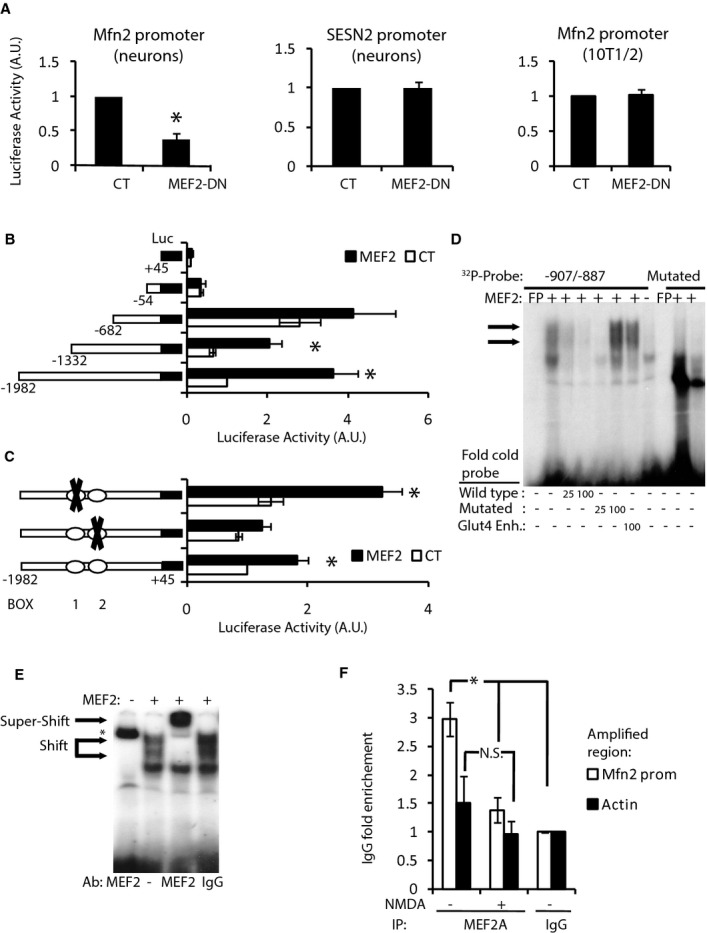
A Luciferase-based reporter of Mfn2 promoter in cortical neurons (left) or 10T1/2 cell line (right) and SESN2 promoter in cortical neurons (center) in control or MEF2-DN-overexpressing cells (n = 3–9). *P < 0.05 compared to control, two-tailed t-test.
B Deletion analysis of a luciferase-based reporter of the Mfn2 promoter in 10T1/2 cells co-expressed with control or MEF2 plasmids (n = 3). *P < 0.05, two-tailed t-test.
C Effect of putative MEF2 binding site mutations on MEF2-dependent induction of the Mfn2 promoter in 10T1/2 cells (n = 3). *P < 0.05 compared to control, two-tailed t-test.
D EMSA performed with nuclear extracts of HeLa cells overexpressing MEF2 and radiolabeled probe encoding BOX 2 from Mfn2 promoter. Retardation complexes are indicated with arrows. Excess of cold oligonucleotide, mutated oligonucleotide or oligonucleotide of the Glut4 gene that contains a MEF2 binding site was used to compete. Radiolabeled probe containing mutated BOX 2 did not produce retardation complexes.
E Supershift was performed using anti-MEF2 polyclonal antibody. Pre-immune serum was used as a negative control. * indicates non-specific binding of the probe to anti-MEF2 antibody.
F ChIP on cortical neurons untreated or treated with NMDA (30 μM) for 4 h using the indicated antibodies (n = 4). *P < 0.05, one-way ANOVA followed by Bonferroni’s post hoc test.
Data information: Results are presented as mean ± SEM.
Next, we determined the cis-elements involved in the effects of MEF2A on the transcriptional activity of the Mfn2 promoter. In agreement with the low expression of MEF2A in 10T1/2 cells, over-expression of MEF2A strongly activated the Mfn2 promoter (Fig 8B). Deletion analysis of the Mfn2 promoter identified the MEF2A activated region as being between −1,332 and −668 relative to the transcription start site (Fig 8B). The MEF2 transcription factors bind DNA in A/T rich sequences (Black & Olson, 1998). The sequence −1,332/−862 contains two putative MEF2 binding sites (Supplementary Fig S8B). In order to show the functional role of these putative MEF2 binding sites, we disrupted the A/T-rich sequence, introducing C and G by directed mutagenesis (Supplementary Fig S8B). Mutation of BOX 1 did not modify the MEF2A-induced promoter activation, but mutation of BOX 2 cancelled the effect of MEF2A (Fig 8C). These results indicated that BOX 2 is the cis-element required for MEF2A mediated activation of the Mfn2 promoter.
Since MEF2A was able to trans-activated the Mfn2 promoter, EMSA experiments were performed in order to ascertain whether MEF2A was bound to BOX 2. A DNA fragment encompassing BOX 2 was radioactively labeled and incubated with nuclear extracts of HeLa cells overexpressing MEF2A. MEF2A bound BOX 2-containing probe, inducing the typical double band mobility shift (Santalucia et al, 2001), but did not bind the mutated probe (Fig 8D). The retarded bands were competed with a 25- or 100-fold excess of unlabeled oligonucleotide probe or 100-fold excess of a probe of the Glut4 promoter that has previously been shown to bind MEF2 (Santalucia et al, 2001), whereas the cold mutated probe was unable to compete with the retarded bands. In addition, an antibody against MEF2 supershifted the complex (Fig 8E).
Bioinformatic analysis of 2.5 kb of the rat Mfn2 promoter identified that there were four putative MEF2 binding sites in the region spanning −2,313/−1,983 (Supplementary Fig S8C). We designed primers to amplify within this region and performed ChIP with anti-MEF2A antibody in rat cortical neurons unstimulated or stimulated with NMDA for 4 h. We found that under basal conditions, MEF2A was bound to this region, but after NMDA stimulation, there was no enrichment of this region with the chromatin immunoprecipitate (Fig 8F). These observations support the hypothesis that MEF2A regulates basal transcription of Mfn2 in neurons and as a consequence of MEF2A degradation Mfn2 transcription is downregulated in excitotoxicity (Fig 9).
Figure 9. Proposed model for the changes in mitochondrial morphology and vulnerability in excitotoxic conditions.
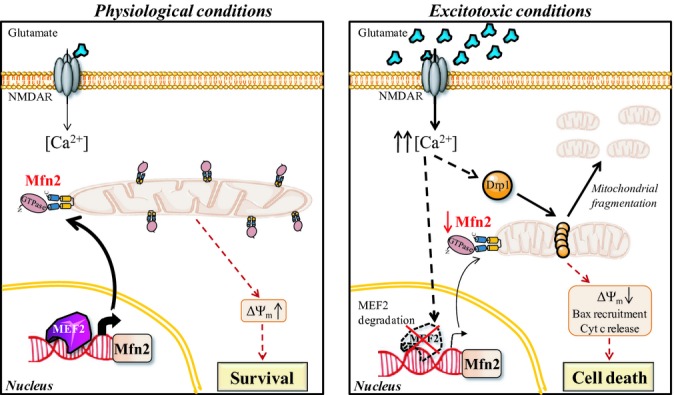
In cortical neurons under physiological conditions, MEF2 binds Mfn2 promoter and regulates its basal gene expression resulting in mainly tubular mitochondria. In excitotoxic conditions, Drp1 translocates to mitochondria and mediates rapid, reversible mitochondrial fission. An increase in cytosolic Ca2+ produces MEF2 degradation, and Mfn2 gene expression is consequently downregulated, producing a delayed long-term effect on mitochondrial morphology. Reduced expression of Mfn2 impairs mitochondrial function, which causes dysregulation of Ca2+ homeostasis, facilitates Bax translocation to mitochondria, release of cytochrome c, and enhances delayed excitotoxic death.
Discussion
Mitochondria are dynamic organelles that continuously fuse and divide. Changes in mitochondrial dynamics have profound effects on mitochondrial function and therefore mitochondrial viability. Here, we have studied mitochondrial dynamics in excitotoxicity. Our findings show that most of the mitochondrial fragmentation occurs within the first hour following the excitotoxic insult. This early phase depends partially on Drp1 mitochondrial translocation and lasts as long as the stimulus is present. Four hours after initiation of the insult, and regardless of its removal, delayed mitochondrial fragmentation that correlates with the reduction in Mfn2 protein levels due to its transcriptional downregulation occurs. Loss of Mfn2 impairs mitochondrial function and participates in delayed excitotoxic damage.
Mechanisms of mitochondrial fragmentation in excitotoxicity
Drp1 is subjected to several posttranslational modifications that regulate its fission activity. Neuronal depolarization activates CaMKI, which phosphorylates Drp1 at Ser600, promoting mitochondrial fragmentation that is reversible once neurons repolarize (Han et al, 2008). The reversibility of Drp1-mediated mitochondrial fragmentation has also been observed in pathological conditions. Mitochondrial fragmentation induced by NO donors is a rapid and reversible process (Barsoum et al, 2006). Ca2+ influx through the NMDA receptor produces NOS activation, which has been implicated in nitrosative stress and cell death (Sattler et al, 1999). Nitrosative stress causes S-nitrosylation of Drp1 at Cys644, which enhances mitochondrial fission (Cho et al, 2009). In agreement with previous reports (Barsoum et al, 2006), we show that NOS inhibition partially blocks NMDA-mediated mitochondrial fragmentation. The degree of inhibition of the mitochondrial fragmentation achieved by NOS inhibitors is similar to that achieved using genetic and pharmacological inhibitors of Drp1. That suggests the involvement of a mechanism other than Drp1 in the early phase of mitochondrial fragmentation. Opa1 is cleaved in a mitochondrial membrane potential-dependent manner, promoting mitochondrial fission (Ishihara et al, 2006; Griparic et al, 2007; Song et al, 2007), but we were unable to detect changes in the proportion of short and long forms of Opa1 in excitotoxicity, either in vitro or in vivo. Another possibility is that excitotoxicity could promote the modification of lipids, which has been shown to affect mitochondrial dynamics (Choi et al, 2006). The possibility that as yet unknown posttranscriptional modifications inhibit the fusion machinery in excitotoxic conditions cannot be ruled out either.
Our data also indicate that Mfn2 intervenes in the delayed phase of mitochondrial fragmentation. Cultured rat primary cortical neurons exposed to excitotoxic doses of NMDA or subjected to OGD, and in vivo middle cerebral artery occlusion in rats show reduced expression of Mfn2 but no changes in Mfn1 or Opa1. During the preparation of the manuscript, two independent studies confirmed the downregulation of Mfn2 in excitotoxicity. Primary rat cortical neurons exposed to 3 h of OGD also produced a reduction in Mfn2 protein with no changes in Opa1 and an increase in Mfn1 expression (Wappler et al, 2013). In an in vivo study, a reduction in Mfn2, and also of Opa1 expression, was observed in mice subjected to MCAO (Kumari et al, 2012).
Dependence of neurons on Mfn2
Mfn1 can compensate for some but not all of the functions of Mfn2. Analysis of Mfn2 and Mfn1 knockout mice indicates that they have both redundant and distinct functions (Chen et al, 2003, 2007). Unlike Mfn1, a lack of or defects in Mfn2 have a great impact on neuronal viability. Mfn2 mutations have been found to cause the dominantly inherited neurological disease Charcot-Marie-Tooth type 2A (CMT2A) (Zuchner et al, 2004). Defects in mitochondrial mobility and fusion have been proposed as the mechanism by which Mfn2 mutations cause CMT2A but this has not been fully established (Baloh et al, 2007). Mfn2 is required for postnatal development of the cerebellum (Chen et al, 2007). Conditional dopaminergic Mfn2 knockout neurons show that Mfn2, but not Mfn1, is essential for striatal projections and proper nigrostriatal circuit function (Lee et al, 2012; Pham et al, 2012).
As a consequence of excitotoxicity-mediated Ca2+ overload neurons die initially by necrosis, surviving neurons can undergo delayed death in a manner dependent on the mitochondrial function (Ankarcrona et al, 1995; Luetjens et al, 2000; D’Orsi et al, 2012). We show here that, unlike Mfn1 expression, exogenous expression of Mfn2 protects neurons against NMDA-induced cell death. Mfn2 expression interferes with Bax recruitment to mitochondria and subsequent release of cytochrome c, as has been observed in other cell types (Sugioka et al, 2004; Neuspiel et al, 2005). The precise mechanism by which Mfn2 interferes with Bax activation is unknown. One possibility is that loss of Mfn2 facilitates OMM remodeling by Drp1 that has been shown to stimulate Bax oligomerization (Montessuit et al, 2010). Mfn2 interacts with different members of the Bcl-2 family, both pro- and anti-apoptotic (Suen et al, 2008). It cannot be ruled out the possibility that by changing Mfn2 levels, it affects the equilibrium of interactions between pro- and anti-apoptotic Bcl-2 proteins to favor Bax assembly and MOMP formation.
The relation between autophagy and cell death is controversial (Baehrecke, 2005; Galluzzi et al, 2012). Although in most known cases, autophagy constitutes a cytoprotective response activated by dying cells in an attempt to cope with stress (Galluzzi et al, 2012), cell death with and by autophagy has been reported in focal cerebral ischemia (Puyal et al, 2009, 2013). Mfn2 KO fibroblasts show reduced autophagy in response to ER stress (Muñoz et al, 2013). Interestingly, it has been reported that phosphorylated Mfn2 by PINK1 acts as a receptor for Parkin (Chen & Dorn, 2013), which is recruited to damaged mitochondria to promote mitophagy (Narendra et al, 2008; Chen & Dorn, 2013). How the reduction of Mfn2 in excitotoxicity affects mitophagy and autophagy-mediated cell death awaits further investigations.
Mfn2 is transcriptionally downregulated in excitotoxicity
During recent years, several groups have established the role of the ubiquitin/proteasomal system in stress-mediated Mfn2 degradation. Mitochondrial depolarization promotes Mfn2 ubiquitinylation and proteasomal degradation in a Parkin-dependent manner (Tanaka et al, 2010; Chan et al, 2011). In stress conditions, JNK phosphorylates Mfn2 at Ser 27, which recruits the E3 ubiquiting ligase Huwe 1, leading to its ubiquitination and degradation (Leboucher et al, 2012). Although excitotoxicity causes both mitochondrial depolarization and JNK activation (Borsello et al, 2003; Soriano et al, 2006b, 2008; Nicholls, 2009), the reduction in Mfn2 protein levels was not blocked by inhibiting the proteasome. We found that the main mechanism implicated in Mfn2 reduction acts at the transcriptional level and does not seem to depend on enhanced proteolysis since inhibiting transcription or translation does not further reduce Mfn2 levels. Interestingly, microarray analysis has showed more than 50% reduction in Mfn2 mRNA in the penumbra of rats subjected to MCAO (Ramos-Cejudo et al, 2012). We have shown that transcription factor MEF2 regulates basal Mfn2 transcription in neurons and that excitotoxicity-mediated MEF2 degradation (Li et al, 2001; Okamoto et al, 2002) results in transcriptional downregulation of Mfn2 (Fig 9). MEF2 transcription factors play a pivotal role in brain development, synapse development, and neuronal survival. The genetic program regulated by MEF2 that controls synaptic remodeling has been characterized (Flavell et al, 2008), but although many studies have pointed to the importance of MEF2 in supporting neuronal viability (Mao et al, 1999; Li et al, 2001; Okamoto et al, 2002), the precise target genes are poorly defined. Here, we demonstrate that Mfn2 is a novel MEF2 target gene that can mediate its pro-survival function.
Mitochondrial dynamics in neurodegeneration
Mitochondrial dysfunction and excitotoxicity are common features of adult-onset neurodegenerative disorders such as Alzheimer’s, Parkinson’s, and Huntington’s diseases, some of them occurring primarily in the absence of genetic linkage. Alterations in mitochondrial dynamics have been reported in all these diseases (Itoh et al, 2013). Mfn2 is dynamically regulated; thus, its misregulation could intervene in the pathogenesis of neurodegenerative disorders. Further studies are required to clarify the role of Mfn2 in the progression of chronic late-onset neurodegenerative disorders. In this study, we have shown that neurons with reduced Mfn2 have dysfunctional mitochondria, altered Ca2+ homeostasis that could sensitize neurons to additional insults. In our excitotoxicity model, Mfn2 expression protects against delayed excitotoxic cell death by interfering with Bax recruitment to mitochondria. This suggests that Mfn2 downregulation could determine the fate of neurons in the penumbra area, the most clinically relevant therapeutic target against ischemic stroke (Lo, 2008).
Thereby, Mfn2 is a potential therapeutic target against excitotoxicity in acute episodes and chronic neurodegenerative diseases.
Materials and Methods
Cell culture, transfection, and determination of cell death
Cortical neurons from E21 Sprague–Dawley rats were cultured as described previously (Soriano et al, 2008). Experiments were performed after a culturing period of 10–11 days during which cortical neurons develop a rich network of processes, express functional NMDA-type and AMPA/kainate-type glutamate receptors, and form synaptic contacts. Prior to stimulations and transfections, neurons were transferred from growth medium to TMo medium containing 10% MEM (Invitrogen), 90% salt-glucose-glycine (SGG) medium (SGG: 114 mM NaCl, 0.219% NaHCO3, 5.292 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM HEPES, 1 mM glycine, 30 mM glucose, 0.5 mM sodium pyruvate, 0.1% phenol red; osmolarity 325 mosm/l). Transfections were performed with Lipofectamine 2000 (Invitrogen). For cell death determination, neurons transfected with GFP plus the indicated expression vectors were treated with NMDA for 6 h and fixed. At this time point, damaged neurons show pyknotic nuclei, but GFP signal has not been lost yet. Nuclei were stained with DAPI, and cell death was determined by counting the number of DAPI-stained pyknotic nuclei as a percentage of the total transfected neurons. NMDA-dependent cell death was calculated subtracting basal cell death to the cell death after NMDA treatment.
10T1/2 and HeLa cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum in subconfluent cultures.
Transfections were performed with either Lipofectamine 2000 (Invitrogen) or Fugene (Roche). For Bax knockdown experiments, 25 ng of Rat Bax (24887) siRNA-SMART pool (Thermo Scientific) containing a pool of 4 siRNAs targeting rat Bax were transfected.
Focal ischemia model
All animal procedures were authorized by the veterinary office of the Canton of Vaud and were in accordance with the directives of the Swiss Academy of Medical Science. After anesthesia (2.5% isoflurane), middle cerebral artery (MCA) occlusion was performed in 12-day-old (P12) male Sprague–Dawley rats as previously described (Vaslin et al, 2009). Briefly, first, the main (cortical) branch of the left MCA was electrocoagulated just below its bifurcation into the parietal and frontal branches. Then, the left common carotid artery was transiently occluded with a clamp for 90 min. Rat pups were maintained at 37°C in the induction chamber (2.5% isoflurane) while the left common carotid artery was clamped. The arterial clamp was then removed, and the rat pups were transferred back to their mother until sacrifice.
Rat pups were decapitated, and the brains were removed in PBS containing 1 mmol/l MgCl2 on ice. The cortex were dissected and collected in protein hypotonic lysis buffer [20 mmol/l HEPES, pH 7.4, 10 mmol/l NaCl, 3 mmol/l MgCl2, 2.5 mmol/l EGTA, 0.1 mmol/l dithiothreitol, 50 mmol/l NaF, 1 mmol/l Na3VO4, 1% Triton X-100, and a protease inhibitor cocktail (Roche)]. Tissues were homogenized and sonicated, and protein concentration was determined using the Bradford assay.
Oxygen and glucose deprivation
Neurons were transferred from TMo, washed once in a glucose-free, balanced salt solution (SGG): 114 mM NaCl, 0.219% NaHCO3, 5.292 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM HEPES, 1 mM glycine, 0.5 mM sodium pyruvate, 0.1% phenol red; which had previously been degassed by flushing the solution with 95% N2/5% CO2 for 5 min. Cells were placed in degassed glucose-free SGG and put in a modular incubator chamber, which was flushed with 95% N2/5% CO2 for 4 min at a flow rate of 20 l/min, according to manufacturer’s instructions (Stemcell Technologies, Grenoble, France), in order to fully expel any remaining oxygen within the chamber. The chambered cells were then left in OGD at 37°C for 1 h, before being returned to normoxic conditions and glucose-containing media (TMo). No OGD control cells were placed in SGG and maintained in normoxic conditions for 1 h before also being returned to TMo. All cells were left in TMo until protein and RNA isolation for the time point indicated.
Plasmids and virus generation
The following plasmids have been described previously: the −1,982/+45 Mfn2-Luciferase vector and its 5′ deletion constructs (Sorianello et al, 2012), SESN2-Luc (Papadia et al, 2008), and mtRFP (Legros et al, 2002). HA-Mfn2 was subcloned into the pEF vector at the BamHI/XbaI sites. The MEF2A expression vector was a gift from Pilar Ruiz-Lozano (Stanford University, USA), GFP-Drp1 was a gift from AM van der Bliek (UCLA, USA), Drp1-K38A-myc was Addgene plasmid 26049, deposited by Dr. Chan, and Mfn2-myc was Addgene plasmid 23213, deposited by Dr. Chan (Chen et al, 2003).
The vectors used to construct and package recombinant adeno-associated viruses (rAAVs) were provided by Dr. Bading [U. Heidelberg, Germany (Zhang et al, 2007)]. For construction of pAAV-MEF2-DN, the first 252 bp of the mouse MEF2D cDNA (a gift from E. Olson; U. Texas South Western, USA) were amplified using the primers: forward 5′- ATA GGA TCC ATG GGG CGA AAG AAG ATA CAA ATC ACA CGC ATA ATG GAT G -3′ and reverse 5′- ATA AAG CTT TCA CAG ATC TTC TTC AGA AAT AAG TTT TTG TTA GGA GGG CCT CGT TTG AAA ATA AAA TC -3′. The amplified product contains sequences that produce BamHI and HindIII restriction sites at the 5′ and 3′, respectively (italics) and also for inserting a myc tag into the C terminus (bold). GFP in the rAAV-GFP vector was removed by BamHI/HindIII digestion, and the MEF2-DN PCR product was cloned into the rAAV vector to express the first 84 amino acids of the N-terminus of MEF2, which contains the DNA binding domain but not the transactivation domain. rAAV for shRNA expression contains the U6 promoter for shRNA expression and a CMV/chicken beta-actin hybrid promoter driving hrGFP expression. rAAV-shRNA targeting Mfn2 were made by swapping the sh-sc sequence of rAAV-sh-sc (gift from H. Bading) for the following sequences of the rat Mfn2 into the BamHI and HindIII sites: shMfn2: 5′- AGA GGG CCT TCA AGC GCC AGT-3′, shMfn2.2: 5′- GGG AAG AGC ACC GTG ATC AAT-3′, shMfn2.3: 5′- TCC TCA AGG TTT ATA AGA ATG-3′. All newly generated constructs were confirmed by sequencing.
Neurons were infected with rAAV at DIV4. Infection efficiencies were determined at DIV 10–11 by analyzing GFP fluorescence or immunocytochemical analysis; they ranged from 80 to 90% of the viable neurons.
Site-directed mutagenesis
Site-directed mutagenesis was performed using the QuikChange® Site-Directed Mutagenesis kit (Stratagene) according to the manufacturer’s instructions. The mutated sequences are shown in Supplementary Fig S8B.
Luciferase assay
Firefly luciferase-based reporter gene was transfected along with a Renilla expression vector (pTK-RL; Promega), and also, where relevant, MEF2-DN expression vector. Forty hours after transfection luciferase assays were performed using the Dual Glo assay kit (Promega) with Firefly luciferase-based reporter gene activity normalized to the Renilla control (pTK-RL plasmid) in all cases.
RNA isolation, RT-PCR, and qPCR
RNA was isolated using an RNA extraction kit (Life Technologies). For qPCR, cDNA was synthesized from RNA using the SuperScript® III First-Strand Synthesis SuperMix (Life Technologies) according to the manufacturer’s instructions. qPCR was performed in a StepOne Real-Time PCR System (Applied Biosystem) using GoTaq QPCR Master Mix (Promega) according to the manufacturer’s instructions. The primers used were Mfn2 -F: 5′- ATG TCA AAG GGT ACC TGT CCA-3′, -R: 5′- CAA TCC CAG ATG GCA GAA CTT-3′; SESN2 -F: 5′- GGA TTA TAC CTG GGA AGA CC -3, -R: 5′- CGC AGT GGA TGT AGT TCC -3′; Mfn1 –F: 5′-CAA ACT GCA GCC ACC AAG T-3′, -R: 5′- GTT GGC ACA GTC GAG CAA-3′; 18S -F: 5′-GTG GAG CGA TTT GTC TGG TT-3′, -R: 5′-CAA GCT TAT GAC CCG CAC TT-3′. Expression of the gene of interest was normalized to 18S, a commonly used control.
Western blotting and antibodies
Total cell lysates were boiled at 100°C for 5 min in 1.5× sample buffer (1.5 M Tris pH 6.8; glycerol 15%; SDS 3%; β-mercaptoethanol 7.5%; bromophenol blue 0.0375%). Gel electrophoresis was performed using 9% polyacrylamide gels. The gels were blotted onto PVDF membranes, which were then blocked for 1 h at room temperature with 5% (w/v) non-fat dried milk in PBS with 0.1% Tween 20. The membranes were then incubated at 4°C overnight with the primary antibodies diluted in blocking solution: anti-Mfn2 (1:2,000; Abcam), Mfn1 (1:250, Santa Cruz), Opa1 (1:1,000; BD Biosciences), Drp1 (1:1,000, BD Biosciences), MEF2A (1:750, Santa Cruz), Jun (1:1,000, BD Biosciences), Actin (1:10,000, Sigma), and Porin (1:100,000, Abcam). For visualization of Western blots, HRP-based secondary antibodies were used followed by chemiluminescent detection on Kodak X-Omat film. Western blots were analyzed by digitally scanning the blots, followed by densitometric analysis (ImageJ). All analyses involved normalizing to a loading control, actin and porin.
Imaging studies
Cells were visualized using a TCS SP2 Leica confocal laser scanning microscope (Leica Lasertechnick GmbH, Mannheim, Germany) adapted to an inverted Leitz DMIRBE microscope at 37°C in a controlled 5% CO2 atmosphere (Life Imaging Services). Pictures were acquired using a 63× (1.32 NA) Leitz Plan-Apochromatic objective. Images were analyzed using ImageJ software (Abramoff et al, 2004).
To quantify mitochondrial membrane, potential neurons were loaded with 20 nM tetramethylrhodamine methylester (TMRM; Sigma) in SGG medium without phenol red. Transfected cells were identified by co-transfecting GFP expression plasmid.
Single cells were monitored, TMRM was excited at 540 nm, and emission was measured using a 570-nm filter. The mitochondrial membrane potential was compared to that observed in surrounding untransfected neurons and subsequently expressed as a percentage of that observed in untransfected cells before NMDA stimulation.
For mitochondrial Ca2+ analysis, neurons were loaded with 5 μM Rhod-2 (Life Technologies) for 30 min at 4°C followed for extensive washout of the dye and incubation for 20 additional minutes with SGG without phenol red at 37°C. Single cells were monitored. Rhod-2 was excited at 540 nm, and emission was measured using a 570-nm filter.
Cytoplasmic Ca2+ was monitored with Fluo-4 (Life Technologies). Neurons were loaded 5 μM Fluo-4 for 30 min at 37°C, after 2 washes with PBS were incubated for additional 15 min and excited at 488 nm and emission captured with a 516-nm filter.
For mitochondrial morphology analysis, neurons were transfected with mitochondrially targeted RFP. After treatment, neurons were fixed, and nuclei were stained with DAPI. The number of live neurons with tubular or fragmented mitochondria was counted.
Drp1 or Bax localization was determined by transfecting cortical neurons with GFP-Drp1 or Bax-GFP, respectively, and mtRFP. Forty-eight hours after transfection neurons were treated when appropriate with 30 μM NMDA and fixed.
For cytochrome c immunofluorescence, neurons were fixed with 4% paraformaldehyde, permeabilized, blocked, and incubated over-night at 4°C with anti-cytochrome c antibody (Millipore). Antibody binding was visualized using biotinylated secondary antibody/Cy3-conjugated streptavidin.
Measurement of cytoplasmic Ca2+ with Indo-1
AAV-sh-sc- or AAV-sh-Mfn2-transduced neurons were loaded with 5 μM of the ratiometric Ca2+ indicator Indo-1 for 30 min at 37°C in SGG medium without phenol red. After two washes with PBS, neurons were incubated for 15 additional minutes in order to allow de-esterification of Indo-1. Measurements were made on Infinite M200PRO (TECAN). Neurons were excited at 350 nm, and the ratio of the emitted fluorescence values at 405 (Indo-1 Ca2+ bound) and 485 nm (Indo-1 Ca2+ free) was used as an index of [Ca2+].
ATP measurements
Thirty minutes before NMDA addition, neurons were incubated with medium without glucose and with pyruvate. After 30 min of NMDA treatment (15 μM), ATP was measured using the ATPlite Luminescence Assay System (Perkin-Elmer) on Infinite M200PRO (TECAN).
NAD+/NADH measurements
NAD+ and NADH levels were measured individually taking advantage of the different stabilities of the different forms in acidic or basic buffer. We employed NAD+/NADH-Glo assay (Promega) following manufacturer’s instructions.
Oxygen consumption measurements
Cortical neurons were plated in SeaHorse Bioscience XF24 plates and transduced with AAV producing shRNA-scr or shRNA-Mfn2. Seahorse Bioscience XF24 extracellular flux analyzer was used to measure oxygen consumption in these cells. The instrument was calibrated the day before the experiment, following the manufacturer’s instructions. On the day of the experiment, the injection ports on the sensor cartridge were loaded with 1.25 μM oligomycin (oli; complex V inhibitor) to distinguish the percentage of oxygen consumption devoted to ATP synthesis and the percentage of oxygen consumption needed to overcome the natural proton leak across the inner mitochondrial membrane, 3 μM FCCP to calculate the ‘spare’ respiratory capacity of cells, 1 μM rotenone (complex I inhibitor) and 1 μM antimycin A (complex III inhibitor) to calculate the remaining respiration due to residual oxygen consumption (rox). During the sensor calibration, neurons remained in a 37 °C incubator without CO2 in 700 μl of artificial cerebrospinal fluid (aCSF) assay medium (120 mM NaCl, 3.5 mM KCl, 1.3 mM CaCl2, 0.4 mM KH2PO4, 1 mM MgCl2, 15 mM glucose, 1.0 mM pyruvate, and 4 mg/ml fatty acid-free bovine serum album, pH 7.2.). Plates were immediately placed into the calibrated Seahorse XF24 flux analyzer for mitochondrial bioenergetic analysis. OCR was normalized for total protein/well and calculated as OCR–OCRrox to subtract non-mitochondrial respiration. Spare respiratory capacity was calculated as OCRCCCP/OCRrutine. Respiratory control ratio was calculated as OCRCCCP/OCRoli.
Chromatin immunoprecipitation (ChIP)
Two 35-mm dishes were used for each treatment (approximately 4.5 × 106 cells). Medium was removed, and treated cells were washed with pre-warmed medium and incubated for 10 min at room temperature with 1% para-formaldehyde in pre-warmed medium, to crosslink proteins to DNA. The reaction was stopped by adding glycine to a final concentration of 125 mM for 5 min. Cells were washed twice with ice-cold PBS and harvested on ice in swelling buffer [25% HEPES pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.1% NP-40 plus protease inhibitor cocktail set III (Calbiochem)]. Nuclei were isolated using a douncer and centrifuging at 1,500 g for 5 min. Nuclei were resuspended in 200 μl sonication buffer (50 mM HEPES pH 7.9, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.25% sodium deoxycholate, 0.1% SDS plus protease inhibitors). The nuclei were sonicated using a Diagenode Bioruptor (Liege, full power 30 s on, 30 s off, in an ice bath for 20 min) to produce fragments < 500 bp. Sonicated chromatin was pre-cleared with protein-A agarose beads/salmon sperm DNA for 1 h at 4°C with agitation, beads were collected by centrifugation, and supernatants were collected and subjected to immunoprecipitation. Eight micrograms of anti-MEF2A (Santa Cruz) or anti-rabbit IgG (control, Sigma) was added for overnight incubation at 4°C with agitation. After chromatin immunoprecipitation, DNA was purified using Qiagen DNA purification columns. Input (1% of total immunoprecipitated) and immunoprecipitated DNA were subjected to qPCR analysis with primers amplifying the Mfn2 promoter (-2,204/-2,136 of the rat Mfn2 promoter) 5′-TGG AGA TGG AAT TCA AGT TGG-3′ forward and 5′-TGGTCACAAAATGGCTCAGT-3′ reverse. As negative controls, we used primers for amplification of the actin gene 5′-AGC CAT GTA CGT AGC CAT CC-3′ forward and 5′-CTC TCA GCT GTG GTG GTG AA-3′ reverse.
Electrophoretic mobility shift assays
HeLa cells were transfected with expression vector for MEF2A and 48 h later, nuclear extracts were prepared as described previously (Santalucia et al, 2001). Radiolabeled double-strand oligonucleotide probe containing the human Mfn2 promoter sequence (containing the MEF2 binding site: 5′-ATT TTT GTA TTT TTA GTA CAG-3′ (MEF2wt), the mutated MEF2 binding site (in bold), which differs from the wild type in the same three nucleotides used in the luciferase assay: 5′-ATT TTT GGA TCC TTA GTA CAG-3′ (MEF2mut), and the MEF2 binding site in the Glut4 promoter: 5′- CGT GGG AGC TAA AAA TAG CCA-3′ (MEF2Glut4) was incubated with 10 μg nuclear extract in a final volume of 20 μl and electrophoresis was performed as described previously (Santalucia et al, 2001). Dried gel was exposed to Kodak film. Competitor mutated oligonucleotide differed from wild-type EMSA sequence by the same base substitution used in the functional experiments. For supershift assays, MEF2 antibody was added to the corresponding binding reactions after incubation with the radiolabeled probe and incubated for a further 10 min at room temperature before loading onto gels.
Statistical analysis
Statistical testing involved two-tailed Student’s t-tests. For any multiple comparisons within data sets, we used a one-way ANOVA followed by the Bonferroni’s post hoc test. All data are presented as mean ± SEM of at least three independent experiments (n).
Acknowledgments
We thank G.E. Hardingham for critically reading the manuscript. This work was supported by the Fundació La Marató de TV3 (111210; FXS) and Spanish Ministerio de Ciencia e Innovación (SAF2011-30283; FXS). FXS is a researcher from the Programa Ramón y Cajal funded by the Ministerio de Ciencia e Innovación (RYC-2009-05407).
Author contributions
AM-R designed and performed most of the experiments and analyzed data; MS-M, JPM, VG, JO, and JP-C designed and performed experiments and analyzed data; MP and MR analyzed data; JP and AZ designed experiments, analyzed data, and had critical input into the manuscript preparation; FXS conceived, designed and performed experiments, analyzed data, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Abramoff MD, Magalhaes PJ, Ram SJ. Image processing with ImageJ. Biophotonics Int. 2004;11:36–42. [Google Scholar]
- Almeida A, Bolanos JP, Medina JM. Nitric oxide mediates glutamate-induced mitochondrial depolarization in rat cortical neurons. Brain Res. 1999;816:580–586. doi: 10.1016/s0006-8993(98)01240-2. [DOI] [PubMed] [Google Scholar]
- Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- Arundine M, Tymianski M. Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium. 2003;34:325–337. doi: 10.1016/s0143-4160(03)00141-6. [DOI] [PubMed] [Google Scholar]
- Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, Daugaard JR, Lloberas J, Camps M, Zierath JR, Rabasa-Lhoret R, Wallberg-Henriksson H, Laville M, Palacín M, Vidal H, Rivera F, Brand M, Zorzano A. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. J Biol Chem. 2003;278:17190–17197. doi: 10.1074/jbc.M212754200. [DOI] [PubMed] [Google Scholar]
- Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol. 2005;6:505–510. doi: 10.1038/nrm1666. [DOI] [PubMed] [Google Scholar]
- Baloh RH, Schmidt RE, Pestronk A, Milbrandt J. Altered axonal mitochondrial transport in the pathogenesis of charcot-marie-tooth disease from mitofusin 2 mutations. J Neurosci. 2007;27:422–430. doi: 10.1523/JNEUROSCI.4798-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Graber S, Kovacs I, Lee WD, Waggoner J, Cui J, White AD, Bossy B, Martinou J-C, Youle RJ, Lipton SA, Ellisman MH, Perkins GA, Bossy-Wetzel E. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006;25:3900–3911. doi: 10.1038/sj.emboj.7601253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black BL, Olson EN. Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu Rev Cell Dev Biol. 1998;14:167–196. doi: 10.1146/annurev.cellbio.14.1.167. [DOI] [PubMed] [Google Scholar]
- Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF, Bogousslavsky J, Bonny C. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med. 2003;9:1180–1186. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]
- Breckenridge DG, Stojanovic M, Marcellus RC, Shore GC. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J Cell Biol. 2003;160:1115–1127. doi: 10.1083/jcb.200212059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan AM, Won Suh S, Joon Won S, Narasimhan P, Kauppinen TM, Lee H, Edling Y, Chan PH, Swanson RA. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat Neurosci. 2009;12:857–863. doi: 10.1038/nn.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008a;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- de Brito OM, Scorrano L. Mitofusin 2: a mitochondria-shaping protein with signaling roles beyond fusion. Antioxid Redox Signal. 2008b;10:621–634. doi: 10.1089/ars.2007.1934. [DOI] [PubMed] [Google Scholar]
- Brooks C, Wei Q, Feng L, Dong G, Tao Y, Mei L, Xie Z-J, Dong Z. Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc Natl Acad Sci USA. 2007;104:11649–11654. doi: 10.1073/pnas.0703976104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho A, Massieu L. Role of glutamate transporters in the clearance and release of glutamate during ischemia and its relation to neuronal death. Arch Med Res. 2006;37:11–18. doi: 10.1016/j.arcmed.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Cereghetti GM, Stangherlin A, de Brito OM, Chang CR, Blackstone C, Bernardi P, Scorrano L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA. 2008;105:15803–15808. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RLJ, Hess S, Chan DC. Broad activation of the ubiquitin–proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20:1726–1737. doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Chen Y, Dorn GW. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340:471–475. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho D-H, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA. S-Nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho D-H, Nakamura T, Lipton S. Mitochondrial dynamics in cell death and neurodegeneration. Cell Mol Life Sci. 2010;67:3435–3447. doi: 10.1007/s00018-010-0435-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SY, Huang P, Jenkins GM, Chan DC, Schiller J, Frohman MA. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat Cell Biol. 2006;8:1255–1262. doi: 10.1038/ncb1487. [DOI] [PubMed] [Google Scholar]
- Cook DJ, Teves L, Tymianski M. Treatment of stroke with a PSD-95 inhibitor in the gyrencephalic primate brain. Nature. 2012;483:213–217. doi: 10.1038/nature10841. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- D’Orsi B, Bonner H, Tuffy LP, Düssmann H, Woods I, Courtney MJ, Ward MW, Prehn JHM. Calpains are downstream effectors of bax-dependent excitotoxic apoptosis. J Neurosci. 2012;32:1847–1858. doi: 10.1523/JNEUROSCI.2345-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavell SW, Kim TK, Gray JM, Harmin DA, Hemberg M, Hong EJ, Markenscoff-Papadimitriou E, Bear DM, Greenberg ME. Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron. 2008;60:1022–1038. doi: 10.1016/j.neuron.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, Kepp O, Knight RA, Kumar S, Lipton SA, Lu X, Madeo F, Malorni W, et al. Molecular definitions of cell death subroutines: recommendations of the nomenclature committee on cell death 2012. Cell Death Differ. 2012;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandre-Babbe S, van der Bliek AM. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell. 2008;19:2402–2412. doi: 10.1091/mbc.E07-12-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griparic L, Kanazawa T, van der Bliek AM. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J Cell Biol. 2007;178:757–764. doi: 10.1083/jcb.200704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X-J, Lu Y-F, Li S-A, Kaitsuka T, Sato Y, Tomizawa K, Nairn AC, Takei K, Matsui H, Matsushita M. CaM kinase Iα–induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol. 2008;182:573–585. doi: 10.1083/jcb.200802164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmstrom KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. 2014;15:411–421. doi: 10.1038/nrm3801. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002;1:383–386. doi: 10.1016/s1474-4422(02)00164-3. [DOI] [PubMed] [Google Scholar]
- Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006;25:2966–2977. doi: 10.1038/sj.emboj.7601184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Nakamura K, Iijima M, Sesaki H. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 2013;23:64–71. doi: 10.1016/j.tcb.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James DI, Parone PA, Mattenberger Y, Martinou J-C. hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem. 2003;278:36373–36379. doi: 10.1074/jbc.M303758200. [DOI] [PubMed] [Google Scholar]
- Karbowski M, Lee Y-J, Gaume B, Jeong S-Y, Frank S, Nechushtan A, Santel A, Fuller M, Smith CL, Youle RJ. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol. 2002;159:931–938. doi: 10.1083/jcb.200209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Norris KL, Cleland MM, Jeong S-Y, Youle RJ. Role of Bax and Bak in mitochondrial morphogenesis. Nature. 2006;443:658–662. doi: 10.1038/nature05111. [DOI] [PubMed] [Google Scholar]
- Kumari S, Anderson L, Farmer S, Mehta SL, Li PA. Hyperglycemia alters mitochondrial fission and fusion proteins in mice subjected to cerebral ischemia and reperfusion. Transl Stroke Res. 2012;3:296–304. doi: 10.1007/s12975-012-0158-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leboucher GP, Tsai YC, Yang M, Shaw KC, Zhou M, Veenstra TD, Glickman MH, Weissman AM. Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol Cell. 2012;47:547–557. doi: 10.1016/j.molcel.2012.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Sterky FH, Mourier A, Terzioglu M, Cullheim S, Olson L, Larsson N-G. Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Hum Mol Genet. 2012;21:4827–4835. doi: 10.1093/hmg/dds352. [DOI] [PubMed] [Google Scholar]
- Legros F, Lombès A, Frachon P, Rojo M. Mitochondrial fusion in human cells is efficient, requires the Inner membrane potential, and is mediated by mitofusins. Mol Biol Cell. 2002;13:4343–4354. doi: 10.1091/mbc.E02-06-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Linseman DA, Allen MP, Meintzer MK, Wang X, Laessig T, Wierman ME, Heidenreich KA. Myocyte enhancer factor 2A and 2D undergo phosphorylation and caspase-mediated degradation during apoptosis of rat cerebellar granule neurons. J Neurosci. 2001;21:6544–6552. doi: 10.1523/JNEUROSCI.21-17-06544.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesa M, Palacín M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev. 2009;89:799–845. doi: 10.1152/physrev.00030.2008. [DOI] [PubMed] [Google Scholar]
- Lo EH. A new penumbra: transitioning from injury into repair after stroke. Nat Med. 2008;14:497–500. doi: 10.1038/nm1735. [DOI] [PubMed] [Google Scholar]
- Luetjens CM, Bui NT, Sengpiel B, Münstermann G, Poppe M, Krohn AJ, Bauerbach E, Krieglstein J, Prehn JHM. Delayed mitochondrial dysfunction in excitotoxic neuron death: cytochrome c release and a secondary increase in superoxide production. J Neurosci. 2000;20:5715–5723. doi: 10.1523/JNEUROSCI.20-15-05715.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Bonni A, Xia F, Nadal-Vicens M, Greenberg ME. Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science. 1999;286:785–790. doi: 10.1126/science.286.5440.785. [DOI] [PubMed] [Google Scholar]
- Misko AL, Sasaki Y, Tuck E, Milbrandt J, Baloh RH. Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J Neurosci. 2012;32:4145–4155. doi: 10.1523/JNEUROSCI.6338-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montessuit S, Somasekharan SP, Terrones O, Lucken-Ardjomande S, Herzig S, Schwarzenbacher R, Manstein DJ, Bossy-Wetzel E, Basañez G, Meda P, Martinou J-C. Membrane remodeling induced by the dynamin-related protein Drp1 stimulates bax oligomerization. Cell. 2010;142:889–901. doi: 10.1016/j.cell.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir KW. Glutamate-based therapeutic approaches: clinical trials with NMDA antagonists. Curr Opin Pharmacol. 2006;6:53–60. doi: 10.1016/j.coph.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Muñoz JP, Ivanova S, Sánchez-Wandelmer J, Martínez-Cristóbal P, Noguera E, Sancho A, Díaz-Ramos A, Hernández-Alvarez MI, Sebastián D, Mauvezin C, Palacín M, Zorzano A. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 2013;32:2348–2361. doi: 10.1038/emboj.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen D-F, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuspiel M, Zunino R, Gangaraju S, Rippstein P, McBride H. Activated mitofusin 2 signals mitochondrial fusion, interferes with bax activation, and reduces susceptibility to radical induced depolarization. J Biol Chem. 2005;280:25060–25070. doi: 10.1074/jbc.M501599200. [DOI] [PubMed] [Google Scholar]
- Nicholls DG. Mitochondrial calcium function and dysfunction in the central nervous system. Biochim Biophys Acta. 2009;1787:1416–1424. doi: 10.1016/j.bbabio.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oettinghaus B, Licci M, Scorrano L, Frank S. Less than perfect divorces: dysregulated mitochondrial fission and neurodegeneration. Acta Neuropathol. 2012;123:189–203. doi: 10.1007/s00401-011-0930-z. [DOI] [PubMed] [Google Scholar]
- Okamoto S-I, Li Z, Ju C, Schölzke MN, Mathews E, Cui J, Salvesen GS, Bossy-Wetzel E, Lipton SA. Dominant-interfering forms of MEF2 generated by caspase cleavage contribute to NMDA-induced neuronal apoptosis. Proc Natl Acad Sci USA. 2002;99:3974–3979. doi: 10.1073/pnas.022036399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. 2010;191:1141–1158. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadia S, Soriano FX, Leveille F, Martel MA, Dakin KA, Hansen HH, Kaindl A, Sifringer M, Fowler J, Stefovska V, McKenzie G, Craigon M, Corriveau R, Ghazal P, Horsburgh K, Yankner BA, Wyllie DJ, Ikonomidou C, Hardingham GE. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci. 2008;11:476–487. doi: 10.1038/nn2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham AH, Meng S, Chu QN, Chan DC. Loss of Mfn2 results in progressive, retrograde degeneration of dopaminergic neurons in the nigrostriatal circuit. Hum Mol Genet. 2012;21:4817–4826. doi: 10.1093/hmg/dds311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pich S, Bach D, Briones P, Liesa M, Camps M, Testar X, Palacín M, Zorzano A. the charcot–marie–tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum Mol Genet. 2005;14:1405–1415. doi: 10.1093/hmg/ddi149. [DOI] [PubMed] [Google Scholar]
- Puyal J, Vaslin A, Mottier V, Clarke PGH. Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann Neurol. 2009;66:378–389. doi: 10.1002/ana.21714. [DOI] [PubMed] [Google Scholar]
- Puyal J, Ginet V, Clarke PGH. Multiple interacting cell death mechanisms in the mediation of excitotoxicity and ischemic brain damage: a challenge for neuroprotection. Prog Neurobiol. 2013;105:24–48. doi: 10.1016/j.pneurobio.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Ramos-Cejudo J, Gutierrez-Fernandez M, Rodriguez-Frutos B, Exposito Alcaide M, Sanchez-Cabo F, Dopazo A, Diez-Tejedor E. Spatial and temporal gene expression differences in core and periinfarct areas in experimental stroke: a microarray analysis. PLoS ONE. 2012;7:e52121. doi: 10.1371/journal.pone.0052121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds I, Hastings T. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J Neurosci. 1995;15:3318–3327. doi: 10.1523/JNEUROSCI.15-05-03318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rintoul GL, Filiano AJ, Brocard JB, Kress GJ, Reynolds IJ. Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J Neurosci. 2003;23:7881–7888. doi: 10.1523/JNEUROSCI.23-21-07881.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santalucia T, Moreno H, Palacin M, Yacoub MH, Brand NJ, Zorzano A. A novel functional co-operation between MyoD, MEF2 and TRalpha1 is sufficient for the induction of GLUT4 gene transcription. J Mol Biol. 2001;314:195–204. doi: 10.1006/jmbi.2001.5091. [DOI] [PubMed] [Google Scholar]
- Sattler R, Xiong Z, Lu W-Y, Hafner M, MacDonald JF, Tymianski M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science. 1999;284:1845–1848. doi: 10.1126/science.284.5421.1845. [DOI] [PubMed] [Google Scholar]
- Shelat PB, Plant LD, Wang JC, Lee E, Marks JD. The membrane-active tri-block copolymer pluronic F-68 profoundly rescues rat hippocampal neurons from oxygen–glucose deprivation-induced death through early inhibition of apoptosis. J Neurosci. 2013;33:12287–12299. doi: 10.1523/JNEUROSCI.5731-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova E, Shurland D-L, Ryazantsev SN, van der Bliek AM. A human dynamin-related protein controls the distribution of mitochondria. J Cell Biol. 1998;143:351–358. doi: 10.1083/jcb.143.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. 2007;178:749–755. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorianello E, Soriano FX, Fernández-Pascual S, Sancho A, Naon D, Vila-Caballer M, González-Navarro H, Portugal J, Andrés V, Palacín M, Zorzano A. The promoter activity of human Mfn2 depends on Sp1 in vascular smooth muscle cells. Cardiovasc Res. 2012;94:38–47. doi: 10.1093/cvr/cvs006. [DOI] [PubMed] [Google Scholar]
- Soriano FX, Liesa M, Bach D, Chan DC, Palacín M, Zorzano A. Evidence for a mitochondrial regulatory pathway defined by peroxisome proliferator–activated receptor-γ coactivator-1α, estrogen-related receptor-α, and mitofusin 2. Diabetes. 2006a;55:1783–1791. doi: 10.2337/db05-0509. [DOI] [PubMed] [Google Scholar]
- Soriano FX, Papadia S, Hofmann F, Hardingham NR, Bading H, Hardingham GE. Preconditioning doses of NMDA promote neuroprotection by enhancing neuronal excitability. J Neurosci. 2006b;26:4509–4518. doi: 10.1523/JNEUROSCI.0455-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano FX, Martel M-A, Papadia S, Vaslin A, Baxter P, Rickman C, Forder J, Tymianski M, Duncan R, Aarts M, Clarke PGH, Wyllie DJA, Hardingham GE. Specific targeting of pro-death NMDA receptor signals with differing reliance on the NR2B PDZ ligand. J Neurosci. 2008;28:10696–10710. doi: 10.1523/JNEUROSCI.1207-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout AK, Raphael HM, Kanterewicz BI, Klann E, Reynolds IJ. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat Neurosci. 1998;1:366–373. doi: 10.1038/1577. [DOI] [PubMed] [Google Scholar]
- Suen D-F, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev. 2008;22:1577–1590. doi: 10.1101/gad.1658508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugioka R, Shimizu S, Tsujimoto Y. Fzo1, a protein involved in mitochondrial fusion, inhibits apoptosis. J Biol Chem. 2004;279:52726–52734. doi: 10.1074/jbc.M408910200. [DOI] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen D-F, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaslin A, Puyal J, Clarke PGH. Excitotoxicity-induced endocytosis confers drug targeting in cerebral ischemia. Ann Neurol. 2009;65:337–347. doi: 10.1002/ana.21584. [DOI] [PubMed] [Google Scholar]
- Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148:228–243. doi: 10.1016/j.cell.2011.11.030. [DOI] [PubMed] [Google Scholar]
- Wappler EA, Institoris A, Dutta S, Katakam PV, Busija DW. Mitochondrial dynamics associated with oxygen-glucose deprivation in rat primary neuronal cultures. PLoS ONE. 2013;8:e63206. doi: 10.1371/journal.pone.0063206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11:872–884. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- Xiang H, Kinoshita Y, Knudson CM, Korsmeyer SJ, Schwartzkroin PA, Morrison RS. Bax involvement in p53-mediated neuronal cell death. J Neurosci. 1998;18:1363–1373. doi: 10.1523/JNEUROSCI.18-04-01363.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol. 2003;23:5409–5420. doi: 10.1128/MCB.23.15.5409-5420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young KW, Piñon LGP, Bampton ETW, Nicotera P. Different pathways lead to mitochondrial fragmentation during apoptotic and excitotoxic cell death in primary neurons. J Biochem Mol Toxicol. 2010;24:335–341. doi: 10.1002/jbt.20343. [DOI] [PubMed] [Google Scholar]
- Yu S-W, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(ADP-Ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- Yuan H, Gerencser AA, Liot G, Lipton SA, Ellisman M, Perkins GA, Bossy-Wetzel E. Mitochondrial fission is an upstream and required event for bax foci formation in response to nitric oxide in cortical neurons. Cell Death Differ. 2006;14:462–471. doi: 10.1038/sj.cdd.4402046. [DOI] [PubMed] [Google Scholar]
- Zhang SJ, Steijaert MN, Lau D, Schutz G, Delucinge-Vivier C, Descombes P, Bading H. Decoding NMDA receptor signaling: identification of genomic programs specifying neuronal survival and death. Neuron. 2007;53:549–562. doi: 10.1016/j.neuron.2007.01.025. [DOI] [PubMed] [Google Scholar]
- Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, Parman Y, Evgrafov O, Jonghe PD, Takahashi Y, Tsuji S, Pericak-Vance MA, Quattrone A, Battologlu E, Polyakov AV, Timmerman V, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause charcot-marie-tooth neuropathy type 2A. Nat Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.