

Abstract
Uracil-DNA glycosylases (UDGs) are evolutionarily conserved DNA repair enzymes that initiate the base excision repair pathway and remove uracil from DNA. The UDG superfamily is classified into six families based on their substrate specificity. This review focuses on the family I enzymes since these are the most extensively studied members of the superfamily. The structural basis for substrate specificity and base recognition as well as for DNA binding, nucleotide flipping and catalytic mechanism is discussed in detail. Other topics include the mechanism of lesion search and molecular mimicry through interaction with uracil-DNA glycosylase inhibitors. The latest studies and findings detailing structure and function in the UDG superfamily are presented.
Keywords: DNA repair, uracil-DNA glycosylase, base excision repair, DNA mimicry
Introduction
Virtually all organisms employ DNA repair pathways to identify and correct damages in DNA molecules. Damages to DNA may be caused by endogeneous (e.g., DNA polymerase infidelity, reactive oxygen species produced from normal metabolic byproducts) and/or exogeneous agents (such as UV radiation, X-rays, gamma rays, plant toxins, human-made mutagenic chemicals, viruses, cancer chemotherapy, and radiotherapy). Some of the observed lesions are listed in Figure 1. Bulky DNA lesions such as thymine dimers or 6,4-photoproducts caused by UV radiation or by environmental mutagens are repaired by the nucleotide excision repair (NER) pathway. NER provides an important defense mechanism for both prokaryotic and eukaryotic organisms. Repair of cyclobutane thymine dimers and 6,4-photoproducts in prokaryotes can also be performed by the enzyme photolyase through an enzymatic process named DNA photoreactivation, which depends on a non-covalently bound cofactor, flavin adenine dinucleotide. Photolyase has been found in organisms from all kingdoms of life, except placental mammals. Repair of cyclobutane pyrimidine dimers (CPDs) by photolyase is faster than removal by NER in prokaryotes and archaea. Structure and function of this enzyme have been reviewed.1 As part of NER in prokaryotes and archaea these same lesions and other structurally unrelated DNA damages (such as single base modifications, intra- and interstrand cross-links, DNA backbone modifications) are identified and repaired by the UvrABC system. The proteins in this system recognize and cleave damaged DNA in a multistep ATP-dependent reaction. A detailed review of prokaryotic nucleotide excision repair featuring the UvrABC system is available.2 NER in eukaryotes involves a multiprotein complex of nine major proteins (XPA, XPB, XPC, XPD, XPE, XPF, XPG, CSA, and CSB) and a number of additional proteins including ERCC1, RPA, RAD23A, RAD23B. Various aspects of NER in eukaryotes have been reviewed.3,4
Figure 1.
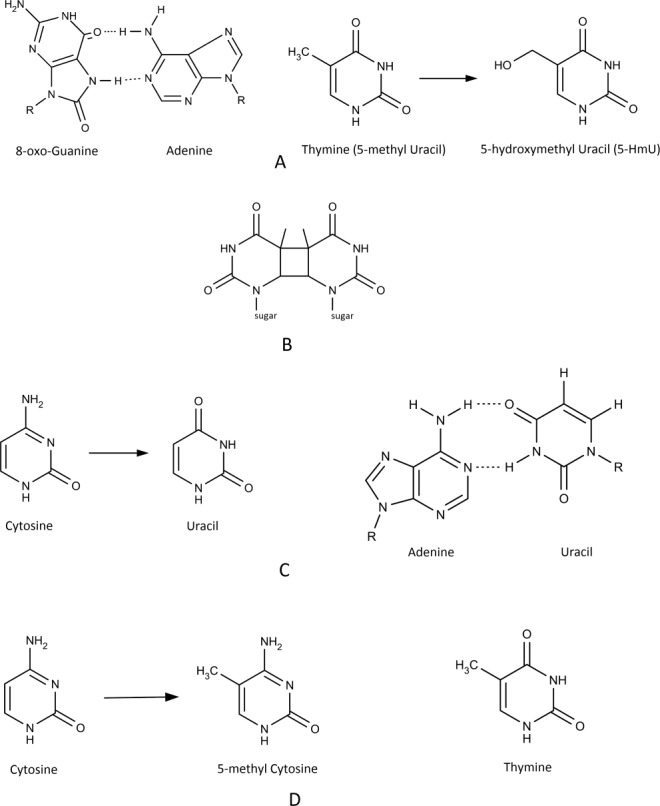
Types of lesions found in DNA. A. Oxidation of bases: 8-oxo Guanine forms a Hoogsteen base with Adenine [left]; oxidation of Thymine leads to 5-hydroxymethyl Uracil (5-HmU) [right]. B. Indirect DNA damage (UV-B light): Cyclobutane dimer. C. Hydrolysis of bases: Cytosine deamination leads to Uracil [left] that forms a base pair with Adenine instead of Guanine [right]. D. Alkylation (methylation): 5-methyl Cytosine is an epigenetic modification of DNA methyltransferases [left]; Cytosine deamination forms Uracil [Fig. 1(C)] while deamination of 5-methyl Cytosine forms Thymine (resulting in a transition mutation: C → T).
Modifications of DNA bases through oxidation, deamination or alkylation are repaired by the base excision repair (BER) pathway.5 BER is initiated by DNA glycosylases, which remove cytotoxic and mutagenic bases from DNA. DNA glycosylases are classified into monofunctional DNA glycosylases and bifunctional DNA glycosylases/AP lyases. The monofunctional enzymes cleave the N-glycosidic bond between the target base and deoxyribose using water as a nucleophile, thereby releasing a free base and leaving an apurinic/apyrimidinic (AP) site (also called abasic site). Bifunctional DNA glycosylases/AP lyases utilize an amino group of the enzyme as a nucleophile to form a Schiff's base intermediate. The amine nucleophile is generated by deprotonation of a conserved lysine residue by a conserved aspartic acid.6 The covalent intermediate then goes through an enzyme-catalyzed β-elimination step, which leads to cleavage of the phosphodiester bond 3' from the abasic site.6 Because abasic sites are highly cytotoxic, the action of monofunctional glycosylase is followed by other enzymes to complete the repair. If left unrepaired, AP sites can lead to mutation during semiconservative replication, and they can also cause replication fork stalling. In the “short-patch” BER, after removal of the abnormal base an AP-endonuclease (APE1 in humans) generates a single-strand break by strand cleavage 5′ to the resulting abasic site.7 Eukaryotic DNA polymerase β (Pol β) can catalyze DNA synthesis during BER, and the enzyme attaches a single nucleotide to the newly generated 3′-OH to displace the abasic sugar-phosphate.8 The intermediate product, a 5′-terminal deoxyribose phosphate (ORP) residue, is subsequently removed by the enzyme's AP-lyase activity.6,8 The “nick” is sealed by DNA ligase III, which interacts with Pol β through the XRCC1 protein, and the combined enzymatic actions restore the original DNA sequence.6,9
Uracil-DNA glycosylase families—Overview
Uracil-DNA glycosylases (UDG) are monofunctional glycosylases and they remove uracil from DNA. Uracil (Ura) in DNA may result from spontaneous deamination of cytosine (Cyt) or incorporation of dUMP during DNA synthesis.10,11 UDG enzymes have been identified in archaea (e.g., Sulfolobus solfataricus, Pyrococcus furiosis, Methanococcus jannaschii), eubacteria (e.g., Escherichia coli, Bacillus subtilis, Neisseria meningitidis, Helicobacter pylori, Mycoplasma pneumonia, Campylobacter jejuni, Deinococcus radiodurans, Mycobacterium tuberculosis), eukaryotes (e.g., Homo sapiens, Saccharomyces cerivisiae, Caenorhabditis elegans, Drosophila melanogaster, Arabidopsis thaliana) and large DNA viruses (herpes viruses, poxviruses). On the basis of substrate specificity UDGs are classified into six families.10,12 The sequence alignment of characteristic motifs in the six UDG families is shown in Figure 2(A). For families I–V only the N-terminal (Motif A) and the C-terminal (Motif B) motifs that correspond to the extended regions of the “water-activating” loop and the “Leu-intercalation” loop are highlighted. For family VI the extended region of the “water-activating loop” in the helix-hairpin-helix (HhH) motif is displayed together with the [4Fe-4S] cluster binding motif. Based on the sequence alignment of all displayed family I-VI UDG sequences in Figure 2(A) a phylogenetic tree was generated that emphasizes the distinct separation between the six families [Fig. 2(B)].
Figure 2.

A. Characteristic motifs in family I–V and family VI. Displayed are characteristic motifs (Motifs A and B) for family I–V and family VI UDG enzymes (the distinct HhH Motif and the Motif for the Iron-Sulfur Cluster). Highlighted (shaded) are active site residues of the catalytic water-activating loop as well as the Leu-intercalation loop for families I–V, and the catalytic water-activating loop in the HhH Motif as well as the conserved Cys residues of the Iron-Sulfur Cluster for family VI. The conserved catalytic Asp residue in the HhH Motif of family VI enzymes is also shaded. Except for family VI amino acid sequences are taken from structures supplemented by other sequences in the same family with secondary assignments based on the first listed structure in family I-V UDGs. The shown sequence alignment is an updated version of the sequence alignment described by Chung et al. with additional sequences, secondary structure assignments and structures provided. Structures and sequences are discussed in the subsections for each family.12 B. Phylogenetic tree of sequence alignment [Fig. 2(A)] for the UDG superfamily. Families I–VI (includes subfamilies in family VI) are labeled. Sequence IDs or PDB IDs are shown at each branch.
Family I UDGs (also referred to as UNGs) such as the Escherichia coli (E. coli) and human UNG proteins are the most extensively studied enzymes within the UDG superfamily. UNGs are highly specific for uracil (Ura) and except for 5-fluorouracil (5-fU), which is cleaved at a reduced rate, UDGs do not even excise 5-substituted Ura.13 Their major biological function is to remove from DNA the uracil produced by cytosine deamination.14 The UNG enzymes excise Ura from both single-stranded DNA (ssDNA) and double-stranded DNA (dsDNA) with the preference ssU > U:G > U:A.
Family II UDGs such as E. coli mispair-specific uracil glycosylase (MUG) and human thymine DNA glycosylase (TDG) are mismatch-specific enzymes for dsDNA. They are active against the U:G mispair but exhibit only weak or no activity against the U:A base pair.15 TDG excises thymine (Thy) from T:G mispairs. Although originally thought to be specific for removing Ura from U:G mispairs, E. coli MUG has subsequently been found to be active against DNA substrates carrying a variety of modified DNA bases.16 At high enzyme concentrations, MUG can also excise Thy from T:G mispairs.15
Family III UDGs (sMUGs) are only found in higher eukaryotes.17,18 Although it was initially identified to be selective for ssDNA, the sMUG enzyme not only uses dsDNA as substrate but has a higher affinity for uracil-containing dsDNA substrates.18 Interestingly, in addition to removing Ura from U:G mispairs and U:A base pairs, the sMUG enzymes can also excise 5-hydroxymethyluracil (5-HmU) but not 5-methyluracil (Thy).
The remaining three families of UDG are found in thermophilic and hyperthermophilic eubacteria and archaea. These enzymes have four conserved cysteine residues that act as ligands for four Fe atoms in the cubic iron-sulfur (4Fe-4S) cluster. The UDG enzymes in these families can remove Ura from uracil-mismatched dsDNA. Families IV and VI but not family V can also excise Ura from ssDNA substrates. The family V UDG enzyme from Thermus thermophilus HB8 lacks a polar residue at the active site but can still excise Ura.10 In contrast, the family IV enzyme from Thermus thermophilus HB8 removes Ura from dsDNA and ssDNA and discriminates against Thy.19 Human UNG has a similar substrate specificity.
UDG families I through V show <10% overall sequence homology but they share a common fold of the core domains and utilize common motifs, one at the N-terminus for pyrimidine binding and the other at the C-terminus for glycosidic bond hydrolyis.6 The sixth UDG family contains an iron-sulfur (4Fe-4S) cluster as families IV and V. Members of this family share a central α-helical domain, which consists of a helix-hairpin-helix (HhH) motif. A conserved Asp residue in the HhH motif is essential for the catalytic activity. Uracil-DNA glycosylase from Methanococcus jannaschii (MjUDG), and mismatch-specific glycosylases (MIG) from Pyrobaculum aerophilum (PaMIG) and Methanobacterium thermoautotrophicum (MthMIG) belong to this family.12,20 The two MIG enzymes are specific for U:G and T:G mismatches, and can process mismatches of Ura and Thy with 7,8-dihydro-oxoguanine, but are not active on ssDNA. Howver, MjUDG is similar to family I UDG in that it catalyzes the removal of Ura from both ssDNA and dsDNA, but unlike any other UDGs, MjUDG can also excise 8-oxoguanine.12 Family I UDGs have been most extensively studied. Approximately 75% of all UDG structures deposited in the Protein Data Bank are for family I enzymes.
Family I uracil-DNA glycosylases (UNGs)
Properties (sequence, fold, motifs, activity)
Family I UDGs (UNGs) are found in a variety of organisms including DNA viruses such as poxviruses and herpes viruses. Overall, the C-terminal 200 amino acid residues that include the catalytic domain are conserved. These proteins contain diverse N-terminal extensions that are associated with subcellular localization, regulation and protein-protein interactions.11 For example, alternative promoter usage and splicing of the gene leads to two mammalian isoforms, mitochondrial UNG1 and nuclear UNG2; the nuclear form contains an N-terminal extension that shows a proliferating cell nuclear antigen (PCNA) binding motif and two replication protein A (RPA) motifs.
Figure 3(A) shows the structure-based sequence alignment of UNGs from various organisms. Overall, amino acid sequences, motifs and structures in these proteins are well conserved while the largest deviations are observed for poxvirus UNGs. When compared with UNGs of other organisms only 9 out of 24 residues in the motifs for DNA binding and catalytic activity are conserved in poxvirus UNGs. On the other hand, 5 of the 6 active site residues are identical to the conserved residues in UNGs of other organisms (Table1). Human UNG (hUNG) shares 56, 54, 49, and 40% amino acid identity with E. coli, yeast, B. subtilis and Herpes simplex virus (HSV) UNG, while the sequence identity with Vaccinia virus UNG (vUNG) is only 20%. When conservative amino acid substitutions are included, the sequence homology increases by 10–20% for E. coli UNG (eUNG), human, Herpes simplex virus 1 (HSV-1) UNG and vUNG.21 Structural superimposition of eUNG with hUNG and HSV-1 UNG shows root-mean-squared (rms) distances for Cα positions of 0.9 and 1.5 Å, respectively, while rms distances for Cα positions between hUNG and HSV-1 UNG are 1.3 Å. On the other hand, only the core domain of vUNG (∼140–150 residues) superimposes reasonably well with hUNG, eUNG and HSV-1 UNG with rms deviations of 2.0, 2.1, and 2.0 Å, respectively; the overall sequence identity for this domain is ∼21%.
Figure 3.

A. Structure-based sequence alignment for UNG subfamily. The secondary structure assignment is based on hUNG [1AKZ]. Residue numbering in this alignment is arbitrary (the first residue in 1AKZ is set to 1). Highlighted (*) are the six active site residues. Identical residues are shaded in red while similar residues are shaded in yellow. The largest differences are observed for vUNG [2OWQ]. B. Topology of UNG. The topology diagram highlights the basic fold: three layers a/b/a (two α-helices on either side of the core of four parallel β-strands in the order 2134). C. Active site (tertiary structure) in UNG. Active site in eUNG [2EUG] highlighting the observed interactions (dashed lines in black) of uracil (Ura) with active site residues. Hydrogen-bonding and van der Waals distances (in Å) are shown. Active site residues and Ura are represented as stick models (C grey, O red, N blue) and labeled. A conserved water molecule in the active site is shown as a red sphere. D. Schematic active site interactions in UNG. Schematic of uracil (Ura) interactions (hydrogen-bonding network) in the binding pocket of UNG (eUNG). Comparison with cytosine (Cyt) in the same orientation illustrates that the same type of hydrogen bonds with Asn123 as seen for Ura are not possible.
Table I.
Motifs in UNG Enzymes in Different Species
| 2EUG (eUNG) | 1AKZ (hUNG) | 4DOF (vUNG) | ||
|---|---|---|---|---|
| Catalytic water- | 62-GQDPYH-67 | 143-GQDPYH-148 | 66-GIDPYP-71 | |
| activating-loop | ||||
| Pro-rich loop | 84-AIPPS-88 | 165-PPPPS-169 | 84-FTKKS-88 | |
| Uracil specificity | 120-LLLN-123 | 201-LLLN-204 | 117-IPWN-120 | |
| Gly-Ser loop | 165-GS-166 | 246-GS-247 | 160-KT-161 | |
| Leu intercalation loop | 187-HPSPLSAHR-195 | 268-HPSPLSVYR-276 | 181-HPAARDR-187 | |
| Active site residues | D64, Y66, F77, N123, H187, L191 | D145, Y147, F158, N204, H268, L272 | D68, Y70, F79, N120, H181, R185 | |
The core of the UNG structure consists of a parallel β-sheet of four strands (order 2134) sandwiched between two pairs of α-helices [Fig. 3(B)].22 The structures of DNA complexes reveal a specific uracil-binding pocket located in a DNA binding groove. Structural data on DNA substrate recognition and the catalytic mechanism of uracil excision have been gathered from the studies of hUNG, eUNG, and HSV-1 UNG in complex with DNA [PDBIDs: 1SSP, 2SSP, 4SKN, 1EMH, 1EMJ, 1FLZ, 1LAU].
UNGs contain five conserved motifs: (1) the catalytic water-activating loop (143-GQDPYH-148 in hUNG); (2) the Pro-rich loop, which compresses the DNA backbone 5′ to the lesion (165-PPPPS-169 in hUNG); (3) the Ura-binding motif (201-LLLN-204 in hUNG); (4) the Gly-Ser loop that compresses the DNA backbone 3′ to the lesion (246-GS-247 in hUNG); (5) the Leu-intercalation loop, which penetrates the minor groove (268-HPSPLS-273 in hUNG). For comparison, these motifs for eUNG, hUNG and vUNG are listed in Table1.
Structural basis for substrate specificity and base recognition
In UNGs amino acid residues from several motifs (described above) combine to form the Ura-binding pocket. These include residues Gln, Asp and Tyr of the catalytic water-activating loop (63, 64, and 66 in eUNG, respectively), a Phe (77 in eUNG) residue at the active site, a Ser residue (88 in eUNG) of the Pro-rich loop, the Asn residue (123 in eUNG) of the Ura-binding motif, and the His residue (187 in eUNG) of the Leu-intercalation loop. Size and shape of the pocket are determined by a number of hydrophobic residues (Phe, Tyr, Leu, Ala) near the active site that include the aromatic residues, which stack against the DNA base. The size of the Ura-binding pocket excludes the larger purine bases and thus contributes to the specificity. Discrimination against Thy and other 5-substituted pyrimidines is achieved by the conserved Tyr residue (66 in eUNG), which presses against C5 of the pyrimidine nucleotide [Fig. 3(C)]. In eUNG [2EUG] the shortest distance of Tyr66 (atom CE1) to C5 in Ura is 3.4 Å, and the addition of a methyl group or another bulky substituent would lead to a clash. Importance of this conserved Tyr in defining the substrate specificity is demonstrated by the observation that a mutant version of hUNG (Y147A), in which the corresponding residue is altered to an Ala, can excise Thy.6,23 Discrimination against cytosine (Cyt) is based on a set of specific hydrogen bonds formed by protein residues with O2, N3 and O4 of Ura [Fig. 3(D)]. Asn123 (eUNG) forms hydrogen bonds through the amide side chain with N3 and O4 of Ura that are specific for Ura and cannot occur with Cyt [Fig. 3(D)]. On the other hand, a N204D mutant in hUNG can also excise Cyt.6,23 Thermodynamic studies suggest that UNGs bind any DNA (with or without damage) with comparable affinity. Although the affinity for damaged Ura-containing DNA is 10–30 fold higher than for undamaged DNA, this increased affinity cannot account for the specificity of UNG for Ura, since the reaction rate for specific Ura-containing oligodeoxynucleotides (ODNs) is at least five orders of magnitude higher than for non-specific ODNs.11 A study using modified DNA analogs established some key findings for substrate recognition by UDGs.24 The preference of hUNG for ssDNA over dsDNA was twofold. This is consistent with data from another study of hUNG that showed a threefold higher activity of hUNG for a ssDNA substrate as compared to dsDNA.14 Stable hairpin and dumbbell duplex DNAs were poor substrates for UNG indicating that UNG activity is higher when the double helix is less stable, and local separation of the DNA double helix into two strands is a possible first step of UNG action. There is no major contribution of the bases to substrate recognition by UNG but UNG interaction with some internucleotide phosphate groups is necessary for DNA recognition. A substrate containing Ura in an uncharged peptide chain instead of the sugar-phosphate backbone demonstrated insignificant binding. UNG does not discriminate Ura from other heterocyclic bases during the binding step, but the nature of a substituent at the C5 position of Ura affects the catalytic efficiency since UNG does not remove 5-bromouracil (BrU) and Thy from DNA and the excision rate for 5-fU is only 15% of the rate for Ura removal.13 The conformation of the ribose plays a role in the binding of DNA to UNG since the 3′-endo conformation of the sugar moiety (as found in A-form RNA) prevents interaction with UNG. The presence of an NH2 group in 2′-position of the sugar has no influence on UNG binding but leads to inhibition of Ura excision activity.24 These results indicate that C5 (uracil base) and C2′ (deoxyribose sugar) positions of deoxyuridine (dU) play a role in formation of an active enzyme-substrate complex. The biochemical results showed good agreement with X-ray structural data for a complex of mutant (L272R/D145N) hUNG with dU-containing DNA [4SKN].24,25 Despite the active site mutation (D145N) that reduced the activity the double mutant still excised Ura. The DNA backbone of the Ura-containing strand contributes 80% of buried DNA surface area, and protein–DNA interactions are mainly observed along the sugar-phosphate backbone of this strand. Local DNA unwinding takes place during the catalytic step through insertion of residue Leu272 (in hUNG) into the DNA minor groove and compression of the DNA backbone flanking the Ura base resulting in a flipped-out nucleotide. After glycosidic cleavage the flipped-out Ura binds within the UNG specificity pocket through interactions with Gln144, Asn145, Tyr147, Phe158, Asn204, and His268: stacking of Ura with Phe158; hydrogen bonds of Ura O4, N3, and O2 atoms with Gln144, Asn145, Phe158, Asn204, and His268; packing of Tyr147 against Ura near the C5-position. Together with Ser270, His268 also interacts with the 3′-phosphate of the abasic deoxyribose phosphate (ORP), and Ser169 forms two hydrogen bonds with the 5′-phosphate of ORP. Mutations of Ser270 and His268 impair both UDG activity and DNA binding, while mutation of Ser169 only impairs activity.26 This suggests that Ser169 binding to the 5′-phosphate of dU plays a role in nucleotide flipping and catalysis. Hydrogen bonds are formed between O4′ and O1′ atoms of the deoxyribose of ORP and side chain nitrogen atoms of residues His148 and Asn145. Substituents at the 2′-position of deoxyribose and 3′-endo puckering (as in RNA uracil) would block His268 movement and impair substrate hydrolysis. Therefore, C5 and C2′ positions of dU are important and are involved in the formation of active enzyme-substrate complexes.
Mutational studies of hUNG indicated a special role for R276 in the Leu272 loop (268-HPSPLSVYR-276). Structural studies of hUNG with DNA suggested that Arg276 may stabilize the Leu272 loop and the Leu272 side chain either before or after it is inserted into the DNA minor groove.25,27,28 The six R276(C,E,H,L,W,Y) mutations showed reduced affinity for dsDNA but retained affinity for ssDNA.29 These results indicated that the mutations at residue 276 affected the enzyme's interactions with Ura-containing dsDNA, thereby transforming hUNG into a ssDNA-specific enzyme.
Structural basis for DNA binding, nucleotide flipping and catalytic mechanism
Structural and spectroscopic evidences show that non-specific binding of undamaged DNA by UNGs results in an increase in the level of base stacking in the DNA.30 When UNG binds damaged Ura-containing DNA, a similar increase in base stacking also takes place. In addition, specific binding to damaged DNA leads to a substrate-directed conformational change in UNG from an ‘open to a closed’ conformation, consistent with an induced-fit mechanism for damage site recognition.28,29
A multistep reaction scheme for UNG activity has been established from stopped-flow and site-directed mutagenesis studies.31,32 This scheme is generally described as a “pinch, push, plug, and pull” base-flipping mechanism. In the first step UNG binds to DNA non-specifically; this is followed by flipping of dU, either actively (enzyme-assisted flip) or passively (spontaneous flip).11 The combined effect of conserved Ser169 (Pro-rich loop), Ser247 (Gly-Ser loop), Ser270 and Ser273 (Leu-intercalation loop), Pro269 (Leu-intercalation loop), Pro167 and Pro168 (Pro-rich loop) leads to DNA bending by compressing the DNA phosphate backbone (“pinch”). The Leu residue of the Leu-intercalation loop penetrates the minor groove and is inserted into the DNA double helix; penetration of the Leu residue and the loop movement are specific for productive Ura binding (“push”).25 In addition, this action also increases the lifetime of the flipped-out uracil nucleotide in the active site (“plug”). This second step is followed by the N-glycosidic bond breakage, and in the final step, the Leu residue is retracted (“pull”).11 Upon cleavage, the uracil moves deeper into the active site, and the abasic nucleotide relaxes to a more puckered C2′-endo form leading to a less strained product-complex than the uncleaved-substrate complex.33 UNG appears to bind preferentially to its cleaved product, and association of abasic DNA with UNG indicates that the enzyme remains bound to or binds back to its apyrimidinic site (AP-site) products.27 Because AP sites are highly mutagenic and cytotoxic, this may indicate a protective role of UNG in vivo until further action of the next enzyme, AP-endonuclease, in the BER pathway.7 Parikh et al. (2000) demonstrated that human APE1 endonuclease significantly increases the uracil excision efficiency of hUNG, and that through competition for binding to AP sites APE1 may promote AP-site release by UNG.33 APE1 interacts directly with DNA repair polymerase Pol β, and this interaction assembles the polymerase onto an AP-site in DNA. After AP-site cleavage by the endonuclease, APE1-assisted excision of 5′-terminal deoxyribose 5-phosphate by Pol β accelerates the next step in the BER pathway.34
Highlighted in Figure 4 are the motifs from three loops that accomplish compression of the phosphates: the Pro-rich loop on the 5′ side; the Gly-Ser loop and the Leu-intercalation loop on the 3′ side.27 The hydroxyl side chains of three conserved serine residues (Ser169, Ser247, and Ser270 in hUNG), one in each loop, are involved in forming polar contacts with the phosphodiester backbone of damaged DNA (pinching). Hydrophobic contacts are provided by Pro167, Pro168, Pro271 and Ser273. Mutational studies in eUNG of S88A, S189A and S192G “pinching mutations” showed a 360-, 80-, and 21-fold decrease in catalytic efficiency (kcat/Km) for the single mutants and a 8200-fold damaging effect in a double mutant (S88A/S189A).35 The formation of the Ser-phosphodiester interactions therefore seem to be critical for catalytic efficiency of UNGs.35 On the other hand, eUNG excises Ura from 5′-HO-dUAAp-3′, which lacks the 5′-phosphodiester group. This is demonstrated by the presence of the Ura trapped in the active site of eUNG, which was incubated with 5′-HO-dUAAp-3′ prior to crystallization [1FLZ].35 A substrate induced conformational change as noticed for large dsDNA substrates was observed. The catalytic power of UDG, defined by the ratio of the enzymatic kmax value for a tetranucleotide sequence (AUAA) and the rate constant for the non-enzymatic reaction (uncatalyzed hydrolysis of deoxyuridine) at neutral pH and 25°C, was calculated to be ∼1012.35,36 It is unlikely that the serine pinching mechanism alone can account for such an efficiency.
Figure 4.
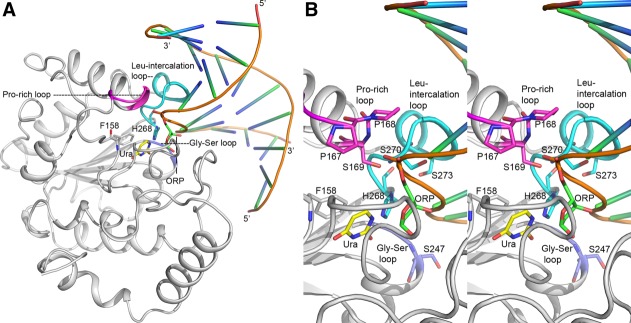
A. Interaction of UNG with DNA. Cartoon diagram showing the binding of a dsDNA to hUNG (grey). This figure captures the UNG–DNA interactions in post uracil-excision state. The cleaved uracil (Ura) in the uracil-binding pocket and the abasic site (ORP) on the DNA strand are shown in stick models and labeled. Phe158 and His268 in the pocket are also shown as stick. Three loops from the UNG structure that are involved in phosphate compression are highlighted: the Gly-Ser loop in blue, the Pro-rich loop in magenta and the Leu intercalation loop in cyan. B. Close-up view of UNG-DNA binding. Stereo-diagram displaying the close-up view of protein–DNA interactions shown in Figure 4(A). The loops are colored similarly as in Figure 4(A) and important residues are shown as stick models and labeled. An interactive view is available in the electronic version of the article.
Features of the catalytic mechanism indicate that the combined action of four loops (water-activating, Pro-rich, Gly-Ser and Leu-intercalation loop) during nucleotide flipping produces a reactive conformation for the sugar ring of dU that leads to bending and subsequent cleavage of the glycosidic bond. In summary, the cleavage of the glycosidic bond produces an intermediate that consists of an oxocarbenium cation and an anionic uracil, and the subsequent attack by an activated water molecule generated by transfer of a proton to the general base Asp145 (in hUNG) leads to the products (Fig. 5). The negative charge of the uracilate anion is stabilized by hydrogen bonding with His268 (in hUNG) that acts as a neutral electrophile. Computational analysis by the Karplus group using a hybrid quantum-mechanical/molecular-mechanical (QM/MM) approach confirmed that the hydrolysis of the glycosidic bond of dU in DNA to yield an apyrimidinic site proceeds in a stepwise dissociative mode of action in contrast to the initially proposed coordinated associative mechanism.37 The primary contribution to lowering the activation energy comes from the substrate through stabilization of the oxocarbenium cation by the phosphate groups of the DNA substrate at nucleotide positions 4–7 (21.9 kcal mol−1), and not from interactions of the enzyme. These four phosphate groups are buried upon binding to UNG (only 11% accessible surface area compared to their accessible surface area in B-form DNA). This event was termed “substrate autocatalysis”.37
Figure 5.
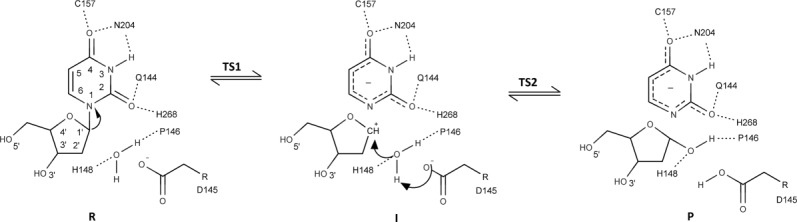
Schematic diagram of the UDG reaction mechanism (R, I, and P are short for reactant, intermediate, and product; TS1 and TS2 indicate the two transition states). The side chains of Asp145 and His268 (in hUNG) stabilize the first transition state (TS1). An intermediate (I) comprised of an oxocarbenium cation and an uracilate anion is generated following cleavage of the N-glycosidic bond. The oxocarbenium cation is stabilized by four phosphate groups of the DNA substrate. A water molecule activated through abstraction of a proton by the conserved aspartate attacks the oxocarbenium cation leading to an abasic site. The leaving group is the uracilate anion, which is stabilized through hydrogen bonding of the conserved His268 that acts as a neutral electrophile.
Mechanism of lesion search
Two groups independently developed a new approach to study the processive cleavage of uracil-containing ODN substrates by UDGs.24,38 This ODN-based assay is used to elucidate the mechanism of lesion search and to explain the efficient location and excision of damaged DNA bases by DNA glycosylases. Site-specific DNA binding proteins such as UDGs must locate their lesion targets to perform their function efficiently. Scanning of long stretches of DNA by one-dimensional (1D) sliding while maintaining continuous contact is an extremely slow and highly repetitive process, which may be suitable only for local DNA sequence.39
A summary of the mechanism of lesion search and recognition by UNG can be formulated based on structural, thermodynamic and kinetic data. The mechanism of lesion search by UNG enzymes is likely a combination of DNA hopping and rapid one-dimensional scanning (short-range sliding) to trap extrahelical Ura bases.11,39,40 Extrahelical bases in dsDNA can emerge spontaneously by thermally induced opening of the corresponding base pairs of Thy or Ura with adenine.41 Parker et al. established for hUNG and eUNG that UNG recognizes these extrahelical bases but does not actively participate in base pair opening.41 Recent studies have shown that UNG locates dU while it is in an extrahelical position during the sliding phase along short stretches of DNA (one helical turn of ∼10 bp).39 In the sliding phase, UNG binds in a random place in DNA and starts scanning it by one-dimensional diffusion, with a partial eversion of the base sampled at the moment. The enzyme is held in place through non-specific contacts with DNA outside of the sampled base but the binding is not strong enough to prevent diffusion-driven translocation along DNA. After inspecting a short stretch of DNA, the enzyme may dissociate from it. However, if an Ura base is found, it can be pulled into the Ura-binding pocket, which increases the residence time at this location and allows for a more precise conformational adjustment of the enzyme-substrate complex. Only dU and other specific UDG-substrates are able to adopt a correct conformation in the enzyme's active site and proceed to breakage of the N-glycosidic bond. For efficient damage repair of long genomic DNA sequences UNG uses hopping in addition to sliding.39 Nonetheless, the sliding mechanism plays an essential function in Ura recognition by UNG since the probability of finding the damaged site by DNA hopping alone is low.39 In their study to characterize the one-dimensional search of DNA by eUNG using the ODN-based assay with ODN substrates that contained two uracil residues at defined positions Mechetin and Zharkov estimated a characteristic one-dimensional search distance of ∼100 nucleotides and a translocation rate constant of ∼2 × 106 s−1.42
Molecular mimicry—interaction with Ugi
Bacteriophage PBS2 uracil-DNA glycosylase inhibitor protein (Ugi) inactivates the host uracil mediated base-excision DNA repair pathway by inhibiting UDG activity. Ugi is an acidic protein of 84 amino acids that inactivates uracil-DNA glycosylase from diverse organisms. Ugi specifically binds and inactivates UNGs from Bacillus subtilis, Escherichia coli, Saccharomyces cerevisae, rat liver, HSV and Homo sapiens, while poxvirus UNGs are not inhibited.43 The secondary structure of Ugi consists of five anti-parallel β-strands and two α-helices. Ugi inhibits eUNG by forming a UNG:Ugi complex with 1:1 stoichiometry. Crystal structures of eUNG, HSV-1 UNG and hUNG with Ugi [1UGH, 1UUG, 2UUG, 1EUI, 1UDI] show that Ugi can successfully mimic DNA backbone interactions by targeting the DNA binding surface of UNG.43–47 Ugi prevents UNG from binding to DNA and can also dissociate UNG from a UNG:DNA complex. Moreover, Ugi forms an irreversible complex with Ugi.45 A hallmark of UNG:Ugi interaction is that the shape, electrostatic and hydrophobic complementarity of Ugi for UNG is defined by their structures and does not involve an induced fit mechanism. The major interactions are defined by hydrogen bonding and packing contacts derived from the complementarity between the conserved Leu-intercalation loop (187-HPSPLS-192) of eUNG and eight hydrophobic residues of Ugi (Met24, Val29, Val32, Ile33, Val43, Met56, Leu58, and Val71), and the electrostatic interactions between acidic residues (Glu20, Glu27, Glu30, Glu31, Asp61 and Glu78) of Ugi with key active site residues (Gln63, Asp64, Tyr66, His67, and His187) of eUNG (Fig. 6). Interaction between UNG and Ugi buries about 2200 Å2 of total accessible surface area. The insertion of Leu191 into the hydrophobic cavity of Ugi alone causes an exclusion of about 250 Å2 of surface area (Fig. 6). A two-step model for the enzyme-inhibitor association was proposed. In the first step, a rapidly reversible loose complex is formed that satisfies long-range electrostatics and a buried hydrophobic surface; followed in the second step by an irreversible “locked” complex that includes the alignment of hydrogen bonding interactions along the interface and a conformational change at Ugi Glu20.45 Mutational studies involving seven acidic residues of Ugi (E20I, E27A, E28L, E30L, E31L, D61G, and E78V mutants) showed that with the exception of the E20I and E28L mutants, which formed reversible complexes, other mutants formed irreversible complexes with eUNG.47 These two mutants seem to interfere with the locking mechanism that causes the complex formation to be irreversible. A mutational analysis of Leu191 (L191G, L191A, L191V, and L191F mutants) within the 187-HPSPLS-192 motif of eUNG demonstrated decreased stability of the UNG-Ugi complex with shortening of the side chain length at position 191 (L191V, L191A, and L191G mutants).44 The larger side chain of the L191F mutant, on the other hand, showed approximately the same stability as wild type UNG. Indeed, in Bacillus subtilis (B. subtilis) UNG, the in vivo target of Ugi, Leu191 of the E. coli enzyme is replaced by Phe. In addition, mutational analysis showed that the L191V and L191F mutants were as efficient as the wild type protein, while the L191A and L191G mutants retained only 10% and 1% of the enzymatic activity of uracil excision from ssDNA and dsDNA.44 A L272A mutation in hUNG, on the other hand, leads to a loss of 99% of the activity.27 Nonetheless, the L272A mutant in complex with AP site dsDNA [2SSP] showed that nucleotide flipping occurred despite the inability of the shorter side chain to reach into the base stack suggesting that Leu272 is not absolutely critical in pushing the target dU into the active site pocket. Overall, Ugi targets the mechanism of uracil flipping by UNG and appears to be a transition-state mimic for flipping of Ura nucleotides from DNA.45 While comparison of structures of free UNGs and UNG:Ugi complexes reveal only minimal conformational changes in UNG upon Ugi binding, binding of substrate DNA induces a substantial “open to closed” conformational change.43–48
Figure 6.
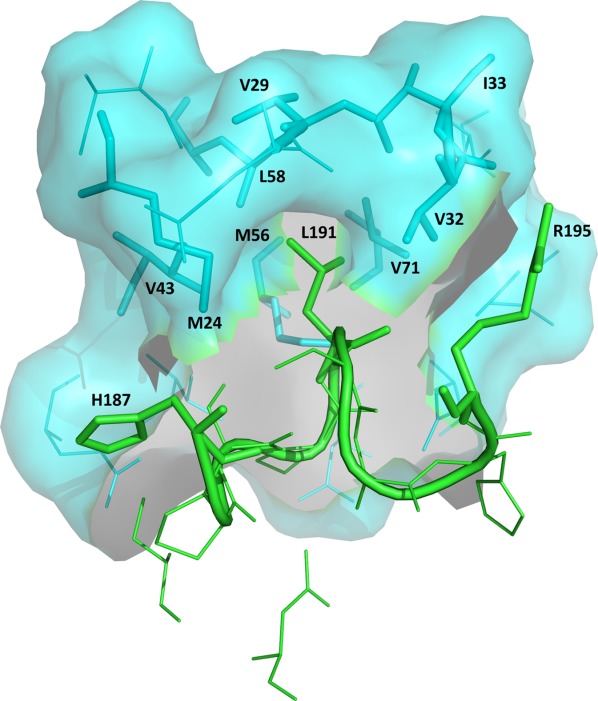
Binding of UNG to Ugi. Close-up view of the protein-Ugi interactions in the eUNG-Ugi complex [1UUG]. The figure emphasizes the insertion of Leu191 in the Leu-intercalation loop (residues 187–195 shown as cartoon in green) of eUNG into the hydrophobic cavity of Ugi (shown as surface in cyan). Residues His187, Leu191, and Arg195 of the Leu–intercalation loop in UNG and residues Met24, Val29, Val32, Ile33, Val43, Met56, Leu58, and Val71 in the hydrophobic cavity of Ugi are represented as stick models. These 11 residues are labeled. The remaining residues in this close-up are shown as thin lines (Ugi in cyan; UNG in green). An interactive view is available in the electronic version of the article.
Recently, a new uracil-DNA glycosylase inhibitor has been reported, and the encoded acidic protein p56 (56 amino acids) from the B. subtilis phage ϕ29 was found to inhibit the DNA-binding ability of the host (B. subtilis) uracil-DNA glycosylase (BsUNG).49,50 Although the genome of the B. subtilis phage ϕ29 does not contain Ura bases as phage PBS2, host UDG activity during replication could be harmful (by creating mutagenic AP sites) if Ura bases occurred in the intermediates through either cytosine deamination or dUMP incorporation of the DNA polymerase.50,51 The role of the viral p56 protein is to prevent the action of the BER pathway by inhibiting BsUNG. Mutational studies on BsUNG indicated a role for Phe191 (corresponding residues in hUNG and eUNG: Leu272 and Leu191, respectively) in the inhibition by p56.21 The two mutants (F191L, F191R) showed wild-type activity but only the F191L mutant but not the F191R mutant (corresponding residue in vUNG: R185) were inhibited by p56.21 The p56 protein was also able to inhibit eUNG, and Ugi was capable to displace p56 previously bound to UNG.50 Results also showed that the p56 protein of phage ϕ29 failed to block the UDG activity in the extracts of vaccinia-infected cells, but it was capable of inhibiting the UDG activity present in human cell extracts.21
Another DNA mimic protein, which acts as a uracil-DNA glycosylase inhibitor, was found in Staphylococcus aureus (S. aureus).52 This acidic 14 kDa protein (112 residues) named S. aureus uracil-DNA glycosylase inhibitor (SAUGI) binds to the DNA binding region of S. aureus UDG (SAUDG). SAUGI is more similar to Ugi than to p56 with respect to protein folding and charge distribution.52 In vitro SAUGI has also a low nM (KD) affinity for hUNG but the binding affinity for SAUDG is six times greater.52 All three inhibitors, Ugi, p56 and SAUGI, target the protruding residue of UDG (Leu191 in eUNG, Phe191 in BsUNG and Leu184 in SAUDG) by forming a hydrophobic pocket.
Special features of UNG enzymes and divergence of poxvirus UNGs
Vaccinia virus is the prototypic poxvirus. Sequence identity for many Vaccinia virus proteins including those in the replication machinery such as D4 (UNG), A20 and E9 with those from orthopoxviruses (such as variola and cowpox virus) is on the order of 98–99%. However, the identity is considerably lower when compared with members of other poxvirus subfamilies. For example, UNGs of Vaccinia virus (vUNG) and crocodile poxvirus have only 48% identity. Sequence homology of vUNG to hUNG, eUNG and HSV-1 UNG is low with an amino acid identity of only 20–25%. Structural superimposition of vUNG [2OWR] using the iSARST server shows rmsd values of 2.35–2.87Å for an alignment of ∼180 residues to Deinococcus radiodurans [2BOO], human [2SSP], HSV-1 [1LAU], Vibrio cholerae [2JHQ], E. coli [1EUI], Atlantic cod [1OKB], Mycobacterium tuberculosis [2ZHX], Epstein-Barr [2J8X] and Leishmania naifii [3CXM] UNGs (in this order), while rmsd values for an alignment size of ∼120 residues to family V [2D3Y] and family IV [1UI1] UDG enzymes from Thermus thermophilus are 3.78–4.0 Å, respectively. Structurally, main differences between vUNG and other members of the family I UDG enzymes can be seen at the N-terminus (additional antiparallel β-sheet) and C-terminus (additional antiparallel β-sheet, pairing of two small α-helices).53 Crystal structures in different space groups [2OWQ, 2OWR, 4DOG, 4DOF, 4LZB] showed a predominant dimeric packing arrangement, in which the central β-sheet is extended by antiparallel pairing of β-strand nine from neighboring subunits.53,54 Interface residues include those in α-helix nine as well as residues in the loops between β-strand 8 and β-strand 9, and between β-strand 9 and α-helix 9. Catalytic properties of D4 compared to hUNG, eUNG and HSV-1 UNG emphasize similar binding affinities (Km) for ssDNA and dsDNA but several orders of magnitude lower turnover numbers (kcat) and catalytic efficiencies (kcat/Km).55–58 In this regard, the enzymatic activity of D4 is most similar to that of UNG from human cytomegalovirus (CMV).59 On the other hand, catalytic efficiency (kcat/Km) of UNG from Epstein-Barr virus (EBV) is similar to hUNG.60 According to a structure-based sequence alignment of UNGs poxvirus UNGs show differences in a number of motifs that are generally highly conserved in UNGs [Table1; Fig. 3(A)]. Although 5 of the 6 active site residues are conserved, only 9 out of 24 residues in the five motifs that define DNA binding and catalytic activity in the UNG family are identical. With the exception of Ser88 in the Pro-rich loop the conserved Ser residues in the Gly-Ser loop and Leu-intercalation loop, are replaced by other residues. Pro residues in the Pro-rich loop are replaced by Lys residues. The motif of the Leu-intercalation loop emphasizes several changes in poxvirus UNGs, especially the replacement of the conserved Leu residue by Arg185 (in vUNG). This Leu residue is not only critical for the catalytic mechanism of UNGs but it plays an important role in the inhibition of various UNG enzymes by Ugi. A remarkable feature of vUNG is that it is not inhibited by Ugi.55 As pointed out by Ellison et al., mutational analysis (R185L mutant) in vUNG highlighted not just the importance of this residue, but also emphasized a difference in the entire loop structure; this mutant had lower catalytic activity and its activity was not inhibited by Ugi.61 Indeed, D4 crystal structures [2OWQ, 2OWR, 4DOG, 4DOF, 4LZB] show that the orientation of the “Leu-intercalation” loop is different from other UNGs.53,54 The structural differences result not only from variations in the amino acid sequence (hUNG: -HPSPLSVYR-; D4: -HPAARDR-), but also from the difference in the number of residues in this motif (hUNG: nine residues; D4: seven residues). In addition, the conserved Gln residue in the catalytic water-activating loop (-GQDPYH-) is replaced by Ile in D4. This conserved Gln plays an important role in stabilizing the UNG-Ugi complexes as its side chain atoms OE1 and NE2 form hydrogen bonds with the peptide backbone atoms of Leu23 in Ugi. The importance of this Gln residue in UNG:Ugi interaction is also demonstrated by the insensitivity of MUG/TUG enzymes, which contain a GINPGL-motif, to Ugi, although they have the conserved Leu residue in the Leu-intercalation loop.15 Therefore, the observed resistance of D4 to Ugi-mediated inhibition may be related to the altered sequence of the “Leu-intercalation” loop and the replacement of the conserved Gln residue in the catalytic water-activating loop.
Eukaryotic (human, yeast) and herpes virus (CMV, HSV-1, EBV) UNGs have been identified as components of the replication fork, although their main function during DNA replication seems to be related to their catalytic uracil-excision activity.59,60,62–67 Of these, hUNG2, yeast UNG, EBV UNG, and CMV UNG interact with the processivity factor of DNA polymerase while HSV-1 UNG (UL2) shows an association with the catalytic subunit of the DNA polymerase. On the other hand D4 is an absolutely essential component of the viral processivity factor, and this newly adopted function is independent of its glycosylase activity.62,63,68,69 Studies by the Traktman laboratory determined a stoichiometry of 1:1 for the viral processivity factor (D4:A20) and 1:1:1 for the DNA polymerase holoenzyme (D4:A20:E9).62,63 Low resolution solution structures derived from small-angle X-ray scattering for the viral processivity factor and the DNA polymerase holoenzyme complex have been reported.70 The interaction of D4 and A20 (N-terminal 50 residue domain) has been recently structurally characterized [4ODA; 4OD8].71
Family II uracil-DNA glycosylases (mismatch-specific)
The bacterial family II mismatch-specific uracil-DNA glycosylases (MUG) and the eukaryotic family II thymine-DNA glycosylases (TDG) are structurally related to the family I UDG enzymes (UNGs) but they use a different mechanism of substrate recognition and their sequence homology is low.15 The catalytic efficiency is similar to other DNA glycosylases and is much lower than for UNG enzymes. Moreover, interactions of MUG with the complementary DNA strand of the ‘lesion’ are quite different from UNG. Nonetheless, MUG/TDGs show a common “flipping” mechanism for the recognition of Ura and Thy as UNG enzymes for uracil.
Compared to eUNG, E. coli MUG [1MUG] shows substantial differences in active site residues [Fig. 7(A)]. Furthermore, the overall fold emphasizes an extra fifth antiparallel β-strand and two additional α-helices on one side of the central β-sheet [Fig. 7(B)]. In the MUG/TDG binding pocket the conserved “-GQDPY–” sequence motif of UNG enzymes is replaced by a conserved “-GINPG-” sequence motif. The replacement of the Tyr residue, which provides the exquisite specificity of UNGs for Ura and sterically excludes the 5-methyl group of Thy, by a Gly residue explains thymine glycosylase activity in the MUG/TDG family.15 Indeed, mutation of this Tyr residue to Ala in hUNG confers the ability to excise Thy.23 As observed in UNGs discrimination against Cyt in the MUG/TDG binding pocket is provided by a specific pattern of hydrogen-bonding interactions by the conserved active site Asn residue. The replacement of the conserved catalytic Asp residue in UNGs by Asn in this family (-GINPG- motif instead of -GQDPY-) has a major impact on the catalytic mechanism of MUG/TDG enzymes. In UNG enzymes the role of the catalytic Asp residue is in binding of a water molecule and its activation by abstraction of a proton for nucleophilic attack on the N-glycosidic bond of dU. No active general-base catalysis as described for UNG enzymes can be provided by the amide side chain of Asn in MUG, although a water molecule is observed here in an equivalent position. Therefore, the N-glycosidic bond in this family is attacked by a water molecule that is weakly nucleophilic instead of a hydroxyl ion as in UNG enzymes.25 In the catalytic mechanism established for UNG enzymes a conserved His residue (His268 in hUNG) of the Leu-intercalation loop (-HPSPLSVYR-) acts as a neutral electrophile forming a hydrogen bond with the Ura O2 atom of the bound dU in the transition state.48,72 This interaction helps to stabilize the negative charge of the generated uracilate anion after glycosidic cleavage. The catalytic His residue of UNGs is replaced by Asn in bacterial MUGs and a Met residue in the mammalian TDGs.15 Catalytic efficiency and mechanism for MUG/TUGs are quite different from the UNG enzymes.25,48,58,72–75 The lack of a general acid and a general base in MUGs results in a several orders of magnitude slower rate of uracil excision than for UNGs. Unlike UNGs, MUGs can only act on dsDNA. While UNG's interaction with the second strand of a dsDNA substrate does not contribute to the stability and specificity of the enzyme-DNA complex, in MUG enzymes interaction with the complementary strand of the duplex provides a significant contribution to substrate recognition.15 MUG shows little affinity for the Ura of dsDNA substrates (U:G mismatch) but displays nanomolar affinity for the abasic site produced by the base excision reaction. The enzyme makes three specific hydrogen bonds (main-chain of residues Gly143 and Ser145) that mimic Watson–Crick base pairing with the unpaired guanine (Gua) of the U:G mismatch after the mispaired pyrimidine is flipped-out into the active site pocket of the enzyme. While other UDGs establish specific contacts with the substrate base, MUG uses the complementary base for substrate discrimination.15,76 For base excision the MUG enzymes employ a “push” mechanism that is made possible by insertion of the intercalation wedge formed by residues Gly143, Leu144, and Arg146 within the catalytic pocket into the minor groove of the duplex thereby occupying the space of the substrate base.15
Figure 7.
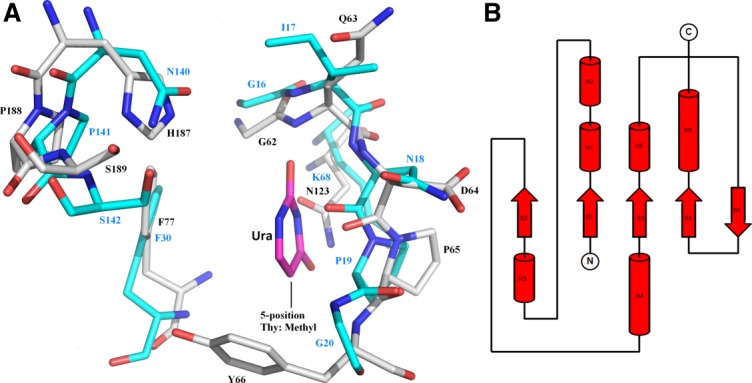
A. Active site comparison between E. coli UNG and MUG enzymes. Superimposition of E. coli UNG and MUG (rmsd of 1.42Å for 62 aligned atoms between 2EUG and 1MUG). Active site residues and uracil (Ura) are represented as stick models (1MUG: C cyan, O red, N blue; 2EUG: C grey, O red, N blue; URA: C magenta, O red, N blue) and labeled (1MUG in blue; 2EUG in black; Ura in magenta). The figure shows that MUG does not exclude Thy (or other 5-substituted pyrimidine bases) from the binding pocket since the conserved Tyr residue in UNG enzymes (Tyr66 in 2EUG) is replaced by Gly (Gly20 in 1MUG). In addition, no discrimination against Cyt is observed since the conserved Asn residue in UNG enzymes (Asn123 in 2EUG) is replaced by Lys (Lys68 in 1MUG). Other major changes are replacement of the conserved His residue in UNG (His187 in 2EUG) by Asn (Asn140 in 1MUG), replacement of the conserved Asp residue in UNG (Asp64 in 2EUG) by Asn (Asn18 in 1MUG) and replacement of Gln in UNG (Gln63 in 2EUG) by Ile (Ile17 in 1MUG). B. Topology diagram of MUG. Notice the extra fifth antiparallel β-strand compared to UNG and the two extra α-helices.
The discovery of eukaryotic TDGs as members of the family II uracil-DNA glycosylases attracted attention because of their ability to remove Thy, a normal DNA base, from T:G mispairs, although the U:G mispair in dsDNA is the common, most efficiently processed substrate.76 Nonetheless, the implication was that BER has a possible function in the restoration of C:G base pairs following the deamination of 5-methylcytosine (5meC) that would induce a C→T mutation [Fig. 1(D)]. A specific role is proposed for the non-conserved N-terminus of TDG that distinguishes eukaryotic TDGs from E. coli MUG. Studies suggested that the N-terminus allows non-specific DNA binding thereby allowing processing of energetically less than optimal substrates such as G:T or G:5meC.76 In contrast to E. coli MUG, which shows only minor conformational changes upon DNA-binding, TDG undergoes a major conformational change upon DNA binding, which involves the N-terminal domain.76 In the proposed model the N-terminal domain forms a flexible clamp holding the glycosylase onto the DNA, which allows sliding of TDG along the DNA in search of a Gua mismatched substrate. Compared to human TDG, which only excises deaminated bases from U:G base pairs, and, to a much lower extent, U:A and I:G base pairs, Schizosaccharomyces pombe (Spo) TDG exhibits glycosylase activity on all deaminated bases in both dsDNA and ssDNA in the descending order of X > I > U_O.77
Family III uracil-DNA glycosylases (single strand selective)
A new class of UDG activity, distinct from UNG enzymes, was identified in mammalian cells, and enzymes in this family initially appeared to be single-strand specific monofunctional uracil-DNA glycosylases (sMUGs). In their study of Xenopus sMUG, Wibley et al. discovered surprisingly that the enzyme was actually 700-fold more active against double-stranded than single-stranded uracil-containing DNA substrates with a preference for the U:G mispair over the U:A base pair.18 In addition, activity against 5-HmU is observed.17,18 In contrast to the MUG/TDG family the family III sMUG enzymes essentially show no activity toward Thy in T:G mispairs.17,18 The explanation for the substrate specificity of sMUGs is based on the observed hydrogen-bonding network of active site residues Gly95, Gly98, Met102, and Asn174 with either 5-HmU or Ura and the conserved water molecule (Fig. 8). As seen in the active site of MUG, sMUG has an Asn residue but not the conserved Asp residue of UNG enzymes. On the other hand, in the Leu-intercalation loop a His residue (as seen in UNGs) instead of an Asn (as in MUG) is observed. These family III sMUG enzymes are also found in insects and amphibians but not in bacteria or viruses. As demonstrated for human sMUG protein, which has an Arg residue instead of the conserved Leu residue in the Leu-intercalation loop, sMUG enzymes are not inhibited by Ugi.18 The structure of Xenopus sMUG in complex with DNA shows a greater disruption and distortion of the DNA duplex than the distortion seen in UNG and MUG complexes with DNA.18 In addition, more extensive protein-DNA interactions are observed here compared to UNG-DNA and MUG-DNA complexes. The 251–260 segment [Motif B in Fig. 2(A)], which is equivalent to the Leu-intercalation loop in UNGs, acts as a wedge penetrating the DNA duplex.18 This segment in sMUG is unique since it contains a sMUG specific short α-helix (residues 256–260) that follows a short loop (residues 251–255). The role of the short helix is to support the loop that includes the catalytic Arg254 in its interaction with DNA.
Figure 8.
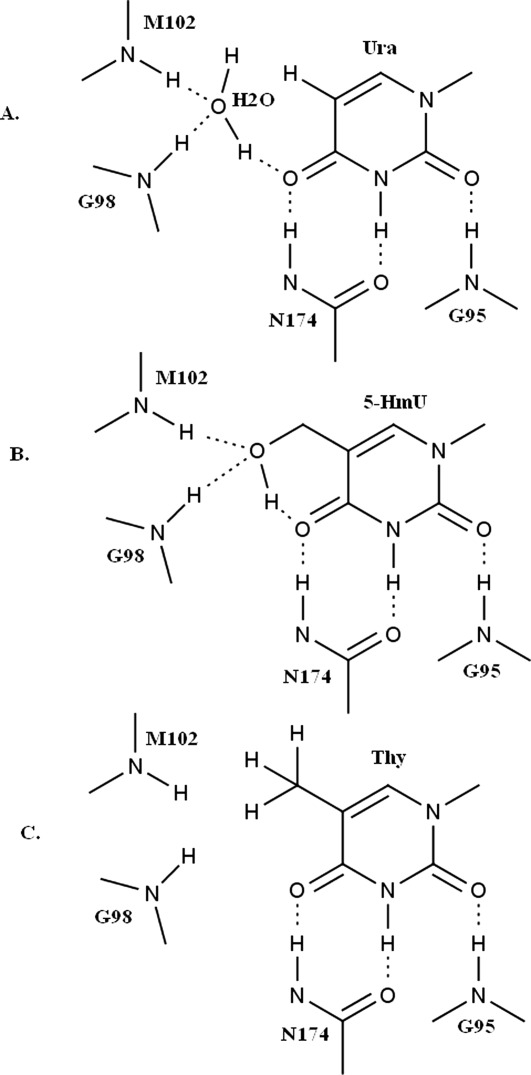
Schematic active site interactions of sMUG with different substrates. Schematic diagram highlighting sMUG specificity for 5-hydroxymethyluracil (5-HmU) and uracil (Ura). A. Characteristic hydrogen-bonding network in sMUG for Ura that includes a conserved active site water molecule. Residues Gly98 and Met102 are hydrogen bonded to the water molecule while hydrogen bonds with Ura at O2, N3, and O4 are provided by Gly95 and Asn174. The water molecule is in hydrogen bonding contact with O4. Gly98 corresponds to Gly20 in 1MUG and Tyr66 in 2EUG while for Asn174 the corresponding residues in 1MUG and 2EUG are Lys68 and Asn123, respectively. B. The hydroxyl group in 5-HmU displaces the water molecule but a similar hydrogen bonding network as for Ura is preserved. C. The Thy methyl group is too large to allow hydrogen bonding that includes an active site water molecule, and the side chain cannot provide hydrogen bonding interactions as seen for the 5-hydroxymethyl group in 5-HmU.
Family IV and family V uracil-DNA glycosylases (structural Fe-S cluster)
A family IV uracil-DNA glycosylase from Sulfolobus solfataricus and its interaction with one of the subunits, PCNA3, of the heterotrimeric PCNA sliding clamp was characterized.78 The enzyme contains a structural [4Fe-4S] cluster and shows preference for dsDNA substrates containing U:G mispairs over U:A base pairs or Ura-containing ssDNA substrates. But so far no crystal structure of UDG from an archaeon is available. Recently, a family IV UDG enzyme from the hyperthermophilic archaeon Sulfolobus tokodaii strain 7 has been crystallized in apo form and in complex with uracil but coordinates for the crystal structure are not available.79 A crystal structure of a family IV UDG may explain the uracil-excision repair mechanism in hyperthermophilic archaea. On the other hand, a structure for a family IV uracil-DNA glycosylase from the Gram negative eubacterium Thermus thermophilus HB8 in apo form and in complex with uracil has been determined.19 This enzyme (TthUDG) excises Ura from ssDNA as well as from dsDNA regardless of the opposing base but it is inactive against T:G mismatched DNA. The active site shows a similar arrangement of residues interacting with Ura as seen in UNG that explains the observed specificity [Fig. 9(A)]. The enzyme possesses a [4Fe-4S] cluster, which does not seem to be essential for activity since it is distant from the active site, but may be important for stabilization because of its interaction with some loop structures.
Figure 9.
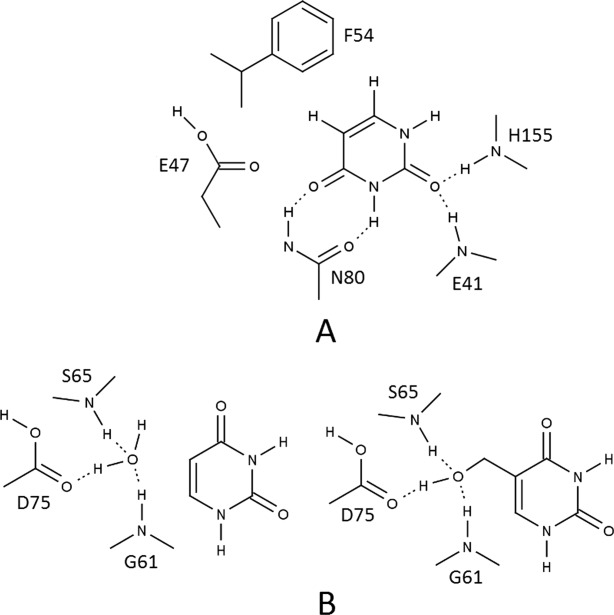
Schematic interactions in active site of family IV and family V UDG enzymes. A. Active site of family IV UDG. The figure shows interactions in the active site of family IV UDG. Stacking of Ura is provided by a conserved Phe residue (as in UNG), and a similar hydrogen-bonding network with Ura as in UNG is observed (Asn residue as in UNG). Residue Glu47 is analogous to Tyr in UNG and discriminates against five-substituted uracil analogs. B. Active site of family V UDG. The figure highlights the hydrogen-bonding pattern in the active site of family V UDG [2D3Y] for substrates Ura and 5-HmU. On the left side: a conserved water molecule takes part in hydrogen-bonding with active site residues Gly61, Ser65, and Asp75. No hydrogen bonds are observed with Ura. On the right side: the 5-hydroxymethyl group of 5-HmU displaces the water molecule and establishes the same type of hydrogen bonds with the difference that now the substrate is hydrogen-bonded to active site residues. This explains the low affinity of family V UDG for Ura. This scheme shows similarity with the situation in sMUG.
A report of the structure of a family V uracil-DNA glycosylase from Thermus thermophilus HB8 in complex with DNA highlighted the first DNA complex structure of a thermostable UDG belonging to families IV and V.10 The iron-sulfur [4Fe-4S] cluster in these two families seems to have only a structural role since it makes no direct contact with DNA and is distant to the DNA-binding surface. Although the family V enzyme (TthUDGb) shows a conserved water molecule in the active site and lacks a polar residue in the active site motif (catalytic water-activating loop), it can still excise Ura but at a lower rate than family I–IV UDGs [Fig. 9(B)]. The enzyme (TthUDGb) removes Ura from dsDNA regardless of the opposing base but not from ssDNA.10 In addition to Thy excision from T:G mismatched DNA, the enzyme removes analogs of Ura (such as 5-HmU or fU) from DNA.
Family VI uracil-DNA glycosylases (helix-hairpin-helix sequence motif)
The uracil-DNA glycosylase enzyme from Methanococcus jannaschii belongs to a novel UDG family termed family VI UDG. MjUDG (MJ1434) showing a helix-hairpin-helix motif and a [4Fe-4S]-binding cluster removes uracil both from ssDNA and dsDNA like other thermophilic UDGs, and in addition the enzyme also catalyzes the excision of 8-oxoguanine from dsDNA.12 Activity for uracil excision follows the order: U:T > U:C > U:G > U:A.12 Homologous members of MjUDG (Q58829) have been identified in Aquifex aeolicus (AAC06526), Thermoplasma volcanium (BAB60438) and Sulfolobus solfataricus (AAK42620). Although MJ1434 possesses the HhH motif and a [4Fe-4S] cluster as members of the EndoIII family and shares 21% sequence identity with E. coli EndoIII, it is instead a monofunctional glycosylase lacking the lysine residue within the HhH motif critical for the catalytic reaction of AP lyases (Fig. 0).12
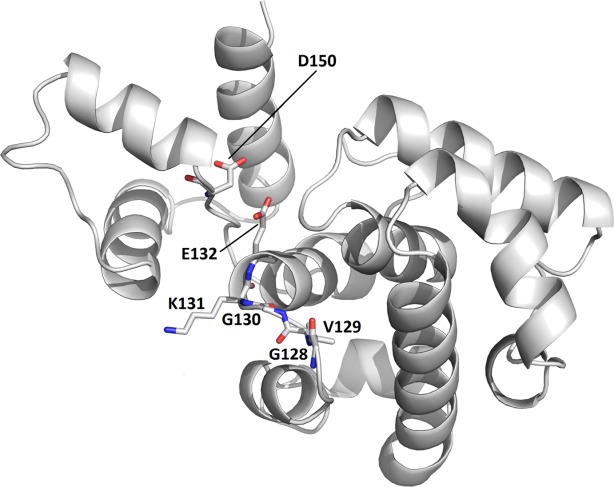
Family VI UDG enzymes and helix-hairpin-helix motif. This figure displays a model (ModWeb: Modeller) for the Helix-hairpin-Helix (HhH) motif in family VI (MjUDG) that is based on sequence homology (39% sequence identity for residues 24–212) with H. pylori 3-methyladenine DNA glycosylase [1PU6]. The protein is shown as a cartoon drawing in grey. Active site residues (water-activating loop 128-GVGKE-132 and the catalytic Asp150) are labeled and represented as stick models (C grey, O red, N blue).
A thermostable DNA glycosylase from the hyperthermophilic archaeon Pyrobaculum aerophilum that belongs to the MIG subfamily (termed Pa-MIG; AAF37270) of Family VI UDG enzymes has been described.20 As the MjUDG subfamily the MIG subfamily features a helix-hairpin-helix motif and it contains an iron-sulfur cluster. The enzyme is specific for U:G and T:G mismatches, and it is also capable of processing 8-oxoguanine (GO) mismatches with adenine (A:GO) and Thy (T:GO).20
Acknowledgments
Schematic Diagrams [Figs. 1, 3(D), 5, 8, 9] were generated with ChemAxon (http://www.chemaxon.com). The listing of Motifs and the Phylogenetic Tree for Families I-VI in Figure 2 are based on sequence alignments using STRAP (http://www.bioinformatics.org/strap/). The structure based alignment of UNG enzymes [Fig. 3(A)] was created by a combination of ALSCRIPT (http://www.compbio.dundee.ac.uk/papers/alscript/alps2.html) and STRAP (www.bioinformatics.org/strap/). Topology Diagrams [Figs. 3(B) and 7(B)] were generated in TOPDRAW (http://www.ccp4.ac.uk/html/topdraw.html). Structural Figures [Figs. 3(C), 4(A,B), 6, 7(A), and 10] were produced using PyMol (http://www.pymol.org).
References
- Weber S. Light-driven enzymatic catalysis of DNA repair: a review of recent biophysical studies on photolyase. Biochim Biophys Acta. 2005;1707:1–23. doi: 10.1016/j.bbabio.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Truglio JJ, Croteau DL, Houten BV, Kisker C. Prokaryotic nucleotide excision repair. Chem Rev. 2006;106:233–252. doi: 10.1021/cr040471u. [DOI] [PubMed] [Google Scholar]
- Rechkunova NI, Lavrik OI. Nucleotide excision repair in higher eukaryotes: mechanism of primary damage recognition in global genome repair. Subcell Biochem. 2010;50:251–277. doi: 10.1007/978-90-481-3471-7_13. [DOI] [PubMed] [Google Scholar]
- Rechkunova NI, Krasikova YS, Lavrik OI. Nucleotide excision repair: DNA damage recognition and preincision complex assembly. Biochemistry (Moscow) 2011;76:24–35. doi: 10.1134/s0006297911010056. [DOI] [PubMed] [Google Scholar]
- Nilsen H, Krokan HE. Base excision repair in a network of defense and tolerance. Carcinogenesis. 2001;22:987–998. doi: 10.1093/carcin/22.7.987. [DOI] [PubMed] [Google Scholar]
- Schärer OD, Jiricny J. Recent progress in the biology, chemistry and structural biology of DNA glycosylases. BioEssays. 2001;23:270–281. doi: 10.1002/1521-1878(200103)23:3<270::AID-BIES1037>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Carey DC, Strauss PR. Human apurinic/apyrimidinic endonuclease is processive. Biochemistry. 1999;38:16553–16560. doi: 10.1021/bi9907429. [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Kim K. Excision of deoxyribose phosphate residues by DNA polymerase beta during DNA repair. Science. 1995;269:699–702. doi: 10.1126/science.7624801. [DOI] [PubMed] [Google Scholar]
- Kubota Y, Nash RA, Klugland A, Schar P, Barnes DE, Lindahl T. Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase beta and the XRCC1 protein. EMBO J. 1996;15:6662–6670. [PMC free article] [PubMed] [Google Scholar]
- Kosaka H, Hoseki J, Nakagawa N, Kuramitsu S, Masui R. Crystal structure of family 5 uracil-DNA glycosylase bound to DNA. J Mol Biol. 2007;373:839–850. doi: 10.1016/j.jmb.2007.08.022. [DOI] [PubMed] [Google Scholar]
- Zharkov DO, Mechetin GV, Nevinsky GA. Uracil-DNA glycosylase: structural, thermodynamic and kinetic aspects of lesion search and recognition. Mutat Res. 2010;685:11–20. doi: 10.1016/j.mrfmmm.2009.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung JH, Im EK, Park H-Y, Kwon JH, Lee S, Oh J, Hwang K-C, Lee JH, Jang Y. A novel uracil-DNA glycosylase family related to the helix-hairpin-helix DNA glycosylase superfamily. Nucleic Acids Res. 2003;31:2045–2055. doi: 10.1093/nar/gkg319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Burdzy A, Sowers LC. Substrate recognition by a family of uracil-DNA Glycosylases: UNG, MUG and TDG. Chem Res Toxicol. 2002;15:1001–1009. doi: 10.1021/tx020030a. [DOI] [PubMed] [Google Scholar]
- Slupphaug G, Eftedal I, Kavli B, Bharati S, Helle NM, Haug T, Levine DW, Krokan HE. Properties of a recombinant human uracil-DNA glycosylase from the UNG gene and evidence that UNG encodes the major uracil-DNA glycosylase. Biochemistry. 1995;34:128–138. doi: 10.1021/bi00001a016. [DOI] [PubMed] [Google Scholar]
- Barrett TE, Sawa R, Panayotou G, Barlow T, Brown T, Jiricny J, Pearl LH. Crystal structure of a G:T/U mismatch-specific DNA glycosylase: mismatch recognition by complementary-strand interactions. Cell. 1998;92:117–129. doi: 10.1016/s0092-8674(00)80904-6. [DOI] [PubMed] [Google Scholar]
- O'Neill RJ, Vorob'eva OV, Shabakhti H, Zmuda E, Bhagwat AS, Baldwin GS. Mismatch uracil Glycosylase from Escherichia coli: general mismatch or a specific DNA glycosylase? J Biol Chem. 2003;278:20526–20532. doi: 10.1074/jbc.M210860200. [DOI] [PubMed] [Google Scholar]
- Kavli B, Sundheim O, Akbari M, Otterlei M, Nilsen H, Skorpen F, Aas PA, Hagen L, Krokan HE, Slupphaug G. hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J Biol Chem. 2002;277:29926–29936. doi: 10.1074/jbc.M207107200. [DOI] [PubMed] [Google Scholar]
- Wibley JE, Waters TR, Haushalter K, Verdine GL, Pearl LH. Structure and specificity of the vertebrate anti-mutator uracil-DNA glycosylase SMUG1. Mol Cell. 2003;11:1647–1659. doi: 10.1016/s1097-2765(03)00235-1. [DOI] [PubMed] [Google Scholar]
- Hoseki J, Okamoto A, Masui R, Shibata T, Inoue Y, Yokoyama S, Kuramitsu S. Crystal structure of a family 4 uracil-DNA glycosylase from Thermus thermophilus HB8. J Mol Biol. 2003;333:515–526. doi: 10.1016/j.jmb.2003.08.030. [DOI] [PubMed] [Google Scholar]
- Yang H, Fitz-Gibbon S, Marcotte EM, Tai JH, Hyman EC, Miller JH. Characterization of a thermostable DNA glycosylase specific for U/G and T/G mismatches from the hyperthermophilic archaeon Pyrobaculum aerophilum. J Bacteriol. 2000;182:1272–1279. doi: 10.1128/jb.182.5.1272-1279.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Lago L, Serrano-Heras G, Baños B, Lázaro JM, Alcorlo M, Villar L, Salas M. Characterization of Bacillus subtilis uracil-DNA glycosylase and its inhibition by phage ϕ29 protein p56. Mol Microbiol. 2011;80:1657–1666. doi: 10.1111/j.1365-2958.2011.07675.x. [DOI] [PubMed] [Google Scholar]
- Aravind L, Koonin EV. The α/β fold uracil DNA glycosylases: a common origin with diverse fates. Genome Biol. 2000;1:1–8. doi: 10.1186/gb-2000-1-4-research0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavli B, Slupphaug G, Mol CD, Arvai AS, Petersen SB, Tainer JA. Excision of cytosine and thymine from DNA by mutants of human uracil-DNA glycosylase. EMBO J. 1996;15:3442–3447. [PMC free article] [PubMed] [Google Scholar]
- Sudina AE, Volkov EM, Kubareva EA. The repair enzyme uracil-DNA glycosylase: study of the mechanism of functioning using modified analogues of DNA. Biocatalysis. 2000;41:121–123. [Google Scholar]
- Slupphaug G, Mol CD, Arvai AS, Kavli B, Alseth J, Krokan HE, Tainer JA. A nucleotide-flipping mechanism from the structure of human uracil-DNA glycosylase bound to DNA. Nature. 1996;384:87–92. doi: 10.1038/384087a0. [DOI] [PubMed] [Google Scholar]
- Mol CD, Arvai AS, Slupphaug G, Kavil B, Alseth I, Krokan HE, Tainer JA. Crystal structure and mutational analysis of human uracil-DNA glycosylase: structural basis for specificity and catalysis. Cell. 1995;80:869–878. doi: 10.1016/0092-8674(95)90290-2. [DOI] [PubMed] [Google Scholar]
- Parikh SS, Mol CD, Slupphaug G, Bharati S, Krokan HE, Tainer JA. Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J. 1998;17:5214–5226. doi: 10.1093/emboj/17.17.5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh SS, Putnam CD, Tainer JA. Lessons learned from structural results on uracil-DNA glycosylase. Mutation Res. 2000;460:183–199. doi: 10.1016/s0921-8777(00)00026-4. [DOI] [PubMed] [Google Scholar]
- Chen C-Y, Mosbaugh DW, Bennett SE. Mutations at arginine 276 transforms human uracil-DNA glycosylase into a single-stranded DNA-specific uracil-DNA glycosylase. DNA Repair. 2005;4:793–805. doi: 10.1016/j.dnarep.2005.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Drohat AC, Stivers JT, Pankiewicz KW, Carey PR. Raman spectroscopy of uracil DNA glycosylase-DNA complexes: insights into DNA damage recognition and catalysis. Biochemistry. 2000;39:13241–13250. doi: 10.1021/bi001437m. [DOI] [PubMed] [Google Scholar]
- Jiang YL, Stivers JT. Mutational analysis of the base-flipping mechanism of uracil DNA glycosylase. Biochemistry. 2002;41:11236–11247. doi: 10.1021/bi026226r. [DOI] [PubMed] [Google Scholar]
- Wong I, Lundquist AJ, Bernards AS, Mosbaugh DW. Presteadystate analysis of a single catalytic turnover by Escherichia coli uracil-DNA glycosylase reveals a “pinch-pull-push” mechanism. J Biol Chem. 2002;277:19424–19432. doi: 10.1074/jbc.M201198200. [DOI] [PubMed] [Google Scholar]
- Parikh SS, Walcher G, Jones GD, Slupphaug G, Krokan HE, Blackburn GM, Tainer JA. Uracil-DNA glycosylase-DNA substrate and product structures: conformational strain promotes catalytic efficiency by coupled stereoelectronic effects. Proc Natl Acad Sci USA. 2000;97:5083–5088. doi: 10.1073/pnas.97.10.5083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett RAO, Wilson DM, III, Wong D, Demple B. Interaction of human apurinic endonuclease and DNA polymerase β in the base excision repair pathway. Proc Natl Acad Sci USA. 1997;94:7166–7169. doi: 10.1073/pnas.94.14.7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner RM, Jiang YL, Gordley RG, Jagadeesh GJ, Ladner JE, Xiao G, Tordova M, Gilliland GL, Stivers JT. Stressing-out DNA? The contribution of serine-phosphodiester interactions in catalysis by uracil DNA glycosylase. Biochemistry. 2000;39:12585–12594. doi: 10.1021/bi001532v. [DOI] [PubMed] [Google Scholar]
- Shapiro R, Kang S. Uncatalyzed hydrolysis of deoxyuridine, thymidine, and 5-bromodeoxyuridine. Biochemistry. 1969;8:1806–1810. doi: 10.1021/bi00833a004. [DOI] [PubMed] [Google Scholar]
- Dinner AR, Blackburn GM, Karplus M. Uracil-DNA glycosylase acts by substrate autocatalysis. Nature. 2001;413:752–755. doi: 10.1038/35099587. [DOI] [PubMed] [Google Scholar]
- Sidorenko VS, Mechetin GV, Nevinsky GA, Zharkov DO. Correlated cleavage of single- and double-stranded substrates by uracil-DNA glycosylase. FEBS Lett. 2008;582:410–414. doi: 10.1016/j.febslet.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Porecha RH, Stivers JT. Uracil DNA glycosylase uses DNA hopping and short-range sliding to trap extrahelical uracils. Proc Natl Acad Sci USA. 2008;105:10791–10796. doi: 10.1073/pnas.0801612105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedglin M, O'Brien PJ. Hopping enables a DNA repair glycosylase to search both strands and bypass a bound protein. ACS Chem Biol. 2010;5:427–436. doi: 10.1021/cb1000185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JB, Bianchet MA, Krosky DJ, Friedman JI, Amzel LM, Stvers JT. Enzymatic capture of an extrahelical thymine in the search for uracil in DNA. Nature. 2007;449:433–438. doi: 10.1038/nature06131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechetin GV, Zharkov DO. Mechanism of translocation of uracil-DNA glycosylase from Escherichia coli between distributed lesions. Biochem Biophys Res Commun. 2011;414:425–430. doi: 10.1016/j.bbrc.2011.09.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mol CD, Arvai AS, Sanderson RJ, Slupphaug G, Kavli B, Krokan HE, Mosbaugh DW, Tainer JA. Crystal structure of human uracil-DNA glycosylase in complex with a protein inhibitor: protein mimicry of DNA. Cell. 1995;82:701–708. doi: 10.1016/0092-8674(95)90467-0. [DOI] [PubMed] [Google Scholar]
- Handa P, Roy S, Varshney U. The role of leucine 191 of Escherichia coli uracil DNA glycosylase in the formation of a highly stable complex with the substrate mimic, Ugi, and in uracil excision from the synthetic substrates. J Biol Chem. 2001;20:17324–17331. doi: 10.1074/jbc.M011166200. [DOI] [PubMed] [Google Scholar]
- Putnam CD, Shroyer MJN, Lundquist AJ, Mol CD, Arvai AS, Mosbaugh DW, Tainer JA. Protein mimicry of DNA from crystal structures of the uracil-DNA glycosylase inhibitor protein and its complex with Escherichia coli uracil-DNA glycosylase. J Mol Biol. 1999;287:331–346. doi: 10.1006/jmbi.1999.2605. [DOI] [PubMed] [Google Scholar]
- Savva R, Pearl LH. Nucleotide mimicry in the crystal structure of the uracil-DNA glycosylase-uracil glycosylase inhibitor protein complex. Nat Struct Biol. 1995;2:752–757. doi: 10.1038/nsb0995-752. [DOI] [PubMed] [Google Scholar]
- Lundquist AJ, Beger RD, Bennett SE, Bolton PH, Mosbaugh DW. Site-directed mutagenesis and characterization of uracil-DNA glycosylase inhibitor protein. J Biol Chem. 1997;272:21408–21419. doi: 10.1074/jbc.272.34.21408. [DOI] [PubMed] [Google Scholar]
- Savva R, McAuley-Hecht K, Brown T, Pearl L. The structural basis of specific base-excision repair by uracil-DNA glycosylase. Nature. 1995;373:487–493. doi: 10.1038/373487a0. [DOI] [PubMed] [Google Scholar]
- Serrano-Heras G, Salas M, Bravo A. A uracil-DNA glycosylase inhibitor encoded by a non-uracil containing viral DNA. J Biol Chem. 2006;281:7068–7074. doi: 10.1074/jbc.M511152200. [DOI] [PubMed] [Google Scholar]
- Serrano-Heras G, Ruiz-Maso JA, del Solar G, Espinosa M, Bravo A, Salas M. Protein p56 from the Bacillus subtilis phage ϕ29 inhibits DNA-binding ability of uracil-DNA glycosylase. Nucleic Acids Res. 2007;35:5393–5401. doi: 10.1093/nar/gkm584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Heras G, Bravo A, Salas M. Phage phi29 protein p56 prevents viral DNA replication impairment caused by uracil excision activity of uracil-DNA glycosylase. Proc Natl Acad Sci USA. 2008;105:19044–19049. doi: 10.1073/pnas.0808797105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H-C, Hsu K-C, Yang J-M, Wu M-L, Ko T-P, Lin S-R, Wang AH-J. Staphylococcus aureus protein SAUGI acts as a uracil-DNA glycosylase inhibitor. Nucleic Acids Res. 2014;42:1354–1364. doi: 10.1093/nar/gkt964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schormann N, Grigorian A, Samal A, Krishnan R, DeLucas L, Chattopadhyay D. Crystal structure of vaccinia virus uracil-DNA glycosylase reveals dimeric assembly. BMC Struct Biol. 2007;7:45. doi: 10.1186/1472-6807-7-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schormann N, Banerjee S, Ricciardi R, Chattopadhyay D. Structure of the uracil complex of Vaccinia virus uracil DNA glycosylase. Acta Cryst. 2013;F69:1328–1334. doi: 10.1107/S1744309113030613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaramozzino N, Sanz G, Crance JM, Saparbaev M, Drillien R, Laval J, Kavli B, Garin D. Characterisation of the substrate specificity of homogeneous vaccinia virus uracil-DNA glycosylase. Nucleic Acids Res. 2003;31:4950–4957. doi: 10.1093/nar/gkg672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duraffour S, Ishchenko AA, Saparbaev M, Crance J-M, Garin D. Substrate specificity of homogeneous monkeypox virus uracil-DNA glycosylase. Biochemistry. 2007;46:11874–11881. doi: 10.1021/bi700726a. [DOI] [PubMed] [Google Scholar]
- Krusong K, Carpenter EP, Bellamy SR, Savva R, Baldwin GS. A comparative study of uracil-DNA glycosylases from human and herpes simplex virus type 1. J Biol Chem. 2006;281:4983–4992. doi: 10.1074/jbc.M509137200. [DOI] [PubMed] [Google Scholar]
- Xiao G, Tordova M, Jagadeesh J, Drohat AC, Stivers JT, Gilliland GL. Crystal structure of Escherichia coli uracil DNA glycosylase and its complexes with uracil and glycerol: structure and glycosylase mechanism revisited. Proteins. 1999;35:13–24. [PubMed] [Google Scholar]
- Ranneberg-Nilsen T, Dale HA, Luna L, Slettebakk R, Sundheim O, Rollag H, Bjørås M. Characterization of human cytomegalovirus uracil DNA glycosylase (UL114) and its interaction with polymerase processivity factor (UL44) J Mol Biol. 2008;381:276–288. doi: 10.1016/j.jmb.2008.05.028. [DOI] [PubMed] [Google Scholar]
- Lu CC, Huang HT, Wang JT, Slupphaug G, Li TK, Wu MC, Chen YC, Lee CP, Chen MR. Characterization of the uracil-DNA glycosylase activity of Epstein-Barr virus BKRF3 and its role in lytic viral DNA replication. J Virol. 2007;81:1195–1208. doi: 10.1128/JVI.01518-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison KS, Peng W, McFadden G. Mutations in active-site residues of the uracil-DNA glycosylase encoded by vaccinia virus are incompatible with virus viability. J Virol. 1996;70:7965–7973. doi: 10.1128/jvi.70.11.7965-7973.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanitsa ES, Arps L, Traktman P. Vaccinia virus uracil DNA glycosylase interacts with the A20 protein to form a heterodimeric processivity factor for the viral DNA polymerase. J Biol Chem. 2006;281:3439–3451. doi: 10.1074/jbc.M511239200. [DOI] [PubMed] [Google Scholar]
- Boyle KA, Stanitsa ES, Greneth MD, Lindgren JK, Traktman P. Evaluation of the role of the vaccinia virus uracil DNA glycosylase and A20 proteins as intrinsic components of the DNA polymerase holoenzyme. J Biol Chem. 2011;286:24702–24713. doi: 10.1074/jbc.M111.222216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogani F, Corredeira I, Fernandez V, Sattler U, Rutvisuttinunt W, Defais M, Boehmer PE. Association between the Herpes Simplex Virus-1 DNA polymerase and uracil DNA glycosylase. J Biol Chem. 2010;285:27664–27672. doi: 10.1074/jbc.M110.131235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prichard MN, Duke GM, Mocarski ES. Human cytomegalovirus uracil DNA glycosylase is required for the normal temporal regulation of both DNA synthesis and viral replication. J Virol. 1996;70:3018–3025. doi: 10.1128/jvi.70.5.3018-3025.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prichard MN, Lawlor H, Duke GM, Mo C, Wang Z, Dixon M, Kemble G, Kern ER. Human cytomegalovirus uracil DNA glycosylase associates with ppUL44 and accelerates the accumulation of viral DNA. Virol J. 2005;2:55. doi: 10.1186/1743-422X-2-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strang BL, Coen DM. Interaction of the human cytomegalovirus uracil DNA glycosylase UL114 with the viral DNA polymerase catalytic subunit UL54. J Gen Virol. 2010;91:2029–2033. doi: 10.1099/vir.0.022160-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Silva FS, Moss B. Vaccinia virus uracil DNA glycosylase has an essential role in DNA synthesis that is independent of its glycosylase activity: catalytic site mutations reduce virulence but not virus replication in cultured cells. J Virol. 2003;77:159–166. doi: 10.1128/JVI.77.1.159-166.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druck Shudofsky AM, Silverman JEY, Chattopadhyay D, Ricciardi RP. Identification of vaccinia D4 point mutants that are defective in processivity yet retain UDG catalytic activity and are functional in A20 and DNA binding. J Virol. 2010;84:12325–12335. doi: 10.1128/JVI.01435-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sèle C, Gabel F, Gutsche I, Ivanov I, Burmeister WP, Iseni F, Tarbouriech N. Low-resolution structure of vaccinia virus DNA replication machinery. J Virol. 2013;87:1679–1689. doi: 10.1128/JVI.01533-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contesto-Richefeu C, Tarbouriech N, Brazzolotto X, Betzi S, Morelli X, Burmeister WP, Iseni F. Crystal structure of the vaccinia virus DNA polymerase holoenzyme subunit D4 in complex with the A20 N-terminal domain. PLOS Pathogens. 2014;10:e1003978. doi: 10.1371/journal.ppat.1003978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson ML, Michaels ML, Lloyd RS. Unified catalytic mechanism for DNA glycosylases. J Biol Chem. 1994;269:32709–32712. [PubMed] [Google Scholar]
- Drohat AC, Jagadeesh J, Ferguson E, Stivers JT. Role of electrophilic and general base catalysis in the mechanism of Escherichia coli uracil DNA glycosylase. Biochemistry. 1999;38:11866–11875. doi: 10.1021/bi9910878. [DOI] [PubMed] [Google Scholar]
- Stivers JT, Pankiewicz KW, Watanabe KA. Kinetic mechanism of damage site recognition and uracil flipping by Escherichia coli uracil DNA glycosylase. Biochemistry. 1999;38:952–963. doi: 10.1021/bi9818669. [DOI] [PubMed] [Google Scholar]
- Mol CD, Parikh SS, Putnam CD, Lo TP, Tainer JA. DNA repair mechanisms for the recognition of damaged DNA bases. Ann Rev Biophys Biomol Struct. 1999;28:101–128. doi: 10.1146/annurev.biophys.28.1.101. [DOI] [PubMed] [Google Scholar]
- Cortázar D, Kunz C, Saito Y, Steinacher R, Schär P. The enigmatic thymine DNA glycosylase. DNA Repair. 2007;6:489–504. doi: 10.1016/j.dnarep.2006.10.013. [DOI] [PubMed] [Google Scholar]
- Dong L, Mi R, Glass RA, Barry JN, Cao W. Repair of deaminated base damage by Schizosaccharomyces pombe thymine DNA glycosylase. DNA Repair. 2008;7:1962–1972. doi: 10.1016/j.dnarep.2008.08.006. [DOI] [PubMed] [Google Scholar]
- Dionne I, Bell SD. Characterization of an archaeal family 4 uracil DNA glycosylase and its interaction with PCNA and chromatin proteins. Biochem J. 2005;387:859–863. doi: 10.1042/BJ20041661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai A, Higuchi S, Tsunoda M, Nakamura KT, Miyamoto S. Purification, crystallization and preliminary X-ray analysis of uracil-DNA glycosylase from Sulfolobus tokodaii strain 7. Acta Cryst. 2012;F68:1102–1105. doi: 10.1107/S1744309112030278. [DOI] [PMC free article] [PubMed] [Google Scholar]