

Abstract
Normal cellular function is maintained by coordinated proteome machinery that performs a vast array of activities. Helping the proteome in such roles is the chaperome, a network of molecular chaperones and folding enzymes. The stressed cell contains, at any time, a complex mixture of chaperome complexes; a majority performs “housekeeping functions” similarly to non-stressed, normal cells, but a finely-tuned fraction buffers the proteome altered by chronic stress. The stress chaperome is epigenetically distinct from its normal, housekeeping counterpart, providing a basis for its selective targeting by small molecules. Here we discuss development of chaperome inhibitors, and how agents targeting chaperome members in stressed cells are in fact being directed towards chaperome complexes and their effect is therefore determined by their ability to sample and engage such complexes. A new approach is needed to target and implement chaperome modulators in the investigation of diseases, and we propose that the classical thinking in drug discovery needs adjustment when developing chaperome-targeting drugs.
Keywords: molecular chaperone, HSP90, HSP70, HSP60, chemical tools, epigenetic regulation
The housekeeping and the stress chaperome
Normal cellular function is maintained by coordinated proteome machinery that performs a vast array of housekeeping activities. These include transmission of signaling impulses, performing metabolic activities, replicating DNA, regulating cell motility and transporting molecules from one location to another. Helping the proteome in such diversity of roles is the chaperome. The chaperome is composed of an interconnected network of molecular chaperones as well as co-chaperones and folding enzymes that assist in their function [1]. We refer to this machinery as the housekeeping chaperome. Together with the protein degradation machinery, these molecular machines maintain cellular homeostasis [2–4].
Stresses imposed on the cell by specific imbalances and alterations in the proteome may lead to disease, which in this context, is a dysfunction of normal homeostasis. Stress on human cells, and in turn disease, is complex in nature and may result from genetic dysfunctions, invasion by a pathogen, an environmental cause or a combination of such factors. What these have in common is that they manifest by altering the proteome, which in turn perturbs the chaperome [5–10]. While often attributed to changes in molecular chaperone expression, it is now evident that expression alone is insufficient to explain the ability of the chaperome system to buffer such large variety of alterations as is characteristic of each stress [11–14]. As we are starting to appreciate, mainly from studies in cancer, chemical modifications such as post-translational modifications (PTMs) [15–18] but also biochemical modifications such as by co-chaperone and adapter protein recruitment [8, 19–24] physically alter the chaperome to modify its function. In addition to activating the chaperome, these epigenetic modifications contribute to the altered cellular location of the chaperome members, as noted in numerous diseases [21, 25–32].
We will discuss below how these epigenetic modifications provide a basis for the specific pharmacologic targeting of the chaperome in the treatment of diseases. We will also make a case that the distinct epigenetic and thermodynamic nature of the chaperome in stressed cells require a new approach into how we target and implement chaperome modulators to the investigation of diseases. We will provide lessons learned from the discovery and development of heat shock protein 90 (HSP90) inhibitors, to serve as a guideline for these efforts. Given the large array of pathologies associated with an altered function of the chaperome, we will make a case that the numerous current efforts towards the discovery of ligands, chemical tools and drugs that target chaperome members should provide both an investigational toolset for the study of the altered proteome, and in turn of diseases, and drugs to be implemented in their treatment (Figure 1).
Figure 1.
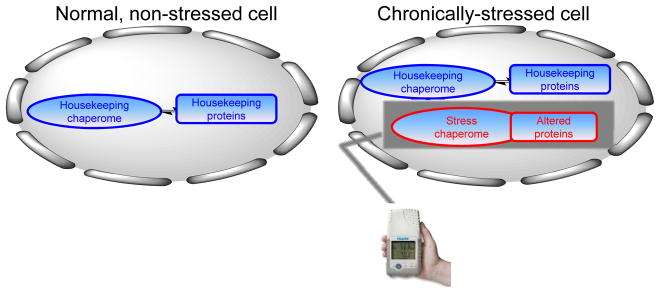
The stress-associated cell contains at any time a mixture of chaperome complexes. Some retain the nature of the “housekeeping” complexes of non-stressed, normal cells, in that they are characterized by a dynamic association between the chaperome and the proteome it regulates. Others, epigenetically and thermodynamically distinct from the “housekeeping” species, are characterized by an increased association between the chaperome and the altered proteins. Such distinct features of the stress chaperome provide the basis for their selective targeting by small molecules. Pharmacologic (chemical) tools may therefore be designed to specifically target the stress chaperome. These stress chaperome-specific chemical tools act as “sensors” of stress (depicted by the hand-held sensor device) and therefore enable to investigate, identify and treat stress-associated cellular states.
The major chaperome members
The human chaperome is composed of a large number of proteins, encoded by as many as 169 genes [1]. While a majority of the chaperome members are referred to as heat shock protein (HSP), and categorized by their molecular size, such as HSP90, HSP70, HSP60 and small HSPs [2, 3], it is estimated that only a fifth of the human chaperome (33/169 genes) is heat inducible [1]. The most studied chaperome member to date is HSP90. It is one of the most conserved HSPs found in bacteria and all eukaryotes, and is widely expressed in all cells. In humans, there are four known HSP90 members, HSP90α, HSP90β, Glucose-Regulated Protein 94 (GRP94) and Tumor Necrosis Factor Receptor-Associated Protein 1 (TRAP1) [33–35]. In normal cells, HSP90α and HSP90β reside mainly in the cytosol with minor localization in the nucleus, whereas GRP94 is found in the endoplasmic reticulum (ER) and TRAP1 in the mitochondria. Another widely expressed HSP family is the HSP70s [36–38]. The human HSP70 family contains at least eight homologous chaperone proteins. Endoplasmic reticulum (ER) and mitochondria have specific HSP70 proteins, GRP78 (also known as BIP or HSP70-5) and HSP70-9 (mtHSP70, mortalin or GRP75), respectively. The remaining members, such as HSP70-1a, HSP70-1b, HSP70-2 and Heat Shock Cognate protein 70 (HSC70), reside mainly in the cytosol and nucleus. HSP60 (chaperonin 60 or Cpn60) typically located in the mitochondria, can also be found in the cytoplasm under normal physiological conditions. It appears to function as an oligomer composed of monomers that form a complex arranged as heptameric rings [39]. For small HSPs, the current view is that they bind unfolding “client” proteins, and then these small HSP-client complexes interact with the large ATP-dependent HSPs [40, 41]. Other HSPs, such as the large HSP40 family, or DnaJs [42], and HSP110 [43] appear to serve a co-regulatory role, in that they participate in protein client regulation together with the major HSPs, and regulate several steps in their activity. These, however, also have independent functions [44, 45]. To perform their function, the chaperones, in turn, are aided by a large number of function- and complex-specific co-chaperones and adapter proteins [46–48].
Targeting the stress chaperome lessons from HSP90
The contribution of epigenetics to altering chaperome function is best understood for HSP90. In human cells, HSP90 exists in equilibrium between three conformational states, the apo-, the ADP and the ATP-bound states [35, 49]. Whereas nucleotide binding provides a modest stabilizing energy biasing HSP90 towards a particular conformation, it is now believed that the conformational fate of the chaperone is regulated by the presence of its co-partners and by the extent of PTMs [15, 50–52]. Together, these provide in the cell a balanced number of HSP90 species that are best primed to assist the need of a specific proteome. Indeed over 20 co-chaperones and a multitude of PTMs such as phosphorylation, sumoylation, methylation and acetylation have been identified that regulate HSP90 function in human cells [15, 52]. Furthermore, it is known that co-chaperones may undergo PTMs that alter their interaction with HSP90 [52]. The chaperoning of each client protein by HSP90 may require a distinct subset of these epigenetic modifiers, each driven by its specific thermodynamic need [53–55]. These epigenetic modifiers may guide HSP90 to sample multiple distinct conformations that ultimately determine the fate of bound substrate (i.e. proper folding and activation or degradation), and as we are now appreciating, the interaction of a small molecule with HSP90 [8, 56, 57].
While a limitation for classical genetic and biochemical approaches (Box 1), the complex presentation of the chaperome in disease represents an advantage for pharmacologic approaches (Figure 1). There are numerous ligands that over the years have been serendipitously or rationally discovered to interact with members of the chaperome (Figure 2). Perhaps a turning point in appreciating the importance of small molecule chemical tools in the investigation of the chaperome came from the serendipitous discovery that the natural product geldanamycin (GM) inhibited HSP90 [58]. Although the HSP90 protein has been known since the 1980s, initially drew little interest, especially as a target in disease. After all it is abundantly (~1–3% of total cellular protein) and ubiquitously expressed in most, if not all human cells, and is not particularly variable in expression between normal and cancer cells [14]. Knock-down of even 50% of HSP90 in cancer cells has little effect on their viability, whereas knock-out of at least the HSP90β paralog is embryonically lethal [59]. Such findings match poorly with the belief that a good therapeutic target has to be crucial to the malignant phenotype and be of low expression in vital organs and tissues [60]. It is GM that changed our thinking. Found in a screen searching for compounds able to revert the phenotype of cells transfected with the v-src oncogene, GM was demonstrated to do so by specifically binding to the N-terminal regulatory pocket of HSP90 and by this, to inhibit its function [58]. Low concentrations of GM were active on many cancer cells and induced differentiation, reduced cell proliferation and/or induced death, while showing no significant toxicity to normal cells [58]. Subsequent crystal structures of HSP90 in complex with GM and the regulatory nucleotides have uncovered the unique characteristics of this pocket and its high potential for druggability, further spurring the interest in HSP90 as a drug target [61]. The low toxicity seen with HSP90 inhibitors on normal cells remained unexplained for more than a decade, until Kamal et al proposed that, although no specific mutations differentiated HSP90 in normal and cancer cells, in cancer cells, the chaperone was found entirely in complexes of high affinity to small molecule inhibitors [62]. In normal cells, by contrast, a dynamic complex of HSP90, with low affinity for small molecule inhibitors was present. This mechanism provided a satisfactory explanation for the distinct sensitivity of normal and cancer cells to GM and other HSP90 inhibitors. It however fell short of explaining other observations, such as the little effect 50% reduction in HSP90 levels had on cancer cells. An explanation came eight years later when Moulick et al showed that HSP90 in cancer cells was not comprised entirely of the high affinity form, but rather it was composed of a “housekeeping HSP90” species, which had low affinity to certain small molecule inhibitors, similar to the HSP90 found in normal cells, but also of a distinct HSP90, defined as the “oncogenic HSP90” species [8]. This epigenetically distinct HSP90 comprises a functionally distinct HSP90 pool, enriched or expanded in cancer cells; cells use it to maintain the altered proteins and protein networks that are needed to drive the malignant phenotype. In this view, small molecules by their ability to interact specifically with the “oncogenic HSP90”, will primarily and selectively affect these complexes, and will act on the “housekeeping HSP90” only at higher or at saturating concentrations. By contrast, genetic targeting of HSP90 will equally reduce the expression of both “oncogenic” and “housekeeping” HSP90 pools, and thus it is conceivable that more than 50% reduction of HSP90 levels would be necessary to lower HSP90 to the threshold level required for cell survival.
Box 1. Limitations of classical approaches in the study of the chaperome in disease.
The complex presentation of the chaperome species in stressed cells helps explain the limitations of classical approaches towards understanding stress, both as it relates to the chaperome and to the proteome it regulates. Most such methods, i.e. genetic and biochemical, treat the chaperome as a monolithic entity and thus, are unable to tackle the acknowledged contribution of epigenetics to the activity of these proteins. By not differentiating between the housekeeping and the stress chaperome species, genetic manipulations silencing the HSPs are also, often, lethal [37, 59]. Alternatively, due to feedback synthesis of one HSP family member after the knock-down of another, such studies may often lead to no observable phenotypes [32, 37, 122]. Cellular manipulations that are often conducted to investigate the function of a protein and its potential interactors, i.e. by transfection of mutants, tagged proteins, or overexpression systems, are also bound to lead to “false positives” for HSPs; this is of no surprise as the chaperome is the “buffer” of cellular stress, and such manipulations, which lead to proteome stress, are likely to impose artificial interactions on HSPs with the transfected proteins. Furthermore, these chaperome complexes are likely to be cell- and type-specific, and in addition, subject to the profound implications induced by post-translational modifications. Depending on the particular cellular context, each HSP may display distinct functions such that the phenotype observed following perturbation by genetic knockdown versus small-molecule probe can be significantly different. Together, these facts help explain why information ensuing from such studies is sometimes conflicting.
Figure 2.

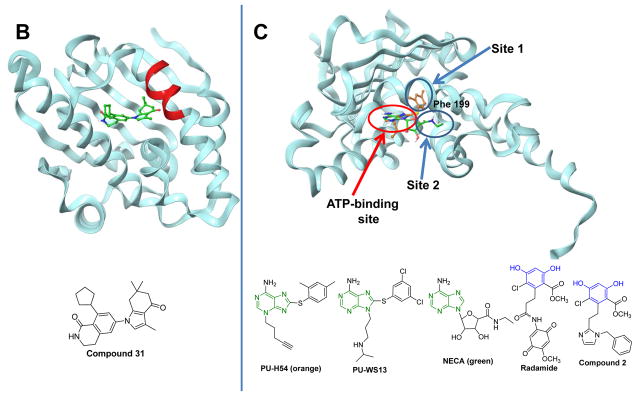


Representative HSP binders and their demonstrated or proposed mode of interaction with the protein. A. Modulators of HSP90. The ribbon representation of the full-length monomer HtpG (E. coli Hsp90, PDB: 2IOQ) is shown. Chemical structures: red, the benzoquinone/hydroquinone core; blue, the resorcinol core; green, the purine and purine-like core. *These compounds have advanced to clinical studies. B. Ribbon view of the N-terminal domain of human Hsp90α in complex with the selective modulator compound 31 (PDB: 4O0B). C. Ribbon view of the N-terminal domain of GRP94 (Canis lupus familiaris, PDB: 3O2F) in complex with the selective binder PU-H54 (Site 1). The binding mode of NECA (Site 2) and the structure of other proposed GRP94 ligands are also shown. For NECA, its complex with GRP94 (PDB: 1U2O) was overlaid with that of PU-H54 to indicate the distinction between the two binding sites. D. Various HSP70 modulators and their proposed sites of interaction with HSP70. The ribbon view of the HSP70 homolog DnaK (E. coli, PDB: 4B9Q) is shown. E. Ribbon view of GroEL (E. coli, PDB: 1XCK) and the proposed binding domains for reported HSP60 modulators. Cysteine residues (e.g. Cys442) are present only in human HSP60; Cys442 was added to indicate the potential interaction site of the ligand with the human protein.
The stressed cell (stress defined as a disease characterized by proteome alteration) therefore contains, at any time, a complex mixture of chaperome complexes; a majority performs “housekeeping functions” similarly to non-stressed, normal cells, but a finely-tuned fraction buffers the proteome altered in the process of chronic stress (Figure 1). These stress-induced and stress-associated species are epigenetically and thermodynamically different from the housekeeping chaperome. Such distinct feature provides the basis for their selective targeting by small molecules. Thus in a stressed cell, the functionally altered proteome becomes regulated by the stress chaperome; this in turn, can be selectively “sensed” by small molecules (Figure 1, depicted as a hand-held device). By chemically sensing the stress chaperome one may investigate its associated, disease-causing, proteome and thus investigate mechanisms associated with and causing the stress state [8, 28, 31, 32, 40]. By using small molecules selectively targeting the stress chaperome species as affinity-purification baits [8, 63, 64] or as global perturbers of the altered proteome [65], one may inquire into its nature, i.e. identify. By attacking specifically the stress chaperome, one may inactivate the altered proteome, and thus revert or slow the disease phenotype, i.e. treat [66–69]. A properly tailored pharmacologic toolset directed at the major stress-associated chaperome members, could enable such mechanistic (i.e. investigate), proteomic (i.e. identify) and therapeutic (i.e. treat) investigations in a large number of diseases such as cancer [41, 67, 69, 70], neurodegeneration [71–74], pathogen-induced diseases [75–79] but also, as it has recently been reported, in polycystic ovarian syndrome [80], alcoholism [81], obesity [82], psoriasis [83] and cystic fibrosis [84] among others.
Ligands targeting the major chaperones
HSP90 binders
To date, most known HSP inhibitors target HSP90. The most amenable domain of HSP90 to inhibitors has been the nucleotide binding pocket located in the N-terminal domain (Figure 2A). This nucleotide site is targeted by numerous inhibitors based on various chemotypes such as ansamycin (e.g. GM, 17-DMAG, 17-AAG, IPI-504), resorcinol (e.g. Radicicol, NVP-AUY922, AT13387, STA-9090/Ganetespib, KW-2478), purine (e.g. PU3, PU-H71, MPC-3100, CUDC-305/Debio 0932, BIIB021) and other interesting scaffolds (e.g. SNX-5422/SNX-2112, HSP990, XL888) [41, 66, 67] and some of these agents have reached clinical trials [61]. These inhibitors bind to the ATP binding pocket of HSP90 and prevent HSP90 from cycling between the ADP and ATP bound conformations thus impairing the chaperone activity [41]. Though N-terminal HSP90 inhibitors share a similar binding site their exact mode of binding may be different, and the result is the wide ranging effects on biology [8, 56]. The specific binding mode as well as a discussion on the residues that many of these compounds interact with has recently been analyzed in detail [85].
While these ligands bind with comparable affinity to the four HSP90 paralogs [85], conformational differences in the HSP90s have recently enabled the discovery of ligands selective for individual paralogs (Figure 2B,C) [32, 86, 87]. Such chemical tools with a high degree of selectivity for HSP90α/β [86, 88] and GRP94 [32] may pave the way for a better understanding of the biological role of these paralogs in specific disease contexts, as they enable parsing out their function in the context of an un-engineered human cell [32]. They may also enable the identification of disease states where inhibition of a specific HSP90 paralog may be more therapeutically advantageous than inhibition of all HSP90s.
For the less targeted C-terminal domain there are a few inhibitors reported in the literature such as celastrol and gedunin, novobiocin and derivatives such as KU174 (Figure 2A) [89]. These interact with HSP90 at non-overlapping sites as demonstrated by their ability to capture HSP90 in distinct conformational states [90].
HSP70 binders
HSP70 inhibitors reported to date target the ATP binding site, the allosteric sites in the nucleotide binding domain (NBD), the substrate binding domain (SBD) or their exact binding is still elusive (Figure 2D) [38, 41]. Adenosine-derived inhibitors were designed and synthesized using modeling and X-ray crystallographic structures leading to VER-155008 and compound 14, that, at least at biochemical level, interact selectively with HSP70 and GRP78, respectively [91, 92]. These compounds target the ATP-binding site of HSP70s, thus inhibiting their chaperone activity (blue circle, Figure 2D). Apoptozole is another compound which inhibits HSP70 activity, presumably via binding to the ATP-binding site, and this was rationalized via modeling studies to suggest that apoptozole can adopt a conformation that overlays with ATP (Figure 2D) [84]. Additionally, MKT-077 and YK5 are two compounds reported to bind to two different allosteric sites in the NBD of HSP70 [68, 93]. The allosteric site 1 (orange circle, Figure 2D) where the cationic rhodacyanine dye MKT-077 binds is negatively charged due to the presence of the side chains of Glu175, Asp199 and Asp206 [68]. On the other hand, YK5 and derivatives were rationally designed to interact with the allosteric site 2 on the NBD (green circle, Figure 2D). Additionally, myricetin [94] is also reported to bind to an allosteric site in the NBD which is adjacent to site 2. Another HSP70 domain that has been targeted is the SBD. Phenylacetylenylsulfonamide (PES, also known as pifithrin-μ) is believed to interact with HSP70 in the SBD (red circle, Figure 2D) and inhibit chaperone activity [41, 95].
HSP60 binders
There are no crystallographic studies on human HSP60, and hence the domain identification and characterization has been thus far based on the structure of GroEL, the bacterial homolog (Figure 2E). The development of HSP60 inhibitors focused primarily on the inhibition of ATP binding or targeting of the HSP60s cysteine residues [96]. As seen for HSP90s and HSP70s, inhibition of ATP binding and hydrolysis impacts the ATP-dependent conformational changes of HSP60 that are essential for its protein folding function [96]. Compounds believed to target the ATPase activity of HSP60 are the imidazole nucleoside mizoribine and the pyrazolopyrimidine derivative EC3016 reported to block ATP binding and hydrolysis, and hence affect the folding function of HSP60 [97]. Covalent binders that target the reactive cysteines of HSP60 also inhibit the chaperone activity, and one illustrative example is epolactaene which covalently binds to Cys442 (Figure 2E, equatorial domain) [39, 98].
No two HSP inhibitors are the same
Agents that are directed towards HSPs in stressed cells are in fact being directed towards HSP complexes and their effect is therefore determined by their ability to sample and engage such complexes. As mentioned above, there are numerous variables that determine the constituency of these complexes at any time. One is conformation (Figure 3). All major HSPs are known to undergo conformational changes associated with substantial rearrangements in their structure [35, 49, 50, 99]. The pocket available for ligand binding may change during these conformational states. By interacting with the protein at different sites, inhibitors sample non-overlapping conformations of the protein or freeze the protein in distinct conformations. For example, this can manifest at the local level, such as capturing changes in the structure of the protein by rearrangements of the protein in a particular domain, or more major in nature such as reflected in the major conformations noted for the HSP90s and HSP70s. Ligand-induced or captured local changes are numerous and best understood for the HSP90s and HSP70s [32, 85, 86, 100, 101]. For example, the N-terminal domain pan-HSP90 inhibitors insert themselves into the same pocket (Figure 2A). However, they take advantage of the flexibility of the residues surrounding such pocket to result in slightly distinct interaction modes. Taldone et al performed a study of the major clinical inhibitors to show that three sub-pockets, presented distinctly as the amino acid residues move during lid opening and closing, are available for these agents and that these agents differ by the sub-pockets they extend into [85]. Because these sub-pockets and their presentation are distinct among the four HSP90 paralogs, this study also provided a basis to understand the paralog binding profile observed for the numerous pan-HSP90 inhibitors. Taking advantage of these distinctions, especially in the conformational flexibility of an amino acid sequence (Figure 2B, the amino acid sequence is INNLGTIA shown in red) in the N-terminal binding pocket of HSP90α, Ernst et al developed a ligand selective for HSP90α/β [86]. This sequence is identical for HSP90α and β but differs in the first two amino acids in GRP94 (VK) and TRAP1 (VS) [88]. Testing a diverse set of HSP90 inhibitor chemotypes they noted that the compounds that use the α-helical binding conformation of HSP90α (i.e. BIIB021 and SNX-2112 analogs) show the highest degree of selectivity against GRP94 and TRAP1 [88]. Using a structure-based approach they prepared novel benzolactam derivatives that were able to access this conformation (Figure 2B) of HSP90α resulting in selective low nanomolar Hsp90 α/β inhibitors (>1000 fold selective vs. GRP94 and TRAP1) [86]. A similar approach was used by Patel et al to identify ligands selective for another HSP90 paralog, GRP94 [32]. To this end, Patel et al. used a strategy that combined library screening and structural studies to discover purine based ligands that were more than 100-fold selective for GRP94 over HSP90α/β and TRAP1 [32]. These ligands took advantage of the conformational flexibility of GRP94 to “freeze” the protein in a state that unveils a pocket possible uniquely in GRP94 due to a 5-amino acid (QEDGQ) insertion that induces a shift in a residue (Phe199) to provide access to this novel allosteric site (Site 1, Figure 2C). The study provided tool compounds such as PU-H54 and PU-WS13. Another reported GRP94 inhibitor is 5′-N-ethylcarboxamidoadenosine (NECA, Figure 2C), an adenosine A2 receptor antagonist. This compound takes advantage of a distinct GRP94 conformation which opens up another cavity that is accessed by the 5′-N-ethylcarboxamido moiety of NECA (Site 2, Figure 2C) but cannot be accessed in HSP90 [100]. Other compounds reported to bind to GRP94 in a similar manner to NECA are the resorcinol based compounds Radamide [87] and the N-benzylimidazole, Compound 2 (Site 2, Figure 2C) [102] for which selectivity was proposed using docking studies.
Figure 3.


Features that influence the efficacy (A) and the therapeutic index (B) of chaperome inhibitors. Each cell is characterized by a complex mixture of chaperome complexes; normal cells maintain the “housekeeping” chaperome complexes, whereas stressed cells are enriched in “stress” chaperome species. The constituency of these complexes is driven by the proteome, resulting in a plethora of chaperome species differentiated by affinity, conformation, number and cellular location. The conformation the HSPs adapt and the epigenetics that they display (defined as both chemical modifications by PTMs and biochemical modifications by co-chaperones and adapter proteins) results into chaperome complexes of distinct thermodynamic nature, i.e. affinity for the small molecule ligand. The proteome also dictates the number of chaperome complexes present in a cells, as well as their cellular location; these change as needed, in order to appease (or buffer) alterations as they occur in the proteome. Small molecules sample and engage these complexes over the time they spend in the cell. These characteristics help explain the two major factors that may determine the efficacy (A) and the therapeutic index (B) of chaperome inhibitors. Efficacy is regulated by how well the small molecule interacts with the relevant active stress chaperome species over the time it spends in the stressed cell (A). Therapeutic index is determined by how selective the small molecule is for the stress over the housekeeping chaperome species (B).
Interestingly, these local conformational preferences of HSP90 ligands translate toward global conformational preferences, where for example some ligands were found to prefer an open conformation of the HSP90 while others a client-protein bound, closed conformation [8, 103]. As each conformation is characterized by a distinct epigenetic profile (i.e. may harbor distinct PTMs and bind different co-chaperones and adapters), it may interact with a client protein at a distinct maturation state or may carry no protein interactor at all, the inhibitor will therefore encounter a plethora of target-containing complexes [24, 104], each having a different binding affinity for the small molecule inhibitor.
Interacting with HSP90 at sites distinct from the nucleotide-binding pocket again elicits a distinct profile, as for example seen for certain C-terminal binders such as the novobiocin-derived KU174 and celastrol (Figure 2A), which elicit and/or prefer an HSP90 conformation distinct from each other and also distinct from that induced by GM [90]. A similar view is now emerging for HSP70 where, for example, certain inhibitors such as those that act through insertion into allosteric sites located in the N-terminal domain (i.e. YK5 and MKT-077, Figure 2D), appear to sample preferentially the ADP-bound conformation [93, 101, 105, 106]. These allosteric pockets become preferentially available in this conformational state, where binding of a nucleotide exchange factor such as HSP110 stabilizes a more open N-terminal domain of HSP70 [101]. Interestingly, depending on their mode of interaction with HSP70, inhibitors also preferentially act on specific complexes, as measured by their ability to inhibit the ATPase activity of HSP70 [93, 107]. Whereas, direct ATP-pocket binders such as VER-155008 inhibit the intrinsic and the HDJ1 (an HSP40) stimulated activity of HSP70 [107], others such as YK5 appear to prefer the HSP110/HSP40 stimulated ATPase activity [93], potentially because its binding pocket is only available in the HSP110-bound conformation.
Another variable that influences HSP inhibitor activity is its sampling capacity (Figure 3). For HSP90, it has been shown that chemically distinct inhibitors that target the same binding pocket are able to select for overlapping but not identical subpopulations of HSP90 [8, 56]. GM and PU-H71 are both ATP competitive inhibitors that bind to the N-terminal nucleotide binding pocket (Figure 2A), yet PU-H71 is less affected by HSP90 phosphorylation and is capable of accessing a broader range of stress HSP90 conformations, and thus complexes [56].
A yet another important variable is cellular location (Figure 3). Altered cellular location of chaperome members has been reported in numerous diseases, although it has been mainly studied in the context of cancer [21, 25–30, 32, 108]. The ability of small molecules to engage any such complexes is highly determined by their ability to permeate cellular membranes and thus access such distinct cellular locations. Unfortunately, no study was performed to address this question. On the other hand, knowledge of the altered mitochondrial location of HSP90 in certain cancers, led the Altieri group to link GM to a mitochondria-targeting sequence [109]. This compound, gamitrinib, quickly transferred into mitochondria where it engaged both HSP90 found translocated in the mitochondria in certain cancers, and its paralog TRAP1 [109, 110].
Such complexity in the cellular presentation of HSPs helps explain why, although the majority of HSP90 inhibitors interact with its nucleotide binding pocket, the phenotype that is observed in cells [56, 66] and moreover, their in vivo properties as measured by efficacy and therapeutic index, hardly overlap among these inhibitors [66, 111–119]. There are two factors that one may analyze to understand such paradigm [66] (Figure 3). One is how well the small molecule interacts over the time it spends in the cancer cell with the relevant active HSP90 complexes (Figure 3A). Another is how selective the small molecule is for the active HSP90 complexes over those with housekeeping functions (Figure 3B). The first concept is not obvious; after all, these inhibitors occupy the same pocket. This may imply that the sampling capacity of a ligand for the stress complexes is a direct measure of its potential biological activity, i.e. potency. Evidence for this concept comes from tumor pharmacokinetics (PK) and pharmacodynamics (PD) studies. For example, while retention in the tumor mass was observed for several HSP90 inhibitors that ultimately progressed toward clinical testing, the tumor residence time was distinct among these inhibitors [111–117]. Similarly, while an extended tumor PK was noted for most such inhibitors, this did not always correlate with the extent and duration of HSP90 client downregulation, i.e. with the PD effect. The interwoven relationship between tumor PK and PD effects for an HSP90 inhibitor has long been appreciated [120, 121]. These two factors are likely affected by the HSP90 species targeted by the inhibitor. As such, the kinetics of association (kon) and more critically dissociation (koff) can vary depending on the particular complexes that the inhibitor is bound to; this, in turn, has a direct influence on the resulting PD profile and ultimately, efficacy. While in studies in tissue culture one may fully saturate the HSP90 sites in the cancer cell by adding more inhibitor, this is hardly the case in mice or humans. Human patients are not test tubes where the inhibitor is added at a constant concentration over a set period of time. The residence time of a compound is influenced by its rate of biodistribution (i.e. how fast does it reach the tumor), rate of clearance (i.e. how fast is cleared from the plasma and surrounding normal tissue) and as mentioned above, its target engagement (i.e. sampling of global HSP complexes present in the cell).
Another implication of such distinct HSP species relates to the therapeutic index of HSP targeting compounds (Figure 3B). The advantage of selectively targeting the stress chaperome versus the housekeeping chaperome is two-fold in the development of drugs. First, the better the selectivity, the less the agent will be interfering with HSP functions in normal cells, and ergo it will be less toxic. Lower toxicity means more drug can be administered to the patient, and thus, a better engagement of the target at the site of disease will be possible, ergo the better the efficacy of the agent in treating disease. Indeed, evidence from in vivo studies supports a distinct interaction of HSP90 ligands with the stress versus the housekeeping target. A general property of most HSP90 inhibitors that have shown promising in vivo activity and translated to clinic is that they are retained in tumors for prolonged periods of time while being rapidly cleared from normal tissues and plasma [111–116]. This results in long residence time of the drug selectively in the tumor and suggests that the off rate (koff) of drug bound to HSP90 in tumor tissues and normal tissues is very different and that it is much lower in tumor HSP90 (tumor koff ≪ nontumor koff). The reason for the significant difference in the koff rate between tumor and normal tissues is likely due to the stress HSP90 species that are found only in cancer cells (and other stressed cells).
Concluding remarks
The recent realization that the stress chaperome is epigenetically distinct from its normal, housekeeping counterpart has opened the door for its selective targeting by small molecules. The prospect of selectively targeting the ‘bad’ side of stress chaperones offers a novel and exciting therapeutic strategy for a range of diseases that are dependent upon their aberrant functions. This prospect is becoming more of a reality through our increasing understanding of the biochemical makeup of the HSPs that direct these two opposing functions and by ligands which have been shown to be able to discriminate between the two. A greater understanding of how epigenetic factors direct the function of HSPs and more pointedly, how they coerce them to function in a manner which is ultimately harmful to the organism (human) will continue to be a major avenue for research in the field.
Recently, there has been a multitude of ligands that have been reported to modulate various HSPs. These small molecule probes have played a vital role in teaching us the biology that we currently know. In fact, probes such as GM have enabled a chemical approach to our current understanding of the dual nature of HSP90 in a manner which genetic approaches are not able to. Although an HSP ligand may be defined as a chemical that interacts with an HSP and modulates its function, an HSP-directed chemical tool requires a more stringent selection, and criteria related to selectivity for the stress chaperome species are key in its use in dissecting mechanisms related to disease and in investigating the potential of the HSPs as targets in disease. The goal is now to translate these agents into clinically useful drugs. While no drugs directly targeting the HSPs are currently approved by the FDA, efforts during the last decade or so in the development of HSP90 inhibitors for cancer have taught us many valuable lessons that can serve as a platform for the development of molecules targeting other HSPs (Box 2). We now know that as a result of the heterogeneous presentation of HSPs in disease a more nuanced approach is required to drug development, one that allows for a clearer understanding of the potential of the ligand to target the aberrant species while minimally affecting HSPs engaged in normal proteostasis. Pre-clinically, this entails extensive testing in various transformed and nontransformed systems in order to gauge the potential of the molecule to discriminate between the various HSP species. Extensive pharmacokinetic evaluation of these agents is required in order to determine distribution into diseased tissues (e.g. tumor) as well as clearance from normal tissues (e.g. noncancerous) and to get a sense of the degree of target engagement at the site of disease and to gauge the potential for toxicity at normal tissues. The translation of these agents into the clinic will require a rethinking as to how drugs are currently developed where plasma PK is often used as a surrogate for tumor drug concentrations. Therefore, alternative noninvasive methods are required in order to determine levels of drug and target engagement at the site of HSP-drug action.
Box 2. Limitations in the use of classical medicinal chemistry approaches to the development of HSP inhibitors.
The normal paradigms of drug discovery and development do not necessarily apply towards the development of HSP agents. Traditionally, drug candidates have been optimized by improving affinity for the target (largely measured in a biochemical assay) and by modifying pharmaceutical properties (e.g. lipophilicity, permeability, metabolic stability, plasma protein binding, etc.) that seek to optimize in vivo plasma PK. Because the content-specific epigenetic modifications of HSPs cannot be recapitulated in biochemical assays, HSP drug discovery may benefit from a more elaborate approach, in which feedback between structural information, biochemical data and phenotypic assays informs on both selective and high-affinity targeting of the stress complexes. Another important factor that needs re-examining is the plasma PK. In current practice, it serves as a surrogate for tissue distribution and concentrations since it is more practical to obtain plasma samples for determination of drug concentrations. This optimization paradigm results in compounds with relatively high plasma half-life, meaning compounds remain in circulation for a long time and as such are exposed to all tissues. This is contrary to what is necessary and observed for HSP agents (HSP90 direct ATP-pocket interactors), where rapid clearance from plasma (low plasma half-life) and from normal tissues while concentrating into the tumor and engaging the tumor HSP species for prolonged period of time (high tumor half-life) are desired. Such PK profiles at the site of disease (i.e. tumors, neurodegenerative neurons) have implications for clinic, especially when it comes to the choice of dose and schedule of administration. So far, only HSP90 inhibitors that act via the ATPase pocket have moved to clinic in cancers. In preclinical tumor models, these successful candidates each show the property of area-under-the-curve (AUC)tumor ≫ AUCtissue, meaning the time residence of the inhibitor is much higher at the site of action than it is in the surrounding tissues. Such a property may obviate the need for continuous dosing and indicates that intermittent administration ensures that the tumor is continuously exposed to drug. Thus the therapeutic index of such agents may be improved by the choice of a schedule that prioritizes exposure of target at the site of action. How can stress HSPs be targeted selectively in practice so as to maximize the therapeutic benefit while minimizing the potential toxicity? An answer may be the residence time of a drug at the site of action, understood not as a ratio of AUCtumor/AUCtissue but rather as the absolute value of AUCtumor that is high and an AUCtissue that is low. As described earlier, successful HSP90 inhibitors in general exhibit high AUCtumor, however, they are likely distinguished by their relative exposures to other tissues. In fact, this variance has been attributed as the cause for ocular toxicity in some inhibitors but not others [123].
There has never been a stronger impetus towards the identification of HSPs modulators as in the past decade. Still, despite the numerous ligands discovered we are still limited by the reduced number of chemical tools and lack of approved drugs. This landscape is poised to change in the future, should we implement our recent insights into the distinct epigenetic and thermodynamic nature of the chaperome in stressed cells. Lessons learned from the development of HSP90 inhibitors have taught us that a one size fits all approach does not apply, rather a ‘personalized’ approach is required in order to decide on the best course of action. With these lessons in mind, agents that target the stress chaperome could be poised to become the next generation of drugs used to treat various diseases.
Highlights.
The stress and the housekeeping chaperome are epigenetically distinct
Small molecules may “sense” these epigenetic and thermodynamic differences
Epigenetic modifications of the stress chaperome provide the basis for selective targeting
Development of stress chaperome inhibitors require a new, stress-specific, paradigm
Acknowledgments
This work was partially funded by U01AG032969, R01CA172546, R01CA155226, R01CA187490, R21AI090501 and R21CA158609.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Finka A, Goloubinoff P. Proteomic data from human cell cultures refine mechanisms of chaperone-mediated protein homeostasis. Cell Stress Chaperones. 2013;18(5):591–605. doi: 10.1007/s12192-013-0413-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, Hartl FU. Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem. 2013;82:323–355. doi: 10.1146/annurev-biochem-060208-092442. [DOI] [PubMed] [Google Scholar]
- 3.Saibil H. Chaperone machines for protein folding, unfolding and disaggregation. Nat Rev Cancer. 2013;13:630–642. doi: 10.1038/nrm3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- 5.Hipp MS, Park SH, Hartl FU. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends in cell biology. 2014 doi: 10.1016/j.tcb.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 6.Marino-Enriquez A, Ou WB, Cowley G, Luo B, Jonker AH, Mayeda M, Okamoto M, Eilers G, Czaplinski JT, Sicinska E, et al. Genome-wide functional screening identifies CDC37 as a crucial HSP90-cofactor for KIT oncogenic expression in gastrointestinal stromal tumors. Oncogene. 2014;33(14):1872–1876. doi: 10.1038/onc.2013.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruckova E, Muller P, Nenutil R, Vojtesek B. Alterations of the Hsp70/Hsp90 chaperone and the HOP/CHIP co-chaperone system in cancer. Cellular & molecular biology letters. 2012;17(3):446–458. doi: 10.2478/s11658-012-0021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moulick K, Ahn JH, Zong H, Rodina A, Cerchietti L, Gomes DaGama EM, Caldas-Lopes E, Beebe K, Perna F, Hatzi K, et al. Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nature chemical biology. 2011;7(11):818–826. doi: 10.1038/nchembio.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaplan KB, Li R. A prescription for ‘stress’--the role of Hsp90 in genome stability and cellular adaptation. Trends in cell biology. 2012;22(11):576–583. doi: 10.1016/j.tcb.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Polier S, Samant RS, Clarke PA, Workman P, Prodromou C, Pearl LH. ATP-competitive inhibitors block protein kinase recruitment to the Hsp90-Cdc37 system. Nature chemical biology. 2013;9(5):307–312. doi: 10.1038/nchembio.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shipp C, Watson K, Jones GL. Associations of HSP90 client proteins in human breast cancer. Anticancer research. 2011;31(6):2095–2101. [PubMed] [Google Scholar]
- 12.McDowell CL, Bryan Sutton R, Obermann WM. Expression of Hsp90 chaperone [corrected] proteins in human tumor tissue. International journal of biological macromolecules. 2009;45(3):310–314. doi: 10.1016/j.ijbiomac.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 13.Muller P, Ruckova E, Halada P, Coates PJ, Hrstka R, Lane DP, Vojtesek B. C-terminal phosphorylation of Hsp70 and Hsp90 regulates alternate binding to co-chaperones CHIP and HOP to determine cellular protein folding/degradation balances. Oncogene. 2013;32(25):3101–3110. doi: 10.1038/onc.2012.314. [DOI] [PubMed] [Google Scholar]
- 14.Sahu D, Zhao Z, Tsen F, Cheng CF, Park R, Situ AJ, Dai J, Eginli A, Shams S, Chen M, et al. A potentially common peptide target in secreted heat shock protein-90alpha for hypoxia-inducible factor-1alpha-positive tumors. Molecular biology of the cell. 2012;23(4):602–613. doi: 10.1091/mbc.E11-06-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walton-Diaz A, Khan S, Bourboulia D, Trepel JB, Neckers L, Mollapour M. Contributions of co-chaperones and post-translational modifications towards Hsp90 drug sensitivity. Future medicinal chemistry. 2013;5(9):1059–1071. doi: 10.4155/fmc.13.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jakobsson ME, Moen A, Bousset L, Egge-Jacobsen W, Kernstock S, Melki R, Falnes PO. Identification and characterization of a novel human methyltransferase modulating Hsp70 protein function through lysine methylation. The Journal of biological chemistry. 2013;288(39):27752–27763. doi: 10.1074/jbc.M113.483248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cloutier P, Lavallee-Adam M, Faubert D, Blanchette M, Coulombe B. A newly uncovered group of distantly related lysine methyltransferases preferentially interact with molecular chaperones to regulate their activity. PLoS genetics. 2013;9 (1):e1003210. doi: 10.1371/journal.pgen.1003210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cho HS, Shimazu T, Toyokawa G, Daigo Y, Maehara Y, Hayami S, Ito A, Masuda K, Ikawa N, Field HI, et al. Enhanced HSP70 lysine methylation promotes proliferation of cancer cells through activation of Aurora kinase B. Nature communications. 2012;3:1072. doi: 10.1038/ncomms2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lorenz OR, Freiburger L, Rutz DA, Krause M, Zierer BK, Alvira S, Cuellar J, Valpuesta JM, Madl T, Sattler M, et al. Modulation of the Hsp90 chaperone cycle by a stringent client protein. Molecular cell. 2014;53(6):941–953. doi: 10.1016/j.molcel.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 20.Echtenkamp FJ, Freeman BC. Expanding the cellular molecular chaperone network through the ubiquitous cochaperones. Biochimica et biophysica acta. 2012;1823(3):668–673. doi: 10.1016/j.bbamcr.2011.08.011. [DOI] [PubMed] [Google Scholar]
- 21.Li J, Richter K, Reinstein J, Buchner J. Integration of the accelerator Aha1 in the Hsp90 co-chaperone cycle. Nature structural & molecular biology. 2013;20(3):326–331. doi: 10.1038/nsmb.2502. [DOI] [PubMed] [Google Scholar]
- 22.Eckl JM, Rutz DA, Haslbeck V, Zierer BK, Reinstein J, Richter K. Cdc37 (cell division cycle 37) restricts Hsp90 (heat shock protein 90) motility by interaction with N-terminal and middle domain binding sites. The Journal of biological chemistry. 2013;288(22):16032–16042. doi: 10.1074/jbc.M112.439257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith MC, Scaglione KM, Assimon VA, Patury S, Thompson AD, Dickey CA, Southworth DR, Paulson HL, Gestwicki JE, Zuiderweg ER. The E3 ubiquitin ligase CHIP and the molecular chaperone Hsc70 form a dynamic, tethered complex. Biochemistry. 2013;52(32):5354–5364. doi: 10.1021/bi4009209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rauch JN, Gestwicki JE. Binding of human nucleotide exchange factors to heat shock protein 70 (Hsp70) generates functionally distinct complexes in vitro. The Journal of biological chemistry. 2014;289(3):1402–1414. doi: 10.1074/jbc.M113.521997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cappello F, Conway de Macario E, Marasa L, Zummo G, Macario AJ. Hsp60 expression, new locations, functions and perspectives for cancer diagnosis and therapy. Cancer biology & therapy. 2008;7(6):801–809. doi: 10.4161/cbt.7.6.6281. [DOI] [PubMed] [Google Scholar]
- 26.Campanella C, Bucchieri F, Merendino AM, Fucarino A, Burgio G, Corona DF, Barbieri G, David S, Farina F, Zummo G, et al. The odyssey of Hsp60 from tumor cells to other destinations includes plasma membrane-associated stages and Golgi and exosomal protein-trafficking modalities. PloS one. 2012;7(7):e42008. doi: 10.1371/journal.pone.0042008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Butler GS, Overall CM. Proteomic identification of multitasking proteins in unexpected locations complicates drug targeting. Nature reviews Drug discovery. 2009;8(12):935–948. doi: 10.1038/nrd2945. [DOI] [PubMed] [Google Scholar]
- 28.Alarcon SV, Mollapour M, Lee MJ, Tsutsumi S, Lee S, Kim YS, Prince T, Apolo AB, Giaccone G, Xu W, et al. Tumor-intrinsic and tumor-extrinsic factors impacting hsp90- targeted therapy. Current molecular medicine. 2012;12(9):1125–1141. doi: 10.2174/156652412803306729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hua Y, White-Gilbertson S, Kellner J, Rachidi S, Usmani SZ, Chiosis G, Depinho R, Li Z, Liu B. Molecular chaperone gp96 is a novel therapeutic target of multiple myeloma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(22):6242–6251. doi: 10.1158/1078-0432.CCR-13-2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shu CW, Huang CM. HSP70s: From Tumor Transformation to Cancer Therapy. Clinical medicine Oncology. 2008;2:335–345. doi: 10.4137/cmo.s475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee AS. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nature reviews Cancer. 2014;14(4):263–276. doi: 10.1038/nrc3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patel PD, Yan P, Seidler PM, Patel HJ, Sun W, Yang C, Que NS, Taldone T, Finotti P, Stephani RA, et al. Paralog-selective Hsp90 inhibitors define tumor-specific regulation of HER2. Nature chemical biology. 2013;9(11):677–684. doi: 10.1038/nchembio.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marzec M, Eletto D, Argon Y. GRP94: An HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochimica et biophysica acta. 2012;1823(3):774–787. doi: 10.1016/j.bbamcr.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rasola A, Neckers L, Picard D. Mitochondrial oxidative phosphorylation TRAP(1)ped in tumor cells. Trends in cell biology. 2014 doi: 10.1016/j.tcb.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krukenberg KA, Street TO, Lavery LA, Agard DA. Conformational dynamics of the molecular chaperone Hsp90. Quarterly reviews of biophysics. 2011;44(2):229–255. doi: 10.1017/S0033583510000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stricher F, Macri C, Ruff M, Muller S. HSPA8/HSC70 chaperone protein: structure, function, and chemical targeting. Autophagy. 2013;9(12):1937–1954. doi: 10.4161/auto.26448. [DOI] [PubMed] [Google Scholar]
- 37.Daugaard M, Rohde M, Jaattela M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS letters. 2007;581(19):3702–3710. doi: 10.1016/j.febslet.2007.05.039. [DOI] [PubMed] [Google Scholar]
- 38.Murphy ME. The HSP70 family and cancer. Carcinogenesis. 2013;34(6):1181–1188. doi: 10.1093/carcin/bgt111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pace A, Barone G, Lauria A, Martorana A, Piccionello AP, Pierro P, Terenzi A, Almerico AM, Buscemi S, Campanella C, et al. Hsp60, a novel target for antitumor therapy: structure-function features and prospective drugs design. Current pharmaceutical design. 2013;19(15):2757–2764. doi: 10.2174/1381612811319150011. [DOI] [PubMed] [Google Scholar]
- 40.Arrigo AP, Gibert B. HspB1, HspB5 and HspB4 in Human Cancers: Potent Oncogenic Role of Some of Their Client Proteins. Cancers. 2014;6(1):333–365. doi: 10.3390/cancers6010333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jego G, Hazoume A, Seigneuric R, Garrido C. Targeting heat shock proteins in cancer. Cancer letters. 2013;332(2):275–285. doi: 10.1016/j.canlet.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 42.Sterrenberg JN, Blatch GL, Edkins AL. Human DNAJ in cancer and stem cells. Cancer letters. 2011;312(2):129–142. doi: 10.1016/j.canlet.2011.08.019. [DOI] [PubMed] [Google Scholar]
- 43.Shaner L, Morano KA. All in the family: atypical Hsp70 chaperones are conserved modulators of Hsp70 activity. Cell Stress Chaperones. 2007;12:1–8. doi: 10.1379/CSC-245R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamamoto S, Subedi GP, Hanashima S, Satoh T, Otaka M, Wakui H, Sawada K, Yokota S, Yamaguchi Y, Kubota H, et al. ATPase activity and ATP-dependent conformational change in the co-chaperone HSP70/HSP90-organizing protein (HOP) The Journal of biological chemistry. 2014;289(14):9880–9886. doi: 10.1074/jbc.M114.553255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mattoo RU, Sharma SK, Priya S, Finka A, Goloubinoff P. Hsp110 is a bona fide chaperone using ATP to unfold stable misfolded polypeptides and reciprocally collaborate with Hsp70 to solubilize protein aggregates. The Journal of biological chemistry. 2013;288(29):21399–21411. doi: 10.1074/jbc.M113.479253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Knapp RT, Wong MJ, Kollmannsberger LK, Gassen NC, Kretzschmar A, Zschocke J, Hafner K, Young JC, Rein T. Hsp70 cochaperones HspBP1 and BAG-1M differentially regulate steroid hormone receptor function. PloS one. 2014;9 (1):e85415. doi: 10.1371/journal.pone.0085415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eckl JM, Richter K. Functions of the Hsp90 chaperone system: lifting client proteins to new heights. International journal of biochemistry and molecular biology. 2013;4(4):157–165. [PMC free article] [PubMed] [Google Scholar]
- 48.Smith DF. Dynamics of heat shock protein 90-progesterone receptor binding and the disactivation loop model for steroid receptor complexes. Molecular endocrinology. 1993;7(11):1418–1429. doi: 10.1210/mend.7.11.7906860. [DOI] [PubMed] [Google Scholar]
- 49.Li J, Buchner J. Structure, function and regulation of the hsp90 machinery. Biomedical journal. 2013;36(3):106–117. doi: 10.4103/2319-4170.113230. [DOI] [PubMed] [Google Scholar]
- 50.Prodromou C. The ‘active life’ of Hsp90 complexes. Biochim Biophys Acta. 2012;1823:614–623. doi: 10.1016/j.bbamcr.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li J, Soroka J, Buchner J. The Hsp90 chaperone machinery: conformational dynamics and regulation by co-chaperones. Biochim Biophys Acta. 2012;1823:624–635. doi: 10.1016/j.bbamcr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 52.Mollapour M, Neckers L. Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim Biophys Acta. 2012;1823:648–655. doi: 10.1016/j.bbamcr.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kancha RK, Bartosch N, Duyster J. Analysis of conformational determinants underlying HSP90-kinase interaction. PloS one. 2013;8(7):e68394. doi: 10.1371/journal.pone.0068394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taipale M, Krykbaeva I, Whitesell L, Santagata S, Zhang J, Liu Q, Gray NS, Lindquist S. Chaperones as thermodynamic sensors of drug-target interactions reveal kinase inhibitor specificities in living cells. Nature biotechnology. 2013;31(7):630–637. doi: 10.1038/nbt.2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lambert JP, Ivosev G, Couzens AL, Larsen B, Taipale M, Lin ZY, Zhong Q, Lindquist S, Vidal M, Aebersold R, et al. Mapping differential interactomes by affinity purification coupled with data-independent mass spectrometry acquisition. Nature methods. 2013;10(12):1239–1245. doi: 10.1038/nmeth.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beebe K, Mollapour M, Scroggins B, Prodromou C, Xu W, Tokita M, Taldone T, Pullen L, Zierer BK, Lee MJ, et al. Posttranslational modification and conformational state of Heat Shock Protein 90 differentially affect binding of chemically diverse small molecule inhibitors. Oncotarget. 2013;4(7):1065–1074. doi: 10.18632/oncotarget.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mollapour M, Bourboulia D, Beebe K, Woodford MR, Polier S, Hoang A, Chelluri R, Li Y, Guo A, Lee MJ, et al. Asymmetric Hsp90 N domain SUMOylation recruits Aha1 and ATP-competitive inhibitors. Molecular cell. 2014;53(2):317–329. doi: 10.1016/j.molcel.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Neckers L, Schulte TW, Mimnaugh E. Geldanamycin as a potential anti-cancer agent: its molecular target and biochemical activity. Invest New Drugs. 1999;17(4):361–373. doi: 10.1023/a:1006382320697. [DOI] [PubMed] [Google Scholar]
- 59.Voss AK, Thomas T, Gruss P. Mice lacking HSP90beta fail to develop a placental labyrinth. Development. 2000;127(1):1–11. doi: 10.1242/dev.127.1.1. [DOI] [PubMed] [Google Scholar]
- 60.Ross JS, Schenkein DP, Pietrusko R, Rolfe M, Linette GP, Stec J, Stagliano NE, Ginsburg GS, Symmans WF, Pusztai L, et al. Targeted therapies for cancer 2004. American journal of clinical pathology. 2004;122(4):598–609. doi: 10.1309/5CWP-U41A-FR1V-YM3F. [DOI] [PubMed] [Google Scholar]
- 61.Jhaveri K, Ochiana SO, Dunphy MP, Gerecitano JF, Corben AD, Peter RI, Janjigian YY, Gomes-DaGama EM, Koren J, 3rd, Modi S, et al. Heat shock protein 90 inhibitors in the treatment of cancer: current status and future directions. Expert opinion on investigational drugs. 2014;23(5):611–628. doi: 10.1517/13543784.2014.902442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, Burrows FJ. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425(6956):407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 63.Nayar U, Lu P, Goldstein RL, Vider J, Ballon G, Rodina A, Taldone T, Erdjument-Bromage H, Chomet M, Blasberg R, et al. Targeting the Hsp90-associated viral oncoproteome in gammaherpesvirus-associated malignancies. Blood. 2013;122(16):2837–2847. doi: 10.1182/blood-2013-01-479972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rodina A, Taldone T, Kang Y, Patel PD, Koren J, 3rd, Yan P, DaGama Gomes EM, Yang C, Patel MR, Shrestha L, et al. Affinity Purification Probes of Potential Use To Investigate the Endogenous Hsp70 Interactome in Cancer. ACS chemical biology. 2014 doi: 10.1021/cb500256u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sharma K, Vabulas RM, Macek B, Pinkert S, Cox J, Mann M, Hartl FU. Quantitative proteomics reveals that Hsp90 inhibition preferentially targets kinases and the DNA damage response. Molecular & cellular proteomics : MCP. 2012;11(3):M111 014654. doi: 10.1074/mcp.M111.014654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Patel HJ, Modi S, Chiosis G, Taldone T. Advances in the discovery and development of heat-shock protein 90 inhibitors for cancer treatment. Expert opinion on drug discovery. 2011;6(5):559–587. doi: 10.1517/17460441.2011.563296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jhaveri K, Taldone T, Modi S, Chiosis G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochimica et biophysica acta. 2012;1823(3):742–755. doi: 10.1016/j.bbamcr.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rousaki A, Miyata Y, Jinwal UK, Dickey CA, Gestwicki JE, Zuiderweg ER. Allosteric drugs: the interaction of antitumor compound MKT-077 with human Hsp70 chaperones. Journal of molecular biology. 2011;411(3):614–632. doi: 10.1016/j.jmb.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang X, Chen M, Zhou J, Zhang X. HSP27, 70 and 90, anti-apoptotic proteins, in clinical cancer therapy (Review) International journal of oncology. 2014;45(1):18–30. doi: 10.3892/ijo.2014.2399. [DOI] [PubMed] [Google Scholar]
- 70.Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18(1):64–76. doi: 10.1158/1078-0432.CCR-11-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Carman A, Kishinevsky S, Koren J, 3rd, Luo W, Chiosis G. Regulatory chaperone complexes in neurodegenerative diseases: a perspective on therapeutic intervention. Current Alzheimer research. 2014;11(1):59–68. doi: 10.2174/1567205010666131119233044. [DOI] [PubMed] [Google Scholar]
- 72.Silva-Fernandes A, Duarte-Silva S, Neves-Carvalho A, Amorim M, Soares-Cunha C, Oliveira P, Thirstrup K, Teixeira-Castro A, Maciel P. Chronic treatment with 17-DMAG improves balance and coordination in a new mouse model of Machado-Joseph disease. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics. 2014;11(2):433–449. doi: 10.1007/s13311-013-0255-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McFarland NR, Dimant H, Kibuuka L, Ebrahimi-Fakhari D, Desjardins CA, Danzer KM, Danzer M, Fan Z, Schwarzschild MA, Hirst W, et al. Chronic treatment with novel small molecule Hsp90 inhibitors rescues striatal dopamine levels but not alpha-synuclein-induced neuronal cell loss. PloS one. 2014;9(1):e86048. doi: 10.1371/journal.pone.0086048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Blair LJ, Nordhues BA, Hill SE, Scaglione KM, O’Leary JC, 3rd, Fontaine SN, Breydo L, Zhang B, Li P, Wang L, et al. Accelerated neurodegeneration through chaperone-mediated oligomerization of tau. The Journal of clinical investigation. 2013;123(10):4158–4169. doi: 10.1172/JCI69003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gao J, Xiao S, Liu X, Wang L, Zhang X, Ji Q, Wang Y, Mo D, Chen Y. Inhibition of HSP90 attenuates porcine reproductive and respiratory syndrome virus production in vitro. Virology journal. 2014;11:17. doi: 10.1186/1743-422X-11-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sun X, Bristol JA, Iwahori S, Hagemeier SR, Meng Q, Barlow EA, Fingeroth JD, Tarakanova VL, Kalejta RF, Kenney SC. Hsp90 inhibitor 17-DMAG decreases expression of conserved herpesvirus protein kinases and reduces virus production in Epstein-Barr virus-infected cells. Journal of virology. 2013;87(18):10126–10138. doi: 10.1128/JVI.01671-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Anderson I, Low JS, Weston S, Weinberger M, Zhyvoloup A, Labokha AA, Corazza G, Kitson RA, Moody CJ, Marcello A, et al. Heat shock protein 90 controls HIV-1 reactivation from latency. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(15):E1528–1537. doi: 10.1073/pnas.1320178111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Geller R, Taguwa S, Frydman J. Broad action of Hsp90 as a host chaperone required for viral replication. Biochimica et biophysica acta. 2012;1823(3):698–706. doi: 10.1016/j.bbamcr.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.O’Meara TR, Cowen LE. Hsp90-dependent regulatory circuitry controlling temperature-dependent fungal development and virulence. Cellular microbiology. 2014;16(4):473–481. doi: 10.1111/cmi.12266. [DOI] [PubMed] [Google Scholar]
- 80.Seeger-Nukpezah T, Proia DA, Egleston BL, Nikonova AS, Kent T, Cai KQ, Hensley HH, Ying W, Chimmanamada D, Serebriiskii IG, et al. Inhibiting the HSP90 chaperone slows cyst growth in a mouse model of autosomal dominant polycystic kidney disease. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(31):12786–12791. doi: 10.1073/pnas.1301904110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ambade A, Catalano D, Lim A, Kopoyan A, Shaffer SA, Mandrekar P. Pharmacological Inhibition of Heat Shock Protein 90 Alleviates Steatosis and Macrophage Activation in Murine Acute and Chronic Alcoholic Liver Injury. Journal of hepatology. 2014 doi: 10.1016/j.jhep.2014.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Desarzens S, Liao WH, Mammi C, Caprio M, Faresse N. Hsp90 blockers inhibit adipocyte differentiation and fat mass accumulation. PloS one. 2014;9(4):e94127. doi: 10.1371/journal.pone.0094127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stenderup K, Rosada C, Gavillet B, Vuagniaux G, Dam TN. Debio 0932, A New Oral Hsp90 Inhibitor, Alleviates Psoriasis in a Xenograft Transplantation Model. Acta dermato-venereologica. 2014 doi: 10.2340/00015555-1838. [DOI] [PubMed] [Google Scholar]
- 84.Cho HJ, Gee HY, Baek KH, Ko SK, Park JM, Lee H, Kim ND, Lee MG, Shin I. A small molecule that binds to an ATPase domain of Hsc70 promotes membrane trafficking of mutant cystic fibrosis transmembrane conductance regulator. Journal of the American Chemical Society. 2011;133(50):20267–20276. doi: 10.1021/ja206762p. [DOI] [PubMed] [Google Scholar]
- 85.Taldone T, Patel PD, Patel M, Patel HJ, Evans CE, Rodina A, Ochiana S, Shah SK, Uddin M, Gewirth D, et al. Experimental and structural testing module to analyze paralogue-specificity and affinity in the Hsp90 inhibitors series. J Med Chem. 2013;56(17):6803–6818. doi: 10.1021/jm400619b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ernst JT, Neubert T, Liu M, Sperry S, Zuccola H, Turnbull A, Fleck B, Kargo W, Woody L, Chiang P, et al. Identification of novel HSP90alpha/beta isoform selective inhibitors using structure-based drug design. demonstration of potential utility in treating CNS disorders such as Huntington’s disease. Journal of medicinal chemistry. 2014;57(8):3382–3400. doi: 10.1021/jm500042s. [DOI] [PubMed] [Google Scholar]
- 87.Immormino RM, Metzger LEt, Reardon PN, Dollins DE, Blagg BS, Gewirth DT. Different poses for ligand and chaperone in inhibitor-bound Hsp90 and GRP94: implications for paralog-specific drug design. Journal of molecular biology. 2009;388(5):1033–1042. doi: 10.1016/j.jmb.2009.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ernst JT, Liu M, Zuccola H, Neubert T, Beaumont K, Turnbull A, Kallel A, Vought B, Stamos D. Correlation between chemotype-dependent binding conformations of HSP90alpha/beta and isoform selectivity-Implications for the structure-based design of HSP90alpha/beta selective inhibitors for treating neurodegenerative diseases. Bioorganic & medicinal chemistry letters. 2014;24(1):204–208. doi: 10.1016/j.bmcl.2013.11.036. [DOI] [PubMed] [Google Scholar]
- 89.Gomez-Monterrey I, Sala M, Musella S, Campiglia P. Heat shock protein 90 inhibitors as therapeutic agents. Recent patents on anti-cancer drug discovery. 2012;7(3):313–336. doi: 10.2174/157489212801820066. [DOI] [PubMed] [Google Scholar]
- 90.Matts RL, Brandt GE, Lu Y, Dixit A, Mollapour M, Wang S, Donnelly AC, Neckers L, Verkhivker G, Blagg BS. A systematic protocol for the characterization of Hsp90 modulators. Bioorganic & medicinal chemistry. 2011;19(1):684–692. doi: 10.1016/j.bmc.2010.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Williamson DS, Borgognoni J, Clay A, Daniels Z, Dokurno P, Drysdale MJ, Foloppe N, Francis GL, Graham CJ, Howes R, et al. Novel adenosine-derived inhibitors of 70 kDa heat shock protein, discovered through structure-based design. Journal of medicinal chemistry. 2009;52(6):1510–1513. doi: 10.1021/jm801627a. [DOI] [PubMed] [Google Scholar]
- 92.Macias AT, Williamson DS, Allen N, Borgognoni J, Clay A, Daniels Z, Dokurno P, Drysdale MJ, Francis GL, Graham CJ, et al. Adenosine-derived inhibitors of 78 kDa glucose regulated protein (Grp78) ATPase: insights into isoform selectivity. Journal of medicinal chemistry. 2011;54(12):4034–4041. doi: 10.1021/jm101625x. [DOI] [PubMed] [Google Scholar]
- 93.Rodina A, Patel PD, Kang Y, Patel Y, Baaklini I, Wong MJ, Taldone T, Yan P, Yang C, Maharaj R, et al. Identification of an allosteric pocket on human hsp70 reveals a mode of inhibition of this therapeutically important protein. Chemistry & biology. 2013;20(12):1469–1480. doi: 10.1016/j.chembiol.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chang L, Miyata Y, Ung PM, Bertelsen EB, McQuade TJ, Carlson HA, Zuiderweg ER, Gestwicki JE. Chemical screens against a reconstituted multiprotein complex: myricetin blocks DnaJ regulation of DnaK through an allosteric mechanism. Chemistry & biology. 2011;18(2):210–221. doi: 10.1016/j.chembiol.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Leu JI, Pimkina J, Frank A, Murphy ME, George DL. A small molecule inhibitor of inducible heat shock protein 70. Molecular cell. 2009;36(1):15–27. doi: 10.1016/j.molcel.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cappello F, Marino Gammazza A, Palumbo Piccionello A, Campanella C, Pace A, Conway de Macario E, Macario AJ. Hsp60 chaperonopathies and chaperonotherapy: targets and agents. Expert opinion on therapeutic targets. 2014;18(2):185–208. doi: 10.1517/14728222.2014.856417. [DOI] [PubMed] [Google Scholar]
- 97.Chapman E, Farr GW, Furtak K, Horwich AL. A small molecule inhibitor selective for a variant ATP-binding site of the chaperonin GroEL. Bioorganic & medicinal chemistry letters. 2009;19(3):811–813. doi: 10.1016/j.bmcl.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nagumo Y, Kakeya H, Shoji M, Hayashi Y, Dohmae N, Osada H. Epolactaene binds human Hsp60 Cys442 resulting in the inhibition of chaperone activity. Biochem J. 2005;387(Pt 3):835–840. doi: 10.1042/BJ20041355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kityk R, Kopp J, Sinning I, Mayer MP. Structure and dynamics of the ATP-bound open conformation of Hsp70 chaperones. Molecular cell. 2012;48(6):863–874. doi: 10.1016/j.molcel.2012.09.023. [DOI] [PubMed] [Google Scholar]
- 100.Soldano KL, Jivan A, Nicchitta CV, Gewirth DT. Structure of the N-terminal domain of GRP94. Basis for ligand specificity and regulation. The Journal of biological chemistry. 2003;278(48):48330–48338. doi: 10.1074/jbc.M308661200. [DOI] [PubMed] [Google Scholar]
- 101.Zuiderweg ER, Bertelsen EB, Rousaki A, Mayer MP, Gestwicki JE, Ahmad A. Allostery in the Hsp70 chaperone proteins. Topics in current chemistry. 2013;328:99–153. doi: 10.1007/128_2012_323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Duerfeldt AS, Peterson LB, Maynard JC, Ng CL, Eletto D, Ostrovsky O, Shinogle HE, Moore DS, Argon Y, Nicchitta CV, et al. Development of a Grp94 inhibitor. Journal of the American Chemical Society. 2012;134(23):9796–9804. doi: 10.1021/ja303477g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nishiya Y, Shibata K, Saito S, Yano K, Oneyama C, Nakano H, Sharma SV. Drug-target identification from total cellular lysate by drug-induced conformational changes. Analytical biochemistry. 2009;385(2):314–320. doi: 10.1016/j.ab.2008.11.034. [DOI] [PubMed] [Google Scholar]
- 104.Ebong IO, Morgner N, Zhou M, Saraiva MA, Daturpalli S, Jackson SE, Robinson CV. Heterogeneity and dynamics in the assembly of the heat shock protein 90 chaperone complexes. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(44):17939–17944. doi: 10.1073/pnas.1106261108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kang Y, Taldone T, Patel HJ, Patel PD, Rodina A, Gozman A, Maharaj R, Clement CC, Patel MR, Brodsky JL, et al. Heat shock protein 70 inhibitors. 1. 2,5′-thiodipyrimidine and 5-(phenylthio)pyrimidine acrylamides as irreversible binders to an allosteric site on heat shock protein 70. Journal of medicinal chemistry. 2014;57(4):1188–1207. doi: 10.1021/jm401551n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Taldone T, Kang Y, Patel HJ, Patel MR, Patel PD, Rodina A, Patel Y, Gozman A, Maharaj R, Clement CC, et al. Heat shock protein 70 inhibitors. 2. 2,5′-thiodipyrimidines, 5-(phenylthio)pyrimidines, 2-(pyridin-3-ylthio)pyrimidines, and 3-(phenylthio)pyridines as reversible binders to an allosteric site on heat shock protein 70. Journal of medicinal chemistry. 2014;57(4):1208–1224. doi: 10.1021/jm401552y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schlecht R, Scholz SR, Dahmen H, Wegener A, Sirrenberg C, Musil D, Bomke J, Eggenweiler HM, Mayer MP, Bukau B. Functional analysis of Hsp70 inhibitors. PloS one. 2013;8(11):e78443. doi: 10.1371/journal.pone.0078443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lee AS. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer. 2014;14(4):263–276. doi: 10.1038/nrc3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kang BH, Plescia J, Dohi T, Rosa J, Doxsey SJ, Altieri DC. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell. 2007;131:257–270. doi: 10.1016/j.cell.2007.08.028. [DOI] [PubMed] [Google Scholar]
- 110.Altieri DC, Stein GS, Lian JB, Languino LR. TRAP-1, the mitochondrial Hsp90. Biochimica et biophysica acta. 2012;1823(3):767–773. doi: 10.1016/j.bbamcr.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Caldas-Lopes E, Cerchietti L, Ahn JH, Clement CC, Robles AI, Rodina A, Moulick K, Taldone T, Gozman A, Guo Y, et al. Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(20):8368–8373. doi: 10.1073/pnas.0903392106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bao R, Lai CJ, Qu H, Wang D, Yin L, Zifcak B, Atoyan R, Wang J, Samson M, Forrester J, et al. CUDC-305, a novel synthetic HSP90 inhibitor with unique pharmacologic properties for cancer therapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15(12):4046–4057. doi: 10.1158/1078-0432.CCR-09-0152. [DOI] [PubMed] [Google Scholar]
- 113.Lundgren K, Zhang H, Brekken J, Huser N, Powell RE, Timple N, Busch DJ, Neely L, Sensintaffar JL, Yang YC, et al. BIIB021, an orally available, fully synthetic small-molecule inhibitor of the heat shock protein Hsp90. Mol Cancer Ther. 2009;8(4):921–929. doi: 10.1158/1535-7163.MCT-08-0758. [DOI] [PubMed] [Google Scholar]
- 114.Chandarlapaty S, Sawai A, Ye Q, Scott A, Silinski M, Huang K, Fadden P, Partdrige J, Hall S, Steed P, et al. SNX2112, a synthetic heat shock protein 90 inhibitor, has potent antitumor activity against HER kinase-dependent cancers. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14(1):240–248. doi: 10.1158/1078-0432.CCR-07-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jensen MR, Schoepfer J, Radimerski T, Massey A, Guy CT, Brueggen J, Quadt C, Buckler A, Cozens R, Drysdale MJ, et al. NVP-AUY922: a small molecule HSP90 inhibitor with potent antitumor activity in preclinical breast cancer models. Breast Cancer Res. 2008;10(2):R33. doi: 10.1186/bcr1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shimamura T, Perera SA, Foley KP, Sang J, Rodig SJ, Inoue T, Chen L, Li D, Carretero J, Li YC, et al. Ganetespib (STA-9090), a nongeldanamycin HSP90 inhibitor, has potent antitumor activity in in vitro and in vivo models of non-small cell lung cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18(18):4973–4985. doi: 10.1158/1078-0432.CCR-11-2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Graham B, Curry J, Smyth T, Fazal L, Feltell R, Harada I, Coyle J, Williams B, Reule M, Angove H, et al. The heat shock protein 90 inhibitor, AT13387, displays a long duration of action in vitro and in vivo in non-small cell lung cancer. Cancer Sci. 2012;103(3):522–527. doi: 10.1111/j.1349-7006.2011.02191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Massey AJ, Schoepfer J, Brough PA, Brueggen J, Chene P, Drysdale MJ, Pfaar U, Radimerski T, Ruetz S, Schweitzer A, et al. Preclinical antitumor activity of the orally available heat shock protein 90 inhibitor NVP-BEP800. Mol Cancer Ther. 2010;9(4):906–919. doi: 10.1158/1535-7163.MCT-10-0055. [DOI] [PubMed] [Google Scholar]
- 119.Ono N, Yamazaki T, Nakanishi Y, Fujii T, Sakata K, Tachibana Y, Suda A, Hada K, Miura T, Sato S, et al. Preclinical antitumor activity of the novel heat shock protein 90 inhibitor CH5164840 against human epidermal growth factor receptor 2 (HER2)-overexpressing cancers. Cancer Sci. 2012;103(2):342–349. doi: 10.1111/j.1349-7006.2011.02144.x. [DOI] [PubMed] [Google Scholar]
- 120.Workman P. Auditing the pharmacological accounts for Hsp90 molecular chaperone inhibitors: unfolding the relationship between pharmacokinetics and pharmacodynamics. Mol Cancer Ther. 2003;2(2):131–138. [PubMed] [Google Scholar]
- 121.Vilenchik M, Solit D, Basso A, Huezo H, Lucas B, He H, Rosen N, Spampinato C, Modrich P, Chiosis G. Targeting wide-range oncogenic transformation via PU24FCl, a specific inhibitor of tumor Hsp90. Chem Biol. 2004;11(6):787–797. doi: 10.1016/j.chembiol.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 122.Havik B, Bramham CR. Additive viability-loss following hsp70/hsc70 double interference and Hsp90 inhibition in two breast cancer cell lines. Oncology reports. 2007;17(6):1501–1510. [PubMed] [Google Scholar]
- 123.Zhou D, Liu Y, Ye J, Ying W, Ogawa LS, Inoue T, Tatsuta N, Wada Y, Koya K, Huang Q, et al. A rat retinal damage model predicts for potential clinical visual disturbances induced by Hsp90 inhibitors. Toxicology and applied pharmacology. 2013;273(2):401–409. doi: 10.1016/j.taap.2013.09.018. [DOI] [PubMed] [Google Scholar]