

Abstract
It will soon be 50 years since the first MHC associations with human disease were described. These seminal studies opened a flourishing area of research, yet much remains to be discovered. Genome-wide association studies of autoimmune diseases have demonstrated that the MHC region has effect sizes that supersede those for any non-MHC locus for most diseases. Thus, an understanding of how particular MHC alleles confer susceptibility will be essential for a comprehensive understanding of autoimmune disease pathogenesis. Here we review recent exciting findings in this important field.
Part I: Genetics
Associations with MHC class I, class II or both
A striking feature is that autoimmune diseases with characteristic autoantibodies are typically associated with MHC class II alleles, whereas seronegative diseases most often are associated with MHC class I alleles. Still many diseases demonstrate multiple MHC associations, in part due to the high degree of linkage disequilibrium in the MHC locus. For instance, diseases like celiac disease, type 1 diabetes and autoimmune thyroid diseases are associated with the HLA-DR3-DQ2 haplotype yet also have associations to the class I alleles encoding HLA-B8 and HLA-A1 which are part of an extended, conserved haplotype. This has given rise to the notion of primary and secondary disease associations, with interest focused on the primary associations. However, by studying large cohorts of patients and ethnically matched controls and carefully controlling for linkage disequilibria, it has become clear that there are often multiple independent associations with alleles of different HLA loci. This has been demonstrated for type 1 diabetes where, in addition to the established association with the MHC class II genes, independent contributions were mapped to the MHC class I genes HLA-B and HLA-A, in particular HLA-B*39[1]. In multiple sclerosis it was shown that variation in the HLA-A, gene is esponsible for an independent protective effect [2]. This protective effect was mapped to HLA-A*02:01, and evidence for an independent signal in a region that does not contain any MHC class I or II genes points to involvement of non-HLA risk allele(s) within the MHC *[3]. Analysis of SLE similarly showed that the best model for SLE association includes both classical loci HLA-DRB1*03:01, HLA-DRB1*08:01, and HLA-DQA1*01:02, as well as two SNPs, one of which is located in the class III region [4]. Notably, it common that alleles conferring susceptibility and protection are both detected, illustrating the complex role of MHC genes in human diseases.
Epistatic interaction of MHC with aminopeptidease genes in class I associated diseases
For several MHC class I associated diseases a new avenue of research has been opened involving aminopeptidases. Genome wide association studies have revealed association between polymorphisms in the aminopeptidase gene ERAP1 and ankylosing spondylitis which is restricted to HLA-B27–positive disease [5]. Recent analysis using the so-called Immunochip further revealed association of ankylosing spondylitis with three additional aminopeptidase genes (LNPEP and ERAP2 located in the same region as ERPA1 as well NPEPPS which is located on another chromosome) [6]. Analysis of the impact of non-synonymous substitutions in has revealed clear effects on enzyme activity for generation of ligands that can bind HLA-B27, although the effects appear to be complex and depend on many factors including the sequence of the peptide substrates (reviewed in [7,8]). Notwithstanding, it seems reasonable that genetic variation in these aminopeptidases impact susceptibility to ankylosing spondylitis by affecting the peptide ligands of HLA-B27 either quantitatively or qualitatively, thereby impacting antigen presentation to CD8 T cells. It could also affect folding and stability of HLA-B27 which is ligand dependent. Impaired folding/stability could eventually lead to increased production of IL-23 and IL-17 by endoplasmic stress responses [9] or recognition of HLA-B27 heavy chain homodimers on the cell surface by KIR receptors [10].
The ERAP1 gene is also associated with susceptibility to psoriasis and Behçet's disease, and in both diseases there is evidence for similar epistatic interactions between MHC and ERAP1 as observed in ankylosing spondylitis. In psoriasis, the epistatic interaction is with HLA-C (particularly HLA-C**06:02)[11]and in Behçet's disease with HLA-B*51 *[12]. In psoriasis Immunochip analysis revealed that ERAP1 and ERAP2 polymorphisms give independent signals of association [13]. Importantly, the epistatic interaction between ERAP1 and MHC suggests that these genes act along the same pathogenic pathway underscoring that generation of peptide ligands for binding to MHC class I proteins is a key step in the pathogenesis of these diseases.
HLA dependent autoantibodies
Several studies have highlighted the importance of HLA class II polymorphisms with emergence of autoantibodies. This is well known in celiac disease where antibody formation to the autoantigen transglutaminase 2 is strictly dependent on the subject being positive for HLA-DQ2 or HLA-DQ8 [14]. Studies of HLA association in seropositive and seronegative rheumatoid arthritis patients have demonstrated that anti-citrullinated protein antibody (ACPA) positive and negative rheumatoid arthritis are genetically distinct [15]. Similarly, anti-neutrophil cytoplasmic antibody (ANCA) associated vasculitis seems to consist of two genetically distinct subsets as myeloperoxidase-specific ANCAs are primarily associated with HLA-DQ polymorphisms whereas proteinase 3-specific ANCAs are primarily associated with HLA-DP polymorphisms [16]. In SLE the association of HLA-DRB1* 03:01 with anti-Ro and anti-La antibody-positive SLE is much stronger than in SLE without these autoantibodies [17]. Analyzing for association of islet autoantibodies in type 1 diabetes with HLA polymorphisms, it was found that particular antibody specificities are associated with different HLA genes [18]. Specifically, development of antibodies to GAD65 is primarily associated with HLA-DQB1, while antibodies to IA-2A are most strongly associated with HLA-DRB1. These data suggest that distinct T cell specificities account for association of different HLA alleles with particular autoantibodies. Also, there is disease heterogeneity with qualitative different immune responses contributing to the pathogenesis.
Part II: Effect of key polymorphisms on MHC structure and peptide specificity
In many autoimmune diseases, susceptibility is associated with particular HLA allele(s), but not closely related alleles that differ only at a few positions in the peptide binding groove. Such comparisons among disease-associated and non-associated alleles have enabled definition of key structural features, with a classical example being the involvement of a non-aspartic acid at HLA-DQ β57 in type 1 diabetes [19]. More recently, massive SNP typing has allowed analysis of associations with disease not only by allele, but also by comparing individual amino acids at each position of a classical MHC molecule.
In 1987, Gregersen and Winchester proposed the ‘shared epitope’ hypothesis for HLA-DR alleles associated with susceptibility to rheumatoid arthritis, which highlighted the importance of residues on the DRβ chain helix, including DRβ 71-74 [20]. A recent study examined genome-wide SNP data on large patient populations to identify key polymorphic MHC residues in this disease *[21]. Three amino acid positions in DRβ1 (11, 71 and 74) as well as singleamino acid polymorphisms in HLA-B (pos. 9) and HLA-DPβ1 (pos. 9) were concluded to almost completely explain the MHC association. Interestingly, the most significant association was observed for DRB1 codon 11, for which aliphatic residues (Val and Leu) are associated with risk to rheumatoid arthritis, while a polar residue (Ser) is highly protective. Position 11 is in tight linkage disequilibrium with position 13, but conditioning on position 11 eliminated with effect of position 13 (but not vice versa). Significant associations were also observed for position 71 (Lys, Arg increase, Glu reduces risk), as well as position 74 (only Ala increases risk). DRβ 71 and 74 shape the P4 pocket of the groove, while DRβ 11 and 13 are located in the neighboring P6 pocket (Figure 1a-c). The polymorphisms in the P4 pocket are relevant for the binding of citrullinated peptides, as described below. The functional role of polymorphisms in the P6 pocket remains to be determined.
Figure 1. HLA-DR polymorphisms associated with the pathogenesis of RA and MS.

(a-c) Key polymorphic DRβ chain residues associated with RA localize to two neighboring pockets, P4 (DRβ Lys71 and Ala74) and P6 (DRβ Val11 and His13) (PDB ID 1J8H). The P4 peptide side chain forms a salt bridge with DRβ Arg71 (c). (d-f) Key polymorphisms of the MS-associated HLA-DRB1*15:01 molecule are located to the P4 pocket of the peptide binding groove, DRβ Ala71 and Ala74 (PDB ID 1BX2). Smaller contributions are made by polymorphic residues in the P1 and P9 pocket. Comparison of RA and MS-associated HLA-DR proteins highlights the important contribution of DRβ 71 to the shape and charge of the P4 pocket: In the MSassociated DRB1*15:01 molecule, the P4 is large and hydrophobic (DRβ Ala71), while this pocket is smaller and positively charged in the RA-associated DRB1*04:01 protein (DRβ Lys71). DR molecules are shown as surfaces (a, d) or ribbon diagrams (b, c, e, f).
A similar approach was used to identify key polymorphic HLA-DR residues for multiple sclerosis [3]. The most significant variant in the MHC was the HLA-DRB1* 15:01 allele, consistent with many prior studies [22,23]. Statistically independent effects for five other HLA-DRB1 alleles (*03:01, *13:03, *04:04, *04:01, *14:01) were also identified. Interestingly, the most significant amino acid position in DRβ1 chain mapped to position 71, and there was a smaller signal contributed by polymorphisms at positions 74, 57 and 86 (which explained most but not all of the HLA-DRB1 effect). These results are interesting from a structural perspective because both DRβ 71 and 74 are located in the P4 pocket of the groove (Figure 2d-f). The HLA-DRB1* 15:01 allele that is most strongly associated with multiple sclerosis has a small side chain (Ala) at DRβ 71 while the majority of alleles have a charged amino acid at this position (Lys, Arg, Glu). The P4 pocket of DRB1*15:01 protein is therefore larger and more hydrophobic, and a crystal structure demonstrated occupancy of this pocket by an aromatic peptide residue [24] (Figure 2f). These results will be valuable for defining key peptide ligands in this disease.
Figure 2. Binding of citrulline versus arginine peptide side chains in the P4 pocket of HLA-DR proteins.
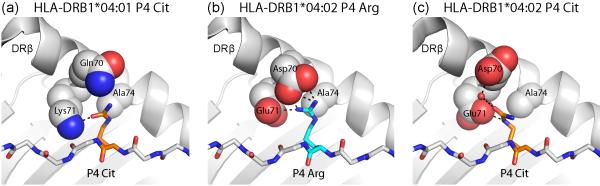
Arginine can be modified enzymatically to citrulline, resulting in loss of its positive charge. (a) The P4 pocket of the RA-associated DRB1*04:01 molecule can accommodate citrulline, which forms a hydrogen bond to DRβ Lys71. In contrast, arginine cannot be accommodated, due to the positive charge of DRβ Lys71 (PDB ID 4MCY). (b, c) The DRB1*04:02 protein is not associated with susceptibility to RA and differs at positions 70 and 71 from RA-associated HLA-DR molecules (PDB ID 4MDJ and 4MDI). Both arginine (b) and citrulline (c) can be accommodated in the P4 pocket. Arginine forms salt bridges with DRβ Glu71 and Asp70 (b), and citrulline forms hydrogen bonds with these two DR residues (c).
It is important to keep in mind that a given amino acid may not have the same affect across allotypes due to different neighboring polymorphic amino acids. This is illustrated by the effect of a non-aspartic acid residue at position 57 of HLA-DQβ, which is the case for both HLA-DQ8 and HLA-DQ2. The P9 pocket of HLA-DQ8 has a strong preference for binding of negatively charged anchors, yet this is not the case for HLA-DQ2 [25].
Part III: Mechanisms for induction of autoimmunity to self-peptide/MHC complexes
Once of the most interesting – and challenging – questions is how autoimmunity is induced. Significant progress has been made in delineating several important molecular mechanisms.
Post-translational modification of peptides
There are several examples how post-translational modifications can be involved in the pathogenesis of autoimmune disease by interplaying with predisposing HLA molecules. The importance of this mechanism was first delineated in celiac disease where deamidation of glutamine to glutamic acid by transglutaminase 2 (gain of negative charge) creates gluten- derived peptides that bind with substantially higher affinity and with slower off-rate to disease-associated HLA-DQ2 and HLA-DQ8 molecules [26]. Another important example is RA where modification of arginine to citrulline (loss of positive charge) plays a role in disease pathogenesis [27,28]. Autoantibodies specific for citrulline can be detected years before clinical symptoms of RA, but only in subjects with RA-associated HLA-DR risk alleles (such as HLA-DEB1*04:01 and *04:04) [29,30]. Smoking is an important environmental risk factor that increases protein citrullination in the lung, providing an intriguing example for how an environmental risk factor is related to MHC-associated disease susceptibility at a molecular level [31,32].
Crystal structures demonstrated how citrullinated peptides are bound by RA-predisposing DRB1*04:01 and *04:04 proteins, as well as the RA non-associated DRB1*04:02 protein **[33](Figure 2). The citrulline side chain forms a hydrogen bond with a key residue of the ‘shared epitope’, DRβ 71 Lys (DRB1*04:01) or Arg (DRB1*04:04)(Figure 2a). Interestingly, a citrulline side chain is also accommodated in the P4 pocket of the non-associated DRB1*04:02 molecule where it forms salt bridges with DRβ 71 Glu and DRβ 70 Asp (Figure 2c). Peptide elution studies demonstrated that DRB1*04:02 binds peptides with unmodified arginine at P4, while the disease-associated DRB1*04:01 and *04:04 proteins do not bind such peptides. These data indicate that the absence of presentation of an unmodified peptide is a critical feature, suggesting differential negative selection of T cells in the thymus by the different DR4 allotypes.
The most straightforward mechanism for post-translational modification to explain MHC association with disease is by creation of neoepitopes which only bind to the susceptibility alleles and to which T cell have not been negative selected against in the thymus. The two examples mentioned above, celiac disease and rheumatoid arthritis, do not fit this simple concept. In the case of celiac disease the modified antigen is foreign whereas in the case of rheumatoid arthritis the post-translationally modified peptides bind both to the susceptibility and resistant HLA alleles. This simple concept should however not be ruled out as for most autoimmune diseases the true autoantigens have not yet been defined. Interestingly, in type 1 diabetes improved binding of several candidate epitopes to HLA-DQ susceptibility allotypes was observed after deamidation by transglutaminase 2 [34]. Yet more complex mechanisms may apply, like in rheumatoid arthritis, where differential thymic selection of T cells by unmodified peptides seems to be involved. Further, post-translational modification can be implicated in the pathogenesis of autoimmune diseases by affecting uptake of antigen into antigen presenting cells. This might be the mechanism underlying improved antigenicity of a chromagranin A epitope relevant to type 1 diabetes after transglutaminase 2 treatment [35]. Antibodies to post-translationally modified residues, like in celiac disease (anti-deamidated gluten antibodies) and rheumatoid arthritis (anti-ACPA), should in particular promote antigen uptake and presentation via involvement of Fc-receptors. Other mechanisms include interference with antigenic processing as shown for citrullination of a vimentin peptide and cathepsin L digestion **[33] and improved T cell recognition of antigen. Two recent reviews discuss in depth how post-translational modification of antigens can be implicated in autoimmune diseases [36,37].
Small-molecule based modification of peptide presentation to T cells
Recent work has provided compelling examples for modification of peptide presentation by small molecules in important clinical settings, including adverse drug reactions and chronic beryllium disease. The best-studied example of adverse drug reactions involves abacavir, a reverse transcriptase inhibitor used for treatment of HIV infection. This drug reaction occurs exclusively in individuals carrying HLA-B* 57:01 (relative risk of >1,000) [38]. In such individuals abacavir induces a strong CD8 T cell response [39]. Crystal structures showed that abacavir binds non-covalently to B*57:01 across the bottom of the peptide binding groove where it alters the selectivity of the F-pocket that accommodates the C-terminal peptide side chain **[40,41] (Figure 3). Abacavir thus induces drastic changes in the peptide repertoire, exposing normally tolerant T cells to previously unseen neo-self epitopes that induce abacavirdependent T cell responses **[40,41]. Substantial changes in the peptide repertoire were also seen for a second drug, carbamazepine, that can cause severe bullous skin disease in subjects with the HLA-B* 15:02 allele **[40].
Figure 3. Induction of T cell mediated immunopathology by binding of a small molecule drug in the MHC class I groove.

Crystal structure of a HLA-B57:01 – peptide complex with bound abacavir, an anti-retroviral drug (PDB ID 3VRJ). Top view (a) and side view (b) of the peptide binding groove, showing how abacavir (red) is bound underneath the peptide (blue). In the side view, the HLA-B α2 helix has been removed to aid visualization of abacavir. The α1 and α2 helices of the HLA-B heavy chain are indicated as well as the N and C terminus of the bound peptide (N and C, respectively).
Chronic beryllium disease is a granulomatous lung disorder which develops in some individuals after exposure to beryllium (Be). The disease has an association with DPβ 69 Glu expressing DP alleles, of which DPB1*02:01 is the most prevalent [42]. The patients have Bespecific CD4 T cells in the lungs [43] which are restricted by disease-associated MHC molecules [44,45]. The crystal structure of a Be2+ specific T cell receptor (TCR) bound to a complex of HLA-DP2, self-peptide and Be2+ was recently solved **[46]. Surprisingly, the TCR does not contact the Be2+ cation which is buried in a highly acidic pocket of DP2 created by negatively charged DP2 and peptide residues. The Be2+ cation alters the local conformation of the DP2 – self-peptide complex and thereby induces T cell responses to DP2 bound self-peptides. Thus, there are important conceptual similarities to abacavir and carbamazepine hypersensitivity where MHC-peptide complexes are altered by small molecule ligands. It is also possible that Be2+ modifies the repertoire of DP2-bound peptide (as observed for abacavir), but this question remains to be addressed. In chronic beryllium disease a restricted, public TCR repertoire of Be2+ specific T cells has been characterized [47], similar to a public TCR repertoire of gluten-reactive T cells in celiac disease [48,49]. Gluten-reactive public TCRs which are specific for deamidated gluten peptides do not contact the Glu anchor residue of the gluten epitopes in the P4 or P6 pockets *[50]. Thus, a common theme emerges: small molecule ligands (metal cations, drugs) or post-translational modifications (citrullination, deamidation) important in human disease frequently do not interact with the TCR. Rather, the T cell response is caused by changes in the peptide repertoire and/or the conformation of selfpeptide – MHC complexes. The previously sharply defined distinction between autoimmunity and allergy is also becoming blurred. An exciting unanswered question is whether cellular metabolites, in particular those preferentially synthesized in particular tissues, can modify selfpeptide presentation and cause autoimmune disease.
Outlook
Major progress has thus been made in defining critical molecular events through which MHC proteins confer susceptibility to human autoimmune diseases. In the future, it will be important to use our growing understanding of the molecular mechanisms of MHC-linked disease susceptibility to develop innovative strategies that benefit patients with autoimmune diseases.
Highlights.
The MHC is the most important genetic risk factor in common autoimmune diseases
For one disease there can be independent contributions by several distinct MHC loci
Presentation of peptide ligands by MHC can be modified by small molecules
T cells recognize post-translationally modified peptides and peptides with modified presentation
Acknowledgments
This work was supported by grants from the Research Council of Norway, the European Research Council and the South-East Norway Regional Health Authority to (L.M.S.), a NIH grant (PO1 AI045757 to K.W.W.) and a postdoctoral fellowship award by the American Diabetes Association (to W.P.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nejentsev S, Howson JM, Walker NM, Szeszko J, Field SF, Stevens HE, Reynolds P, Hardy M, King E, Masters J, et al. Localization of type 1 diabetes susceptibility to the MHC class I genes HLA-B and HLA-A. Nature. 2007;450:887–892. doi: 10.1038/nature06406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.International Multiple Sclerosis Genetics C. Wellcome Trust Case Control C. Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA, Moutsianas L, Dilthey A, Su Z, et al. Genetic risk and a primary role for cellmediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–219. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3*.Patsopoulos NA, Barcellos LF, Hintzen RQ, Schaefer C, van Duijn CM, Noble JA, Raj T, Imsgc Anzgene, Gourraud PA, et al. Finemapping the genetic association of the major histocompatibility complex in multiple sclerosis: HLA and non-HLA effects. PLoS Genet. 2013;9:e1003926. doi: 10.1371/journal.pgen.1003926. [This study indicates that the P4 pocket of the HLA-DR15 binding groove plays an important role in HLA-associated susceptibility to multiple sclerosis. These genetic data are consistent with a previously determined crystal structure of HLA-DR15 with a bound myelin peptide.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morris DL, Taylor KE, Fernando MM, Nititham J, Alarcon-Riquelme ME, Barcellos LF, Behrens TW, Cotsapas C, Gaffney PM, Graham RR, et al. Unraveling multiple MHC gene associations with systemic lupus erythematosus: model choice indicates a role for HLA alleles and non-HLA genes in Europeans. Am J Hum Genet. 2012;91:778–793. doi: 10.1016/j.ajhg.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Evans DM, Spencer CC, Pointon JJ, Su Z, Harvey D, Kochan G, Oppermann U, Dilthey A, Pirinen M, Stone MA, et al. Interaction between ERAP1 and HLA-B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA-B27 in disease susceptibility. Nat Genet. 2011;43:761–767. doi: 10.1038/ng.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.International Genetics of Ankylosing Spondylitis C. Cortes A, Hadler J, Pointon JP, Robinson PC, Karaderi T, Leo P, Cremin K, Pryce K, Harris J, et al. Identification of multiple risk variants for ankylosing spondylitis through highdensity genotyping of immunerelated loci. Nat Genet. 2013;45:730–738. doi: 10.1038/ng.2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stratikos E, Stern LJ. Antigenic peptide trimming by ER aminopeptidasesinsights from structural studies. Mol Immunol. 2013;55:212–219. doi: 10.1016/j.molimm.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alvarez-Navarro C, Lopez de Castro JA. ERAP1 structure, function and pathogenetic role in ankylosing spondylitis and other MHCassociated diseases. Mol Immunol. 2014;57:12–21. doi: 10.1016/j.molimm.2013.06.012. [DOI] [PubMed] [Google Scholar]
- 9.Colbert RA, Tran TM, Layh-Schmitt G. HLA-B27 misfolding and ankylosing spondylitis. Mol Immunol. 2014;57:44–51. doi: 10.1016/j.molimm.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bowness P, Ridley A, Shaw J, Chan AT, Wong-Baeza I, Fleming M, Cummings F, McMichael A, Kollnberger S. Th17 cells expressing KIR3DL2+ and responsive to HLA-B27 homodimers are increased in ankylosing spondylitis. J Immunol. 2011;186:2672–2680. doi: 10.4049/jimmunol.1002653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Genetic Analysis of Psoriasis C. the Wellcome Trust Case Control C. Strange A, Capon F, Spencer CC, Knight J, Weale ME, Allen MH, Barton A, Band G, et al. A genomewide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet. 2010;42:985–990. doi: 10.1038/ng.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12*.Kirino Y, Bertsias G, Ishigatsubo Y, Mizuki N, Tugal-Tutkun I, Seyahi E, Ozyazgan Y, Sacli FS, Erer B, Inoko H, et al. Genomewide association analysis identifies new susceptibility loci for Behcet's disease and epistasis between HLA-B* 51 and ERAP1. Nat Genet. 2013;45:202–207. doi: 10.1038/ng.2520. [This paper demonstrates that in Behcet's disease, like previously demonstarted for ankylosing spondylitis and psorisis, there is epitatic interaction between disease associated MHC class I alleles and ERAP1.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsoi LC, Spain SL, Knight J, Ellinghaus E, Stuart PE, Capon F, Ding J, Li Y, Tejasvi T, Gudjonsson JE, et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet. 2012;44:1341–1348. doi: 10.1038/ng.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bjorck S, Brundin C, Lorinc E, Lynch KF, Agardh D. Screening detects a high proportion of celiac disease in young HLA-genotyped children. J Pediatr Gastroenterol Nutr. 2010;50:49–53. doi: 10.1097/MPG.0b013e3181b477a6. [DOI] [PubMed] [Google Scholar]
- 15.Han B, Diogo D, Eyre S, Kallberg H, Zhernakova A, Bowes J, Padyukov L, Okada Y, Gonzalez-Gay MA, Rantapaa-Dahlqvist S, et al. Fine mapping seronegative and seropositive rheumatoid arthritis to shared and distinct HLA alleles by adjusting for the effects of heterogeneity. Am J Hum Genet. 2014 doi: 10.1016/j.ajhg.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lyons PA, Rayner TF, Trivedi S, Holle JU, Watts RA, Jayne DR, Baslund B, Brenchley P, Bruchfeld A, Chaudhry AN, et al. Genetically distinct subsets within ANCAassociated vasculitis. N Engl J Med. 2012;367:214–223. doi: 10.1056/NEJMoa1108735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morris DL, Fernando MM, Taylor KE, Chung SA, Nititham J, Alarcon-Riquelme ME, Barcellos LF, Behrens TW, Cotsapas C, Gaffney PM, et al. MHC associations with clinical and autoantibody manifestations in European SLE. Genes Immun. 2014 doi: 10.1038/gene.2014.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Howson JM, Stevens H, Smyth DJ, Walker NM, Chandler KA, Bingley PJ, Todd JA. Evidence that HLA class I and II associations with type 1 diabetes, autoantibodies to GAD and autoantibodies to IA2, are distinct. Diabetes. 2011;60:2635–2644. doi: 10.2337/db11-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Todd JA, Bell JI, McDevitt HO. HLA-DQ beta gene contributes to susceptibility and resistance to insulindependent diabetes mellitus. Nature. 1987;329:599–604. doi: 10.1038/329599a0. [DOI] [PubMed] [Google Scholar]
- 20.Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987;30:1205–1213. doi: 10.1002/art.1780301102. [DOI] [PubMed] [Google Scholar]
- 21*.Raychaudhuri S, Sandor C, Stahl EA, Freudenberg J, Lee HS, Jia X, Alfredsson L, Padyukov L, Klareskog L, Worthington J, et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet. 2012;44:291–296. doi: 10.1038/ng.1076. [A large cohort of RA patients was investigated to identify key HLA polymorphisms associated with susceptibility to RA. Polymorphisms in the P4 and P6 pocket of HLA-DR proteins were shown to be most important.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hillert J. Human leukocyte antigen studies in multiple sclerosis. Ann Neurol. 1994;36(Suppl):S15–17. doi: 10.1002/ana.410360706. [DOI] [PubMed] [Google Scholar]
- 23.Caillier SJ, Briggs F, Cree BA, Baranzini SE, Fernandez-Vina M, Ramsay PP, Khan O, Royal W, 3rd, Hauser SL, Barcellos LF, et al. Uncoupling the roles of HLA-DRB1 and HLA-DRB5 genes in multiple sclerosis. J Immunol. 2008;181:5473–5480. doi: 10.4049/jimmunol.181.8.5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith KJ, Pyrdol J, Gauthier L, Wiley DC, Wucherpfennig KW. Crystal structure of HLA-DR2 (DRA*0101, DRB1*1501) complexed with a peptide from human myelin basic protein. J Exp Med. 1998;188:1511–1520. doi: 10.1084/jem.188.8.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quarsten H, Paulsen G, Johansen BH, Thorpe CJ, Holm A, Buus S, Sollid LM. The P9 pocket of HLA-DQ2 (nonAspβ57) has no particular preference for negatively charged anchor residues found in other type 1 diabetespredisposing nonAspβ57 MHC class II molecules. Int Immunol. 1998;10:1229–1236. doi: 10.1093/intimm/10.8.1229. [DOI] [PubMed] [Google Scholar]
- 26.Molberg O, McAdam SN, Korner R, Quarsten H, Kristiansen C, Madsen L, Fugger L, Scott H, Noren O, Roepstorff P, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gutderived T cells in celiac disease. Nat Med. 1998;4:713–717. doi: 10.1038/nm0698-713. [DOI] [PubMed] [Google Scholar]
- 27.Schellekens GA, de Jong BA, van den Hoogen FH, van de Putte LB, van Venrooij WJ. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritisspecific autoantibodies. J Clin Invest. 1998;101:273–281. doi: 10.1172/JCI1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Girbal-Neuhauser E, Durieux JJ, Arnaud M, Dalbon P, Sebbag M, Vincent C, Simon M, Senshu T, Masson-Bessiere C, Jolivet-Reynaud C, et al. The epitopes targeted by the rheumatoid arthritisassociated antifilaggrin autoantibodies are posttranslationally generated on various sites of (pro)filaggrin by deimination of arginine residues. J Immunol. 1999;162:585–594. [PubMed] [Google Scholar]
- 29.Moeez S, John P, Bhatti A. Anticitrullinated protein antibodies: role in pathogenesis of RA and potential as a diagnostic tool. Rheumatol Int. 2013;33:1669–1673. doi: 10.1007/s00296-012-2635-6. [DOI] [PubMed] [Google Scholar]
- 30.Klareskog L, Padyukov L, Ronnelid J, Alfredsson L. Genes, environment and immunity in the development of rheumatoid arthritis. Curr Opin Immunol. 2006;18:650–655. doi: 10.1016/j.coi.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 31.Klareskog L, Alfredsson L, Rantapaa-Dahlqvist S, Berglin E, Stolt P, Padyukov L. What precedes development of rheumatoid arthritis? Ann Rheum Dis. 2004;63(Suppl 2):ii28–ii31. doi: 10.1136/ard.2004.028225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klareskog L, Stolt P, Lundberg K, Kallberg H, Bengtsson C, Grunewald J, Ronnelid J, Harris HE, Ulfgren AK, Rantapaa-Dahlqvist S, et al. A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA-DR (shared epitope)restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum. 2006;54:38–46. doi: 10.1002/art.21575. [DOI] [PubMed] [Google Scholar]
- 33**.Scally SW, Petersen J, Law SC, Dudek NL, Nel HJ, Loh KL, Wijeyewickrema LC, Eckle SB, van Heemst J, Pike RN, et al. A molecular basis for the association of the HLA-DRB1 locus, citrullination, and rheumatoid arthritis. J Exp Med. 2013;210:2569–2582. doi: 10.1084/jem.20131241. [Modification of arginine to citrulline was previously shown to be critical in the pathogenesis of rheumatoid arthritis. The crystal structure of several HLA-DR4 proteins was determined with peptides that positioned a citrulline residue in the P4 pocket. Citrulline was shown to form a hydrogen bond to DRβ 71, a polymorphic residue implicated in rheumatoid arthritis.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Lummel M, Duinkerken G, van Veelen PA, de Ru A, Cordfunke R, Zaldumbide A, Gomez-Tourino I, Arif S, Peakman M, Drijfhout JW, et al. Posttranslational modification of HLA-DQ binding islet autoantigens in type 1 diabetes. Diabetes. 2014;63:237–247. doi: 10.2337/db12-1214. [DOI] [PubMed] [Google Scholar]
- 35.Delong T, Baker RL, He J, Barbour G, Bradley B, Haskins K. Diabetogenic T-cell clones recognize an altered peptide of chromogranin A. Diabetes. 2012;61:3239–3246. doi: 10.2337/db12-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dunne JL, Overbergh L, Purcell AW, Mathieu C. Posttranslational modifications of proteins in type 1 diabetes: the next step in finding the cure? Diabetes. 2012;61:1907–1914. doi: 10.2337/db11-1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doyle HA, Yang ML, Raycroft MT, Gee RJ, Mamula MJ. Autoantigens: novel forms and presentation to the immune system. Autoimmunity. 2014;47:220–233. doi: 10.3109/08916934.2013.850495. [DOI] [PubMed] [Google Scholar]
- 38.Mallal S, Phillips E, Carosi G, Molina JM, Workman C, Tomazic J, Jagel-Guedes E, Rugina S, Kozyrev O, Cid JF, et al. HLA-B* 5701 screening for hypersensitivity to abacavir. N Engl J Med. 2008;358:568–579. doi: 10.1056/NEJMoa0706135. [DOI] [PubMed] [Google Scholar]
- 39.Chessman D, Kostenko L, Lethborg T, Purcell AW, Williamson NA, Chen Z, Kjer-Nielsen L, Mifsud NA, Tait BD, Holdsworth R, et al. Human leukocyte antigen class Irestricted activation of CD8+ T cells provides the immunogenetic basis of a systemic drug hypersensitivity. Immunity. 2008;28:822–832. doi: 10.1016/j.immuni.2008.04.020. [DOI] [PubMed] [Google Scholar]
- 40**.Illing PT, Vivian JP, Dudek NL, Kostenko L, Chen Z, Bharadwaj M, Miles JJ, Kjer-Nielsen L, Gras S, Williamson NA, et al. Immune selfreactivity triggered by drugmodified HLA-peptide repertoire. Nature. 2012;486:554–558. doi: 10.1038/nature11147. [DOI] [PubMed] [Google Scholar]
- 41**.Ostrov DA, Grant BJ, Pompeu YA, Sidney J, Harndahl M, Southwood S, Oseroff C, Lu S, Jakoncic J, de Oliveira CA, et al. Drug hypersensitivity caused by alteration of the MHCpresented selfpeptide repertoire. Proc Natl Acad Sci U S A. 2012;109:9959–9964. doi: 10.1073/pnas.1207934109. [References 40 and 41 both demonstrate that a small molecule drug, abacavir, binds underneath the peptide in a HLA class I protein (HLA-B57:01), changes the binding specificity of the F-pocket and induces profound changes in the peptide repertoire. Abacavir sensitive patients have T cells that recognize such abacavir dependent neo-self epitopes.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richeldi L, Sorrentino R, Saltini C. HLA-DPB1 glutamate 69: a genetic marker of beryllium disease. Science. 1993;262:242–244. doi: 10.1126/science.8105536. [DOI] [PubMed] [Google Scholar]
- 43.Fontenot AP, Canavera SJ, Gharavi L, Newman LS, Kotzin BL. Target organ localization of memory CD4(+) T cells in patients with chronic beryllium disease. J Clin Invest. 2002;110:1473–1482. doi: 10.1172/JCI15846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fontenot AP, Torres M, Marshall WH, Newman LS, Kotzin BL. Beryllium presentation to CD4+ T cells underlies diseasesusceptibility HLA-DP alleles in chronic beryllium disease. Proc Natl Acad Sci U S A. 2000;97:12717–12722. doi: 10.1073/pnas.220430797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lombardi G, Germain C, Uren J, Fiorillo MT, du Bois RM, Jones-Williams W, Saltini C, Sorrentino R, Lechler R. HLA-DP allele-specific T cell responses to beryllium account for DP-associated susceptibility to chronic beryllium disease. J Immunol. 2001;166:3549–3555. doi: 10.4049/jimmunol.166.5.3549. [DOI] [PubMed] [Google Scholar]
- 46**.Clayton GM, Wang Y, Crawford F, Novikov A, Wimberly BT, Kieft JS, Falta MT, Bowerman NA, Marrack P, Fontenot AP, et al. Structural basis of chronic beryllium disease: linking allergic hypersensitivity and autoimmunity. Cell. 2014;158:132–142. doi: 10.1016/j.cell.2014.04.048. [The paper reports the crystal structure of a Be2+ specific TCR of a patient with beryllium hypersensitivity which is bound to a complex of HLA-DP2, self-peptide and Be2+. Surprisingly, the TCR does not interact with the Be2+ itself, but rather with surface changes induced by the firmly bound Be2+ and an accompanying Na+ cation.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bowerman NA, Falta MT, Mack DG, Wehrmann F, Crawford F, Mroz MM, Maier LA, Kappler JW, Fontenot AP. Identification of multiple public TCR repertoires in chronic beryllium disease. J Immunol. 2014 doi: 10.4049/jimmunol.1400007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qiao SW, Christophersen A, Lundin KE, Sollid LM. Biased usage and preferred pairing of α- and β-chains of TCRs specific for an immunodominant gluten epitope in coeliac disease. Int Immunol. 2014;26:13–19. doi: 10.1093/intimm/dxt037. [DOI] [PubMed] [Google Scholar]
- 49.Qiao SW, Raki M, Gunnarsen KS, Loset GA, Lundin KE, Sandlie I, Sollid LM. Posttranslational modification of gluten shapes TCR usage in celiac disease. J Immunol. 2011;187:3064–3071. doi: 10.4049/jimmunol.1101526. [DOI] [PubMed] [Google Scholar]
- 50.Petersen J, Montserrat V, Mujico JR, Loh KL, Beringer DX, van LMearin ML, Schweizer J, Kooy-Winkelaar Y, et al. T-cell receptor recognition of HLA-DQ2-gliadin complexes associated with celiac disease. Nat Struct Mol Biol. 2014;21:480–488. doi: 10.1038/nsmb.2817. [DOI] [PubMed] [Google Scholar]