

Abstract
Vertebrate hedgehog signaling is coordinated by the differential localization of the receptors patched-1 and smoothened in the primary cilium. Cilia assembly is mediated by intraflagellar transport (IFT) and cilia defects disrupt hedgehog signaling, causing many structural birth defects. We generated Ift25 and Ift27 knockout mice and show they have structural birth defects indicative of hedgehog signaling dysfunction. Surprisingly ciliary assembly is not affected, but abnormal hedgehog signaling is observed in conjunction with ciliary accumulation of patched-1 and smoothened. Similarly smoothened accumulates in cilia on cells mutated for BBSome components or the BBS binding protein/regulator Lztfl1. Interestingly, the BBSome and Lztfl1 accumulate to high levels in Ift27 mutant cilia. Since Lztfl1 mutant cells accumulate BBSome but not IFT27 it is likely that Lztfl1 functions downstream of IFT27 to couple the BBSome to the IFT particle for coordinated removal of patched-1 and smoothened from cilia during hedgehog signaling.
Keywords: intraflagellar transport, Hedgehog signaling, cilia
Introduction
The cilium plays a key role in development by coordinating cell physiology with signals coming from the extracellular environment. Defects in ciliary structure or ciliary signaling underlie a diverse class of human diseases that range from adult onset degenerative disorders such as polycystic kidney disease, liver fibrosis and retinal degeneration to structural birth defects of the heart, brain and skeleton to obesity and other metabolic disorders. The cilium functions by creating a cellular microenvironment where receptors and signaling pathways are sequestered and concentrated in a small projection from the surface of the cell. Cilia can detect many types of signals, from photons in rod and cone outer segments, to diverse odorants detected by olfactory cilia, to mechanical stimuli detected by epithelial cilia or morphogenic ligands like sonic hedgehog (Shh) detected by the cilia of the developing embryo (Satir and Christensen, 2007).
Cilia themselves are extremely complex organelles that are composed of at least 500-1000 structural proteins organized around a microtubule based cytoskeleton and an associated membrane domain (Pazour et al., 2005). Many of the structural components of the cilium are known to be encoded by genes involved in human diseases and the number of human disorders or syndromes caused by ciliary defects is rapidly growing (Badano et al., 2006). Understanding how this organelle is assembled and how the signaling environment is created and maintained is critical to understanding how the cilium functions in the etiology of these diseases. The cilium is assembled by the process of intraflagellar transport (IFT) where large protein complexes called IFT particles are carried along the ciliary microtubules by kinesin and dynein motors (Rosenbaum and Witman, 2002; Pedersen and Rosenbaum, 2008). The IFT particles are composed of more than 20 unique proteins organized in two subcomplexes called IFT-A and IFT-B (Cole et al., 1998; Ou et al., 2007; Follit et al., 2009). The IFT particles are thought to function as a cargo adaptor to connect proteins needed to build cilia to the molecular motors so the ciliary structural components can be transported from the cell body into the cilium for assembly. IFT-B appears to be critically important for ciliary assembly as mutations in most IFT-B components block ciliary assembly. IFT-A mutations typically cause less severe defects in ciliary assembly and often result in accumulations of materials in the cilium suggesting that it is more important for retrograde transport (Iomini et al., 2009). In addition to IFT-A and IFT-B, a third complex called the BBSome is connected to the IFT particle. Mutations in BBSome components do not typically block ciliary assembly but prevent the delivery of specific receptors to the cilium (Berbari et al., 2008) and cause accumulations of abnormal membrane-associated proteins in the cilium (Lechtreck et al., 2009).
In this work we examine the function of the IFT-B subunit IFT27 (also known as Rabl4), which was recently shown to be mutated in two siblings with Bardet-Biedl syndrome (BBS). The affected siblings displayed classic BBS phenotypes including obesity, polydactyly, mental disabilities, renal dysfunction, and retinal degeneration (Aldahmesh et al., 2014). IFT27 is a small G protein distantly related to the Rab subfamily of Ras-like GTPases (Qin et al., 2007). The mammalian protein lacks the prenylation sites found in other Rab proteins indicating that it is not membrane associated (at least via these moieties). Instead, IFT27 forms a stable heterodimer with IFT25 that exists both within the IFT-B complex and outside of it. IFT27 binds nucleotide with micromolar affinity but has a very low intrinsic GTPase activity (Bhogaraju et al., 2011). We became interested in IFT25 and IFT27 because unlike most IFT-B proteins these two subunits are missing from several species that assemble cilia. This suggested that IFT25 and IFT27 are not required for ciliary assembly but could be involved in sensory or other functions of cilia (Keady et al., 2012). We recently showed that IFT25 is not required for ciliary assembly but mice homozygous for a null allele died at birth with a variety of structural defects (Keady et al., 2012). The Ift25 phenotype differed from previously studied null alleles of IFT complex B proteins as those alleles caused lethality earlier in development, typically at mid-gestation. In this study, we generated an Ift27 mutant mouse model and showed it has a phenotype overlapping that of the Ift25 mutant. Using these two mutant mouse models, we show that Ift25and Ift27 are dispensable for ciliogenesis, but both are required for BBSome trafficking essential for hedgehog signaling.
Experimental Procedures
Additional methods can be found in Supplemental Experimental Procedures
Mouse Breeding
Ift27tm1a(EUCOMM)Hmgu-targeted ES cell line HEPD0653_7_E10 was obtained from the European Conditional Mouse Mutagenesis Program (EUCOMM) project and injected into C57Bl/6J albino blastocysts to generate chimeric animals. Chimeric mice were mated to C57Bl/6J albino mice (B6(Cg)-Tyrc-2J/J, Jax 000058) or to C57Bl/6J mice (Jax 000664). This allele, Ift27neo was converted to Ift27null1, Ift27flox and Ift27null2 using C57Bl/6 congenic PrmCre (O’Gorman et al., 1997) and FlpE (Farley et al., 2000) (Figure S1). In vivo Gli1 expression was monitored using Gli1-LacZ (Jax 008211) (Bai et al., 2002). Genotyping was carried out as described in Figure S1. All mouse work was carried out at UMMS and was approved by the UMMS IACUC.
Immunofluorescence Microscopy
Cells for immunofluorescence microscopy were grown, fixed, and stained as described (Keady et al., 2012). Primary antibodies used included acetylated tubulin (6-11B-1, Sigma), beta-tubulin (B-5-1-2, Sigma), gamma-tubulin (GTU-88, Sigma), MmIFT20, MmIFT52, MmIFT57, MmIFT88 (Pazour et al., 2002), MmIFT25 (Proteintech), MmIFT140 (Jonassen et al., 2012), Gli1 (V812, Cell Signaling), Gli3 (AF3690, R&D), beta actin (13E5; Cell Signaling), Gli2 (gift of J. Eggenschwiler, Univ. of Georgia), Ptch1, Smo (gifts of R. Rohatgi, Stanford Univ.), Dync2h1 (gift of R. Vallee, Columbia Univ.), Gpr161 (gift of S. Mukhopadhyay, Univ. Texas SW), Arl6 (gift of M. Nachury, Stanford Univ.), BBS9 (Sigma and Proteintech), Lztfl1 and BBS5 (Proteintech), Pax6, Shh, and Olig2 (DSHB, University of Iowa). Anti-MmIFT25 and anti-MmSmo were made by expressing the mouse protein in bacteria as a maltose binding protein fusion and injecting into rabbits (Figure S3). Antibodies were affinity purified against the same fragment expressed as a glutathione S-transferase fusion.
DNA Constructs
Flag-GST (BK35), Glutathione S-transferase from pGex6p1 cloned into JAF113 (Follit et al., 2009), a slightly modified p3xFLAG-myc-CMV-26 (Sigma) (Keady et al., 2011). TE24 is the equivalent construct in lentiviral vector pHAGE_DN_CMV_nucEGFP (gift of D. Nedelcu and A. Salic).
Flag-GFP (JAF 146), EGFP cloned into JAF113.
Flag-IFT25 (JAF143), Mouse Ift25 cloned into JAF113. Flag-IFT25T40R/T42R/S128E (BK61) was made by PCR. TE14 and TE16 are equivalent constructs in the lentiviral vector pHAGE_DN_CMV_nucEGFP.
IFT27-Flag (BK8), Mouse Ift27 cloned into p3XFLAG-myc-CMV-14 (Sigma) (Follit et al., 2009).
IFT27T19N-Flag (BK12) and IFT27K68L-Flag (BK10) were generated by PCR. TE17, TE19 and TE18 are equivalent constructs in pHAGE_DN_CMV_nucEGFP.
Flag-Lztfl1 (TE30), Mouse Lztfl1 cloned into pHAGE_DN_CMV_nucEGFP. Flag was added at N-terminus during PCR amplification.
Smo-M2-mCherry. SmoM2 in pHAGE_DN_CMV vector (gift of D. Nedelcu and A. Salic).
CRIPSR/Cas9 genome editing
Candidate sgRNAs were identified by searching for G(N)20GG motifs 300 bases upstream and 100 bases downstream of the targeting sequence that conform with the nucleotide requirements for U6 Pol III transcription and the spCas9 PAM recognition element (NGG) (Jinek et al., 2012; Mali et al., 2013) using Web-based software ZiFiT targeter 4.2 (Sander et al., 2010). Sequences generated were aligned to mouse genome using nBLAST to search for potential off-target sites. Pairs of oligonucleotides were subsequently annealed together and cloned into pBSK-gRNA (Gift of R.Maehr). Finally, this construct was electroporated into NIH3T3 cells together with pCas9p2a (Gift of R. Maehr) and cells were selected for Cas9 vector expression with appropriate antibiotic.
Results
Ift27 mutants have pleiotropic structural birth defects
To understand the importance of IFT27 to ciliary assembly and mouse development, we used EUCOMM cells to create an Ift27 mutant mouse (Fig 1-4). The Ift27null1 allele (Fig S1) that we analyzed for most studies (all except Fig 4H) contained a β-galactosidase gene in intron 2 and had exons 3-5 deleted. The β-galactosidase gene has a strong splice acceptor at its 5’ end and is expected to capture the upstream exons of Ift27 to prevent their splicing to the downstream exons. It is likely that the Ift27null1 allele is a null or a strong hypomorph as quantitative RT-PCR indicated that transcripts containing the 3’ exons were reduced to less than 0.5% of controls (Fig 3A, 4A) and no protein was detected by western blot (Fig 3A, 4A). The expected Mendelian ratio of genotypes was found in animals harvested the day prior to birth (E18) and on the day of birth (P0) but all homozygous mutants exhibited cyanosis and were stillborn or died shortly after birth (Fig 1A, 1B).
Figure 1. Ift27 null mutants display multiple developmental defects.

A. Images of P0 animals. Ift27null1 genotypes are given below. All animals were alive when photographed.
B. Genotype distribution at the day prior to birth (E18), day of birth (P0) and later (>P0) in offspring of Ift27null1/+ by Ift27null1/+ crosses. Blue, orange and green represent +/+, +/- and -/- genotypes respectively. Homozygous mutant animals were alive on the day prior to birth but died on P0 so the P0 numbers reflect a mix of live and dead animals.
C. Images of embryos at E18.5 (Ca, Cb, Cc) and E15.5 (Cd). Note abnormal facial structure, abnormal lower jaw (arrow) and more closely spaced eyes (hypotelorism).
D. Alcian blue and alizarin red staining of the skull shows abnormal palate development.
E. H&E stained frontal sections of the oral cavity. The mutant lacks the body of the mandible (md) and the tongue (to).
F. H&E stained sections of nasal cavities of E15.5 (Fa, Fb) and E18.5 (Fc, Fd) embryos. The mutant embryos in each case show under-developed nasal structures, particularly the inferior regions including the vomer bone of the nasal septum (ns), although superior cartilaginous parts of the septum appear to develop normally. 1p, primary palate; 2p, secondary palate; ui, upper incisor; vno, vomeronasal organ. Scale bars = 200 μm.
G, H. Alcian blue and alizarin red staining of the skeleton shows abnormal skull shape, curvature of the spine and abnormal rib cage including malaligned sternal vertebrate.
I, J. Alcian blue and alizarin red stained (I) and unstained (J) images of limbs showing a variety of digit defects.
Figure 2. Ift27 mutants have structural heart and lung disease.

A. Lung isomerism. H&E images show the normal arrangement of 4 right lobes and 1 left lobe in the control animals (Aa, Ac). Mutants (Ab, Ad) have a single lobe on both sides indicating a left isomerism. Note the large open sac (TEF balloon) within the thoracic cavity that projects through the diaphragm and connects to the stomach (St). Lg, lung; Lv, liver; Ht, heart. Scale bars are 500 μm. The * marks a crack in the tissue that occurred during processing.
B. Immunofluorescence images of the epithelium lining the TEF balloon (IFT88 green, 6-11B-1 red, DAPI blue). A section adjacent to the one imaged in Ad was stained and the approximate position of the imaged region is marked by an arrow in Ad. Likewise the arrow in Ba marks the cell that was imaged in Bb. Scale bar in Ba is 50 μm and 10 μm in Bb. Ba and Bb are maximum projections of a 16 and 10 layer Z-stacks acquired every 0.5 μm.
C. Surface renderings of ECM image stacks show heart placement defects. The apex of the heart normally points towards the left side of the thoracic cavity (levocardia). Heart orientation in Ift27 mutants is variable with 11/14 showing levocardia, 1/14 mesocardia (apex at midline) and 2/14 dextrocardia (apex pointed to the right side).
D. ECM imaging reveals structural heart defects in Ift27 mutants. Images shown are single planes of reconstructed hearts taken at levels to highlight the ventricular septum (top row) or atrial septum (bottom row). Movies of these 3D reconstructions and additional planes are included in the supplementary material. Left panel is a control heart while the right two panels are from mutants illustrating either partial (middle) or complete (right) atrioventricular septal defects. CA, Common atrium; VSD, ventricular septal defect; AVSD, atrioventricular septal defect; Ao, aorta; dAo, descending aorta; PT, pulmonary trunk; PA, pulmonary artery; LSVC, left superior vena cava; RSVC, right superior vena cava; LA, left atrium; RA, right atrium; LV, left ventricle; RV, right ventricle; TEF, tracheoesophageal fistula; T, trachea; O, esophagus.
Figure 4. IFT27 is required for normal Hedgehog signaling.

A-C. Ift27+/+ and Ift27null1/null1 MEFs were left untreated or treated with SAG, a Shh pathway activator. Cell lines from three different embryos were used for each genotype. A. RNA was isolated from one set of cells and analyzed for gene expression by quantitative real time PCR (left column). Proteins were isolated from another set of cells and analyzed by western blotting. Quantitation of protein levels are listed on the right side of each western blot and compared to γ-tubulin loading control. Groups were compared by ANOVA; only comparisons between SAG treated control and mutant cells are depicted in this figure (**p=<0.01 and ***p=<0.001) but other comparisons are in Supplemental Table 4. Full length (FL) and repressor (R) form of Gli3 protein were analyzed separately.
B, C, D. Cells were fixed and stained for cilia (arrows, 6-11B-1, red) and Smo (green, B), Gpr161 (green C) or Gli2 (green, D). Insets show the green channel (Smo, Gpr161 or Gli2). Ciliary Smo or ciliary tip Gli2 was quantitated in 25 cilia from 3 independent cell lines of each genotype. Ciliary Gpr161 was quantitated in 25 cilia from 1 cell line of each genotype. Groups were compared by ANOVA (*p=<0.05, **p=<0.01 and ***p=<0.001, ns: not significant). Cilia length and percent ciliation for these cells is shown in Fig. 3D. Scale bars are 10 μm and apply to all images in B, C and D.
E. Ift27+/+and Ift27null1/null1 MEFs were transfected with SmoM2-mCherry, fixed and stained for cilia (6-11B-1, red) and Gli2 (green). Note the concentration of Gli2 at the ciliary tip (arrow) of control cells as compared to the broader distribution in the mutant cell. Total Gli2 was quantitated from >100 cilia and was not significantly different in the two groups. The distribution of Gli2 along the cilium was quantitated by determining what percentage of the cilium length had Gli2 fluorescence intensity above background. Scale bars are 2 μm and apply to all images.
F. Neural tube patterning of Ift27null1/null1 embryos. All images are shown with the ventral side (floor plate) on bottom. Cryosections were cut from caudal regions of E9.5 embryos and immunostained for Olig2 (green) and Shh or Pax6 (red). Merged images with DAPI (blue) are shown in the top row. Arrows depict the absence of Pax6 and Olig2 in the wild type floor plate and an expansion of Pax6 and Olig2 into the mutant floor plate. Scale bar is 50 μm and applies to all images in F.
G. Ift27+/+ and Ift27null1/null1 protein extracts from E11.5 hindlimbs were immunoblotted for Gli3 protein (Gli3-FL: full length and Gli3-R: repressor). Quantitation of Gli3 protein levels (ratio of Gli3-FL/Gli3-R) from hindlimbs is shown below the gel (*p=0.032). Number of embryos (n) analyzed is given below genotypes.
H. E12.5 Ift27null2, Gli1-LacZ embryos were fixed and stained for β-galactosidase activity. Ift27null2 genotypes are provided above the embryos. Isolated limb buds are shown on the right side. Additional images are in Supplemental Figure 4. Scale bar is 2 mm and applies to whole embryos. FL, forelimb, HL, hindlimb.
I. Paraxial mesoderm stained with Smo and Ptch1 antibodies. Sections of E10.5 embryos were stained for cilia (6-11B-1, green) and either Smo or Ptch1 (red). Insets are 4X enlargements of the cilium marked with an arrow. Scale bar is 10 μm. Images are maximum projections of 16 layer Z-stacks acquired every 0.25 μm. Graph at bottom shows ciliary Smo and Ptch1 are significantly (*** p<0.001) increased in the mutant embryos.
Figure 3. Ift27 is not required for ciliation.
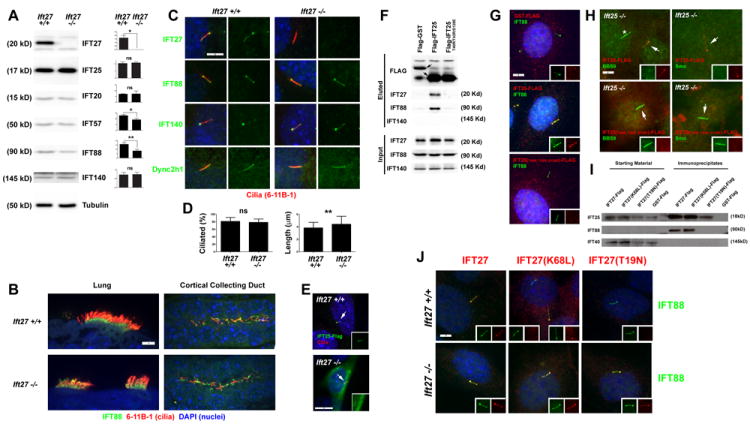
A. Effect of the Ift27null1 mutation on IFT protein stability. Protein extracts from wild-type and mutant MEFs were immunoblotted with the antibodies indicated on the right side of each western blot panel. Approximate molecular weights are listed on the left side. Quantitation of IFT protein levels relative to γ-tubulin loading control are listed on the right side of each western blot (n=3 embryos/MEF lines per genotype) (*p<0.05, **p<0.01).
B. E18.5 lung and kidney sections from Ift27+/+ and Ift27null1/null1 mice immunostained with IFT88 (green) and the acetylated tubulin cilia marker 6-11B-1 (red). Scale bar is 5μm and applies to all images. Images are maximum projections of 10 layer Z-stacks acquired every 0.5 μm
C. Immunofluorescence of control and mutant MEFs immunostained with 6-11B-1 (cilia, red) and IFT27, IFT88, IFT140 or Dync2h1 (green). Note the lack of IFT27 staining of mutant cells (25/25 wild type cells and 0/25 mutant cells showed IFT27 staining). Staining of cilia for the other antibodies were similar in both mutant and control cells (25/25 cells positive for each condition).
D. Quantitation of ciliation and ciliary length in MEF cells. Percentage of ciliated cells and ciliary lengths based on ciliary IFT88 immunostaining in serum starved MEFs (n=3 Ift27+/+ and 3 Ift27null1/null1 embryos/MEF lines for % ciliation and n=50 cilia per cell line for length). Differences for percent ciliation were not significantly different, but mutant cilia were slightly longer (** p<0.01).
E. IFT25-Flag (green) localizes to cilia (6-11B-1, red) in wild type cells but not Ift27null1/null1 cells. Inset shows green (IFT25-Flag) channel. Quantification showed 23/25 transfected wild type cells had ciliary-localized IFT25-Flag while 0/25 transfected mutant cells had ciliary-localized IFT25-Flag.
F, G. Interaction between IFT25 and IFT27 are required for IFT25 to enter cilia. F. Immunoprecipitation from IMCD3 cells transfected with Flag-GST, Flag-IFT25 or Flag-IFT25(T40R/T42R/S128E) demonstrate that wild type but not the mutant form of IFT25 bind to IFT27 and IFT88. G. In IMCD3 cells, wild type Flag-IFT25 (Flag, red) localizes to cilia (IFT88, green) but Flag-IFT25(T40R/T42R/S128E) (Flag, red) does not. Quantification showed that 0/50 Flag-GST, 47/50 Flag-IFT25, 0/50 Flag-IFT25(T40R/T42R/S128E) transfected cells had Flag-positive cilia. Scale bar is 5 μm and applies to all images in G.
H. Wild type Flag-IFT25 (top row, Flag, red) but not Flag-IFT25(T40R/T42R/S128E) (bottom row, Flag, red) rescues the BBS9 (left panel, green) and Smo (right panel green) accumulation phenotypes in Ift25null1/null1 mutant MEFs. Arrows mark cilia on transfected cells which are shown in the insets with separate red and green channels. Quantification showed 25/25 cells transfected with wild type IFT25 showed rescue of the accumulation of ciliary BBS9 and Smo phenotypes while 0/25 transfected with IFT25(T40R/T42R/S128E) showed rescue. The * marks a cilium on a non-transfected cell. Scale bar is 5 μm and applies to all images in H.
I. Immunoprecipitation from MEF cells transfected with IFT27-Flag, IFT27(K68L)-Flag, IFT27(T19N)-Flag or Flag-GST demonstrate that all forms of IFT27 bind to IFT25 but only wild type and the K68L forms of IFT27 bind to IFT88. No binding between any form of IFT27 and IFT140 was detected. Note that with this exposure time, IFT88 is not detectable until after immunoprecipitation.
J. Wild type IFT27-Flag (left column, Flag, red), IFT27(K68L)-Flag (middle column, Flag, red) and Flag-IFT27(T19N) (right column, Flag, red) expressed in Ift27+/+ (top row) and Ift27null1/null1 (bottom row) MEFs. Quantification showed that, 80/100 IFT27-Flag, 24/100 IFT27(K68L)-Flag and 0/100 IFT27(T19N)-Flag transfected Ift27+/+ cells had Flag-positive cilia while 72/100 IFT27-Flag, 86/100 IFT27(K68L)-Flag and 36/100 IFT27(T19N)-Flag transfected Ift27null1/null1 cells had Flag-positive cilia. Scale bar is 5 μm and applies to all images in J.
Fourteen Ift27 mutant E18 embryos were analyzed by MRI (Table S1) and episcopic confocal microscopy (ECM) (Fig 2, Table S2) and 11 other E18 animals were analyzed by necropsy and histology (Fig 1, 2, Table S3). The phenotypes observed in each animal are listed in the tables and summarized here. Many of the same phenotypes observed in the Ift25 mutants (Keady et al., 2012) were also observed in the Ift27 mutant animals, but the phenotypes were more severe and more penetrant in the Ift27 mutants. Like Ift25, Ift27 mutants had a high incidence of omphaloceles or umbilical cord hernias. Polydactyly and other digit defects were observed in the Ift27 mutants. In contrast to the Ift25 mutant in which only preaxial duplication of digit one was observed, the Ift27 animals also showed central polydactyly and syndactyly. Ift27 mutants occasionally exhibited abnormal flexure of the wrists resulting in a clubbing phenotype (talipomanus)(Fig 1J).
A small lower jaw (micrognathia) of varying severity was observed (Fig 1Cb, 1Cc) and most animals had an abnormally shaped nose (Fig 1C, 1E) with closely spaced eyes (hypotelorism) (Fig 1Cd). Morphological assessment of the cranial facial region showed hypoplasia of the midline including nasal structures, maxilla and mandible. Histological analysis indicated that the mutants lacked development of the palate, upper incisor, vomeronasal organ, and body of the mandible with the development of a single anterior nasal aperture (Fig 1F). The latter anomaly appeared to develop as a result of a smaller, malformed nasal septum that failed to fuse with structures at the floor of the nose (Fig 1F). The Ift27 mutants also developed various malformations of the tongue that ranged from aglossia (lack of tongue development) to microglossia (abnormally small tongue) to the tongue being abnormally attached to the floor of the oral cavity (Fig 1E, Table S1).
Like Ift25, Ift27 mutants had malaligned sternal vertebrate and malformed ribs, yielding an abnormally shaped chest cavity (Fig 1G, 1H). Also similar to Ift25 mutants, lung isomerisms (Fig 2A) and other structural respiratory tract abnormalities were prevalent in Ift27 mutants. Fusions between the trachea and esophagus (tracheoesophageal fistulas) were found in many Ift27 mutants. The Ift27 mutant lungs often contained a large abnormal balloon-like cavity (Fig 2Ad). ECM imaging revealed these cavities arise from the bronchial airways after they branch from the fused trachea-esophagus and the balloons then project through the diaphragm to connect with the stomach (Fig 2Ad). Histological analyses showed multi-ciliated cells were present on the lining of the cavity (Fig 2B).
Cardiac malformations (Fig 2C, 2D, Fig S2) were found in all Ift27 mutants. Similar to the Ift25 animals, we observed double-outlet right ventricle (Fig S2A, Movie 1), which was associated with hypoplasia of the pulmonary trunk (Fig S2B, Movie 2). The Ift27 mutants, similar to the Ift25 mutants, showed partial or complete atrioventricular septal defects (AVSD) that reflect defects in development of the endocardial cushions. This resulted in a single orifice with common AV valves formed instead of separate tricuspid and mitral valves required for separation of the right atrium/right ventricle from the left atrium/left ventricle. In mutants with partial AVSD, the atrioventricular orifice is asymmetrically positioned to favor one ventricle (Fig 2D, Movie 1). These defects are also associated with a common atrium due to complete failure in atrial septum formation. In addition, one mutant exhibited a pulmonary artery defect where a long common artery extended from the pulmonary trunk (Fig.S2D, Movie 2) and aortic arch anomalies were also observed, with one mutant exhibiting a double arch forming a vascular ring (Fig.S2F, Movie 3). The latter phenotype was observed in conjunction with trachea-esophageal fistulas. As with the Ift25 mutants, Ift27 mutant animals likely die neonatally from these severe congenital heart defects.
Left lung isomerism was the only laterality phenotype observed in the Ift25 mutants. This was observed in Ift27 mutants but the Ift27 animals also show a variety of left-right patterning defects characterized by heterotaxy with randomization of visceral organ situs. Defects include malpositioning of the stomach (dextrogastria), liver isomerisms and cardiac malformations. Laterality defects of the heart include abnormal positioning of the organ (dextrocardia and mesocardia) (Fig.2C), right atrial isomerism and duplication of the inferior vena cava (Fig.S2F, Movie 4).
IFT27 is not required for IFT25 stability but is required for IFT25 entry into cilia
IFT25 was not required for ciliary assembly or for the stability of any of the IFT particle proteins that we have antibodies against except for IFT27, which is largely depleted when IFT25 was lost (Keady et al., 2012). Similarly IFT27 is not required for ciliary assembly in cultured fibroblasts or in the embryo (Fig 3). As expected IFT27 protein is missing from the Ift27 mutant cells as detected by western blot and by IF staining. The abundance of IFT25 is not affected by the loss of IFT27 (Fig 3A) indicating that while IFT27 requires IFT25 for stability the requirement is not mutual. The levels of some complex B proteins (IFT88, IFT57) were reduced in the Ift27 mutant cells but the significance of this is not clear as ciliation is normal and IFT88 staining of mutant cilia is similar to wild type (Fig 3B-D). In Trypanosoma, Ift27 RNAi caused a reduction of IFT dynein and IFT-A proteins and an increase in IFT-B proteins in the cilia (Huet et al., 2014). However, we did not observe any differences in IFT dynein (Dync2h1), IFT-A (IFT140) or IFT-B (IFT88) distribution in our cells (Fig 3C). We were unable to determine if the endogenous distribution of IFT25 was altered in Ift27 cells as neither a commercial antibody nor any that we generated, worked for immunofluorescence (Fig S3). However, Flag-IFT25 was unable to enter cilia on Ift27 mutant cells but was able to enter wild type cells (Fig 3E) suggesting that while IFT25 is stable in the absence of IFT27, it does not bind the IFT particle. Direct interaction between IFT25 and IFT27 appears to be important for the delivery of IFT25 to cilia as mutation of three residues (T40R/T42R/S128E) in IFT25 that make direct contact with IFT27 (Bhogaraju et al., 2011) disrupts the interaction as detected by IP and prevents the entry of IFT25 into cilia (Fig 3F,G). This mutation also fails to rescue the Smo1 and BBS9 accumulation defects (see below) caused by the loss of IFT25 (Fig 3H).
IFT27 is a small G protein in RAS superfamily and as such is thought to bind and hydrolyze GTP (Qin et al., 2007; Bhogaraju et al., 2011). To understand how the GTP/GDP state of IFT27 affects binding of IFT27 to IFT25 and the IFT particle, we generated IFT27 constructs with the mutations that are expected to mimic the GTP-bound (K68L) and GDP-bound or nucleotide free (T19N) states. These mutations in IFT27 did not affect binding to IFT25 (Fig 3I) indicating that the GTP cycle does not influence the formation of the IFT25/IFT27 heterodimer. The binding of IFT27 to the IFT particle (IFT88) was disrupted by the Ift27T19N mutation but not by the Ift27K68L mutation (Fig 3I). Consistent with this, in wild type cells IFT27T19N was excluded from cilia (Fig 3J top row). However, IFT27T19N was able to enter cilia of Ift27 mutant cells (Fig 3J, bottom row) suggesting that it retains some ability to bind the IFT particle but the affinity is reduced such that it cannot compete with wild type. Furthermore, Ift27T19N was not able to fully rescue the Smo1 and BBS9 accumulation defects (see below) caused by the loss of IFT27 while wild type Ift27 and Ift27K68L were able to rescue (Supplemental Fig 3C). Thus, it is likely that IFT27 is in the GTP-bound state in the cilium and the data further suggests that the IFT particle is an effector of IFT27 since the interaction is affected by the GTP/GDP state of IFT27.
Ift27 mutants have hedgehog signaling defects
It is well established that cilia play a major role in the hedgehog pathway of the developing embryo (Huangfu et al., 2003) and many of the phenotypes observed in the Ift27 mutant mouse are consistent with defects in hedgehog signaling. Thus, we analyzed how hedgehog signaling was affected by the loss of IFT27 in the developing embryo and in vitro cultured fibroblasts. Analysis of cultured MEFs (Fig 4A) showed that while Gli1 is highly upregulated in response to the hedgehog agonist SAG in wild type cells, the response is dampened significantly in the mutant cells. Ptch1 behaved similarly but the high amount of variation in expression precluded reaching statistical significance. This attenuation was similar to what we observed in Ift25 and Ift88 mutant cells (Keady et al., 2012). The response was observed at both the mRNA and protein levels. As expected, other genes (Gli2, Smo, etc.) not transcriptionally activated by the hedgehog pathway were not affected.
When the hedgehog pathway is normally activated, Ptch1 and Gpr161 exit from the cilium and Smo and Gli2 enter. Ift27 mutants are defective in maintaining low ciliary Smo levels when the pathway is off (Fig 4B). Interestingly, additional Smo can enter the cilium when the pathway is activated by SAG. This additional accumulation is likely because SAG activates the pathway by binding Smo directly (Chen et al., 2002) and does not depend on activation of the upstream parts of the pathway. Similar to what we observed in cultured cells, Smo accumulates to high levels in the cilia of the embryo, which is also the case for Ptch1 (Fig 4I). Gpr161 is normally found in unstimulated cilia and exits after pathway activation (Mukhopadhyay et al., 2013). In Ift27 mutants more Gpr161 is present in the cilia than normal at the basal state and the protein is not fully cleared after activation (Fig 4C). Even though SAG is capable of stimulating the entry of additional Smo and partial removal of Gpr161 in Ift27 mutant cells, this is not sufficient to cause Gli2 to accumulate at the ciliary tip in normal levels (Fig 4D). The failure of Gli2 to accumulate at the ciliary tip could be caused by the lack of signal from the upstream components of the pathway or because IFT27 is needed to transport Gli2 into the cilium. To distinguish between these possibilities we transfected wild type and Ift27 mutant cells with SmoM2. SmoM2 is an oncogenic form of Smo that is constitutively activated (Xie et al., 1998). Expression of SmoM2 in wild type cells caused Gli2 accumulation at the ciliary tip similar to what would be observed if the pathway were activated (Fig 4E). Ift27 mutant cells transfected with SmoM2 accumulated ciliary Gli2 to the same level as the control cells. However in the mutant cells, the Gli2 was more broadly distributed along the length of the cilium rather than being concentrated at the tip. This suggests that IFT27 is not needed for entry of Gli2 into the cilium but is needed for its transport to the tip or alternatively in some yet unknown structural aspect of the ciliary tip.
In vivo analysis of the hedgehog pathway in mouse neural tube showed caudal expansions of Olig2- and Pax6-positive cells and a failure of the floor plate to express sonic hedgehog (Fig 4F). These phenotypes are similar to what we saw in the Ift25 mouse, similar to other IFT complex B mutant mice (Keady et al., 2012; Ko et al., 2009) and are consistent with the attenuated hedgehog signaling observed in vitro. The processing of Gli3 into the repressor form was also reduced in the Ift27 mutants (Fig 4G) similar to how it is affected by Ift25 and other IFT complex B mutants (Keady et al., 2012; Haycraft et al., 2005). To understand how the Ift27 mutation affects global hedgehog signaling, Gli1-LacZ expression was monitored in E12.5 embryos. Note that the Ift27 allele (Ift27null2) used in this experiment did not contain a beta-galactosidase gene (Supplemental Fig 1). Alterations in Gli1-LacZ expression were observed in the brain, limb buds and facial regions consistent with these regions being the most highly affected by the lack of IFT27 (Fig 4H and Fig S4).
Ift27 mutants accumulate BBS proteins in cilia
Much the same as what we observe in Ift25 and Ift27 mutant cells, Ptch1 and Smo accumulate in the cilia of Bbs mutant cells (Zhang et al., 2012)(Fig S5) prompting us to examine how the BBSome is affected by the Ift27 mutation. In wild type cells, the BBSome subunit BBS5 is not detectable in cilia with our antibody and the BBSome subunit BBS9 is found primarily at the base of the cilium with very faint label along the ciliary shaft. Interestingly, in Ift27 mutant cells both of these proteins accumulate to high levels in the cilium such that BBS5 is easily detected and BBS9 is redistributed from the base of the cilium into the cilium (Fig 5A). This accumulation also is observed in Ift25 mutant cells (data not shown). Similarly BBS5 and BBS9 are not detectable on cilia in the paraxial mesoderm of wild type embryos but are highly enriched in these cilia on Ift27 mutant embryos (Fig 5B). This data suggests that the BBSome enters cilia independent of IFT25/IFT27 but requires them for removal.
Figure 5. BBSome subunits BBS5, BBS9 and BBS regulators Arl6, Lztfl1 accumulate in Ift27 mutant cilia.

A. Ift27+/+ and Ift27null1/null1 MEFs were stained for cilia (6-11B-1, red) and BBS5 (top row, green), BBS9 (second row, green), Arl6 (third row, green) or Lztfl1 (bottom row, green). Insets show the green channel (BBS5, BBS9, Arl6, Lztfl1). Quantification showed 0% wild type cells had detectable BBS5 in cilia while 88±12% of mutant cells had detectable BBS5 in cilia and 0% wild type cells had strong BBS9 in cilia (all had weak ciliary staining with moderate staining of the centrosomal region) while 95±6% of mutant cells had strong staining of BBS9 in cilia and no staining of the centrosome (n = 25 cilia from 3 cell lines per genotype, p<0.001). Similarly 0% of wild type cells had detectable Arl6 in cilia while 80±3% of mutant cells had detectable Arl6 in cilia and 0% of wild type cells had detectable Lztfl1 in cilia while 97±2% of Ift27 mutant cells had strong Lztfl11 label in cilia (n = >25 cilia per genotype from three experiments, p<0.001). Scale bar is 10 μm and applies to all images in A.
B. Paraxial mesoderm stained with BBS5 and BBS9 antibodies. Sections of E10.5 embryos were stained for cilia (6-11B-1, green) and either BBS5 or BBS9 (red). Insets are 4X enlargements of the cilium marked with an arrow. Scale bar is 10 μm. Images are maximum projections of 16 layer Z-stacks acquired every 0.25 μm.
Ciliary levels of the BBSome are regulated by Lztfl1 and the small GTPase Arl6, which is also known as BBS3. Lztfl1 is a BBSome binding protein that was found in the cell body but not in the cilium. Knockdown cells accumulate high levels of the BBSome in their cilia suggesting that Lztfl1 functions in the cell body to negatively regulate entry of the BBSome into cilia (Seo et al., 2011). Arl6 is required for entry of the BBSome into cilia and knockdown cells have reduced levels of ciliary BBSome (Jin et al., 2010). Interestingly, both Arl6 and Lztfl1 are highly enriched in Ift27 mutant cilia (Fig 5A) suggesting that both proteins cycle through the cilia and that IFT25/IFT27 are needed for their removal. Like Ift25 and Ift27, Lztfl1 is conserved in Chlamydomonas but absent from Drosophila and Caenorhabditis. The Chlamydomonas homolog of Lztfl1 (XP_001696645.1) was found in our flagellar proteome (CrFP C_1570028, three peptides). The Ltzfl1 peptides were found in the membrane and matrix fraction, which is where IFT proteins fractionate (Pazour et al., 2005) suggesting that in contrast to conclusions of prior studies (Seo et al., 2011) Lztfl1 is a ciliary protein. Lztlfl1 mutant mice are not available and so we created Ltzfl1 null NIH3T3 lines using Crispr/Cas9 (Cong et al., 2013). In these cells, Ltzfl1 protein was not detectable by western blotting (Fig 6A) and the cells showed Smo and BBSome accumulation as previously reported in knockdown cells (Seo et al., 2011) (Fig 6C). Ciliary assembly appeared normal and the Lztfl1 mutant cells had normal IFT27 ciliary staining (Fig 6B, C) suggesting that Lztfl1 functions downstream of IFT27. Previously it was shown that a C-terminal fragment of Lztfl1 bound the BBSome and acted as a dominant negative causing accumulation of BBSome components into cilia (Seo et al., 2011). Interestingly, the C-terminal half of Lztfl, which functions as a dominant negative is trafficked to cilia whereas the N-terminal half, which does not function as a dominant negative (Fig 6D) is not found in cilia. Neither fragment affected the distribution of IFT27 (data not shown) supporting a role of Lztfl1 downstream of IFT27.
Figure 6. The BBSome regulator Lztfl1 functions downstream of IFT27 in the removal of the BBSome and Smo from cilia.
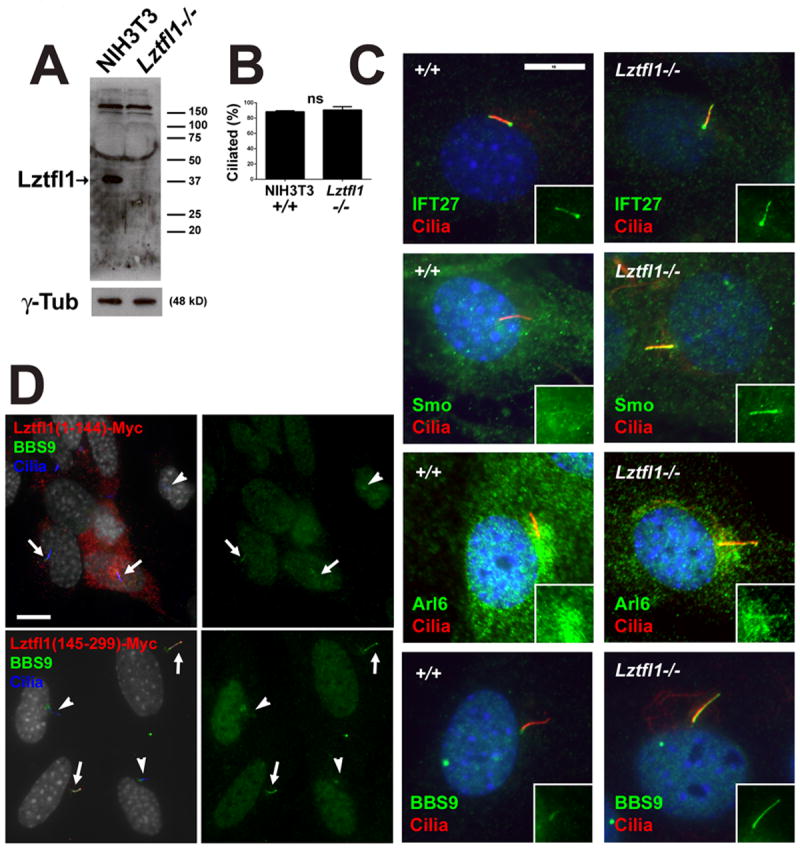
A. NIH3T3 control (+/+) and Lztfl1 mutant (-/-) cells probed for Lztfl1 show that the mutant does not have any detectable protein. γ-tubulin is a loading control. Note the blot is overexposed to ensure that no protein remains.
B. Ciliation is not affected by the loss of Lztfl1 (n=3 experiments on 1 cell line).
C. NIH3T3 control (+/+) and Lztfl1 mutant cells were stained for cilia (6-11B-1, red) and IFT27 (top row, green), Smo (second row, green), Arl6 (third row, green) or BBS9 (bottom row, green). Insets show the green channel (IFT27, Smo, Arl6, BBS9). Scale bar is 10 μm. Quantification showed 100% of control cilia and Lztfl1 mutant cilia had normal IFT27 label (strong peribasal body label with weaker ciliary shaft label)(n=3 experiments on 1 cell line per genotype); 4.0±4% of control and 60±11% of Lztfl1 mutant cells had ciliary Smo label in unstimulated cells. (n=3 experiments on 1 cell line per genotype, p<0.01); 0% of control cilia and 59±26% of Lztfl1 mutant cilia had weak but detectable levels of Arl6 (n=3 experiments on 1 cell line per genotype, p<0.05); 0% of control cilia and 98±2% of Lztfl1 mutant cilia had strong ciliary BBS9 label (n=3 experiments on 1 cell line per genotype, p<0.001).
D. The N-terminal half of Lztfl1 (residues 1-144) does not localize to cilia and does not perturb ciliation or increase the ciliary levels of BBS9. Quantification showed that 0/10 ciliated transfected cells had detectable ciliary BBS9. The C-terminal half of Lztfl1 (residues 145-299) localizes to cilia and causes BBS9 to accumulate in cilia. Quantification showed that 10/10 ciliated transfected cells had detectable ciliary BBS9. Arrows mark cilia on myc-positive cells and arrowheads mark cilia on myc-negative cells. Scale bar is 10 μm.
To further understand the relationship between IFT27 and the BBSome in regulating the ciliary distribution of hedgehog components and the BBSome we examined MEF cell lines mutated for BBSome components BBS2 and BBS7, and the BBS regulator BBS3/Arl6 (Fig S5). As expected, in all lines the BBS components were not enriched in cilia and all lines had increased levels of ciliary Smo. None of these cells lines had detectable alterations in IFT27 or Lztfl1 distribution suggesting that IFT27 and Lztfl1 function upstream of the BBSome. The observations that mutations in Bbs2, Bbs7, Lztfl1 and Ift27 all cause increases in ciliary Smo, while mutations in Lztfl1 and Ift27 cause increases in ciliary BBSome and mutations in Ift27 alone causes increased ciliary Lztfl1 (Fig 7A) suggests a model where Lztfl1 couples the BBSome to IFT25/IFT27 for removal from the cilium by retrograde transport (Fig 7B).
Figure 7. Model for IFT25/IFT27 function.

A. Graphical summary of the effects of Ift and Bbs mutations on ciliary assembly and localization of IFT, BBS and hedgehog components. X means the process or localization was blocked by the mutation in the left column; = means the process or localization is not affected by the mutation and the arrow indicates that the protein is elevated in the cilia on the mutant cells. The crossed out arrow means that we cannot detect the protein in cilia on wild type cells and the ciliary level is not increased in the mutant.
B. Model for function of IFT25/IFT27. Details are provided in the Discussion.
Discussion
Why does the IFT particle need 6 complex A, 16 complex B, and 8 BBSome proteins to carry out its work? The high degree of evolutionary conservation from algae to humans suggests that each has an important function to play in the assembly of the cilium or in the signaling functions of the organelle. Trying to understand the functions of individual subunits has been a challenge as mutations in most subunits disrupt the function of their respective subcomplex. For example, in mouse null mutations in most complex B subunits block ciliary assembly and result in embryonic lethality at mid gestation. In contrast, the IFT25/IFT27 dimer appears to be a sub module of complex B and their loss does not disrupt cilia assembly. In trypanosomes, IFT27 is required for IFT as cells depleted of the protein accumulate complex B proteins in short stumpy flagella (Huet et al., 2014). The difference between IFT27 function in trypanosomes and mouse is unknown, but the fact that Ift27 mutations do not block ciliary assembly in the mouse allowed us to uncouple the function of IFT25/IFT27 from the function of complex B in ciliary assembly. Ift25 and Ift27 mutant mice survive to birth but die soon after with a constellation of phenotypes including heart, lung, skeletal and brain abnormalities. The phenotypes of the Ift27 and Ift25 mutants are similar but the Ift27 phenotypes are stronger and more penetrant. This was unexpected as IFT27 is highly destabilized in the Ift25 mutant and the protein was mostly gone. However, a small amount of IFT27 remains in the Ift25 mutant animals whereas no IFT27 is detected in the Ift27null1 mutant and it is likely the small amount of IFT27 that remains in the Ift25 mutant animals is able to partially function. The mouse Ift27 phenotype suggests weakened hedgehog signaling and we observed dysfunctional hedgehog signaling in both the embryo and in fibroblasts derived from the animals.
Model for the function of IFT25/IFT27
Our finding that the IFT25/IFT27 dimer is not required for ciliary assembly but is needed to remove Smo when hedgehog signaling is off and to remove Ptch1 and Gpr161 when the pathway is activated raises the question about how IFT25/IFT27 function to accomplish this.
Based on our data and data in the literature, we developed the following model for IFT25/IFT27 function (Fig 7). Entry of Ptch1 and Smo into cilia appears to be independent of IFT25, IFT27 and the BBSome as Ptch1 and Smo accumulate in cilia on Ift25, Ift27, Bbs2 and Bbs7 mutant cells. Thus our model focuses on the removal of these hedgehog components. IFT27 is a small G-protein and interactions of G-proteins with other proteins are often regulated by the nucleotide bound to the G-protein. The binding of IFT27 to IFT25 does not appear to depend on the type of guanine nucleotide bound as the dimer formed as well with IFT27T19N form (thought to mimic the GDP-bound or nucleotide free state of IFT27) as it did with the IFT27K68L (thought to reflect the GTP bound state) and the wild type forms. However, the IFT25/IFT27T19N dimer was unable to associate with the rest of complex B when wild type IFT27 was present and the T19N mutant form was not able to fully rescue the Smo and BBS accumulation phenotypes of Ift27 mutant cells. This suggests that while IFT25/IFT27T19N retains some ability to bind to the IFT particle, the affinity is reduced. The binding of IFT25/IFT27 to IFT complex B is likely through interactions with IFT81/IFT74 (Lucker et al., 2010; Bhogaraju et al., 2013).
IFT25/IFT27 could directly bind Smo, Ptch1 and Gpr161 to couple them to the IFT particle for removal from the cilium. However, in vitro Ptch1 and Smo accumulate in BBS-defective cilia and the BBSome interacts directly with the cytoplasmic tail of Smo (Zhang et al., 2012). Our findings that the BBSome and Lztfl1 accumulates in Ift27 mutant cilia along with the data showing that the BBSome accumulates in Lztfl1 mutant cilia suggest that IFT25/IFT27 work with the BBSome through Lztfl1 to actively remove ciliary membrane proteins such as Ptch1, Smo and Gpr161 in a signal-dependent manner. The relationship between Arl6 and IFT25/IFT27 remains to be established but the observation that Arl6 accumulates in Ift27 mutant cilia suggests that Arl6 requires IFT25/IFT27 for removal from cilia.
In addition to the role of IFT25/IFT27 in the regulated removal of Ptch1, Smo and Gpr161 from cilia, IFT25/IFT27 may also play a role in transport of Gli2. Ift25 and Ift27 mutant cells fail to elevate Gli2 at the ciliary tip when the hedgehog pathway is activated. It is likely that this phenotype is largely indirect and due to the failure of the pathway to be activated. However, our SmoM2 results suggest that IFT25/IFT27 do play a role in transporting Gli2 to the ciliary tip. When cells are transfected with this oncogenic form of Smo, the pathway downstream of Smo is activated. In wild type cells, this causes accumulation of Gli2 at the ciliary tip much like would be observed if the pathway were activated by ligand. In Ift27 mutant cells transfected with SmoM2 the total amount of Gli2 in the cilia was similar to what was seen in control cells but the protein was not concentrated at the tip as normally observed. This indicates that IFT25/IFT27 are needed to transport Gli2 to the tip or the tip of Ift27 mutant cilia are abnormal. Live cell imaging will be needed to address this question.
Conclusions
In this work we demonstrate that IFT27, like IFT25 is not required for ciliary assembly but is required for the dynamic movements of Ptch1, Smo and Gpr161 that occur during hedgehog signaling. The BBSome and BBSome regulator Lztfl1 also accumulate in Ift27 mutant cilia. This suggests a functional model whereby Lztfl1 coordinates the interactions between the BBSome and the IFT particle to regulate the removal of Ptch1 and Smo from cilia at the appropriate times during hedgehog signaling.
Supplementary Material
Figure S1: Map of the Ift27 alleles used in this work
Figure S2: Additional planes documenting heart defects observed in the Ift27 mutant mice
Figure S3: Characterization of the Ift25 antibody used in this work
Figure S4: Additional images of Gli1-LacZ animals
Figure S5: Analysis of Bbs2, Arl6 and Bbs7 mutant MEFs
Table S1: MRI phenotypes
Table S2: Detailed heart phenotypes
Table S3: Necropsy phenotypes
Table S4: Statistical analysis of data in Figure 4A
Movie S1: Movie documenting atrioventricular septal defects (AVSD) with double outlet right ventricle (DORV)
Movie S2: Movie documenting pulmonary artery defects
Movie S3: Movie documenting aortic arch anomalies
Movie S4: Movie documenting right atrial isomerism
Acknowledgments
We thank Drs. S. Jones (Transgenic Mouse Core) and P. Furcinitti (Digital Imaging Core) for assistance during this work. We thank Dr. P. Odgren for use of his bright field microscope, Drs. J. Eggenschwiler (Princeton Univ.), R. Vallee (Columbia Univ.), M. Nachury (Stanford Univ.), R. Rohatgi (Stanford Univ.), S. Mukhopadhyay (Univ. Texas SW), and S. Seo (Univ. of Iowa) for reagents. We thank M. Nachury (Stanford Univ.), B. Yoder (Univ. Alabama), Q Zhang (Univ. of Iowa), V Sheffield (Univ. of Iowa), L. Leehy and R. Bortell for mouse fibroblasts. We thank Drs. D. Nedelcu and A. Salic (Harvard Med. Sch.) for assistance with lentiviral constructs, Dr. R. Maher for assistance with genome editing and Dr. E. Lorentzen for advice on IFT25/IFT27 structure. This work was supported by the National Institutes of Health GM060992 to GJP, 5U01HL098180 to CWL, and received funding from the European Community’s Seventh Framework Programme FP7/2009 under grant agreement no: 241955 SYSCILIA to CAJ. YL was supported by a grant from China Scholarship Council (No.2011621077). ZAA was supported by a grant from the Rosetree’s Trust (No. JS16/M279). Core resources supported by the Diabetes Endocrinology Research Center grant DK32520 and the Alabama Recessive Polycystic Kidney Disease Core Center DK074038 were used.
Abbreviations
- Shh
Sonic Hedgehog
- Smo
Smoothened
- Ptch1
Patched 1
- H&E
hematoxylin and eosin
- IFT
intraflagellar transport
- SD
standard deviation
- MEF
mouse embryonic fibroblast
- TEM
transmission electron microscopy
- e
embryonic
- p
postnatal
- ECM
episcopic confocal microscopy
- AVSD
atrioventricular septal defect
- AV
atrioventricular
- VSD
ventricular septal defect
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aldahmesh MA, Li Y, Alhashem A, Anazi S, Alkuraya H, Hashem M, Awaji AA, Sogaty S, Alkharashi A, Alzahrani S, Al Hazzaa SA, Xiong Y, Kong S, Sun Z, Alkuraya FS. IFT27, encoding a small GTPase component of IFT particles, is mutated in a consanguineous family with Bardet-Biedl syndrome. Hum Mol Genet. 2014;23:3307–3315. doi: 10.1093/hmg/ddu044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet. 2006;7:125–148. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- Bai CB, Auerbach W, Lee JS, Stephen D, Joyner AL. Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development. 2002;129:4753–4761. doi: 10.1242/dev.129.20.4753. [DOI] [PubMed] [Google Scholar]
- Berbari NF, Lewis JS, Bishop GA, Askwith CC, Mykytyn K. Bardet-Biedl syndrome proteins are required for the localization of G protein-coupled receptors to primary cilia. Proc Natl Acad Sci U S A. 2008;105:4242–4246. doi: 10.1073/pnas.0711027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhogaraju S, Cajanek L, Fort C, Blisnick T, Weber K, Taschner M, Mizuno N, Lamla S, Bastin P, Nigg EA, Lorentzen E. Molecular basis of tubulin transport within the cilium by IFT74 and IFT81. Science. 2013;341:1009–1012. doi: 10.1126/science.1240985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhogaraju S, Taschner M, Morawetz M, Basquin C, Lorentzen E. Crystal structure of the intraflagellar transport complex 25/27. EMBO J. 2011;30:1907–1918. doi: 10.1038/emboj.2011.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JK, Taipale J, Young KE, Maiti T, Beachy PA. Small molecule modulation of Smoothened activity. Proc Natl Acad Sci U S A. 2002;99:14071–14076. doi: 10.1073/pnas.182542899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole DG, Diener DR, Himelblau AL, Beech PL, Fuster JC, Rosenbaum JL. Chlamydomonas kinesin-II-dependent intraflagellar transport (IFT): IFT particles contain proteins required for ciliary assembly in Caenorhabditis elegans sensory neurons. J Cell Biol. 1998;141:993–1008. doi: 10.1083/jcb.141.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley FW, Soriano P, Steffen LS, Dymecki SM. Widespread recombinase expression using FLPeR (flipper) mice. Genesis. 2000;28:106–110. [PubMed] [Google Scholar]
- Follit JA, Xu F, Keady BT, Pazour GJ. Characterization of mouse IFT complex B. Cell Motil Cytoskeleton. 2009;66:457–468. doi: 10.1002/cm.20346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005;1:e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426:83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- Huet D, Blisnick T, Perrot S, Bastin P. The GTPase IFT27 is involved in both anterograde and retrograde intraflagellar transport. Elife. 2014;3:e02419. doi: 10.7554/eLife.02419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iomini C, Li L, Esparza JM, Dutcher SK. Retrograde intraflagellar transport mutants identify complex A proteins with multiple genetic interactions in Chlamydomonas reinhardtii. Genetics. 2009;183:885–896. doi: 10.1534/genetics.109.101915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H, White SR, Shida T, Schulz S, Aguiar M, Gygi SP, Bazan JF, Nachury MV. The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell. 2010;141:1208–1219. doi: 10.1016/j.cell.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonassen JA, SanAgustin J, Baker SP, Pazour GJ. Disruption of IFT complex A causes cystic kidneys without mitotic spindle misorientation. J Am Soc Nephrol. 2012;23:641–651. doi: 10.1681/ASN.2011080829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keady BT, Le YZ, Pazour GJ. IFT20 is required for opsin trafficking and photoreceptor outer segment development. Mol Biol Cell. 2011;22:921–930. doi: 10.1091/mbc.E10-09-0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keady BT, Samtani R, Tobita K, Tsuchya M, San Agustin JT, Follit JA, Jonassen JA, Subramanian R, Lo CW, Pazour GJ. IFT25 links the signal-dependent movement of Hedgehog components to intraflagellar transport. Dev Cell. 2012;22:940–951. doi: 10.1016/j.devcel.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko HW, Liu A, Eggenschwiler JT. Analysis of hedgehog signaling in mouse intraflagellar transport mutants. Methods Cell Biol. 2009;93:347–369. doi: 10.1016/S0091-679X(08)93017-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechtreck KF, Johnson EC, Sakai T, Cochran D, Ballif BA, Rush J, Pazour GJ, Ikebe M, Witman GB. The Chlamydomonas reinhardtii BBSome is an IFT cargo required for export of specific signaling proteins from flagella. J Cell Biol. 2009;187:1117–1132. doi: 10.1083/jcb.200909183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucker BF, Miller MS, Dziedzic SA, Blackmarr PT, Cole DG. Direct interactions of intraflagellar transport complex B proteins IFT88, IFT52, and IFT46. J Biol Chem. 2010;285:21508–21518. doi: 10.1074/jbc.M110.106997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Wen X, Ratti N, Loktev A, Rangell L, Scales SJ, Jackson PK. The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic hedgehog pathway via cAMP signaling. Cell. 2013;152:210–223. doi: 10.1016/j.cell.2012.12.026. [DOI] [PubMed] [Google Scholar]
- Ou G, Koga M, Blacque OE, Murayama T, Ohshima Y, Schafer JC, Li C, Yoder BK, Leroux MR, Scholey JM. Sensory ciliogenesis in Caenorhabditis elegans: assignment of IFT components into distinct modules based on transport and phenotypic profiles. Mol Biol Cell. 2007;18:1554–1569. doi: 10.1091/mbc.E06-09-0805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ, Agrin N, Leszyk J, Witman GB. Proteomic analysis of a eukaryotic cilium. J Cell Biol. 2005;170:103–113. doi: 10.1083/jcb.200504008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen LB, Rosenbaum JL. Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr Top Dev Biol. 2008;85:23–61. doi: 10.1016/S0070-2153(08)00802-8. [DOI] [PubMed] [Google Scholar]
- Qin H, Wang Z, Diener D, Rosenbaum J. Intraflagellar transport protein 27 is a small G protein involved in cell-cycle control. Curr Biol. 2007;17:193–202. doi: 10.1016/j.cub.2006.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum JL, Witman GB. Intraflagellar transport. Nat Rev Mol Cell Biol. 2002;3:813–825. doi: 10.1038/nrm952. [DOI] [PubMed] [Google Scholar]
- Sander JD, Maeder ML, Reyon D, Voytas DF, Joung JK, Dobbs D. ZiFiT (Zinc Finger Targeter): an updated zinc finger engineering tool. Nucleic Acids Res. 2010;38:W462–W468. doi: 10.1093/nar/gkq319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satir P, Christensen ST. Overview of structure and function of mammalian cilia. Annu Rev Physiol. 2007;69:377–400. doi: 10.1146/annurev.physiol.69.040705.141236. [DOI] [PubMed] [Google Scholar]
- Seo S, Zhang Q, Bugge K, Breslow DK, Searby CC, Nachury MV, Sheffield VC. A novel protein LZTFL1 regulates ciliary trafficking of the BBSome and Smoothened. PLoS Genet. 2011;7:e1002358. doi: 10.1371/journal.pgen.1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Murone M, Luoh SM, Ryan A, Gu Q, Zhang C, Bonifas JM, Lam CW, Hynes M, Goddard A, Rosenthal A, Epstein EH, Jr, de Sauvage FJ. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391:90–92. doi: 10.1038/34201. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Seo S, Bugge K, Stone EM, Sheffield VC. BBS proteins interact genetically with the IFT pathway to influence SHH-related phenotypes. Hum Mol Genet. 2012;21:1945–1953. doi: 10.1093/hmg/dds004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Map of the Ift27 alleles used in this work
Figure S2: Additional planes documenting heart defects observed in the Ift27 mutant mice
Figure S3: Characterization of the Ift25 antibody used in this work
Figure S4: Additional images of Gli1-LacZ animals
Figure S5: Analysis of Bbs2, Arl6 and Bbs7 mutant MEFs
Table S1: MRI phenotypes
Table S2: Detailed heart phenotypes
Table S3: Necropsy phenotypes
Table S4: Statistical analysis of data in Figure 4A
Movie S1: Movie documenting atrioventricular septal defects (AVSD) with double outlet right ventricle (DORV)
Movie S2: Movie documenting pulmonary artery defects
Movie S3: Movie documenting aortic arch anomalies
Movie S4: Movie documenting right atrial isomerism