

Summary
Global patterns of DNA methylation, mediated by the DNA methyltransferases (DNMTs), are disrupted in all cancers by mechanisms that remain largely unknown, hampering their development as therapeutic targets. Combinatorial acute depletion of all DNMTs in a pluripotent human tumor cell line, followed by epigenome and transcriptome analysis, revealed DNMT functions in unprecedented detail. DNMT3B occupancy regulates methylation during differentiation, while an unexpected interplay was discovered in which DNMT1 and DNMT3B antithetically regulate methylation and hydroxymethylation in gene bodies, a finding confirmed in other cell types. DNMT3B mediated nonCpG methylation, while DNMT3L influenced the activity of DNMT3B toward nonCpG versus CpG site methylation. Taken together, these data reveal new functional targets of each DNMT suggesting that isoform selective inhibition would be therapeutically advantageous.
Introduction
DNA methylation (5mC) is an epigenetic modification occurring most commonly at cytosines within CpG dinucleotides, which confers genomic stability and regulates transcription dependent on the cytosine dinucleotide context (CpG vs nonCpG) and genomic locality. The current paradigm in normal cells places a majority (∼80%) of 5mC at repetitive centromeric sequences and transposable elements to maintain chromosomal stability. Conversely, 5mC is largely absent at transcription start sites (TSS) flanked by CpG islands (CGIs), whose methylation status is generally inversely related to transcriptional activity. These patterns undergo significant reversal in cancers in which global hypomethylation of repetitive DNA contributes to genomic instability and tumor suppressor gene CGI promoter hypermethylation effectively extinguishes transcription. Much effort has been devoted to understanding the regulation of gene transcription as a consequence of methylation changes and the subsequent contribution to cancer progression. The past few years have added additional layers of regulatory complexity, including nonCpG methylation and DNA hydroxymethylation (5hmC), to the dynamics of cytosine modification. However, there is still much to be learned about the regulation of 5mC patterning, knowledge that is essential for understanding how the nonrandom patterns of aberrant methylation arise in cancer. Central to understanding the regulation of 5mC is determining how the coordinated efforts of the enzymes responsible for this modification, the DNA methyltransferases (DNMTs), contribute to the 5mC profile genome-wide.
DNMT1, the maintenance methyltransferase, is preferentially recruited to hemimethylated DNA by UHRF1 to maintain 5mC following DNA replication (Sharif et al., 2007). DNMT3A and DNMT3B, the de novo methyltransferases, are crucial for establishing methylation patterns early in development and for initiating celltype specific methylation patterns during differentiation. DNMT3L, a fourth member of the DNMT family that lacks catalytic activity, is crucial for proper development of primordial germ cells and establishment of maternal genomic imprints (Bourc'his et al., 2001). Complete genetic inactivation of the catalytically active DNMTs results in embryonic (DNMT1, DNMT3B) and postnatal (DNMT3A) lethality (Li et al., 1992; Okano et al., 1999), demonstrating that they are essential for development. Indeed, hypomorphic mutations in human DNMT3B lead to a rare, autosomal recessive developmental disorder: Immunodeficiency, Centromeric instability, Facial anomalies (ICF) syndrome (Hansen et al., 1999). DNMT3A is essential for hematopoietic stem cell lineage differentiation through silencing of lineage specific genes (Challen et al., 2012). Taken together, these studies show that DNMT3A and DNMT3B mediate 5mC patterns essential for regulating distinct lineage differentiation pathways, yet the mechanisms regulating their recruitment are perhaps more enigmatic than for DNMT1.
Dysregulation of the DNMTs, including isoforms of DNMT3A and DNMT3B, has been observed in many types of cancer, and is believed to contribute to aberrant 5mC patterns. DNMT1 is required for cell survival and specific promoter hypermethylation in colorectal cancer (Egger et al., 2006). Mutations in DNMT3A occur in over 20% of acute myeloid leukemia (AML) patients, and are associated with poor prognosis (Yamashita et al., 2010). DNMT3B, like DNMT1, promotes and sustains proliferation in colon cancers (Linhart et al., 2007). Alternatively spliced isoforms of DNMT3B, such as DNMT3B7, have opposing roles dependent on the cancer type; expression of this truncated isoform suppresses neuroblastoma growth but enhances lymphomagenesis (Ostler et al., 2012; Shah et al., 2010). Collectively, these studies demonstrate not only that DNMTs directly alter cancer phenotypes in different capacities (tumor suppressor versus proliferator), but also do so in cell-type specific contexts.
The goal of this study was to obtain a comprehensive understanding of the role of each DNMT in regulating global CpG and nonCpG methylation, transcription, differentiation, and interfacing with other epigenetic marks. To accomplish this we assayed 5mC and transcriptional patterns genome-wide in a cancer and differentiation-relevant model after depleting DNMT1, DNMT3A, DNMT3B, and DNMT3L alone and in all combinations using siRNA. To our knowledge, this is the first study to comprehensively and acutely deplete the DNMTs in all combinations. Our approach led to several interesting and unexpected observations. Most notably, depletion of DNMT3B caused global and specific ‘hypermodification’ events in gene bodies and intergenic sequences at CpGs prone to dynamic shifts in cytosine modification patterning. These ‘hotspots’ of cytosine modification were most often targeted for hypermethylation or hyperhydroxymethylation upon DNMT3B depletion. In contrast, DNMT1 and DNMT3A depletion caused moderate to mild global DNA hypomethylation, respectively. CpGs hypomethylated under DNMT1 depletion conditions overlapped significantly with CpGs hypermodified upon DNMT3B depletion, revealing an antithetical regulatory interaction between DNMT1, DNMT3B, and the TETs. Finally, our approach also revealed that DNMT3L regulates CpG versus nonCpG substrate preference of DNMT3A and DNMT3B in vivo. Taken together, our results provide unprecedented insight into the function of each DNMT in regulating cytosine epigenetic modifications that have important implications for the regulation of these marks during differentiation and for developing more specific and effective therapeutic strategies to correct or target aberrant methylation in cancer cells.
Results
Acute DNMT depletion causes differential effects on global 5mC
Using the human embryonic carcinoma (EC) cell line NCCIT as a model system, we depleted the DNMTs using siRNA both individually and in combination (Figure 1A) to identify direct functional target sites in a more comprehensive manner than has been attempted previously (DNMT1+DNMT3L was omitted as no published evidence has linked their function). NCCIT cells are derived from an extragonadal germ cell tumor and demonstrate expression patterns similar to embryonic stem cells including high level DNMT expression (Figure S1A) (Jin et al., 2012). Accordingly, they can be induced to differentiate into the three embryonic germ layers and extraembryonic lineages (Jin et al., 2012; Sperger et al., 2003). Due to its pluripotency and developmental implications, undifferentiated (UD) NCCIT cells serve as a model to study DNMT function in both development and cancer. DNMTs were acutely depleted by siRNA transfection in UD NCCIT cells for 72 hours. After extensive optimization (not shown), this timepoint was chosen to observe the most immediate impact on 5mC and gene expression, and avoid potential compensatory epigenetic changes (Bachman et al., 2003; Egger et al., 2006). mRNA transcripts were depleted by ∼80% among individual DNMT knockdowns (KD) compared to the no-target control (NTC) siRNA and no off-target effects on other DNMTs were observed (Figure S1B). Efficiency of the combination KDs varied. DNMT3B depletion efficiency in combination samples, for example, was typically in the 40-60% range (Figure S1B). However, DNMT3B is the most highly expressed de novo methyltransferase in NCCIT cells (Sperger et al., 2003), so this level of depletion is still sufficient to observe robust methylation changes in combination KDs with DNMT3B, as will be presented later. Depletion of the DNMTs was also confirmed at the protein level (Figure S1C). Housekeeping genes (RPL30, DYNLL1) and pluripotency factors (NANOG, OCT4) showed little change in expression during the 72 hour KD period, whereas markers of different germ layers varied in their expression (Figure S1D) indicating that developmental pathways, especially ectodermal, are affected by DNMT depletion. We also examined TET mRNA expression in our DNMT KDs (Figure S1D); overall, TET1 expression was maintained while expression of TET2 and TET3 was mildly decreased.
Figure 1. Differential impact of single and combinatorial DNMT depletion on genome-wide CpG methylation and expression.
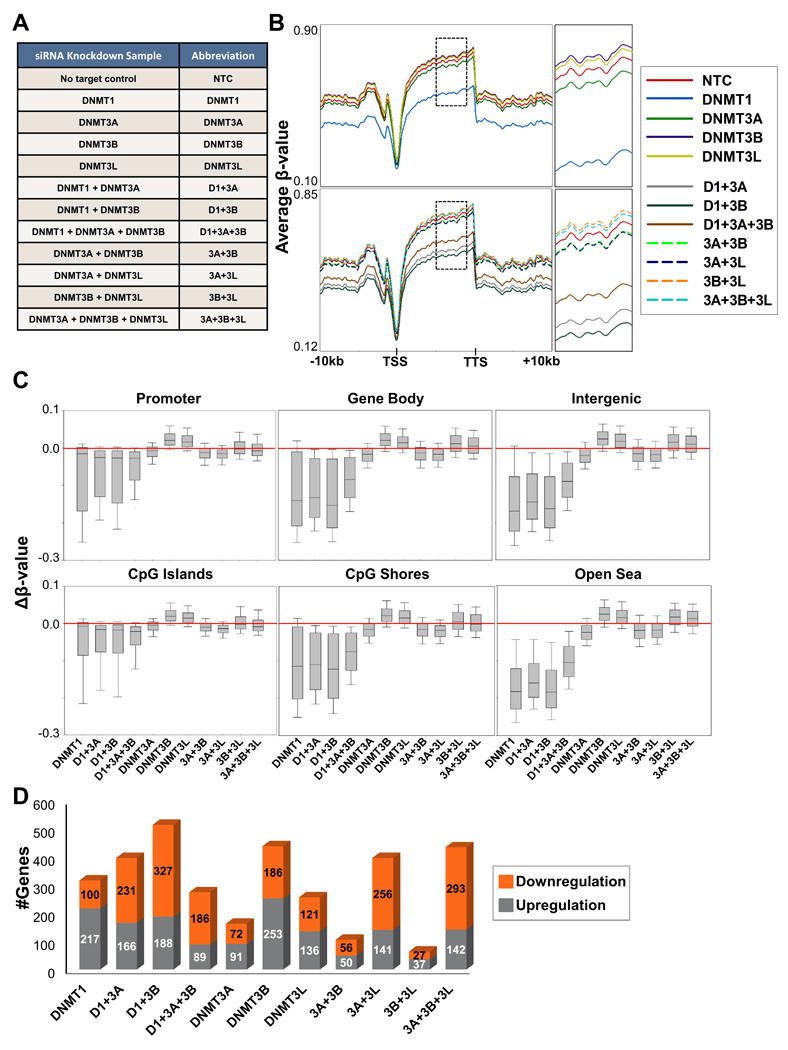
(A) Sample table detailing all siRNA KDs conducted in NCCIT cells with respective abbreviations used throughout the text. (B) Spatial distribution plots across intragenic regions derived from average β-values for 5mC in NCCIT DNMT depleted samples obtained from the 450K array. Top panel: Individual siRNA KDs Bottom panel: Combination siRNA KDs. Dashed black box (to the right) indicates region magnified to more effectively visualize relative methylation levels among samples. (C) Box plots representing all Δβ-values for each sample in the indicated features. Red bar highlights the zero position (no change in methylation). All distributions are statistically significant from NTC (except 3B+3L, 3A+3B+3L in promoter, CpG islands, and shores). (D) Total number of genes upregulated/downregulated (≥ 1.5 foldchange) by microarray analysis for NCCIT DNMT depleted samples. See also Figure S1.
Multiple independent siRNA KD transfections were pooled for each sample in subsequent 5mC and gene transcription analyses. Genome-wide methylation was assayed using Illumina's Infinium HumanMethylation450 BeadChip (450K array). DNMT1 depletion (individual/combination) resulted in global hypomethylation, most notably in gene bodies and intergenic sequences (Figures 1B-C). Consistent with previous reports (Rhee et al., 2002), combined depletion of DNMT1 and DNMT3B results in the most extensive DNA hypomethylation among all DNMT1 depleted samples (Figure S1E). The DNMT3 depletions, however, resulted in more distinctive global changes in methylation. Individual depletion of DNMT3B and DNMT3L resulted in hypermethylation across the genome, while DNMT3A depletion caused a small overall decrease in methylation (Figure 1B). Consistent with these results, combination depletion of DNMT3B with DNMT3L (3B+3L), including the addition of DNMT3A depletion (3A+3B+3L) also induced hypermethylation. In contrast, siRNA KD's involving DNMT3A (3A+3L, 3A+3B) resulted in hypomethylation throughout the genome (Figure 1B). Further examination of methylation changes at specific genomic features revealed that DNMT3B/3L KDinduced hypermethylation is most prevalent in gene bodies and intergenic sequences, and DNMT3A KDinduced hypomethylation (individual/combination) is consistent across all genomic features (Figure 1C). We confirmed reproducibility of the genome-wide 5mC observations by performing two additional independent biological replicates on a subset of the KDs (siDNMT1, siDNMT3B, si3B+3L) followed by 450K array analysis. All siDNMT1 replicate samples displayed hypomethylation across all genomic features, while siDNMT3B replicate samples consistently displayed gains in 5mC in gene bodies and intergenic sequences (Figure S1F). In general, gene expression profiles for each DNMT depleted sample did not correlate with differentially methylated loci (data not shown), however, redundancy in regulation of particular genes was observed. For example, more genes were upregulated in individually depleted DNMT conditions, while combination depletion, in general (with the exception of 3B+3L), resulted in more genes being downregulated (Figure 1D). Accordingly, hierarchical clustering of gene expression profiles grouped the individual DNMT KD samples together, while most of the combination DNMT depleted samples clustered together (Figure S1G). Taken together, these results demonstrate unique and cooperative functions for each of the DNMTs in regulating 5mC across the genome. Importantly, the most immediate changes in 5mC induced by depletion of the DNMTs do not directly alter gene expression, but rather the DNMTs potentially co-regulate expression of genes independent of their 5mC writing function.
Depletion of DNMT3B results in specific hypermethylation events that coincide with DNMT1 depletiondriven hypomethylation events
Next, we examined the most significantly differentially methylated CpG sites (pval < 0.05) in each DNMT depleted sample to define loci most prone to changes in 5mC. Consistent with global methylation patterns, DNMT1 depletion (individual/combination) conferred widespread hypomethylation across the genome, with particular enrichment in gene bodies, 3′UTRs, and intergenic sequences (Figures 2A-B, Figures S2A, S2B upper panels). We independently confirmed siDNMT1 associated hypomethylation using bisulfite genomic sequencing (BGS) for the PTPRN2 (intron), CREBBP (intron/exon), and TACSTD2 (5′UTR) loci (Figure S2C). Consistent with microarray expression analysis, correlating changes in expression and methylation were not observed at these loci (Figure S2D). Overall, hypomethylation across all genomic features significantly overlapped among KD's involving DNMT1 (Figure S3A, left panel). Ontology analysis of the top 3,000 genes containing the most significant hypomethylated CpG sites located in the gene body revealed marked enrichment for pathways associated with development and cancer (Figure 2C).
Figure 2. Analysis of significantly differentially methylated CpG sites reveals distinct effects of DNMT1 vs DNMT3 depletion.
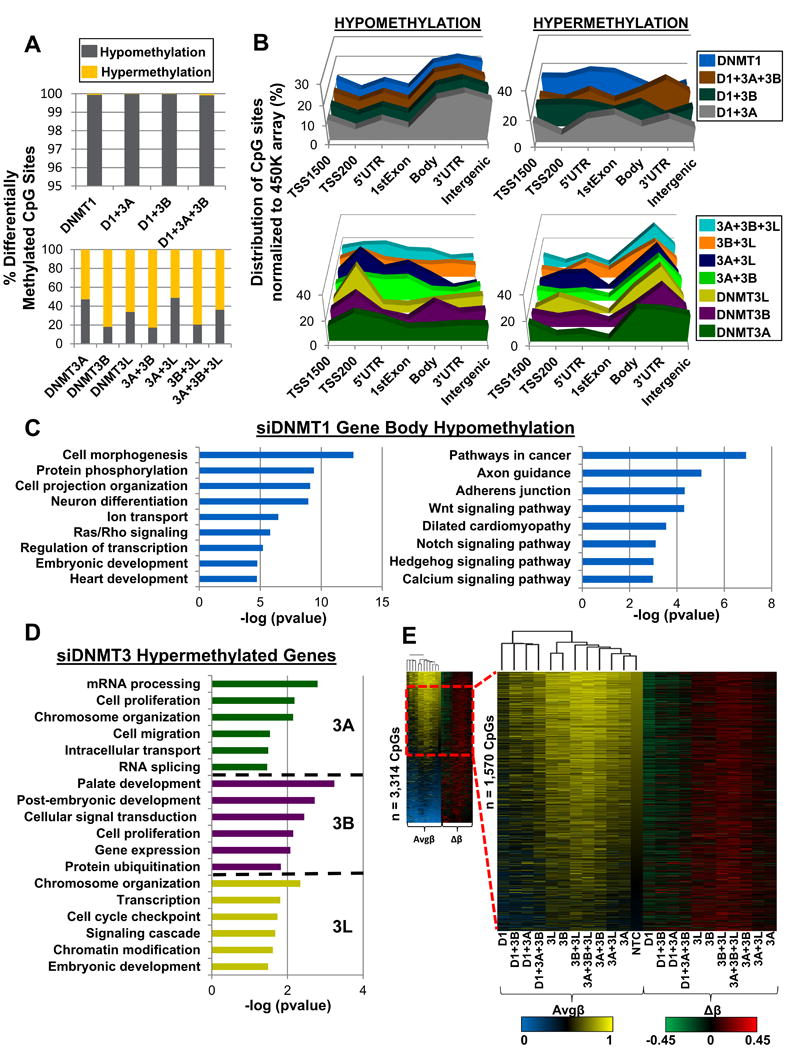
(A) Percentage of significantly differentially methylated CpG sites (pval < 0.05) classified by methylation change upon siRNA KD in NCCIT DNMT siRNA depleted cells. (B) Normalized distribution of significantly differentially methylated CpG sites by genomic feature. Top: DNMT1 depletion (individual/combination) Bottom: DNMT3 depletion (individual/combination). (C) Ontology analysis for the top 3,000 most hypomethylated gene bodies (CpG sites with greatest Δβ-value) upon siRNA depletion of DNMT1. Left panel: Gene ontology analysis Right panel: KEGG pathway analysis. (D) Gene ontology for hypermethylated genes in DNMT3 individual knockdown (at least 1 CpG site displaying hypermethylation within gene). Coloring scheme from 2B is used. (E) Left panel: Clustered heat map (Euclidean distance) of hypermethylated (Δβ ≥ 0.15) CpG sites for each DNMT depleted condition (duplicate CpG sites removed) stratified by NTC Avgβ-value most methylated to least methylated CpG sites. Dashed red box indicates magnified region (Avgβ-value [0.45 - 0.75] in NTC). Right panel: Heat map of Δβ-values for respective CpG sites from the left panel. See also Figure S2, S3.
Unexpectedly, DNMT3 siRNA KDs, particularly those involving DNMT3B, showed more significant hypermethylation than hypomethylation events (Figures 2A-B, Figures S2A, S2B lower panels). Inclusion of two additional independent biological replicates of the siDNMT3B and si3B+3L KD conditions confirmed this observation (Figure S3B). Overwhelmingly, DNMT3 depletion associated hypermethylation events occurred in gene bodies, 3′UTRs, and intergenic sequences, while significantly hypomethylated CpG sites were more evenly distributed among genomic features (Figure 2B; lower panel). Both hypo- and hypermethylated genes significantly overlapped among DNMT3 depleted samples (Figure S3A, left and middle panels) with hypomethylation events overlapping predominately in promoters and hypermethylation events overlapping in gene bodies. Although overlap analysis of genes that gain and lose methylation (Figure S3A) indicates redundancy in target sites among the DNMT3s, ontology analysis of hypermethylated loci showed enrichment of distinct gene sets for the individual DNMT3 depleted samples (Figure 2D).
Since siDNMT1 hypomethylated genes and siDNMT3 hypermethylated genes showed similar enrichment profiles (Figure 2B, compare upper left to lower right), we investigated whether common loci were targeted. Indeed, significant overlap was observed, particularly for siDNMT3B hypermethylated genes and siDNMT1 hypomethylated loci, in 5′UTRs and gene bodies (Figure 2E; Figure S3A, right panel). Next, we focused our analysis on CpG sites that displayed hypermethylation in at least one DNMT depleted sample, and examined the methylation level of these loci across all DNMT depleted samples (Figure 2E). Consistent with our previous analyses, we observed overlap of hypermethylation events among the DNMT3B depleted samples that coincided with hypomethylation events upon DNMT1 depletion (Figure 2E). Independent biological replicates of the siDNMT1, siDNMT3B, and si3B+3L KD condition confirmed the reproducibility of these observations (Figure S3C). Taken together, these results demonstrate distinct and in some cases antithetical roles for the DNMTs in regulating 5mC, particularly in gene bodies.
DNMT3B regulation of nonCpG methylation in human EC cells
NonCpG methylation has recently been shown to comprise up to 25% of all cytosine methylation in ESCs and to accumulate specifically during neuronal maturation (Lister et al., 2013; Lister et al., 2009). Given its potential importance but poorly understood regulation, we analyzed nonCpG methylation levels among our DNMT depleted samples to determine the relative contribution of each DNMT to the writing of this mark. The 450K array interrogates 3,091 nonCpG dinucleotides allowing us to examine the role of the DNMTs in regulating this mark at single nucleotide resolution across the genome, albeit at low density. In NCCIT cells depleted of DNMT3B (alone or in combination), cytosine hypomethylation occurred primarily at nonCpG sites, with the single exception of the 3B+3L depleted sample (Figure 3A). Average methylation across genomic features (Avgβ-value) revealed an interesting pattern, where DNMT3B depletion results in hypomethylation of nonCpG sites across all regions, but depletion of DNMT3L results in hypermethylation of nonCpG sites (Figure 3B). The DNMT3A depletion also follows this trend, although to a lesser extent. DNMT3A siRNA KD combined with depletion of DNMT3L (3A+3L) in contrast results in hypermethylation at nonCpG sites. Indeed, histograms of Δβ-values for all 450K array nonCpG sites reveal that DNMT3A depletion results in a shift toward hypomethylation; however DNMT3L depletion has the opposite effect where nonCpG sites gain methylation (Figure 3C). Combined depletion of DNMT3A and DNMT3L overall results in the creation of more hypermethylated cytosines, however the majority of sites shift toward the baseline (that is, no-change in methylation (Δβ = 0)). A similar pattern was observed with DNMT3B depletion, but to a much larger extent where most nonCpG sites on the 450k array become broadly ([-0.4 – 0.0]) hypomethylated. Depletion of all DNMT3 family members results in the hypomethylation of almost all nonCpG sites (Figure 3C). We verified these observations with clone-based BGS of target genes identified via the 450K array-based analysis for DROSHA, USP14, UBR4, and SIN3A (Figure S4A). For DROSHA, nonCpG hypomethylation is observed in all depletions involving DNMT3B, and hypermethylation is observed with depletion of DNMT3L alone. Expression of these genes was not significantly altered (Figure S4B). Notably, CpG sites within the assayed regions became hypermethylated in the DNMT3B single depletion sample (Figure S4A). Independent biological replicates of the siDNMT1, siDNMT3B, and si3B+3L confirmed that nonCpG hypomethylation specifically occurs upon depletion of DNMT3B (Figure S4C).
Figure 3. Unique impact of DNMT3B and DNMT3L depletion on nonCpG methylation.
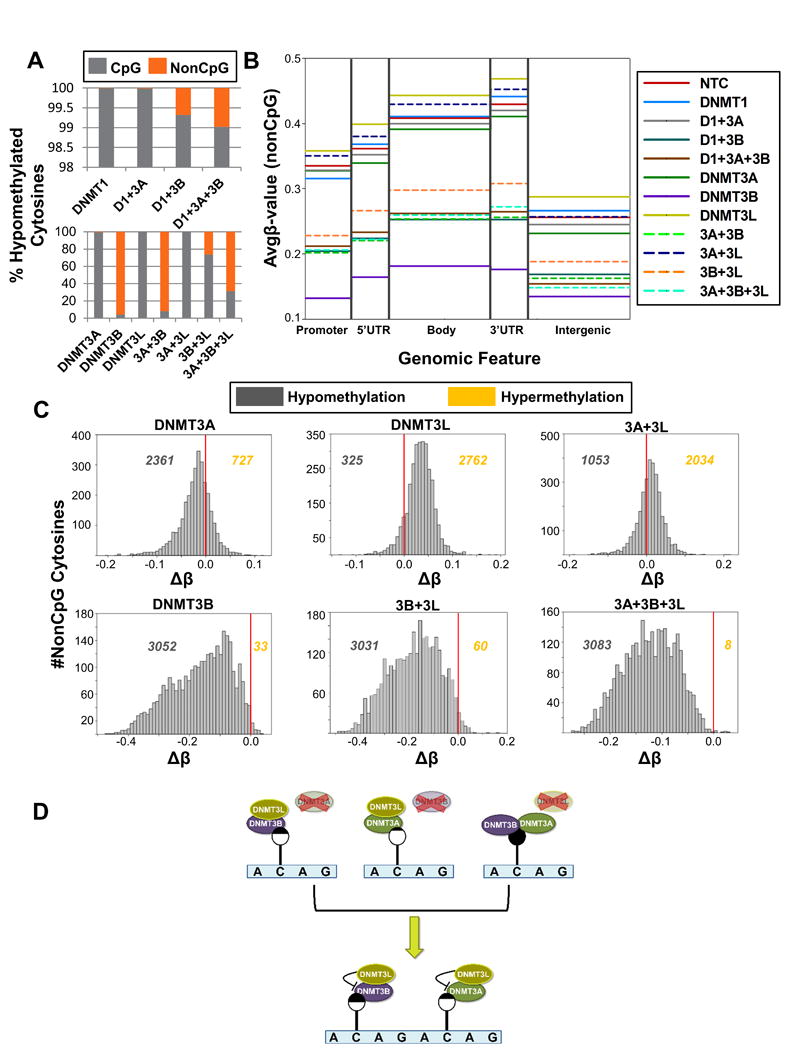
(A) Percent of significantly hypomethylated cytosines (pval < 0.05) classified by dinucleotide type (CpG vs. nonCpG) for NCCIT DNMT depleted samples. (B) Average β-values of methylation at nonCpG sites from the 450K array across genomic features. (C) Histograms of Δβ-values (Bin# = 50) for nonCpG dinucleotides. Red line indicates zero (no change in methylation). Values to the left and right of the red line indicate the number of nonCpG cytosines that are hypomethylated and hypermethylated respectively, compared to the NTC control transfection. (D) NonCpG methylation is regulated by the coordinated efforts of the DNMT3 family. Depletion of DNMT3A or DNMT3B results in nonCpG hypomethylation. Depletion of DNMT3L, however, has the opposite effect where nonCpG sites become hypermethylated, suggesting it restricts the activity of DNMT3A and DNMT3B toward nonCpG sites. Lollipop shading reflects the relative amount of methylation at the indicated cytosine (black = methylated; white = unmethylated). See also Figure S4.
Collectively, these results indicate that DNMT3B, and to a lesser extent DNMT3A, plays an important role in promoting nonCpG methylation at sites that can be interrogated by the 450K array in human EC cells, while DNMT3L plays an opposing role (Figure 3D). Depletion of DNMT3L alone results in hypermethylation of nonCpG cytosines (Figure 3B-C; Figure S4A), most likely due to altered control of DNMT3B and DNMT3A in its absence. DNMT3L depletion in combination with DNMT3A further emphasizes DNMT3L's role in regulating nonCpG methylation, as the combined depletion also displays increased 5mC at nonCpG cytosines (Figure 3C), most likely due to the unrestricted activity of DNMT3B in their absence.
DNMT3B target loci are epigenomic ‘hotspots’ for dynamic regulation of DNA modifications
Given the unexpected result of DNA hypermethylation upon DNMT3B depletion and the fact that standard bisulfite-based methods cannot distinguish between 5mC and 5hmC, we investigated the regulation of DNA modifications (both 5mC and 5hmC), at selected DNMT3B target loci identified from the 450K array analysis at single CpG resolution using BGS and Tet-assisted bisulfite sequencing (TAB-seq) (Yu et al., 2012), respectively. Analysis of cytosine modifications at the HOXA9 locus upon DNMT3B depletion revealed not only an increase of 5mC, but also a gain in 5hmC, notably at an exon-intron junction (Figure 4A). Cytosine modification analysis of the GPX6 locus (Figure 4B), however, revealed increased 5mC only; 5hmC was absent. To further enhance our understanding of 5mC/5hmC dynamics at DNMT3B target loci, we performed MeDIP/hMeDIP-qPCR to specifically assay 5mC/5hmC levels, respectively, in DNMT1 and DNMT3B individually depleted samples. We first validated the specificity of our MeDIP/hMeDIP assay in several ways (Figure S5A-C). MeDIP analysis for the DNMT1 depleted sample displayed reduced 5mC levels at all loci tested (S5D, left panel), consistent with the 450K array (Figure 1B, C; Figure 2A, B) and BGS (Figure S2C, Figure 4) results. 5hmC showed minor changes upon DNMT1 depletion with the assayed loci displaying small increases (APOA4), decreases (HOXA9), or no change (EIF4G3, ZNF311) in levels of the mark (Figure S5D, right panel). DNMT3B KD resulted in dynamic shifts of both 5mC and 5hmC levels (S5E). Some loci revealed that changes in 5mC were dominant (PDZK1, EIF4G3), while at other loci increases in 5hmC (FCGR2A, PDE4DIP, ZNF311) dominated the cytosine modification changes under DNMT3B KD conditions (Figure S5E). At other loci, such as APOA4 and HOXA9, levels of both 5mC and 5hmC increased (Figure S5E), consistent with BGS/TAB-seq results for the HOXA9 locus (Figure 4A). To gain further evidence for the specificity and reproducibility of these effects on cytosine modifications, we performed a ‘rescue’ experiment. Ectopic expression of murine FLAG-tagged Dnmt1 (Figure S5F) or Dnmt3b1 (Figure S5G), coupled with our siRNA transfection protocol, rescued the siRNA-induced changes to 5mC/5hmC levels and returned them to the level of the control sample, as assayed by MeDIP/hMeDIP (Figure 4C,D). Notably, ectopic expression of murine FLAG-Dnmt3b1 in the presence of endogenous DNMT3B resulted in reduced 5mC levels at all assayed loci (Figure 4D), supporting the notion that DNMT3B is recruited to these CpGs and limits access of other DNA modifiers. Taken together, these results indicate that dynamic regulation of cytosine modifications occur at CpG sites targeted by DNMT3B in which DNMT3B appears to protect these loci from modification with 5mC or 5hmC deposition by other DNMT and TET enzymes.
Figure 4. Single CpG resolution analysis of cytosine modifications using bisulfite genomic sequencing (BGS) and TET-assisted BGS (TAB) analysis.
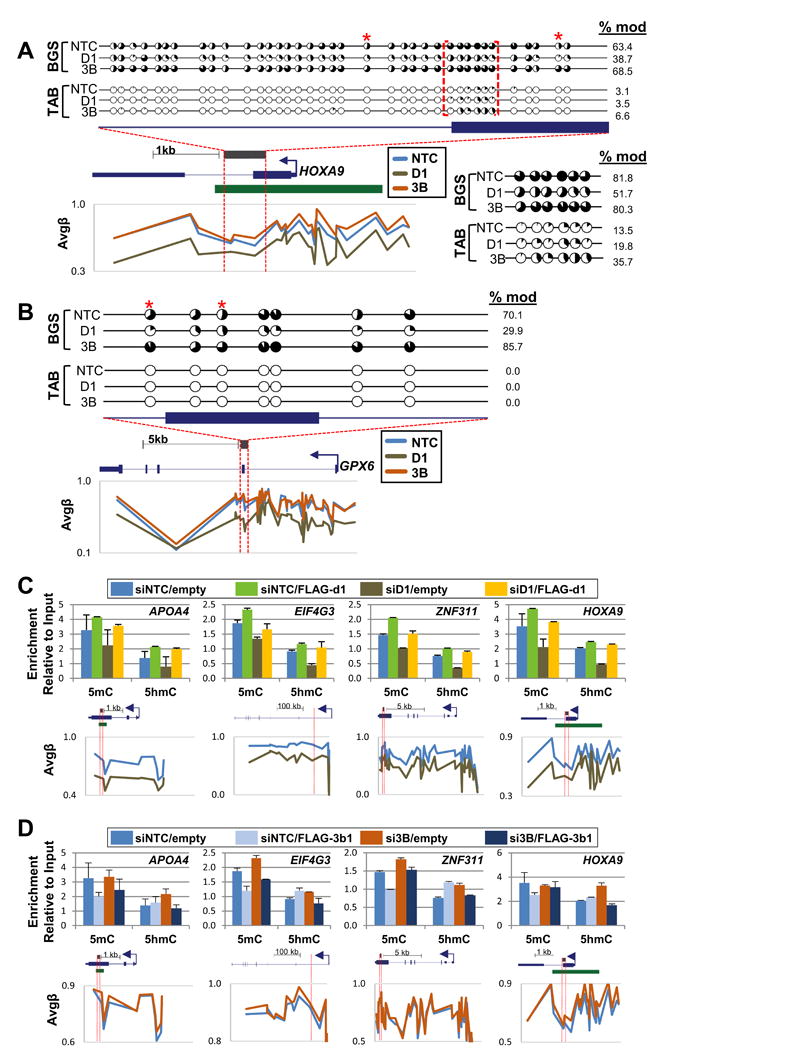
(A) The HOXA9 locus is hypomethylated upon depletion of DNMT1, but CpG hypermethylated (based on 450K array analysis) and hyperhydroxymethylated upon DNMT3B depletion. Red brackets indicate the region of hyperhydroxymethylation, and a blow-up of this region is shown below the BGS plot. Each pie chart indicates the percentage of modified (black) and unmodified (white) CpGs for a single CpG site among at least 10 (15 for TAB) clones (red* = 450K probe). Gene structure for the region analyzed is shown directly below the BGS plots. BGS amplified region is indicated by the gray bar and red dashed lines (bent arrow =TSS, green bar = CGI). β-values of 450K CpG loci for HOXA9 (scaled to length of the gene) are presented below the gene structure for siDNMT1 and siDNMT3B depletion conditions to permit comparison of 450K array and BGS-based results. (B) The GPX6 locus demonstrates hypomethylation upon depletion of DNMT1 and hypermethylation upon DNMT3B depletion, confirming results derived from the 450K array analysis. 5hmC was not detected. At least 10 (25 for TAB) clones were sequenced. (C & D) Rescue experiment. (h)MeDIP analysis of NCCIT siRNA transfected depleted cells transfected with expression vectors encoding murine (C) FLAG-Dnmt1 and (D) FLAG-Dnmt3b1 resistant to the human siRNAs. Gene structures highlighting (h)MeDIP assayed regions (red bars) are shown below each graph. β-values for CpG sites on the 450K array present in each locus (scaled to length of the gene) are presented below the gene schematics in graphical form. See also Figure S5.
MBD-seq analysis following DNMT3B depletion supports key 450K array findings and reveals that hypermethylation events occur at highly expressed genes marked by H3K36me3
Since our observation of significant hypermethylation events in DNMT3B depleted samples was unexpected, we sought to gain a more comprehensive, genome-wide view of 5mC changes under this experimental condition by performing methyl-CpG-binding domain (MBD)-seq on the DNMT3B siKD sample. MBD-seq (utilizing the methyl-binding domain isolated from MBD2) preferentially binds 5mC over 5hmC by a 5-fold margin, providing specificity for analyzing 5mC distribution (Hashimoto et al., 2012). Overall, hypomethylation (≥ 2-fold change in 5mC) was observed across all genomic features (Figure 5A). However, increasing stringency by evaluating larger magnitude changes in 5mC (≥4-fold) revealed features (e.g. introns & gene bodies) in which hypermethylation events were more numerous than hypomethylation events (Figure 5A), consistent with 450K array results. Next, we sought to decipher the functional implications of gene body hypermethylation events upon DNMT3B depletion. As actively transcribed genes are associated with gene body methylation (Lister et al., 2009) and trimethylation of lysine 36 on histone H3 (H3K36me3) (Kolasinska-Zwierz et al., 2009), we compared genes with ≥ 4-fold change in methylation upon DNMT3B depletion with gene expression stratified into three tiers (high, medium, and low expression in the siDNMT3B KD sample) and H3K36me3 localization in NCCIT UD cells previously published by our laboratory (Jin et al., 2012). Indeed, gene body hypermethylation events under DNMT3B KD conditions significantly overlapped with highly expressed H3K36me3 marked genes, while genes with low expression were largely excluded (Figure 5B). Ontology analysis of genes in common with all three parameters revealed marked enrichment for functions in RNA splicing, translation, and protein ubiquitination (Figure 5C). Collectively, these results demonstrate targeted hypermethylation upon DNMT3B depletion to gene bodies of highly expressed genes, and potential functional implications for stabilizing expression of protein fidelity pathways.
Figure 5. Hypermethylation events resulting from DNMT3B depletion correlate with highly expressed, H3K36me3 marked genes.
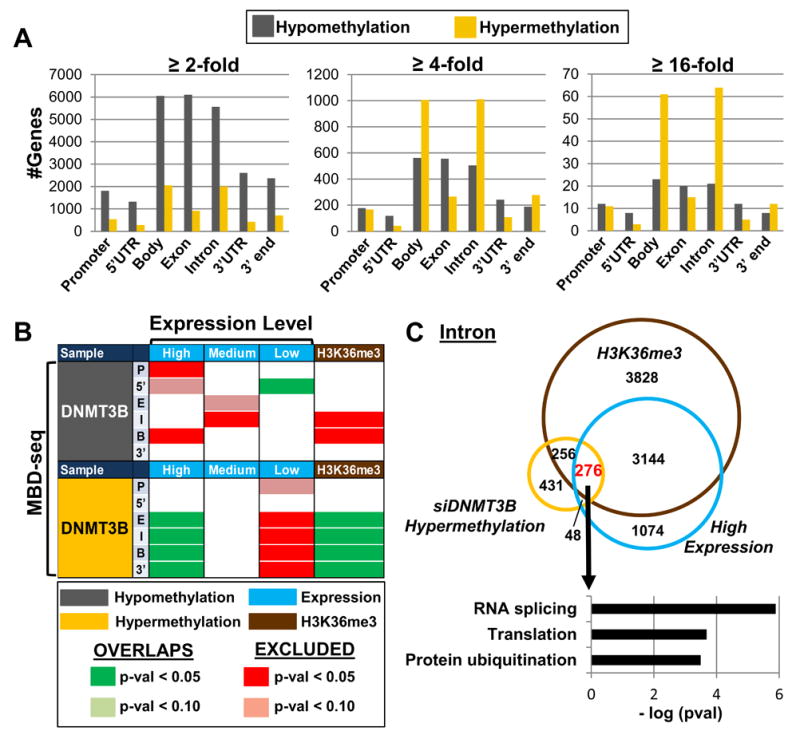
(A) Number of genes with respective fold changes in methylation (using MBD-seq) across genomic features for the NCCIT DNMT3B knockdown. (B) Overlap analysis of genes with ≥ 4-fold 5mC changes (by MBD-seq) in the features indicated (P = promoter, 5′ = 5′UTR, E = exon, I = intron, B = body, 3′ = 3′UTR) with gene expression stratified by level (high, medium, low) and H3K36me3 occupancy in NCCIT UD cells. (C) Venn diagram illustrating overlap between hypermethylated introns (≥ 4-fold by MBD-seq), high expressing genes, and H3K36me3. Color scale from Fig. 5B applies. Ontology analysis of genes that concurrently gain 5mC upon DNMT3B depletion, are marked by H3K36me3, and are highly expressed.
Differentiation induced changes in gene expression and 5mC patterns mirror those observed upon siRNA depletion of DNMT3B
Since it is well-established that the DNMTs are crucial for development (Li et al., 1992; Okano et al., 1999) and NCCIT cells also serve as a model for differentiation (Jin et al., 2012), we next compared gene expression and 5mC patterns from the DNMT depletions with differential gene expression and 5mC data derived from NCCIT cells differentiated with retinoic acid for seven days (DF). A significant percent of genes (pval < 0.0001) upregulated in singly depleted DNMT1, DNMT3B, and DNMT3L conditions overlapped with genes that become upregulated during differentiation (Figure 6A, left). Overlapping genes among DNMT depleted and NCCIT DF datasets are enriched in processes involved in cell proliferation (data not shown). Genes downregulated during NCCIT differentiation significantly overlap among nearly all DNMT depleted samples (Figure 6A, right), and show enrichment for ectoderm differentiation and amino acid biosynthesis and transport pathways (data not shown). Changes in expression of select genes in the siDNMT depleted samples and NCCIT differentiation sample based on microarray analysis were confirmed by qRT-PCR (Figure S6A).
Figure 6. Global changes in 5mC patterns resulting from DNMT3B siRNA depletion mimic methylation changes during differentiation.
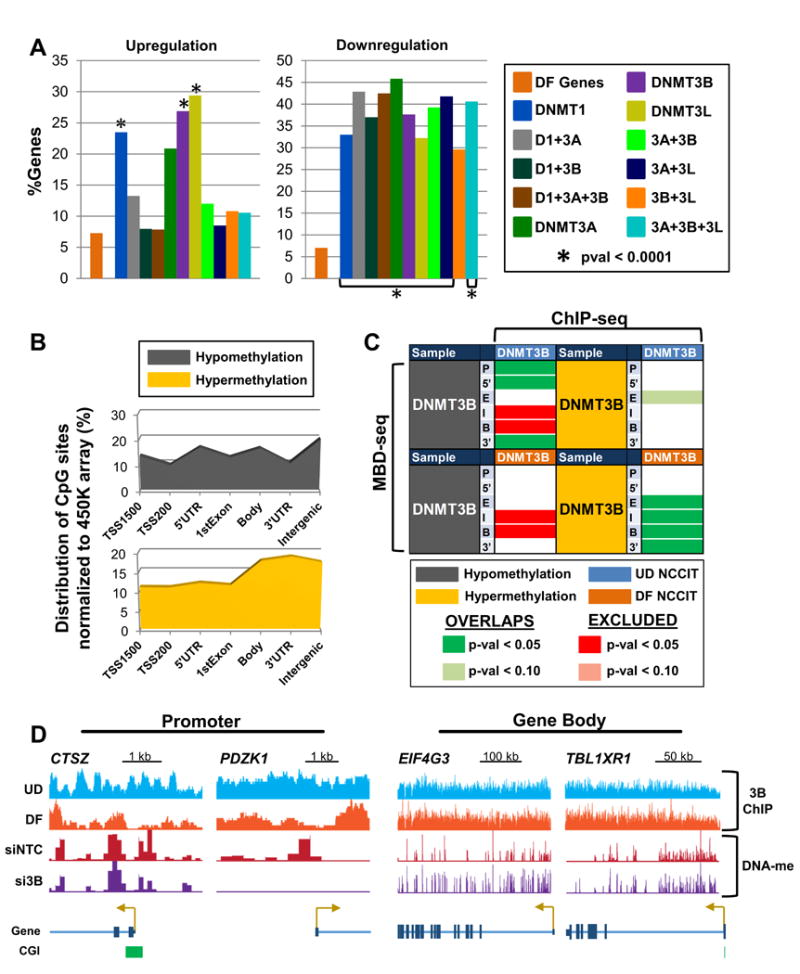
(A) Overlap analysis of DNMT knockdown differentially expressed genes (≥ 1.5 fold-change) with differentially expressed genes in differentiating (DF) NCCIT cells (following 7 days of retinoic acid treatment). ‘DF Genes’ (orange bar) represents the percentage of genes in differentiated NCCIT cells that are differentially expressed. The ‘%Genes’ for siDNMT depleted samples represents the percent overlap of differentially expressed genes among the respective siDNMT depleted sample with genes differentially expressed in differentiated NCCIT cells. (B) Normalized distribution of significantly differentially methylated CpG sites (pval < 0.05) by genomic feature in NCCIT DF cells. (C) Overlap analysis of genes with ≥ 4-fold 5mC changes (by MBD-seq) for the indicated features with DNMT3B binding (ChIP-seq) in NCCIT undifferentiated (UD) and differentiated (DF) cells. (D) Representative browser views of promoters displaying hypomethylation upon DNMT3B depletion, are bound by DNMT3B in UD NCCIT cells, and become hypomethylated upon differentiation (left). Gene body hypermethylation with DNMT3B depletion coincides with sustained binding of DNMT3B in DF NCCIT cells (right). See also Figure S6.
Next, we evaluated the most significant changes (pval < 0.05) in 5mC in DF NCCIT cells for their distribution across the genome. Hypomethylation events did not show enrichment in any particular genomic feature (Figure 6B, top). In contrast, hypermethylation events, which accounted for ∼70% of significant methylation changes (data not shown), predominately occurred in gene bodies, 3′UTRs, and intergenic sequences (Figure 6B, bottom), an enrichment profile that resembles hypermethylation events occurring upon DNMT3 depletion (Figure 2B, bottom right). Overlap analysis of DF NCCIT gene 5mC with 5mC patterns in NCCIT DNMT depleted samples revealed significant overlap (pval < 0.05) of hypomethylation events, particularly in promoter regions, and hypermethylation events in gene bodies (Figure S6B). We next compared the MBD-seq methylation profiles (≥ 4-fold change in methylation) with DNMT3B ChIP-seq from NCCIT UD and DF cells published previously (Jin et al., 2012). Genes that became hypomethylated in promoters and 5′UTRs significantly overlapped with DNMT3B binding in UD NCCIT in these same regions, linking the altered methylation observed upon DNMT3B depletion with the presence of DNMT3B. In contrast, siRNA DNMT3B induced hypermethylation overlapped significantly with DNMT3B binding in DF NCCIT cells in gene bodies and 3′UTRs (Figure 6C). The CTSZ and PDZK1 promoters are representative of loci that demonstrate hypomethylation upon DNMT3B depletion and are occupied by DNMT3B in UD NCCIT cells (Figure 6D). The entire gene body of EIF4G3 and TBL1XR1 display hypermethylation upon DNMT3B depletion that corresponds with DNMT3B occupancy that does not decrease upon differentiation (Figure 6D). Decreased binding of DNMT3B in DF cells is observed primarily in gene promoters, indicating that as DNMT3B expression is reduced during differentiation, the remaining DNMT3B binds strongly to gene bodies to regulate 5mC patterns through development. Taken together, these results show that DNMT3B is a critical regulator of 5mC during early differentiation.
Hypermethylation events resulting from DNMT3B depletion are conserved in other cell types
We next sought to determine whether some of the key observations derived from NCCIT cells were reproducible in other cell lines. To accomplish this, we evaluated 5mC in the HCT116 colorectal carcinoma cell line and its isogenic derivatives in which DNMT1 and/or DNMT3B have been genetically inactivated or over expressed (Rhee et al., 2002; Sun et al., 2008) (Figures S7A). Genome-wide methylation analysis using the 450K array revealed that global 5mC levels for each sample were similar to what has been observed in previous studies, and corresponded well with the known activities of DNMT1 and DNMT3B (Figure 7A) (Rhee et al., 2002).
Figure 7. Knockout and ectopic expression of DNMT1 and DNMT3B in HCT116 colorectal carcinoma cells reveals conserved modes of regulating 5mC among.
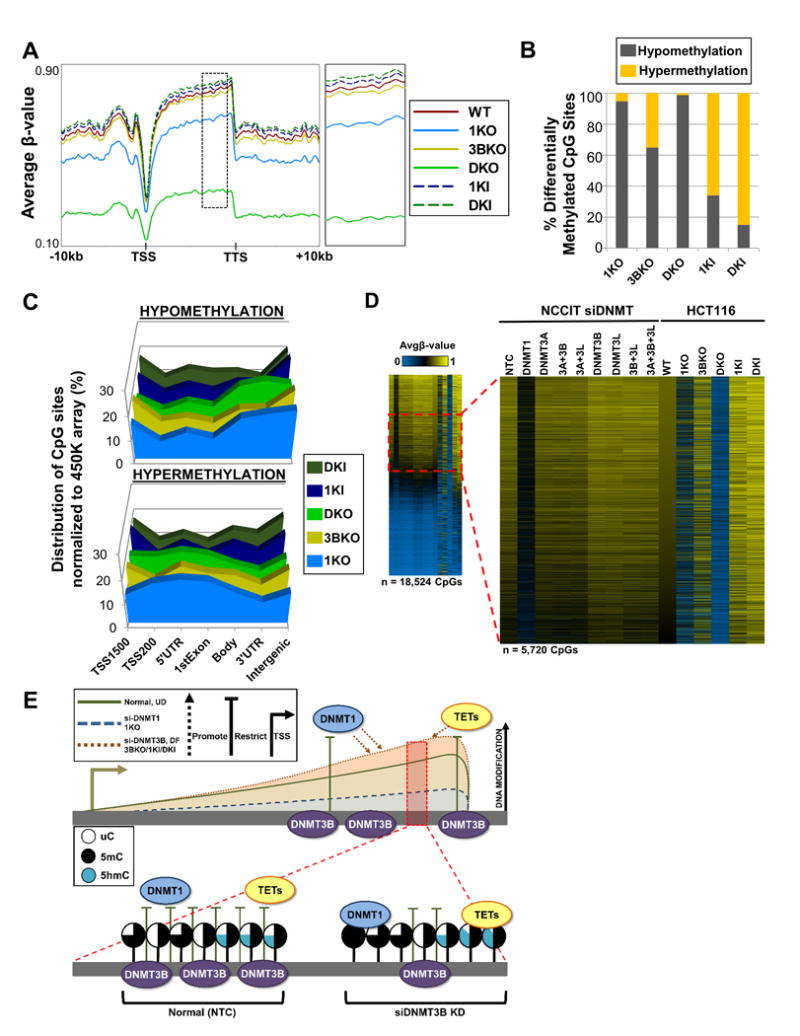
(A) Spatial distribution plots across intragenic regions derived from average β-values for 5mC in HCT116 DNMT1/DNMT3B knockout (KO) and overexpressing (KI) cells derived from the 450K array. Dashed black box (to the right) indicates region magnified. (B) Percent of significantly differentially methylated CpG sites (pval < 0.05) classified by methylation status. (C) Normalized distribution of significantly differentially methylated CpG sites by genomic feature in HCT116 KO/KI cells. (D) Heat map of hypermethylated (Δβ ≥ 0.15) CpG sites for HCT116 KO/KI cells in conjunction with the respective CpG loci in NCCIT DNMT siRNA depleted samples stratified by HCT116 WT most methylated to least methylated CpG sites. Dashed red box indicates magnified region (Avgβ-value [0.50 – 0.70] in WT). Duplicate CpG sites were removed. (E) DNMT3B restricts DNMT1 and TET function at certain loci. Conserved loci were identified in NCCIT DNMT3B depletion, NCCIT differentiation, and HCT116 3BKO, 1KI, and DKI conditions that gain DNA modifications (5mC and 5hmC); these same loci significantly overlap with CpGs that become hypomethylated with DNMT1 depletion, indicating a regulatory interaction. Additionally, in NCCIT cells depleted of DNMT3B, 5hmC increases at these loci indicating that TETs are also recruited. Shaded lollipops indicate the level of each cytosine modification (uC = unmodified cytosine; 5mC = 5-methylcytosine; 5hmC = 5-hydroxymethylcytosine). See also Figure S7.
The majority of significantly differentially methylated CpG sites in the 1KO, 3BKO, and DKO cell lines were hypomethylation, while hypermethylation events were the dominant change in HCT116 cells ectopically expressing DNMT1 and DNMT3B. Interestingly, however, we observed that almost 40% of differentially methylated CpG sites gained methylation in DNMT3B KO cells. The distribution of hypomethylation changes among DNMT1 KO lines revealed enrichment profiles similar to those observed when DNMT1 was acutely depleted (Figure 7C compared to Figure 2B). For 3BKO, 1KI, and DKI HCT116 cells, hypomethylation (relative to the parental cell line) was enriched upstream of the TSS and within intergenic sequences; hypermethylated CpG sites were relatively evenly distributed (Figure 7C). We next analyzed CpG sites that became hypermethylated (Δβ-value > 0.15) in each HCT116 cell line to determine if common sites were targeted, and indeed, 3BKO hypermethylated CpG sites became hypermethylated in 1KI and DKI HCT116 cells (Figure S7B). Finally, we compared the methylation status of HCT116 KO/KI hypermethylated CpG sites (whose methylation levels in parental HCT116 cells were comparable to those of the NCCIT NTC (Δβ-value between NCCIT NTC vs. HCT116 WT [-0.10 - 0.10])), across all DNMT depleted samples (Figure 7D). Sites hypermethylated in 3BKO, IKI, and DKI cells are those that became hypermethylated in NCCIT DNMT3B depleted cells (Figure 7D) indicating that regulation of 5mC at particular CpG sites is conserved across other cell types.
Discussion
In the current manuscript we tackled the important but still unanswered question of why mammalian genomes contain four DNA methyltransferase genes and how each of the encoded proteins function independently and cooperatively to establish and maintain genome-wide patterns of 5mC. This work represents, to our knowledge, the first study to comprehensively address this issue by depleting all four DNMTs individually and in all combinations and assaying the impact of these perturbations on genome-wide patterns of 5mC and transcription. In addition, by employing siRNA to acutely deplete the DNMTs, we focused on the earliest and most direct effect of each DNMT on genomic 5mC patterns. Coupling this system with a cell line that provides information on both pluripotency and differentiation, we observed dynamic changes in 5mC that revealed a number of unexpected findings. For example, siRNA depletion of DNMT3B resulted in numerous DNA hypermodification events (increased 5mC and/or 5hmC) that are not only conserved in other cell types, but also correlate with high-expressing, H3K36me3-marked loci that become hypomethylated when DNMT1 is depleted, revealing an interplay between these two DNMTs within gene bodies. In addition, we identified an important role for DNMT3B in mediating nonCpG methylation and a role for DNMT3L in regulating the choice between CpG and nonCpG methylation activity of DNMT3A/DNMT3B. Finally, we showed that DNMT3B depletion in NCCIT cells mirrored the methylation and expression changes that occur during the normal differentiation program and these changes occur at loci bound by DNMT3B. Taken together, these results provide insight into the differential role of each DNMT in positively and negatively regulating cytosine modifications.
A key finding from this study was that depletion of each DNMT had a distinct impact on different genomic features. Depletion of DNMT3B by siRNA resulted predominately in hypermethylation events throughout the genome, but particularly centered on gene bodies. Not only did 5mC increase, but 5hmC was also dynamically regulated at DNMT3B target sites and contributed to the appearance of DNA hypermethylation as detected using traditional BGS. To date, the role of gene body methylation remains unclear, although intriguing correlations have been identified related to differential promoter use and alternative splicing (Maunakea et al., 2010; Shukla et al., 2011). The recent discovery of dual-use codons (duons) for transcription factor binding generates further possible regulatory roles for cytosine modifications in gene bodies as it could impact transcription factor binding (Stergachis et al., 2013). In this regard our BGS and TAB-BGS results from the HOXA9 locus showing elevated 5hmC focused specifically at an exon-intron junction upon depletion of DNMT3B is intriguing. Although this is only a single example, it raises questions regarding the role of the CpG epigenetic regulatory ‘hotspots’ we identified that are sensitive to DNMT3B levels. To better define loci prone to hypermethylation upon DNMT3B depletion, we performed MBD-seq, and observed that 5mC increased primarily within introns of high expressing, H3K36me3-marked genes. Recent reports identify exonintron boundaries as genomic locations that undergo dynamic regulation of cytosine modifications; Laurent et al. observed sharp peaks of cytosine modification across cell lines in different stages of differentiation (Laurent et al., 2010), and Huang et al. noted that Tet2 maintains 5hmC at exon boundaries of high expressing genes (Huang et al., 2014). Taken together with our results, we propose that DNMT3B target loci are susceptible to dynamic shifts in 5mC and 5hmC levels, and at least a subset of these may contribute to important intragenic regulatory processes including RNA processing.
Further investigation of hypermethylation events induced by DNMT3B depletion led to one of the more unexpected findings to arise from this study: a hitherto unknown 5mC regulatory mechanism coordinated by DNMT1 and DNMT3B. Loci that gained 5mC and/or 5hmC upon depletion of DNMT3B correlated significantly with those that lost methylation upon DNMT1 depletion, suggestive of co-regulation by DNMT1 and DNMT3B. Importantly, these hypermethylated loci were conserved in HCT116 cells with ectopic expression of DNMT1 indicating that the hypermethylation is a result of DNMT1 activity. Taken together, these results allow us to propose a mechanism of 5mC regulation in which DNMT3B occupancy of mildly to moderately methylated intragenic regions hinders DNMT1 and/or TET access (Figure 7E). When DNMT3B is depleted, DNMT1 and/or the TETs gain access to previously DNMT3B-bound sites and subsequently increase methylation or allow TET access and conversion of the 5mC to 5hmC (Figure 7E). Alternatively, DNMT3B has been reported to possess dehydroxmethylase activity in vitro (Chen et al., 2012); potentially 5hmC accumulates in the absence of DNMT3B as it is no longer present to dehydroxymethylate 5hmC to 5C. We showed previously that DNMT3B was the DNMT most highly enriched in transcribed gene bodies (Jin et al., 2012). DNMT1 and DNMT3B directly interact in vitro and co-localize (Kim et al., 2002), however no direct link between DNMTs and TETs has been reported. If coordination between DNMT and TET activity exists, such as through an adaptor protein like UHRF1 that binds 5hmC and can also recruit DNMT1 for maintenance methylation (Frauer et al., 2011; Sharif et al., 2007), then the net effect of reduced DNMT3B might primarily be elevated 5hmC, an idea that will require further testing. Our results therefore add another layer of complexity to the co-regulation of 5mC by DNMT1 and DNMT3B; in addition to the positive effect on 5mC genome-wide by these two enzymes observed by our laboratory and others, an opposing role also exists dependent on genomic context. This antagonistic relationship may also have implications for the creation of aberrant 5mC patterns in cancer where the DNMTs and TETs are often aberrantly expressed or mutated. The particular milieu of DNMT deregulation potentially in the setting of deregulated TET activity, may drive methylation changes differently in distinct regions of the genome (e.g. promoter hyper- and intragenic hypomethylation). Given that stem-cell like characteristics are frequently observed in cancer cells, our finding that DNMT3B depletion recapitulates differentiation-induced 5mC changes suggests that some epigenetic changes contributing to tumor stem cell development may reflect deregulated DNMT activity impacting intragenic regulatory mechanisms such as alternative promoter selection and/or splicing. Future work aimed at elucidating this novel regulatory mechanism will be crucial to fully understanding its contribution to the cancer epigenome.
NonCpG methylation is most abundant in pluripotent embryonic cells (Lister et al., 2009), accumulates during development of the adult brain (Lister et al., 2013) and has thus far been attributed almost exclusively to DNMT3A (Ramsahoye et al., 2000). Our results challenge this paradigm by showing that, at least for sites interrogated on the 450k array, DNMT3B is a major contributor to nonCpG methylation. DNMT3L restricted the activity of DNMT3A and DNMT3B toward nonCpG sites (Figure 3D). Furthermore, within regions in which we confirmed the presence of nonCpG methylation using BGS, CpG sites were highly methylated, consistent with observations that nonCpG methylation occurs adjacent to highly methylated CpG sites (Lister et al., 2009; Ziller et al., 2011). At loci we assayed using BGS, DNMT3B depletion consistently resulted in increased 5mC at CpGs adjacent to hypomethylated nonCpGs, while combined depletion of DNMT3B and DNMT1 resulted in loss of methylation at CpG and nonCpG sites (DROSHA, USP14 in Figure S5A). We therefore propose that DNMT3B occupies certain loci to confer nonCpG methylation, while concurrently restricting access of DNMT1 (and possibly also the TETs) to CpG sites. Depletion of DNMT3B then results in hypomethylation of nonCpG and hypermethylation of CpG sites as DNMT3B occupancy is lost and DNMT1 occupancy is gained (Figure 7E). One caveat to this model is our limited ability to decipher relationships between nonCpG methylation and nearby CpG methylation due to the low density coverage of nonCpG sites on the 450K array. NonCpG methylation correlates positively with expression in ESCs but negatively with expression in developed adult brain (Lister et al., 2013; Lister et al., 2009). Indeed, Lister and colleagues noted that DNMT3A expression correlates with accumulation of nonCpG methylation and synaptogenesis in developing adult brain (Lister et al., 2013). Collectively, these studies demonstrate that both DNMT3A and DNMT3B methylate nonCpG sites, but one particular DNMT3 may predominate in a developmental stage- or cell type-specific manner.
In summary, our results clarify the division of labor among the DNMT family members and reveal previously unknown regulatory interactions, including opposing roles for DNMT1 and DNMT3B dependent on genomic feature and cytosine dinucleotide type, and new evidence for DNMT3L participating in the choice of substrate by DNMT3A and DNMT3B. The only currently FDA-approved DNA methylation inhibitors, 5-aza-2′-deoxycytidine and 5-azacytidine, are effective treatments for acute myeloid leukemia, yet typically responses are transient (Malik et al., 2014). Both agents broadly and non-specifically inhibit the DNMTs and result in significant DNA damage (Palii et al., 2008). Assuming the aza nucleosides act through DNA methylation and not DNA damage, therapeutic efficacy may be enhanced by development of DNMT inhibitors that bind the free enzyme rather than require incorporation into the DNA. Our results add further to this idea by showing that inhibiting each DNMT may have a different consequence on genomic 5mC patterns and broad inhibition may not be the most effective way to normalize gene expression. Depletion of DNMT1 results in broad hypomethylation over the genome with some preference toward gene bodies, which may result in altered intragenic regulatory processes that may or may not increase therapeutic effect. On the other hand, inhibition of DNMT3B will have less effect on total 5mC but cause more specific changes including some promoter hypomethylation and gene body hypermethylation events. Given that the key lesion in the four DNA epigenetic marks now known to exist that drives tumor initiation and promotion is not known (e.g. 5mC or other mark, promoter versus gene body methylation), development and testing of isoform specific inhibitors in an attempt to increase therapeutic efficacy of DNMT inhibitors and reduce their off-target side effects appears warranted.
Experimental Procedures
Cell culture, siRNA transfections, and DNA/RNA extraction
NCCIT and HCT116 parental cells (from ATCC), and the isogenic derivatives of parental HCT116 (Rhee et al., 2002; Sun et al., 2008) were grown in McCoy's 5A medium supplemented with 10% fetal bovine serum and Lglutamine. HCT116 DNMT overexpression lines (Sun et al., 2008) were grown under G418 selection (750 μg/ml). Differentiation of NCCIT was induced by addition of 10 μM all-trans retinoic acid (Sigma) for 7 days. On-TARGETplus and siGENOME siRNA SMARTpools (Dharmacon, Thermo Scientific) targeting a single gene were used against DNMT1 (L-004605-00-0005), DNMT3A (M-006672-03-0005), DNMT3B (L-006395-00-0005), and DNMT3L (L-013637-01-0005) in individual and combination experiments. siRNA transfection with a negative control non-targeting siRNA (D-001206-13-20; Dharmacon, Thermo Scientific) was performed in parallel. siRNA transfection was performed with PepMute transfection reagent (SignaGen) according to the manufacturer protocol. Total RNA was extracted by Trizol homogenization and purified according to the manufacturer protocol (Life Technologies). Genomic DNA was extracted by proteinase K digestion and phenol:chloroform extraction. Refer to Supplemental Experimental Procedures for the full siRNA transfection protocol and siRNA rescue experimental conditions.
Expression analysis by QRT-PCR and microarray
cDNA synthesis and QRT-PCR was performed in triplicate as described (Jin et al., 2012). Primer sequences are listed in Table S1. Gene expression profiling was performed using Affymetrix Human Gene 1.0 ST arrays. All samples were analyzed in duplicate at the Georgia Regents University Cancer Center Genomics Core facility as described (Jin et al., 2012).
Western Blot
Western blots for protein expression were performed using the LI-COR imaging system following the manufacturer protocol. Detailed conditions and antibodies are listed in Supplemental Experimental Procedures.
450K array data analysis
DNA samples were processed on the HumanMethylation450 BeadChip array (Illumina) and analyzed as described in the Supplemental Experimental Procedures.
MBD-seq
MBD-seq experiments were performed as previously described (Jin et al., 2012) and analyzed as described in the Supplemental Experimental Procedures.
MeDIP/hMeDIP pull-down assays
MeDIP experiments were performed as previously described (Putiri et al., 2014) with the 33D3 5mC antibody (Diagenode). hMeDIP experiments followed the same protocol as MeDIP, with the exception that 2.5 μg of 5hmC antibody (in-house) was used for 5hmC capture. Procedures for the generation and validation of the 5hmC antibody are provided in Supplemental Experimental Procedures.
Supplementary Material
Figure S1: Validation of model system used in all main figures, and additional siDNMT1 specific spatial distribution plots and microarray expression profiles, related to main Figure 1.
Validation of the experimental system used for this study, related to Figure 1. (A) mRNA expression by quantitative RT-PCR of DNMTs in undifferentiated (UD) and differentiated (DF) NCCIT cells performed in triplicate relative to GAPDH expression and normalized to UD (set at 1.0). Error bars indicate standard error. NCCIT UD cells were treated with retinoic acid (10μM) for 7 days prior to the RNA harvest for DF cells, and this represented the “differentiated” condition used throughout this study. * = pval < 0.005 (B) Expression of DNMTs in all DNMT siRNA knockdown (KD) samples (single and combination) by quantitative RT-PCR relative to GAPDH expression, normalized to a no-target control (NTC) siRNA transfection. Depletion efficiency was monitored on the same samples used for the HumanMethylation 450K and Affymetrix Gene 1.0 ST arrays. * = pval < 0.005. (C) Expression of DNMTs for individual NCCIT siDNMT KD samples at the protein level confirms the RT-PCR results. PCNA was used as a loading control. (D) Expression of developmental markers, pluripotency factors, housekeeping genes, and TETs by quantitative RT-PCR under conditions of DNMT depletion relative to TUBA1C expression, normalized to NTC. (E) Intragenic spatial distribution plot for DNMT1 siRNA depleted samples derived from average β-values for DNA methylation obtained from the HumanMethylation 450K array. TSS = transcription start site; TTS = transcription termination site. Dashed black box indicates region magnified (to the right) to better visualize relative methylation levels among samples. (F) Feature-based analysis of differential methylation demonstrates reproducibility among independent siDNMT KD biological replicates. Box plots representing all δβ-values for each sample in the indicated genomic features (promoter, gene body, intergenic, islands, shores, open sea) in three independent KD experiments (“0, 1, 2”; 0 = original experiment). Outliers outside of the 5th/95th percentile have been removed. Red bar highlights the zero line (no change in methylation). (G) Clustered heat map of gene expression (Affymetrix Human Gene 1.0 ST array) measured as log2 fold change for all NCCIT DNMT KD samples.
Figure S2: Total number of significantly differentially methylated CpGs and genes to support Figure 2, and validation of identified target sites, related to main Figure 2.
Confirmation of array-based CpG methylation data using bisulfite genomic sequencing, related to Figure 2. (A) Table showing numbers of hypo/hypermethylated CpG sites (including intergenic sites) and genes for each NCCIT siRNA DNMT depletion condition. (B) Normalized distribution of significantly differentially methylated CpG sites by CpG island annotation. Normalization was conducted by comparing the number of significantly differentially methylated CpG sites to the total number of sites for each island annotation present on the 450K array. Top: DNMT1 depletion (individual/combination) Bottom: DNMT3 depletion (individual/combination). (C) Bisulfite genomic sequencing (BGS) of identified DNMT1 target genes from the 450K array analysis. Gene structures with PCR-amplified region indicated by a dashed red box are provided below each CpG plot. Each pie chart indicates the percentage of methylated (black) and unmethylated (white) CpGs for a single CpG site among > 10 clones sequenced with the total percent methylation shown at the right. (D) Expression of PTPRN2 and CREBBP (DNMT1 target genes) by quantitative RT-PCR for all DNMT depleted samples relative to GAPDH expression, normalized to NTC. Error bars indicate standard error.
Figure S3: Overlap analysis of differentially methylated genes and 450K array biological replicates of DNMT siRNA KD, related to main Figure 2.
Overlap of significantly differentially methylated genes across all NCCIT DNMT siRNA depleted samples, related to Figure 2 (A) Overlap analysis of hypo/hypermethylated genes with significantly differentially methylated CpG sites (pval < 0.05) among genomic features (P = promoter, 5′ = 5′UTR, B = body, 3′ = 3′UTR) for all NCCIT DNMT depleted samples. Statistical significance of overlap was calculated using the Fisher's Exact test. Left: hypomethylation vs hypomethylation; Middle: hypermethylation vs hypermethylation; Right: siDNMT1 (single/combo) hypomethylation vs hypermethylation. For example, in the right panel, genes that become hypomethylated in the gene body of DNMT1 depleted samples significantly overlap with genes that become hypermethylated in the gene body upon DNMT3B individual and combination siRNA depletion. (B) Distribution of differentially methylated CpG sites for siDNMT KD biological replicates. Percentage of significantly differentially methylated CpG sites (pval < 0.05) classified by methylation status (hypo/hypermethylation) in three independent NCCIT DNMT depletion experiments demonstrates the reproducibility of our findings. (C) Clustered heat map of hypermethylated CpG sites for each DNMT depleted biological replicate sample to demonstrate reproducibility of the siDNMT3B hypermethylation phenotype at conserved loci. Hypermethylated (Δβ>0.15) CpG loci (duplicate CpG sites removed) were clustered by siDNMT samples stratified by NTC most methylated CpG sites to least methylated CpG sites (left panel). Δβ-values for respective CpG loci are also presented (right panel). Dashed red box indicates magnified region (Avgβ-value [0.45 - 0.75] in NTC).
Figure S4: Independent confirmation of array-based nonCpG methylation data using bisulfite genomic sequencing, related to main Figure 3. (A) Bisulfite genomic sequencing (BGS) analysis of nonCpG hypomethylation events in DNMT3 depleted samples identified using the 450K array data. CpH status is indicated above each BGS plot; the 450K array CpA is denoted by *. Gene structures with amplified region indicated by the dashed red box provided below each CpH plot. Each pie chart indicates the percent methylated (black) and unmethylated (white) CpHs for a single CpH site among at least 10 clones sequenced. The table to the right of each plot indicates the total percent methylation for all CpGs and CpAs shown in each sample. (B) Expression of UBR4, USP14, and SIN3A by quantitative RT-PCR for all DNMT depleted samples relative to GAPDH expression, normalized to NTC. Error bars indicate standard error. (C) Box plots of Δβ-values for nonCpG methylation among three independent, biological replicates of siDNMT1, siDNMT3B, and si3B+3L conditions (“0, 1, 2”; 0 = original experiment). Outliers outside of the 5th/95th percentile have been removed. Red bar highlights the zero line (no change in methylation).
Figure S5: Independent confirmation of array-based CpG methylation and TAB-seq results using MeDIP/hMeDIP pull-downs, related to Figure 4. (A) Validation of MeDIP technique. HCT116 colorectal carcinoma (WT) and DNMT1/DNMT3B knockout (DKO) cells were assayed for 5mC at promoters of selected genes with previously established changes in 5mC levels (selected based on the MBD-seq method – data not shown) by pull-down with the Diagenode 33D3 5mC antibody. WT cells demonstrate enrichment for 5mC while DKO cells show no enrichment for 5mC at these loci. TUBA1C serves as an unmethylated control and is not enriched in either cell line. (B) Representative browser shots demonstrating 5mC profiles for HCT116 WT and DKO cells. Confirmed utility and specificity of the MeDIP technique for 5mC by assaying promoters predicted to have methylation from MBD-seq analysis of HCT116 WT and DKO cells (unpublished data). Red lines indicate the MeDIP PCR amplification region. Green bars represent CpG islands. (C) Validation of in-house produced 5hmC antibody by spike-in hMeDIP. HCT116 WT DNA was spiked with unmodified (Cytosine), methylated (5mC), and hydroxymethylated (5hmC) controls (Diagenode). hMeDIP was performed using the same protocol as for MeDIP, and enrichment for spike-in controls was analyzed by qPCR in triplicate reactions. The 5hmC antibody demonstrated specific enrichment of the hydroxymethylated control sequence, but not the methylated and unmethylated sequences. (D) MeDIP/hMeDIP analysis of siDNMT1 target loci. siRNA depletion of DNMT1 in NCCIT cells followed by (h)MeDIP revealed reduced enrichment for 5mC across all assayed regions relative to NTC. 5hmC enrichment was locus-specific with the assayed regions demonstrating small gains (APOA4), loss (HOXA9), or no change (ZNF311, EIF4G3) in 5hmC. (E) MeDIP/hMeDIP analysis of siDNMT3B target loci. 5mC and 5hmC in NCCIT cells depleted for DNMT3B was locus dependent but consistent with 450K array/MBD-seq-based data. PDZK1 and EIF4G3 (identified from MBD-seq, browser shots in main text Fig. 6D) resulted in loss (PDZK1) and gain (EIF4G3) of 5mC, consistent with MBD-seq results; 5hmC was reduced at these loci. The FCGR2A, PDE4DIP, and ZNF311 loci showed consistent 5mC levels, while 5hmC levels increased in DNMT3B depleted cells. The APOA4 and HOXA9 loci showed elevated enrichment of both 5mC and 5hmC (note the HOXA9 results are consistent with BGS/TAB-sequencing in Figure 4A). Gene structures showing (h)MeDIP assayed regions (red bars) in the FCGR2A and PDE4DIP loci are shown at the right (for other genes, refer to Figure 4C,D). β-values for CpG sites in these two loci that are present on the 450K array (scaled to length of the gene) are presented in graphical form below each gene schematic. Supporting data validating the siRNA rescue experiment for (F) DNMT1 and (G) DNMT3B. (F) Western blot analysis of protein expression demonstrates efficient depletion of endogenous DNMT1 in the presence of transfected empty ectopic expression vector (empty) and efficient ectopic expression of FLAG-Dnmt1 at levels near that of endogenous DNMT1 in NCCIT cells transfected with the latter expression vector. (G) Western blot analysis of protein expression demonstrates efficient depletion of endogenous DNMT3B in the presence of transfected empty ectopic expression vector and efficient expression of FLAG-Dnmt3b1 at levels near that of endogenous DNMT3B in NCCIT cells transfected with the latter expression vector. Note that the gel photo was cut to facilitate comparison with part F because samples were loaded in a different order.
Figure S6: Microarray validation and overlap analysis with NCCIT differentiation methylation analysis, related to main Figure 6.
DNMT siRNA depletion mirrors the regulation of gene expression and DNA methylation observed during differentiation, related to Figure 6. (A) Validation of microarray expression data. QRT-PCR-based validation of differentially expressed genes identified from microarray analysis (blue line) performed in triplicate relative to TUBA1C expression and normalized to the NTC (siDNMT samples). Error bars indicate the standard error. mRNA expression (qPCR) of each gene upon differentiation of NCCIT cells with retinoic acid treatment (orange line) is overlaid to demonstrate concordance of expression profiles between siDNMT and NCCIT DF samples. NCCIT UD cells were treated with retinoic acid (10μM) for 7, 10, 14, and 21 days prior to RNA harvest for DF cells. Microarray fold changes (blue line) are overlaid upon the qRT-PCR results (gray bars) for siDNMT samples to demonstrate the consistency between qPCR and microarray data. R2 = Spearman rank correlation between siDNMT qRT-PCR and microarray data. The secondary axis (right) for DUSP6 refers to fold expression changes for the NCCIT DF series as the increase in expression becomes more substantial (relative to the siDNMT samples) as differentiation proceeds. (B) Overlap analysis of hypo/hypermethylated genes with significantly differentially methylated CpG sites (pval < 0.05) among genomic features (P = promoter, 5′ = 5′UTR, B = body, 3′ = 3′UTR) for all NCCIT DNMT depleted samples versus NCCIT DF significantly hypo/hypermethylated genes.
Figure S7: Validation of HCT116 DNMT1/DNMT3B knockout and overexpression experimental system, related to Figure 7. (A) Expression of DNMT mRNA by quantitative RTPCR for all HCT116 samples relative to GAPDH expression, normalized to WT (parental HCT116 cells). Error bars indicate standard error. * = pval < 0.05. (B) Heat map of hypermethylated (Δβ > 0.15) CpG sites for each HCT116 sample (duplicate CpG sites removed) stratified by WT (parental HCT116 cells) least methylated to most methylated CpG sites.
Related to Experimental Procedures for QRT-PCR, BGS, and MeDIP.
Table S1. Primer pair sequences used in this study for PCR, related to Experimental Procedures for QRT-PCR, BGS, and MeDIP. *indicates primer sequences from Ehrlich et al. (Ehrlich et al., 2006).
Acknowledgments
We thank Dr. B. Volgelstein for the HCT116 DNMT KO cell lines, and Dr. D. Gius for the HCT116 DNMT overexpression cell lines. We also thank Eiko Kitamura (Georgia Regents University Cancer Center) for assistance with expression microarrays, and Joshua Hysong for data analysis support. This work was supported by NIH grants R01 CA114229 (KDR), R01 AA19976 (KDR, CL), F31 CA171727 (RLT), and the Mayo Clinic Cancer Center.
Footnotes
Author Contributions: RLT coordinated design of the study, conducted all experiments and most data/statistical analysis, and drafted the manuscript. ELP, J-HL, RAH, and KK provided valuable input for experimental design/data analysis. J-HL, TO, and ZZ created and optimized the hMeDIP antibody (by the Mayo Clinic Center for Individualized Medicine). CL edited the manuscript and provided intellectual input. J-HC performed the SWAN 450K array normalization, MBD-seq data processing, and coordinated the data/statistical analyses. KDR conceived of the study and its design, coordinated the data/statistical analyses, and edited the manuscript.
Accession Numbers: The NCBI GEO accession number for the microarray, 450K array, and sequencing data reported in this paper is GSE54843.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bachman KE, Park BH, Rhee I, Rajagopalan H, Herman JG, Baylin SB, Kinzler KW, Vogelstein B. Histone modifications and silencing prior to DNA methylation of a tumor suppressor gene. Cancer Cell. 2003;3:89–95. doi: 10.1016/s1535-6108(02)00234-9. [DOI] [PubMed] [Google Scholar]
- Bourc'his D, Xu GL, Lin CS, Bollman B, Bestor TH. Dnmt3L and the establishment of maternal genomic imprints. Science. 2001;294:2536–2539. doi: 10.1126/science.1065848. [DOI] [PubMed] [Google Scholar]
- Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, Bock C, Vasanthakumar A, Gu H, Xi Y, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2012;44:23–31. doi: 10.1038/ng.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Wang KY, Shen CKJ. The mammalian de novo DNA methyltransferases DNMT3A and DNMT3B are also DNA 5-hydroxymethylcytosine dehydroxymethylases. J Biol Chem. 2012;287:33116–33121. doi: 10.1074/jbc.C112.406975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger G, Jeong S, Escobar SG, Cortez CC, Li TWH, Saito Y, Yoo CB, Jones PA, Liang G. Identification of DNMT1 (DNA methyltransferase 1) hypomorphs in somatic knockouts suggests an essential role for DNMT1 in cell survival. Proc Natl Acad Sci U S A. 2006;103:14080–14085. doi: 10.1073/pnas.0604602103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frauer C, Hoffmann T, Bultmann S, Casa V, Cardoso MC, Antes I, Leonhardt H. Recognition of 5-hgydroxymethylcytosine by the Uhrf1 SRA domain. PLoS ONE. 2011;6:e21306. doi: 10.1371/journal.pone.0021306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen RS, Wijmenga C, Luo P, Stanek AM, Canfield TK, Weemaes CMR, Gartler SM. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc Natl Acad Sci U S A. 1999;96:14412–14417. doi: 10.1073/pnas.96.25.14412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin B, Ernst J, Tiedemann RL, Xu H, Sureshchandra S, Kellis M, Dalton S, Liu C, Choi JH, Robertson KD. Linking DNA methyltransferases to epigenetic marks and nucleosome structure genome-wide in human tumor cells. Cell Rep. 2012;2:1411–1424. doi: 10.1016/j.celrep.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim GD, Ni J, Kelesoglu N, Roberts RJ, Pradhan S. Co-operation and communication between the human maintenance and de novo DNA (cytosine-5) methyltransferases. EMBO J. 2002;21:4183–4195. doi: 10.1093/emboj/cdf401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolasinska-Zwierz P, Down T, Latorre I, Liu T, Liu XS, Ahringer J. Differential chromatin marking of introns and expressed exons by H3K36me3. Nat Genet. 2009;41:376–381. doi: 10.1038/ng.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, Low HM, Kin Sung KW, Rigoutsos I, Loring J, Wei CL. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20:320–331. doi: 10.1101/gr.101907.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- Linhart HG, Lin H, Yamada Y, Moran E, Steine EJ, Gokhale S, Lo G, Cantu E, Ehrich M, He T, et al. Dnmt3b promotes tumorigenesis in vivo by gene-specific de novo methylation and transcriptional silencing. Genes Dev. 2007;21:3110–3122. doi: 10.1101/gad.1594007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341 doi: 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik P, Cashen A. Decitabine in the treatment of acute myeloid leukemia in elderly patients. Cancer Manag Res. 2014;6:53–61. doi: 10.2147/CMAR.S40600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D'Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–257. doi: 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- Ostler KR, Yang Q, Looney TJ, Zhang L, Vasanthakumar A, Tian Y, Kocherginsky M, Raimondi SL, DeMaio JG, Salwen HR, et al. Truncated DNMT3B isoform DNMT3B7 suppresses growth, induces differentiation, and alters DNA methylation in human neuroblastoma. Cancer Res. 2012;72:4714–4723. doi: 10.1158/0008-5472.CAN-12-0886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palii SS, Van Emburgh BO, Sankpal UT, Brown KD, Robertson KD. DNA methylation inhibitor 5-aza-2′-deoxycytidine induces reversible genome-wide DNA damage that is distinctly influenced by DNA methyltransferases 1 and 3B. Mol Cell Biol. 2008;28:752–771. doi: 10.1128/MCB.01799-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putiri EL, Tiedemann RL, Liu C, Choi JH, Robertson KD. Impact of human MLL/COMPASS and polycomb complexes on the DNA methylome. Oncotarget. 2014;5:6338–6352. doi: 10.18632/oncotarget.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsahoye BH, Biniszkiewicz D, Lyko F, Clark V, Bird AP, Jaenisch R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci U S A. 2000;97:5237–5242. doi: 10.1073/pnas.97.10.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee I, Bachman KE, Park BH, Jair KW, Yen RWC, Schuebel KE, Cui H, Feinberg AP, Lengauer C, Kinzler KW, et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature. 2002;416:552–556. doi: 10.1038/416552a. [DOI] [PubMed] [Google Scholar]
- Shah MY, Vasanthakumar A, Barnes NY, Figueroa ME, Kamp A, Hendrick C, Ostler KR, Davis EM, Lin S, Anastasi J, et al. DNMT3B7, a truncated DNMT3B isoform expressed in human tumors, disrupts embryonic development and accelerates lymphomagenesis. Cancer Res. 2010;70:5840–5850. doi: 10.1158/0008-5472.CAN-10-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif J, Muto M, Takebayashi Si, Suetake I, Iwamatsu A, Endo TA, Shinga J, Mizutani-Koseki Y, Toyoda T, Okamura K, et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007;450:908–912. doi: 10.1038/nature06397. [DOI] [PubMed] [Google Scholar]
- Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, Oberdoerffer P, Sandberg R, Oberdoerffer S. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011;479:74–79. doi: 10.1038/nature10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperger JM, Chen X, Draper JS, Antosiewicz JE, Chon CH, Jones SB, Brooks JD, Andrews PW, Brown PO, Thomson JA. Gene expression patterns in human embryonic stem cells and human pluripotent germ cell tumors. Proc Nat Acad Sci U S A. 2003;100:13350–13355. doi: 10.1073/pnas.2235735100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stergachis AB, Haugen E, Shafer A, Fu W, Vernot B, Reynolds A, Raubitschek A, Ziegler S, LeProust EM, Akey JM, Stamatoyannopoulos JA. Exonic transcription factor binding directs codon choice and affects protein evolution. Science. 2013;342:1367–1372. doi: 10.1126/science.1243490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Huang L, Nguyen P, Bisht KS, Bar-Sela G, Ho AS, Bradbury CM, Yu W, Cui H, Lee S, et al. DNA Methyltransferase 1 and 3B activate BAG-1 expression via recruitment of CTCFL/BORIS and modulation of promoter histone methylation. Cancer Res. 2008;68:2726–2735. doi: 10.1158/0008-5472.CAN-07-6654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita Y, Yuan J, Suetake I, Suzuki H, Ishikawa Y, Choi YL, Ueno T, Soda M, Hamada T, Haruta H, et al. Array-based genomic resequencing of human leukemia. Oncogene. 2010;29:3723–3731. doi: 10.1038/onc.2010.117. [DOI] [PubMed] [Google Scholar]
- Yu M, Hon GC, Szulwach KE, Song CX, Jin P, Ren B, He C. Tet-assisted bisulfite sequencing of 5-hydroxymethylcytosine. Nat Protocols. 2012;7:2159–2170. doi: 10.1038/nprot.2012.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziller MJ, Müller F, Liao J, Zhang Y, Gu H, Bock C, Boyle P, Epstein CB, Bernstein BE, Lengauer T, et al. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet. 2011;7:e1002389. doi: 10.1371/journal.pgen.1002389. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Validation of model system used in all main figures, and additional siDNMT1 specific spatial distribution plots and microarray expression profiles, related to main Figure 1.
Validation of the experimental system used for this study, related to Figure 1. (A) mRNA expression by quantitative RT-PCR of DNMTs in undifferentiated (UD) and differentiated (DF) NCCIT cells performed in triplicate relative to GAPDH expression and normalized to UD (set at 1.0). Error bars indicate standard error. NCCIT UD cells were treated with retinoic acid (10μM) for 7 days prior to the RNA harvest for DF cells, and this represented the “differentiated” condition used throughout this study. * = pval < 0.005 (B) Expression of DNMTs in all DNMT siRNA knockdown (KD) samples (single and combination) by quantitative RT-PCR relative to GAPDH expression, normalized to a no-target control (NTC) siRNA transfection. Depletion efficiency was monitored on the same samples used for the HumanMethylation 450K and Affymetrix Gene 1.0 ST arrays. * = pval < 0.005. (C) Expression of DNMTs for individual NCCIT siDNMT KD samples at the protein level confirms the RT-PCR results. PCNA was used as a loading control. (D) Expression of developmental markers, pluripotency factors, housekeeping genes, and TETs by quantitative RT-PCR under conditions of DNMT depletion relative to TUBA1C expression, normalized to NTC. (E) Intragenic spatial distribution plot for DNMT1 siRNA depleted samples derived from average β-values for DNA methylation obtained from the HumanMethylation 450K array. TSS = transcription start site; TTS = transcription termination site. Dashed black box indicates region magnified (to the right) to better visualize relative methylation levels among samples. (F) Feature-based analysis of differential methylation demonstrates reproducibility among independent siDNMT KD biological replicates. Box plots representing all δβ-values for each sample in the indicated genomic features (promoter, gene body, intergenic, islands, shores, open sea) in three independent KD experiments (“0, 1, 2”; 0 = original experiment). Outliers outside of the 5th/95th percentile have been removed. Red bar highlights the zero line (no change in methylation). (G) Clustered heat map of gene expression (Affymetrix Human Gene 1.0 ST array) measured as log2 fold change for all NCCIT DNMT KD samples.
Figure S2: Total number of significantly differentially methylated CpGs and genes to support Figure 2, and validation of identified target sites, related to main Figure 2.
Confirmation of array-based CpG methylation data using bisulfite genomic sequencing, related to Figure 2. (A) Table showing numbers of hypo/hypermethylated CpG sites (including intergenic sites) and genes for each NCCIT siRNA DNMT depletion condition. (B) Normalized distribution of significantly differentially methylated CpG sites by CpG island annotation. Normalization was conducted by comparing the number of significantly differentially methylated CpG sites to the total number of sites for each island annotation present on the 450K array. Top: DNMT1 depletion (individual/combination) Bottom: DNMT3 depletion (individual/combination). (C) Bisulfite genomic sequencing (BGS) of identified DNMT1 target genes from the 450K array analysis. Gene structures with PCR-amplified region indicated by a dashed red box are provided below each CpG plot. Each pie chart indicates the percentage of methylated (black) and unmethylated (white) CpGs for a single CpG site among > 10 clones sequenced with the total percent methylation shown at the right. (D) Expression of PTPRN2 and CREBBP (DNMT1 target genes) by quantitative RT-PCR for all DNMT depleted samples relative to GAPDH expression, normalized to NTC. Error bars indicate standard error.
Figure S3: Overlap analysis of differentially methylated genes and 450K array biological replicates of DNMT siRNA KD, related to main Figure 2.
Overlap of significantly differentially methylated genes across all NCCIT DNMT siRNA depleted samples, related to Figure 2 (A) Overlap analysis of hypo/hypermethylated genes with significantly differentially methylated CpG sites (pval < 0.05) among genomic features (P = promoter, 5′ = 5′UTR, B = body, 3′ = 3′UTR) for all NCCIT DNMT depleted samples. Statistical significance of overlap was calculated using the Fisher's Exact test. Left: hypomethylation vs hypomethylation; Middle: hypermethylation vs hypermethylation; Right: siDNMT1 (single/combo) hypomethylation vs hypermethylation. For example, in the right panel, genes that become hypomethylated in the gene body of DNMT1 depleted samples significantly overlap with genes that become hypermethylated in the gene body upon DNMT3B individual and combination siRNA depletion. (B) Distribution of differentially methylated CpG sites for siDNMT KD biological replicates. Percentage of significantly differentially methylated CpG sites (pval < 0.05) classified by methylation status (hypo/hypermethylation) in three independent NCCIT DNMT depletion experiments demonstrates the reproducibility of our findings. (C) Clustered heat map of hypermethylated CpG sites for each DNMT depleted biological replicate sample to demonstrate reproducibility of the siDNMT3B hypermethylation phenotype at conserved loci. Hypermethylated (Δβ>0.15) CpG loci (duplicate CpG sites removed) were clustered by siDNMT samples stratified by NTC most methylated CpG sites to least methylated CpG sites (left panel). Δβ-values for respective CpG loci are also presented (right panel). Dashed red box indicates magnified region (Avgβ-value [0.45 - 0.75] in NTC).
Figure S4: Independent confirmation of array-based nonCpG methylation data using bisulfite genomic sequencing, related to main Figure 3. (A) Bisulfite genomic sequencing (BGS) analysis of nonCpG hypomethylation events in DNMT3 depleted samples identified using the 450K array data. CpH status is indicated above each BGS plot; the 450K array CpA is denoted by *. Gene structures with amplified region indicated by the dashed red box provided below each CpH plot. Each pie chart indicates the percent methylated (black) and unmethylated (white) CpHs for a single CpH site among at least 10 clones sequenced. The table to the right of each plot indicates the total percent methylation for all CpGs and CpAs shown in each sample. (B) Expression of UBR4, USP14, and SIN3A by quantitative RT-PCR for all DNMT depleted samples relative to GAPDH expression, normalized to NTC. Error bars indicate standard error. (C) Box plots of Δβ-values for nonCpG methylation among three independent, biological replicates of siDNMT1, siDNMT3B, and si3B+3L conditions (“0, 1, 2”; 0 = original experiment). Outliers outside of the 5th/95th percentile have been removed. Red bar highlights the zero line (no change in methylation).
Figure S5: Independent confirmation of array-based CpG methylation and TAB-seq results using MeDIP/hMeDIP pull-downs, related to Figure 4. (A) Validation of MeDIP technique. HCT116 colorectal carcinoma (WT) and DNMT1/DNMT3B knockout (DKO) cells were assayed for 5mC at promoters of selected genes with previously established changes in 5mC levels (selected based on the MBD-seq method – data not shown) by pull-down with the Diagenode 33D3 5mC antibody. WT cells demonstrate enrichment for 5mC while DKO cells show no enrichment for 5mC at these loci. TUBA1C serves as an unmethylated control and is not enriched in either cell line. (B) Representative browser shots demonstrating 5mC profiles for HCT116 WT and DKO cells. Confirmed utility and specificity of the MeDIP technique for 5mC by assaying promoters predicted to have methylation from MBD-seq analysis of HCT116 WT and DKO cells (unpublished data). Red lines indicate the MeDIP PCR amplification region. Green bars represent CpG islands. (C) Validation of in-house produced 5hmC antibody by spike-in hMeDIP. HCT116 WT DNA was spiked with unmodified (Cytosine), methylated (5mC), and hydroxymethylated (5hmC) controls (Diagenode). hMeDIP was performed using the same protocol as for MeDIP, and enrichment for spike-in controls was analyzed by qPCR in triplicate reactions. The 5hmC antibody demonstrated specific enrichment of the hydroxymethylated control sequence, but not the methylated and unmethylated sequences. (D) MeDIP/hMeDIP analysis of siDNMT1 target loci. siRNA depletion of DNMT1 in NCCIT cells followed by (h)MeDIP revealed reduced enrichment for 5mC across all assayed regions relative to NTC. 5hmC enrichment was locus-specific with the assayed regions demonstrating small gains (APOA4), loss (HOXA9), or no change (ZNF311, EIF4G3) in 5hmC. (E) MeDIP/hMeDIP analysis of siDNMT3B target loci. 5mC and 5hmC in NCCIT cells depleted for DNMT3B was locus dependent but consistent with 450K array/MBD-seq-based data. PDZK1 and EIF4G3 (identified from MBD-seq, browser shots in main text Fig. 6D) resulted in loss (PDZK1) and gain (EIF4G3) of 5mC, consistent with MBD-seq results; 5hmC was reduced at these loci. The FCGR2A, PDE4DIP, and ZNF311 loci showed consistent 5mC levels, while 5hmC levels increased in DNMT3B depleted cells. The APOA4 and HOXA9 loci showed elevated enrichment of both 5mC and 5hmC (note the HOXA9 results are consistent with BGS/TAB-sequencing in Figure 4A). Gene structures showing (h)MeDIP assayed regions (red bars) in the FCGR2A and PDE4DIP loci are shown at the right (for other genes, refer to Figure 4C,D). β-values for CpG sites in these two loci that are present on the 450K array (scaled to length of the gene) are presented in graphical form below each gene schematic. Supporting data validating the siRNA rescue experiment for (F) DNMT1 and (G) DNMT3B. (F) Western blot analysis of protein expression demonstrates efficient depletion of endogenous DNMT1 in the presence of transfected empty ectopic expression vector (empty) and efficient ectopic expression of FLAG-Dnmt1 at levels near that of endogenous DNMT1 in NCCIT cells transfected with the latter expression vector. (G) Western blot analysis of protein expression demonstrates efficient depletion of endogenous DNMT3B in the presence of transfected empty ectopic expression vector and efficient expression of FLAG-Dnmt3b1 at levels near that of endogenous DNMT3B in NCCIT cells transfected with the latter expression vector. Note that the gel photo was cut to facilitate comparison with part F because samples were loaded in a different order.
Figure S6: Microarray validation and overlap analysis with NCCIT differentiation methylation analysis, related to main Figure 6.
DNMT siRNA depletion mirrors the regulation of gene expression and DNA methylation observed during differentiation, related to Figure 6. (A) Validation of microarray expression data. QRT-PCR-based validation of differentially expressed genes identified from microarray analysis (blue line) performed in triplicate relative to TUBA1C expression and normalized to the NTC (siDNMT samples). Error bars indicate the standard error. mRNA expression (qPCR) of each gene upon differentiation of NCCIT cells with retinoic acid treatment (orange line) is overlaid to demonstrate concordance of expression profiles between siDNMT and NCCIT DF samples. NCCIT UD cells were treated with retinoic acid (10μM) for 7, 10, 14, and 21 days prior to RNA harvest for DF cells. Microarray fold changes (blue line) are overlaid upon the qRT-PCR results (gray bars) for siDNMT samples to demonstrate the consistency between qPCR and microarray data. R2 = Spearman rank correlation between siDNMT qRT-PCR and microarray data. The secondary axis (right) for DUSP6 refers to fold expression changes for the NCCIT DF series as the increase in expression becomes more substantial (relative to the siDNMT samples) as differentiation proceeds. (B) Overlap analysis of hypo/hypermethylated genes with significantly differentially methylated CpG sites (pval < 0.05) among genomic features (P = promoter, 5′ = 5′UTR, B = body, 3′ = 3′UTR) for all NCCIT DNMT depleted samples versus NCCIT DF significantly hypo/hypermethylated genes.
Figure S7: Validation of HCT116 DNMT1/DNMT3B knockout and overexpression experimental system, related to Figure 7. (A) Expression of DNMT mRNA by quantitative RTPCR for all HCT116 samples relative to GAPDH expression, normalized to WT (parental HCT116 cells). Error bars indicate standard error. * = pval < 0.05. (B) Heat map of hypermethylated (Δβ > 0.15) CpG sites for each HCT116 sample (duplicate CpG sites removed) stratified by WT (parental HCT116 cells) least methylated to most methylated CpG sites.
Related to Experimental Procedures for QRT-PCR, BGS, and MeDIP.
Table S1. Primer pair sequences used in this study for PCR, related to Experimental Procedures for QRT-PCR, BGS, and MeDIP. *indicates primer sequences from Ehrlich et al. (Ehrlich et al., 2006).