

Abstract
The BBSome is a coat-like ciliary trafficking complex composed of proteins mutated in Bardet-Biedl syndrome (BBS). A critical step in BBSome-mediated sorting is recruitment of the BBSome to membranes by the GTP–bound Arf-like GTPase ARL6. We have determined crystal structures of C. reinhardtii ARL6–GDP, ARL6–GTP and the ARL6–GTP–BBS1 complex. The structures demonstrate how ARL6–GTP binds the BBS1 β-propeller at blades 1 and 7 and explain why GTP– but not GDP–bound ARL6 can recruit the BBSome to membranes. Single point mutations in the ARL6–GTP–BBS1 interface abolish the interaction of ARL6 with the BBSome and prevent the import of BBSomes into cilia. Furthermore, we show that BBS1 with the M390R mutation, responsible for 30% of all reported BBS disease cases, fails to interact with ARL6–GTP providing a molecular rationale for patient pathologies.
Cilia protrude from eukaryotic cells and serve as signaling hubs regulating important cellular processes such as sensory reception and developmental signaling 1,2. To construct a cilium, the cell utilizes ciliary trafficking pathways that rely on molecular motors 3, intraflagellar transport (IFT) and BBSome complexes for cargo recognition 4–7. Whereas IFT complexes appear to mainly transport precursors of the ciliaryaxoneme8–10, the BBSome is involved in the transport of ciliary membrane proteins such as G protein-coupled receptors and components of the hedgehog-signaling pathway 11–15. The BBSome consists of eight Bardet-Biedl syndrome (BBS) protein subunits 13,16 in which genetic mutations result in BBS, a ciliopathic human genetic disorder characterized by obesity, blindness, mental retardation and kidney failure 17–20. The BBSome travels between the base and tip of cilia in association with intraflagellar transport (IFT) complexes 21–23 and also mediates the ciliary export of several proteins 11–15,24,25. Interestingly, mutations in the IFT complex subunits IFT172 and IFT27 were recently shown to also cause BBS 26,27. The small GTPase IFT27 is of particular interest as it was shown to be required for cilium-mediated hedgehog signaling and the ciliary export of the BBSome28–30. To fulfill its function in membrane protein trafficking, the BBSome is recruited to membranes as the major effector of the small GTPase ARL6 (also known as BBS3) in the active guanosine triphosphate (GTP) –bound form, which is necessary for ciliary entrance of BBSomes13,16,31. Currently, we have only little insight into the mechanism of BBSome recruitment to membranes by ARL6–GTP or the molecular basis for the disease phenotypes of BBS patient mutations.
We set out to gain mechanistic insights into the membrane recruitment of the BBSome by ARL6. It was previously shown that ARL6–GTP binds the BBSome and that the interaction is likely mediated by the BBS1 subunit 31. We purified a stable complex of ARL6–GTP bound to the N-terminal domain of BBS1 and determined the crystal structure of this complex as well as of ARL6 alone bound to either GDP or GTP. These results provide a molecular basis for BBSome effector recruitment by ARL6–GTP. We probed the interaction interface between ARL6–GTP and BBS1 and showed that single point mutations disrupted complex formation and prevented the recruitment of the BBSome into cilia in a cell-based system. Finally, we tested the impact of two BBS1 patient mutations to demonstrate that the very frequent BBS1M390R mutant fails to interact with ARL6–GTP providing a molecular rationale for patient pathologies.
RESULTS
ARL6–GTP binds BBS1N with ~0.5μM affinity
To unravel the molecular basis for membrane recruitment of the BBSome and BBS disease phenotypes, we purified Homo sapiens (Hs) and Chlamydomonasreinhardtii (Cr) versions of ARL6 (full-length or ΔN (residues 1–15 deleted)) in the GDP- or GTP–bound form, the N-terminal domain of BBS1 (BBS1N: HsBBS1N (residues 1–416), CrBBS1N (residues 1–425)) and the ARL6ΔN–GTP–BBS1N complex (Fig. 1a and Supplementary Fig. 1). Both Chlamydomonas and human ARL6ΔN–GTP–BBS1N complexes were stable during size exclusion chromatography (SEC) and eluted in peaks well separated from the peaks of excess ARL6–GTP (Fig. 1a and Supplementary Fig. 1a). To determine the affinity between the two proteins, we carried out isothermal titration calorimetry (ITC) measurements. The results demonstrated that CrBBS1N binds CrARL6ΔN–GTP to form a stoichiometric complex with a dissociation constant (Kd) of 0.35μM (Fig. 1b). Consistently, HsBBS1N bound HsARL6ΔN–GTP with a Kd of 0.54μM to form a stoichiometric complex (Supplementary Fig. 1a). Given that ARL6 and BBS1 proteins are well conserved across species (40–50% conservation between Cr and Hs proteins), we tested if CrBBS1N interacts with HsARL6ΔN–GTP. Indeed, we found that HsARL6ΔN–GTP–CrBBS1N could be purified by SEC and that the Kd for this chimeric complex is 0.30μM (Supplemental Fig. 1d). We conclude that ARL6–GTP forms a stable complex with the N-terminal domain of BBS1 and that the interaction is conserved between the Chlamydomonas and human proteins.
Fig. 1. Structure of Chlamydomonasreinhardtii ARL6ΔN–GTP–BBS1 complex.

(a)(left) Size exclusion chromatography profile of CrBBS1N (blue), CrARL6ΔN–GTP (green) and the CrARL6ΔN–GTP–CrBBS1N complex (black). The complex displays a significant shift in elution volume compared to either of the two individual proteins. (right) SDS PAGE gel of peak fractions stained with Coomassie. (b) Isothermal titration calorimetry with purified CrARL6ΔN–GTP and CrBBS1N protein reveals a 1:1 complex with a dissociation constant of 0.35±0.045μM (S.D. calculated from 3 independent experiments). (c) Crystal structure of the CrARL6ΔN–GTP–CrBBS1N complex shown in cartoon representation. The GTP molecule is shown as sticks, the Mg2+ ion as a yellow ball. N- and C-termini as well as secondary structure elements of ARL6 are indicated. The black dotted line represents an extended loop region without interpretable electron density in our structure. This extension is predicted to contain αhelical structure.(d) Perpendicular view compared to panel (c) with the seven blades of the BBS1 β-propeller labeled 1–7 and the four β-strands in blade 7 labeled A–D
Structures of CrARL6ΔN–GTP and CrARL6ΔN–GTP–CrBBS1N
We determined crystal structures of CrARL6ΔN–GTP and the CrARL6ΔN–GTP–CrBBS1N complex (Figs. 1c–d, Supplementary Figs. 2a–b and Table 1). The CrARL6ΔN–GTP structure was determined at 2.2Å resolution by molecular replacement using the available structure of HsArl6–GTP 32. Crystals of the CrARL6ΔN–GTP–CrBBS1N complex reproducibly diffracted to resolutions of 3.1–3.5Å and we determined the structure using experimental phasing on a mercury derivative (Supplementary Fig. 2f). The structures showed that CrARL6 adopts a classical small GTPase fold and that CrBBS1N adopts the fold of a seven bladed WD40-like β-propeller (Figs. 1c–d). A comparison of the CrARL6ΔN–GTP structure with previously published structures of human and Trypanosomabrucei (Tb) ARL6–GTP 32–33 revealed largely identical structures with RMSDs of 0.7–0.9Å (Supplementary Fig. 2a). The CrARL6ΔN–GTP–CrBBS1N structure revealed an elongated assembly with complex formation mediated by blades 1 and 7 of the CrBBSN1 β-propeller and helix α3, switch2 and the loop preceding helix α1 from the GTP–binding region of CrARL6 (Figs. 1c–d). The finding that BBS1 binds at the switch regions of the GTP-site of ARL6 is compatible with the BBSome being an effector for ARL631. Comparison of ARL6ΔN–GTP and ARL6ΔN–GTP–BBS1N structures revealed that GTP–bound ARL6 is already in a conformation competent for BBS1-binding with no major structural changes occurring upon BBS1N-complex formation (Supplementary Fig. 2b).
Table 1.
Summary of X-ray data collection and refinement statistics
| ARLΔN-BBS1 | ARLΔN-GTP | ARLΔN-GDP CdCl2 | ARLΔN-BBS1 EMP (Hg) | ARLΔN-BBS1 Pb (C2H3O2)2 | |
|---|---|---|---|---|---|
| Data collection | |||||
| Space group | P3121 | P6222 | P3121 | P3121 | P3121 |
| Cell dimensions | |||||
| a, b, c (Å) | 124.8,124.8,441.8 | 119.4,119.4,147.3 | 65.7,65.7,185.6 | 123.6,123.6,443.6 | 123.7,123.7,439.0 |
| α, β, γ(°) | 90.0,90.0,120.0 | 90.1,90.0,60.0 | 90.0,90.0,120.0 | 90.0,90.0,120.0 | 90.0,90.0,120.0 |
| Wavelength | 1.0000 | 1.0332 | 1.0000 | 1.0075 | 0.9497 |
| Resolution (Å) | 50–3.45 (3.55–3.45) * | 50–2.20 (2.32– 2.20)* | 50–1.43 (1.51–1.43)* | 50–3.13 (3.24–3.13)* | 50–3.35 (3.47–3.35)* |
| Rsym | 0.12 (0.82) | 0.034 (0.55) | 0.031 (0.60) | 0.11 (0.79) | 0.11 (0.81) |
| I /σI | 9.1 (1.0) | 21.1 (1.5) | 17.8 (1.8) | 12.6 (1.7) | 8.6 (1.6) |
| Completeness (%) | 99.1 (97.6) | 99.9 (99.1) | 96.2 (89.8) | 99.2 (91.81) | 99.56 (96.76) |
| Redundancy | 6.5 (6.0) | 12.7 (12.2) | 8.8 (8.5) | 13.2 (11.3) | 8.2 (7.5) |
| Refinement | |||||
| Resolution (Å) | 3.45 | 2.20 | 1.43 | 3.13 | 3.35 |
| No. reflections | 51878 (6201) | 29942 (3511) | 83301 (11179) | 70112 (6437) | 56947 (5395) |
| Rwork/Rfree | 0.222/0.277 | 0.183/0.217 | 0.157/0.185 | 0.213/0.249 | 0.221/0.268 |
| No. atoms | |||||
| Protein | 14270 | 2627 | 2467 | 14399 | 14369 |
| Ligand/ion | 132 | 66 | 70 | 189 | 150 |
| Water | 0 | 303 | 474 | 0 | 0 |
| B factors | |||||
| Protein | 80.8 | 47.6 | 25.8 | 52.4 | 26.7 |
| Ligand/ion | 79.9 | 35.4 | 26.1 | 50.1 | 25.3 |
| Water | N.A. | 55.1 | 39.3 | N.A. | N.A. |
| r.m.s deviations | |||||
| Bond lengths (Å) | 0.011 | 0.007 | 0.007 | 0.11 | 0.012 |
| Bond angles (°) | 1.5 | 1.2 | 1.3 | 1.5 | 1.5 |
one crystal was used for each measurement.
The structure of the CrARL6ΔN–GTP–CrBBS1N complex revealed a highly complementary but relatively small interaction interface with only 600Å2 of buried surface area (Supplementary Fig. 2e). The interaction interface is bipartite with one hydrophilic patch closer to the GTP-pocket and one hydrophobic patch farther away (Figs. 2a–b). The hydrophobic patch is formed by L100, V103 and V104 from helix α3 of CrARL6 contacting L41, I415 and M417 from blade 1 and T86 from blade 2 of CrBBS1N (Figs. 2a–b). The hydrophilic patch is made by D26 and N27 from the loop preceding helix α1, R77 from switch2 and R101 and E108 from helix α3 of CrArl6 that interact with R399, E400 and R420 from blade 1 of the CrBBS1N β-propeller (Figs. 2a–b). The ARL6–BBS1N interface is highly conserved between different species (Fig. 2a and Supplementary Fig. 3). Of the eight residues from ARL6 directly involved in BBS1N binding, all are completely conserved between the Chlamydomonas and human proteins (Supplementary Fig. 3). Of the seven BBS1N residues that interact with ARL6, five are completely conserved between Chlamydomonas and human protein sequences whereas one residue represents a conservative hydrophobic substitution (M417 in CrBBS1 and I401 in HsBBS1) and oneresidue constitutes a threonine to proline substitution (T86 in CrBBS1 and P74 in HsBBS1) (Supplementary Fig. 3). These data support the notion of an evolutionarily conserved ARL6-BBS1 complex
Fig. 2. Structural basis of the ARL6–BBS1interaction.

(a) Surface rendering of the CrARL6ΔN–GTP–CrBBS1N structure with non-conserved residues colored white and conserved residues colored red. After rotation of the CrARL6 and BBS1N subunits to reveal the interaction surfaces, the positions of the highly conserved interacting residues are indicated. (b) Zoom-in on the hydrophobic part of the interface (top) and hydrophilic part of the interface (bottom) with the interacting residues displayed as sticks. (c) Capture of native BBSome from bovine retinal extracts using GST-tagged human ARL6 variants. Whereas GST-HsARL6Q73LΔN efficiently pulls down the BBSome (assessed by BBS4 immunoblotting, top), structure-based HsARL6 interface point-mutations prevent interaction with the BBSome. The membrane was post-stained with Coomassie Brilliant Blue (bottom) to reveal equal amounts of ARL6 variants in the eluates. Control capture (last lane) was performed in the absence of GST-HsARL6. (d) Pull-down of recombinantly expressed wild-type or structure-guided interface mutations of human His-BBS1N using GST-ARL6Q73L. As in panel (c), single point-mutations of conserved residues of the ARL6-BBS1N interface prevent the interaction between the two proteins as visualized by anti-His-tag antibody staining.
Mutation analysis of human ARL6ΔN–BBS1N
Based on the structure of CrARL6ΔN–GTP–CrBBS1N described above, we designed and tested single point-mutations of human ARL6 and BBS1N for their ability to form a protein complex. Non-conservative substitutions were introduced in HsARL6 to replace residues in either the hydrophobic or the hydrophilic interaction interface and the recombinant GST-tagged proteins were purified. The HsARL6 variants were then incubated with native BBSome from bovine retinal extract and probed for interaction in pull-down experiments (Fig. 2c). Whereas GST-tagged HsARL6Q73LΔ N without mutations in the interface efficiently captured the BBSome from retinal extracts, R77A, L100E or E108A single point-mutant ARL6 failed to do so (Fig. 2c; all mutants used in this study were tested for proper folding using SEC and for nucleotide binding by NMR in case of ARL6, see Supplementary Fig. 4). Residues of the interaction interface between ARL6 and BBS1N elucidated from the CrARL6ΔN–GTP–CrBBS1N structure are thus required for the efficient binding of the mammalian BBSome to ARL6. To reciprocally probe for interaction, we designed HsBBS1N point-mutations (I399E or R404A) and performed pull-down experiments with GST-tagged HsARL6. The results shown in Fig. 2d demonstrated that a single point-mutation in either the hydrophilic (R404A) or the hydrophobic (I399E) interface is sufficient to abolish complex formation, which is consistent with the relatively small buried surface area within the CrARL6ΔN–GTP–CrBBS1N complex.
ARL6E108A mutation prevents BBSome recruitment to cilia
Previously published data have shown that active GTP–bound ARL6 is required to recruit the BBSome to membranes and allow access of the BBSome complex to the ciliary compartment 31. As we showed that single ARL6 point-mutations were sufficient to abolish the interaction with native BBSome from retinal extracts we hypothesized that ciliary entry of BBSomes might also be compromised. To test the functional implications of the ARL6–GTP–BBS1 interaction in a cellular system, we thus generated clonal RPE-hTERT cell lines that expressed either wild-type 2 or E108A mutant ARL6. After knocking down endogenous ARL6 by siRNA targeting the 3’ UTR, the recruitment of the BBSome to cilia was monitored (Fig. 3). The knockdown of Arl6 reduced the percentage of BBSome positive cilia from above 40% to below 5%, which could be fully rescued by re-introducing wild-type ARL6. In contrast, rescue experiments with the ARL6 (E108A) interface mutant, which abolished BBSome interaction in pull-down experiments (see Fig. 2c), failed to increase the number of BBSome positive cilia (Fig. 3). These data confirmed the structural results and, given that the BBSome is the major effector of ARL6, suggested that the ARL6–GTP–BBS1 interaction is required to recruit the BBSome to cilia.
Fig. 3. Disruption of the ARL6–BBS1 interface prevents recruitment of the BBSome to cilia.
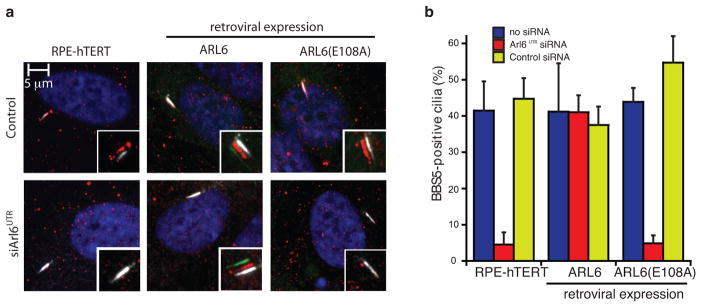
(a) Recruitment of the BBSome to cilia of clonal RPE-hTERT cell lines stably expressing either HsARL6 or HsARL6E108A with either GFP or mNeonGreen C-terminal fluorescent tags (green). Endogenous ARL6 was knocked down with ansiRNA targeting the 3’ UTR. Cells were then serum starved and immunostained for BBS5 (red) and polyglutamylated tubulin (GT335 antibody, white). (b) BBS5-positive cilia in the experiment shown in panel (a) were counted. Both untreated and a control siRNA had similar percentages of BBS5-positive cilia. The error bars represent SE between microscope fields and are technical replicates of three or more experiments.
Structure of CrARL6ΔN–GDP
CrARL6 recombinantly expressed in E. colico-purified with GTP bound at the GTPase site (Supplementary Figs. 2h). Following treatment with EDTA, the GTP was replaced by GDP and the crystal structure of CrARL6ΔN–GDP determined at 1.4Å resolution by experimental phasing using a Cd-derivative (Fig. 4 and Supplementary Fig. 2i). Small GTPases of the Arf-family associate with membranes via an amphipathic N-terminal helix. The exchange from GDP- to GTP–bound Arf results in a shift of two amino acids in the interswitch region, which in turn pushes out and exposes the N-terminal amphipathic helix to allow membrane association 34,35. To examine if ARL6 is likely to use a similar mechanism for membrane-attachment, we superposed the structures of CrARL6ΔN bound to either GDP or GTP to the equivalent structures of Arf1 34,35 revealing similar interswitch conformations (Supplementary Figs. 2c–d). The structural studies of CrARL6 presented here were carried out in absence of the amphipathic N-terminal helix. Nevertheless, our analysis demonstrated a shift of two residues in the interswitch region of ARL6–GTP compared toARL6–GDP, which is a hallmark of Arf proteins (Fig. 4a and Supplementary Figs. 2c–d). The structures thus support a canonical Arf-mechanism of membrane association via the amphipathic N-terminal helix of ARL6.
Fig. 4. Nucleotide-depended conformational changes in CrARL6.
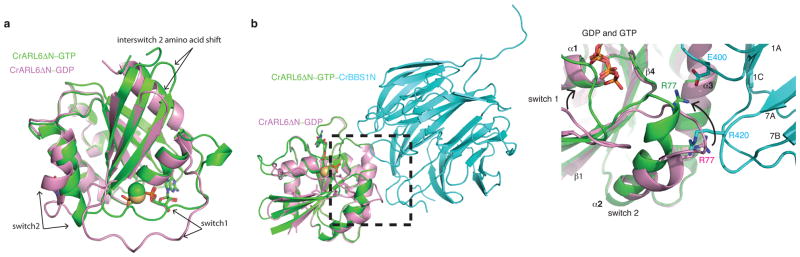
(a) Superpositioning of CrARL6ΔNin the GDP- and GTP–bound forms reveals a canonical 2 residue shift in the interswitch region. (b) Comparison of the switch regions of CrARL6ΔN–GTP in context of the CrARL6ΔN–GTP–BBS1N complex with the CrARL6ΔN–GDP structure (boxed region represents a zoom-in).
Furthermore, in the structure of CrARL6ΔN–GDP, both switch 1 and switch 2 adopted substantially altered conformations when compared to the CrARL6ΔN–GTP structure (Fig. 4a). The reason that GTP– but not GDP–bound CrARL6 can recruit the BBSome to membranes 31 is likely a result of structural changes in switch 2. The conformational change in switch 2 upon hydrolysis of GTP to GDP results in the disruption the ARL6R77–BBS1E400 salt-bridge and would cause ARL6R77to clash with BBS1NR420 (Fig. 4b). These observations explained why GTP–bound but not GDP–bound ARL6 can recruit the BBSome to membranes 21–23,31.
CrBBS1N is not a GTPase activating protein for CrARL6
The crystal structures of CrARL6ΔN–GTP and CrARL6ΔN–GTP–CrBBS1N presented here both have GTP bound at the GTPase site as demonstrated by the well-defined electron densities (Supplementary Figs. 2g–h). As nucleotides were not added to the sample, we conclude that the GTP co-purified with the proteins, which suggested that GTP-hydrolysis by CrARL6ΔN and CrARL6ΔN-BBS1N is slow. This notion was confirmed by GTPase assays (Supplementary Fig. 5) and is compatible with the fact that the catalytic glutamine found in most small GTPases (Q73 in HsARL6) is an alanine in CrARL6. In the CrARL6ΔN–GTP–CrBBS1N structure, we find no residues from the CrBBS1N subunit inserted into the GTP–binding pocket of Arl6ΔN in agreement with the slow hydrolysis rate by the CrARL6ΔN–GTP–CrBBS1N complex. We conclude that CrBBS1N does not act as a GTPase activating protein (GAP) for CrArl6ΔN, which is in contrast to COPII coat recruitment by Sar1, where the COPII component Sec23 serves as a Sar1-GAP by introducing an arginine into the active site thus promoting GTP hydrolysis 36. Membrane recruitment of BBSomes by ARL6–GTP may thus be long-lived in the absence of an external GAP.
BBS1 M390R patient mutation does not interact with ARL6–GTP
Mutational analyses of BBS patients have uncovered several point mutations in BBS proteins including variants that disrupt GTP–binding by ARL6 32,37,38. Interestingly, a single M390R point-mutation in BBS1 represents ~80% of all BBS1 disease mutations and accounts for 18–32% of all BBS disease mutations 39,40. Based on the CrARL6ΔN–GTP–CrBBS1N structure reported here, we modeled the human ARL6ΔN–GTP–BBS1N complex structure and mapped reported BBS1 disease mutations onto the structure (Fig. 5a). This analysis revealed that HsBBS1M390 is located at blade 1 of the β-propeller close to the GTP–binding site of ARL6 (Fig. 5a). The HsBBS1 M390 residue is not directly involved in the interaction interface with ARL6 but its position suggests that mutation to arginine might disrupt the structure of the β-propeller in the region around blade 1 of BBS1 and thus indirectly prevent complex formation with ARL6. To test this notion, we purified the HsBBS1NM390R mutant protein for interaction studies (Supplementary Fig. 1c). Although the HsBBS1NM390R purification resulted in a much lower yield than the wild-type protein, it eluted in SEC as a broad peak well separated from the void volume where aggregated proteins elute (Supplementary Fig. 1c). To assess the folding state of the HsBBS1NM390 protein in more detail, circular dichroism (CD) experiments were carried out. The CD spectrum of HsBBS1NM390R was that of an overall folded protein but with significantly lower β-strand content compared to wild-type HsBBS1N suggesting that the β-propeller of HsBBS1NM390R protein is partly unstructured (Fig. 5b). The HsBBS1NM390R mutant protein failed to interact with GST-tagged HsARL6Q73L and HsARL6Q73LΔN in pull-down experiments (Fig. 5c and Supplementary Fig. 6). Additionally, ITC and SEC experiments did not detect any interaction between HsARL6Q73LΔN and HsBBS1NM390R, which suggested that the affinity is at least two orders of magnitudes lower than for wild-type BBS1N (Fig. 5d and Supplementary Fig. 1c). The main defect of the BBS1M390R patient mutation thus appears to be mis-folding of the β-propeller, which in turn disrupts the association with binding partners such as ARL6. As a positive control, we used the BBS1NE234K mutant protein, which is an infrequent BBS patient mutation 40, as E234 is located at the top of the β-propeller far away from the ARL6 interaction site (Fig. 5a). BBS1NE234K was efficiently pulled down by GST-tagged HsARL6Q73L and HsARL6Q73LΔN and bound untagged HsARL6Q73LΔN with a Kd of 0.35μM in ITC experiments, which is similar to wild-type BBS1N (Figs. 5b–c and Supplementary Fig. 6). These results demonstrated that up to 30% of all BBS patient mutations could result in failure of ARL6-mediated recruitment of BBSomes to membranes, which in turn likely prevents the proper ciliary trafficking of several membrane proteins.
Fig 5. HsBBS1 M390R patient mutant does not bind HsARL6–GTP.

(a) Structural mapping of M390 and E234 residues mutated in BBS onto the model of the HsARL6ΔN–GTP-HsBBS1N complex. M390 is located in blade 1 close to the ARL6-interaction interface whereas E234 is located at the top of the β-propeller far away from the ARL6-interaction interface. (b) Circular dichroism (CD) spectra of WT, E234K and M390R mutants of the HsBBS1N protein. Secondary structure content is tabulated below. (c) GST pull-down of wild-type or mutant HsBBS1N with GST-tagged HsARL6Q73LΔN. (d) Tabulation of dissociation constants (Kd) from ITC titrations of HsARL6Q73LΔN to BBS1N, wild-type, E234K and M390R. (S.D. calculated from 3 independent experiments, see Supplementary Fig. 1 for ITC curves).
Discussion
There are clear similarities in sequence and domain composition of subunits from membrane coating complexes (COPI, COPII and clathrinAP-1) and subunits from ciliary trafficking complexes (IFT and BBSome complexes), suggesting a common evolutionary origin 31,41,42. Another commonality is the recruitment to membranes by Arf and Arf-like proteins as COPI, AP-1 and GGA1-GAT are recruited by Arf1 and COPII by Sar1 36,43–45. While these complexes bind switch1, switch2 and the interswitch region (β-strands 2–3) of Arf1 and Sar1 mainly via α-helical structural elements, the recruitment of BBS1 by ARL6 is quite different (Fig. 6). The β-propeller of BBS1 binds switch2, α helix 3 and residues from the loop preceding helix α1 of ARL6, which positions the β propeller on the opposite side of ARL6 as compared to effector complexes of Arf1 and Sar1 (Fig. 6). The recruitment mode of BBSomes by ARL6 is thus different from that found in other coating complexes and it will be very interesting to see how the additional BBSome subunits related to COP and clathrin AP-1 complex subunits contribute to coat-formation and recognition of membrane proteins for ciliary transport.
Fig 6. Structural comparison of membrane recruitment of coating complexes by Arf, Sar and Arf-like proteins.

Recruitment of Arf, Sar and ArlGTPases (shown in green) to lipid bilayers (top) requires the active GTP–bound state, which exposes an N-terminal amphipathic helix (not shown) and allow for effector-binding. The complexes of Sar1-Sec23, Arf1-γ-COP, Arf1-AP-1 and Arf1-GAT are shown in cartoon representation after structural superpositioning with the ARL6-BBS1 complex (only GTPase domains superposed). Switch regions are labeled similarly to Fig. 1. Affinities of the Arf-effector complexes are indicated below.
Online Methods
Protein purification and crystallization
Untagged or C-terminally His-tagged CrBBS1N (residues1–425) and HsBBS1N (residues 1–417) were cloned into pFL vectors and the proteins recombinantly expressed in High Five insect cells (Invitrogen). Full-length or ΔN (lacking residues 1–15) Cr- and HsArl6 were cloned into pET vectors with cleavable N-terminal His tags and recombinantly over-expressed in E. coli BL21 (DE3). Proteins were purified by Ni-NTA affinity chromatography after lysing cells in buffer A: 20mM Tris*HCl pH 7.5, 150mM NaCl, 10% glycerol, 10mM imidazol and 5mM MgCl2. After elution with 10–500mM imidazole and overnight dialysis and His-tag cleavage by TEV protease, the proteins were passed back onto a Ni-NTA column and the flow-through further purified by anion exchange chromatography on a MonoQ column. As a last purification step, proteins were subjected to size exclusion chromatography on a superdex 75 column in buffer B containing: 10mM Hepes pH7.5, 150mM NaCl and 1mM MgCl2. CrARL6ΔN and CrBBS1N were cloned as a bicistronic construct for co-expression in High Five cells and the CrARL6ΔN–GTP–CrBBS1N complex purified as described above.
Crystallization experiments were done by sitting drop vapour diffusion methods at 18°C. CrArl6ΔN that co-purified with GTP was concentrated to 75mg/ml and crystals obtained by mixing the proteins with an equal volume of precipitant containing 30% v/v pentaerythritolethoxylate, 50mM Bis-Tris pH 6.5 and 50mM ammonium sulfate. To obtain GDP–bound CrArl6ΔN, purified CrArl6ΔN–GTP was incubated with buffer B containing 5mM EDTA and no MgCl2 for 3h at 20°C followed by SEC in buffer B without MgCl2. The eluted nucleotide free protein was mixed with 1mM GDP in buffer B and again subjected to SEC followed by concentration to 15mg/ml and crystallization by mixing with equal volumes of precipitant containing 12% polyethylene glycol 3350, 0.1M Hepes pH7.5, 5mM CdCl2, 5mM MgCl2, 5mM NiCl2 and 5mM CoCl2. The CrARL6–GTP–CrBBS1N complex was crystallized by mixing the protein complex at 10mg/ml with equal volumes of precipitant containing 29% PEG400 and 0.1M Tris pH 8.0. Before flash cooling, crystals were cryo-protected by soaking in mother liquor supplemented with 25% glycerol (ARL6 crystals) or with the PEG400 concentration increased to 35% (CrARL6–GTP–CrBBS1N complex crystals).
X-ray diffraction data collection and structure determination
X-ray diffraction data were acquired at the Swiss Light Source (SLS, Villigen, Switzerland) at beam lines PXII and PXIII. All diffraction data were collected at cryogenic temperatures (100K) at wavelengths of 1.000Å (CrARL6ΔN-CrBBS1N and CrARL6ΔN-GDP –CdCl2), 1.0322Å (CrARL6ΔN-GTP), 1.0075Å (CrARL6ΔN-CrBBS1N-Hg) or 0.9497Å(CrARL6ΔN-CrBBS1N-Pb). The data were indexed with the XDS package46 before scaling with Aimless as part of the CCP4 package 47,48. The structure of CrARL6–GTP was determined at 2.2Å resolution by molecular replacement (MR) with the human ARL6 structure (pdb code 2h57) as a search model in the programPhaser49 as implemented in the PHENIX software package50. Two molecules of CrARL6 were found in the asymmetric unit and the structure was completed by iterative cycles of model building in Coot 51 and refinement in PHENIX. The CrARL6–GDP structure was determined at 1.4Å resolution using single anomalous dispersion on Cd-derivatized crystals followed by autobuilding in PHENIX. For the CrARL6ΔN–GTP–CrBBS1N complex structure, crystals were soaked in mother liquor complemented with 1.3 mM of the Hg-compound EMP for 2h. Crystals were then back-soaked in a cryo-solution without EMP and flash cooled as described above. Anomalous data were recorded at the Hg-peak wavelength of 1.0075Å, which gave diffraction to 3.1Å and a significant anomalous signal to 4.4Å. For structure determination, MR with CrARL6–GTP using Phaser located 4 molecules in the asymmetric unit. This MR solution was then used in an MR-SAD procedure as implemented in Phaser to find 35 Hg sites and produce a high quality electron density map (see Supplementary Fig. 2f) into which the CrBBS1N molecules were modelled. The four copies of CrARL6–GTP–CrBBS1N are very similar and the structure was refined in PHENIX using 4-fold NCS restraints. In addition to the Hg-derivatized CrARL6–GTP–CrBBS1N complex, a structure was refined using native data to 3.45Å resolution and of a Pb-derivatized crystal that diffracted to 3.35Å resolution. The CrARL6–GTP–CrBBS1N Pb-derivatized crystals gave only a weak anomalous signal that did not extend beyond 7Å resolution because of low occupancy Pb-sites and this dataset can thus be considered as near-native. All crystal structures reported here have 90–97% of the residues in the most favoured region of the ramachandran plot with 0.1–0.6% in the disallowed regions. See Table 1 for refinement statistics. The human ARL6ΔN–GTP–BBS1N complex structure shown in Fig. 5a was modelled using the program Modeller 52 based on the experimentally determined CrARL6ΔN–GTP–CrBBS1N crystal structure.
Isothermal Titration Calorimetry (ITC)
ITC was done using an ITC200 instrument (MicroCal, Wolverton Mill, UK) at 25°C using purified proteins in buffer B. Each experiment was independently carried out at least 3 times using different purification batches of protein. The titration protocol consisted of one initial injection of 0.2μl followed by 39 injections of 1μl of the ligand (350–450μM) into the protein sample (35–45μM) with intervals of 150 sec to allow the titration peak to stabilize at the baseline. The data were fitted to titration curves using the program Origin v7.0 (MicroCal).
GST pull-down experiments
For GST pull-downs with recombinant proteins, GST-HsARL6Q73L ΔN–GTP was mixed with wild-type or mutant forms of His-HsBBS1N and incubated with GSH beads for 1h in a buffer containing 10mM Hepes pH 7.5, 150mM NaCl, 1mM MgCl2 and 1mM GTP. After washing 3X with 40X the bead volume, proteins were eluted with 30mM reduced glutathione containing buffer. Analysis was done on an SDS page gel stained with Coomassie. Full-length GST-HsARL6Q73L–GTP was incubated with insect cell extract with over-expressed wild-type or mutant His-HsBBS1N for 1h at 4°C with GSH beads. After washing 3X with 40X the bead volume of buffer, proteins were eluted with buffer containing 30mM reduced glutathione. Visualization was done using SDS-PAGE followed by Western-blot analysis using a His probe antibody that recognizes the hexa-Histidine tag of BBS1N. For bovine retinal extract pull-downs, HsARL6Q73LΔN constructs and bovine retinal extract were prepared as described previously 31. Equal amounts of GST-HsArl6 in E. coli lysate were bound to GSH beads, and incubated with retinal extract at 4°C for 2h. After washing three times with 1X PBS, proteins were eluted in 6M guanidinium*HCl, and analyzed by SDS-PAGE. Western blot analysis was done with anti-BBS4 as described in13. Full size scans of western blots and SDS-PAGE gels are shown in Supplementary Data Set 1.
Stable Cell Lines and Immunofluorescence
RPE clones and immunofluorescence experiments were done as described in 2. In short, retroviruses were produced using pBabe-puro containing Arl6-GFP or Arl6E108A-NeonGreen 53, and used to infect RPE cells. Single cell clones were screened for low expression levels. RPE, RPE [Arl6-GFP], and RPE [Arl6E108A-mNeonGreen] were transfected with 20nM siRNA duplex against the 3’ UTR of endogenous Arl6 for 24 h, and then shifted from 10% to 0.2% serum media for 48h. Cells were fixed by the enhanced immunofluorescence protocol described in 31. Commercial antibodies against BBS5 (14569-1-AP, Proteintech) and Polyglutamylated tubulin (GT335) 54 were used. GTPase assay The GTPase activity of CrARL6ΔN and the CrARL6ΔN-CrBBS1N complex were measured using the EnzCheck Phosphate kit (Invitrogen) by incubating 50μM of the proteins with 1mM GTP at 20°C. GTP hydrolysis was followed by monitoring the conversion of the released inorganic phosphate and 2-amino-6-mercapto-7-methylpurine riboside into ribose 1-phosphate and 2-amino-6-mercapto-7-methyl-purine by absorbance measurements at 360nm. Measurements were recorded every min over a 20 min time course. For the negative control buffer without protein was added and for the positive control 100μM of inorganic phosphate was added.
NMR spectroscopy
Prior to NMR data acquisition, protein samples were buffered exchanged into 20mM NaPO4 pH 6.5,100mM NaCl and 1mM MgCl2 and concentrated to 10–60μM. GTP was titrated to each protein sample in a 1.2 molar excess. NMR data were acquired at 25°C on a Bruker AV600 spectrometer equipped with a room temperature triple-resonance probe. The spectra were processed with Topspin software (http://www.bruker.com/products/mr/nmr/nmr-software/software/topspin/overview.html).
Circular Dichroism
CD spectroscopy was performed with a Jasco J-715 spectropolarimeter. Measurements were performed at 20°C in a 0.1cm quartz cuvette and buffer corrected. Proteins were measure in buffer B at 1.5μM concentration. Data analysis and secondary structure fractions were obtain using the CONTIN method of the CDPro analysis program (http://lamar.colostate.edu/~sreeram/CDPro/main.html). Although we observe no α helices in BBS1N from the CrARL6-GTP–CrBBS1N structure, electron density is missing for the N-terminal 34 residues as well residues 150–212. Residues 150–212 are predicted to adopt the fold of 4 α-helices likely accounting for the α-helical content of HsBBS1N. In case of the M390R mutant, the β-sheet content is significantly decreased suggesting a mostly unfolded β-propeller. The increase in α-helical content may suggest that part of the unfolded β-propeller adopts an α-helical structure in case of the HsBBSN M390R mutant.
Supplementary Material
Acknowledgments
We thank the staff at SLS for guidance with X-ray diffraction data collection, the biochemistry core facility and the crystallization facility of the Max Planck Institute of Biochemistry (MPI-B, Munich) for access to crystallization screening and the Bavarian NMR Center for NMR measurement time. We also thank I.B. Schaefer (MPI-B) for DNA encoding BBS subunits, S. Wachter for assistance with GTPase assays and M. Taschner (MPI-B) for expert advice on matters of protein production from insect cells and for carefully reading the manuscript. This work was funded by an Emmy No ether grant (DGF; LO1627/1-1), the European Research Council (ERC grant 310343) and by the EMBO Young Investigator program. A.R.N was supported by the Fayez Sarofim Fellowship of the Damon Runyon Cancer Research Foundation (DRG 2160-13).
Footnotes
Accession codes
Atomic coordinates have been submitted to the Protein Data Bank (PDB), accession codes 4v0l (CrARL6ΔN–GTP), 4v0k (CrARL6ΔN–GDP), 4v0m (CrARL6ΔN–GTP–CrBBS1N, native data), 4v0n (CrARL6ΔN–GTP–CrBBS1N, mercury soak) and 4v0o (CrARL6ΔN–GTP–CrBBS1N, lead soak).
The authors declare that they have no conflict of interest.
Author Contributions A.M carried out the protein biochemistry and structural biology under the supervision of E.L. A.R.N. carried out the pull-down experiments of native BBsome using WT and mutant ARL6 and the cell biology experiments under the supervision of M.V.N.A.M. and E.L. designed the experiments and wrote the paper with input from A.R.N and M.V.N.
References
- 1.Veland IR, Awan A, Pedersen LB, Yoder BK, Christensen SORT. Primary Cilia and Signaling Pathways in Mammalian Development, Health and Disease. Nephron Physiol. 2009;111:39–53. doi: 10.1159/000208212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sung CH, Leroux MR. The roles of evolutionarily conserved functional modules in cilia-related trafficking. Nature. 2013;15:1387–1397. doi: 10.1038/ncb2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scholey JM. Intraflagellar transport. Annu Rev Cell Dev Biol. 2003;19:423–443. doi: 10.1146/annurev.cellbio.19.111401.091318. [DOI] [PubMed] [Google Scholar]
- 4.Rosenbaum JL, Witman GB. Intraflagellar transport. Nat Rev Mol Cell Biol. 2002;3:813–825. doi: 10.1038/nrm952. [DOI] [PubMed] [Google Scholar]
- 5.Ishikawa H, Marshall WF. Ciliogenesis: building the cell’s antenna. Nature Publishing Group. 2011;12:222–234. doi: 10.1038/nrm3085. [DOI] [PubMed] [Google Scholar]
- 6.Bhogaraju S, Engel BD, Lorentzen E. Intraflagellar transport complex structure and cargo interactions. Cilia. 2013;2:10–10. doi: 10.1186/2046-2530-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taschner M, Bhogaraju S, Lorentzen E. Architecture and function of IFT complex proteins in ciliogenesis. Differentiation. 2012;83:S12–22. doi: 10.1016/j.diff.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hou Y, et al. Functional analysis of an individual IFT protein: IFT46 is required for transport of outer dynein arms into flagella. The Journal of Cell Biology. 2007;176:653–665. doi: 10.1083/jcb.200608041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ishikawa H, et al. TTC26/DYF13 is an intraflagellar transport protein required for transport of motility-related proteins into flagella. eLife. 2014;3:e01566–e01566. doi: 10.7554/eLife.01566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhogaraju S, et al. Molecular basis of tubulin transport within the cilium by IFT74 and IFT81. Science. 2013;341:1009–1012. doi: 10.1126/science.1240985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Domire JS, et al. Dopamine receptor 1 localizes to neuronal cilia in a dynamic process that requires the Bardet-Biedl syndrome proteins. Cell Mol Life Sci. 2011;68:2951–2960. doi: 10.1007/s00018-010-0603-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seo S, et al. A Novel Protein LZTFL1 Regulates Ciliary Trafficking of the BBSome and Smoothened. PLoS Genet. 2011;7:e1002358. doi: 10.1371/journal.pgen.1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nachury MV, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129:1201–1213. doi: 10.1016/j.cell.2007.03.053. [DOI] [PubMed] [Google Scholar]
- 14.Berbari NF, Lewis JS, Bishop GA, Askwith CC, Mykytyn K. Bardet-Biedl syndrome proteins are required for the localization of G protein-coupled receptors to primary cilia. Proceedings of the National Academy of Sciences. 2008;105:4242–4246. doi: 10.1073/pnas.0711027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Q, et al. Bardet-Biedl syndrome 3 (Bbs3) knockout mouse model reveals common BBS-associated phenotypes and Bbs3 unique phenotypes. Proc Natl Acad Sci U S A. 2011;108:20678–20683. doi: 10.1073/pnas.1113220108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loktev AV, et al. A BBSome Subunit Links Ciliogenesis, Microtubule Stability, and Acetylation. Developmental Cell. 2008;15:854–865. doi: 10.1016/j.devcel.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 17.Katsanis N, Lupski JR, Beales PL. Exploring the molecular basis of Bardet-Biedl syndrome. Human Molecular Genetics. 2001;10:2293–2299. doi: 10.1093/hmg/10.20.2293. [DOI] [PubMed] [Google Scholar]
- 18.Tobin JL, Beales PL. Bardet-Biedl syndrome: beyond the cilium. Pediatr Nephrol. 2007;22:926–936. doi: 10.1007/s00467-007-0435-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sheffield VC. The blind leading the obese: the molecular pathophysiology of a human obesity syndrome. Trans Am Clin Climatol Assoc. 2010;121:172–81. discussion 181–2. [PMC free article] [PubMed] [Google Scholar]
- 20.Blacque OE, Leroux MR. Bardet-Biedl syndrome: an emerging pathomechanism of intracellular transport. Cell Mol Life Sci. 2006;63:2145–2161. doi: 10.1007/s00018-006-6180-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lechtreck KF, et al. The Chlamydomonas reinhardtii BBSome is an IFT cargo required for export of specific signaling proteins from flagella. The Journal of Cell Biology. 2009;187:1117–1132. doi: 10.1083/jcb.200909183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosenbaum JL, Witman GB. Intraflagellar transport. Nat Rev Mol Cell Biol. 2002;3:813–825. doi: 10.1038/nrm952. [DOI] [PubMed] [Google Scholar]
- 23.Ou G, Blacque OE, Snow JJ, Leroux MR, Scholey JM. Functional coordination of intraflagellar transport motors. Nature. 2005;436:583–587. doi: 10.1038/nature03818. [DOI] [PubMed] [Google Scholar]
- 24.Lechtreck KF, et al. Cycling of the signaling protein phospholipase D through cilia requires the BBSome only for the export phase. The Journal of Cell Biology. 2013;201:249–261. doi: 10.1083/jcb.201207139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y, et al. BBS mutations modify phenotypic expression of CEP290-related ciliopathies. Human Molecular Genetics. 2014;23:40–51. doi: 10.1093/hmg/ddt394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bujakowska KM, et al. Mutations in IFT172 Cause Isolated Retinal Degeneration and Bardet-Biedl Syndrome. Human Molecular Genetics. 2014 doi: 10.1093/hmg/ddu441. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aldahmesh MA, et al. IFT27, encoding a small GTPase component of IFT particles, is mutated in a consanguineous family with Bardet-Biedl syndrome. Human Molecular Genetics. 2014;23:3307–3315. doi: 10.1093/hmg/ddu044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keady BT, et al. IFT25 Links the Signal-Dependent Movement of Hedgehog Components to Intraflagellar Transport. Developmental Cell. 2012;22:940–951. doi: 10.1016/j.devcel.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liew GM, et al. The Intraflagellar Transport Protein IFT27 Promotes BBSome Exit from Cilia through the GTPase ARL6/BBS3. Dev Cell. 2014 doi: 10.1016/j.devcel.2014.09.004. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eguether T, et al. IFT27 Links the BBSome to IFT for Maintenance of Ciliary Signaling Compartment. Dev Cell. 2014 doi: 10.1016/j.devcel.2014.09.011. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin H, et al. The Conserved Bardet-Biedl Syndrome Proteins Assemble a Coat that Traffics Membrane Proteins to Cilia. Cell. 2010;141:1208–1219. doi: 10.1016/j.cell.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wiens CJ, et al. Bardet-Biedl syndrome-associated small GTPase ARL6 (BBS3) functions at or near the ciliary gate and modulates Wnt signaling. Journal of Biological Chemistry. 2010;285:16218–16230. doi: 10.1074/jbc.M109.070953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hemsworth GR, Price HP, Smith DF, Wilson KS. Crystal structure of the small GTPase Arl6/BBS3 from Trypanosoma brucei. Protein Science. 2013;22:196–203. doi: 10.1002/pro.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amor JC, Harrison DH, Kahn RA, Ringe D. Structure of the human ADP-ribosylation factor 1 complexed with GDP. Nature. 1994:704–708. doi: 10.1038/372704a0. [DOI] [PubMed] [Google Scholar]
- 35.Liu Y, Kahn RA, Prestegard JH. Dynamic structure of membrane-anchored Arf•GTP. Nat Struct Mol Biol. 2010;17:876–881. doi: 10.1038/nsmb.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bi XX, Corpina RAR, Goldberg JJ. Structure of the Sec23/24-Sar1 pre-budding complex of the COPII vesicle coat. Nature. 2002;419:271–277. doi: 10.1038/nature01040. [DOI] [PubMed] [Google Scholar]
- 37.Chiang AP, et al. Comparative genomic analysis identifies an ADP-ribosylation factor-like gene as the cause of Bardet-Biedl syndrome (BBS3) Am J Hum Genet. 2004;75:475–484. doi: 10.1086/423903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fan Y, et al. Mutations in a member of the Ras superfamily of small GTP-binding proteins causes Bardet-Biedl syndrome. Nat Genet. 2004;36:989–993. doi: 10.1038/ng1414. [DOI] [PubMed] [Google Scholar]
- 39.Mykytyn K, et al. Identification of the gene (BBS1) most commonly involved in Bardet-Biedl syndrome, a complex human obesity syndrome. Nat Genet. 2002;31:435–438. doi: 10.1038/ng935. [DOI] [PubMed] [Google Scholar]
- 40.Beales PL, et al. Genetic Interaction of BBS1 Mutations with Alleles at Other BBS Loci Can Result in Non-Mendelian Bardet-Biedl Syndrome. Am J Hum Genet. 2003;72:1187–1199. doi: 10.1086/375178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jékely G, Arendt D. Evolution of intraflagellar transport from coated vesicles and autogenous origin of the eukaryotic cilium. Bioessays. 2006;28:191–198. doi: 10.1002/bies.20369. [DOI] [PubMed] [Google Scholar]
- 42.van Dam TJP, et al. Evolution of modular intraflagellar transport from a coatomer-like progenitor. Proceedings of the National Academy of Sciences. 2013;110:6943–6948. doi: 10.1073/pnas.1221011110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shiba T, et al. Molecular mechanism of membrane recruitment of GGA by ARF in lysosomal protein transport. Nat Struct Biol. 2003;10:386–393. doi: 10.1038/nsb920. [DOI] [PubMed] [Google Scholar]
- 44.Yu X, Breitman M, Goldberg J. A Structure-Based Mechanism for Arf1-Dependent Recruitment of Coatomer to Membranes. Cell. 2012;148:530–542. doi: 10.1016/j.cell.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ren X, Farías GG, Canagarajah BJ, Bonifacino JS, Hurley JH. Structural basis for recruitment and activation of the AP-1 clathrin adaptor complex by Arf1. Cell. 2013;152:755–767. doi: 10.1016/j.cell.2012.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Collaborative Computational Project N4. The CCP4 Suite: Programs for protein crystallography. Acta Crystallogr Sec D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 48.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr D Biol Crystallogr. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 50.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fiser A, Sali A. Modeller: Generation and Refinement of Homology-Based Protein Structure Models. Methods in Enzymology. 2003;374:461–491. doi: 10.1016/S0076-6879(03)74020-8. [DOI] [PubMed] [Google Scholar]
- 53.Shaner NC, et al. A bright monomeric green fluorescent protein derived from Branchiostomalanceolatum. Nat Methods. 2013;10:407–409. doi: 10.1038/nmeth.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wolff A, et al. Distribution of glutamylated alpha and beta-tubulin in mouse tissues using a specific monoclonal antibody, GT335. Eur J Cell Biol. 1992;59:425–432. [PubMed] [Google Scholar]
- 55.Krissinel E, Henrick K. Inference of Macromolecular Assemblies from Crystalline State. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.