

Abstract
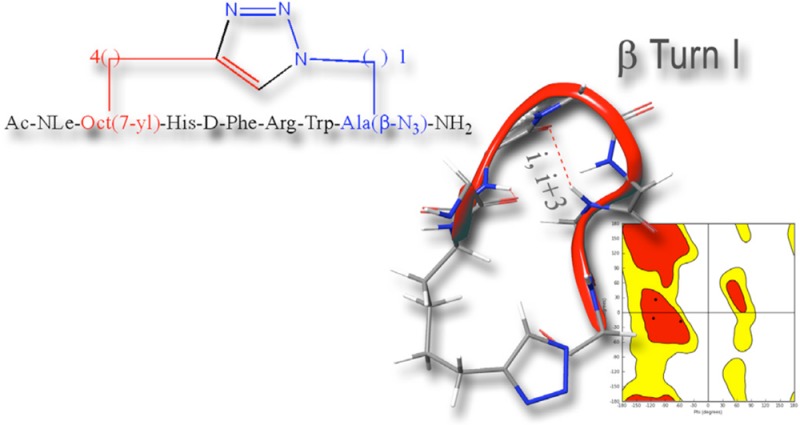
Side chain-to-side chain cyclizations represent a strategy to select a family of bioactive conformations by reducing the entropy and enhancing the stabilization of functional ligand-induced receptor conformations. This structural manipulation contributes to increased target specificity, enhanced biological potency, improved pharmacokinetic properties, increased functional potency, and lowered metabolic susceptibility. The CuI-catalyzed azide–alkyne 1,3-dipolar Huisgen’s cycloaddition, the prototypic click reaction, presents a promising opportunity to develop a new paradigm for an orthogonal bioorganic and intramolecular side chain-to-side chain cyclization. In fact, the proteolytic stable 1,4- or 4,1-disubstituted [1,2,3]triazolyl moiety is isosteric with the peptide bond and can function as a surrogate of the classical side chain-to-side chain lactam forming bridge. Herein we report the design, synthesis, conformational analysis, and functional biological activity of a series of i-to-i+5 1,4- and 4,1-disubstituted [1,2,3]triazole-bridged cyclopeptides derived from MT-II, the homodetic Asp5 to Lys10 side chain-to-side chain bridged heptapeptide, an extensively studied agonist of melanocortin receptors.
Introduction
Melanocortins are a group of peptide hormones including adrenocorticotropin (ACTH) and α-, β-, and γ-melanocyte stimulating hormones (MSHs). To date, five human melanocortin receptors (MC1R-MC5R) have been characterized as belonging to group α of the rhodopsin family of G-protein coupled receptors (GPCR).1,2 The MCRs mediate a plethora of biological functions that include sexual function,3 feeding behavior,4,5 pain modulation,6 thermoregulation, energy homeostasis,7,8 cardiovascular effects, and skin pigmentation,9,10 making them potential drug targets for treating pain, food intake, and body weight as well as erectile dysfunction. Therefore, development of an expansive toolbox of structural modifications that can be used to fine-tune the predominant conformations to achieve modulation of specificity toward receptor subtypes, physicochemical and pharmacological properties continues to be of great interest in the development of peptide-based drugs in general and melanocortinergic drugs in particular. Recently, the growing interest in designing highly selective and potent superagonist of the human melanocortin receptors hMC3R and hMC4R has been driven by their prospects of becoming effective drugs for treating feeding disorders such as the respective obesity and energy homeostasis. Structure–activity relationship studies (SAR) of α-MSH,11 a post-translationally generated tridecapeptide fragment originating from proopiomelanocortin (POMC),12 led to the development of MT-II, Nα-Ac[Nle4,c(Asp5,d-Phe7,Lys10)]αMSH(4–10)NH2.13−15 This homodetic Asp5 to Lys10 side chain-to-side chain bridged lactam stabilizes the pharmacophore containing sequence His6-d-Phe7-Arg8-Trp9 in a type-II β-turn, resulting in a potent long acting nonselective superagonist of MCRs.16 Evidently, the plethora of melanocortin-mediated activities, the five melanocortin receptor subtypes, and the high profile of potential therapeutic targets associated with the melanocortin system underscores the unmet need for highly selective, pharmacokinetically diverse, and bioavailable agonists and antagonists. Intramolecular side chain-to-side chain cyclization of linear peptides is an established approach to achieve stabilization of specific conformations and a recognized strategy to improve resistance toward proteolytic degradation, increasing the metabolic stability in vitro and in vivo.17,18 We have previously reported a novel intramolecular i-to-i+4 side chain-to-side chain [1,2,3]triazolyl-bridged modification that is bioisosteric to the peptide bond and employs the prototypical click reaction, CuI-catalyzed azido-to-alkyne 1,3-dipolar cycloaddition (CuAAC).19 Importantly, the CuAAC does not require synthetic schemes with elaborate orthogonal protection strategies. Our studies explored the relationship between the size of the bridge containing the [1,2,3]triazolyl moiety, the location of this moiety within the bridge, its orientation relative to the peptide backbone, and the predominant conformations displayed by these heterodetic cyclic peptides.
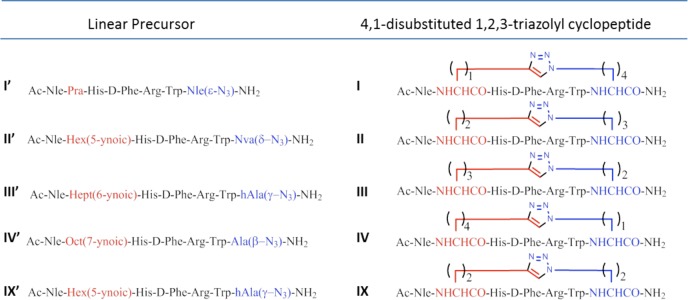
In the current study, we explored the potential of the i-to-i+5 side chain-to-side chain 1,4- and 4,1-disubstituted [1,2,3]triazolyl-containing bridges to stabilize β-turn conformations in the context of the MT-II scaffold and evaluate its potential as a modulator of receptor subtype selectivity. We synthesized a series of heterodetic MT-II related cyclo-heptapeptides that varied in the size of the disubstituted [1,2,3]triazolyl-containing bridge connecting Cαs of residues 4 and 10 and in the location and orientation of the [1,2,3]triazolyl moiety within the bridge (I–X, Schemes 1 and 2). The [1,2,3]triazolyl moiety was flanked on each side by 1 to 4 methylenes totaling in 4 or 5 methylenes. The biological activity of all the 1,4- and 4,1-disubstituted-[1,2,3]triazolyl-containing cyclopeptides (I–X, Table 3) is compared to the prototypic lactam-containing peptide MT-II, as well as to the linear control precursors (I′–X′, Tables 2 and 3).
Scheme 1. Sequences of the Linear Precursors Nα-Ac[Nle4,Yaa5,d-Phe7,Xaa10]αMSH(4-10)NH2 and of the [1,2,3]Triazolyl-Containing Cyclo-peptides,Nα-Ac[Nle4,Yaa5(&1),d-Phe7,Xaa10(&2)]αMSH(4-10)NH2 [{&1(CH2)n-1,4-[1,2,3]triazolyl-(CH2)m&2}]20.
All amino acids except when mentioned otherwise are of the l configuration.
Scheme 2. Sequences of the Linear Precursors Nα-Ac[Nle4,Xaa5,d-Phe7,Yaa10]αMSH(4-10)NH2 and of the [1,2,3]Triazolyl-Containing Cyclopeptides, Nα-Ac[Nle4,Xaa5(&1),d-Phe7,Yaa10(&2)]αMSH(4–10)NH2 [{&1(CH2)m-1,4-[1,2,3]triazolyl-(CH2)n&2}].

All amino acids except when mentioned otherwise are of the l configuration.
Table 3. Agonist Potency (EC50) of the [1,2,3]Triazolyl-Containing Cyclopeptides Nα-Ac[Nle4,Yaa5(&1),d-Phe7,Xaa10(&2)]αMSH(4-10)NH2 [{&1(CH2)n-1,4-[1,2,3]triazolyl-(CH2)m&2}] (I, II, III, IV, and IX) and Nα-Ac[Nle4,Xaa5(&1),d-Phe7,Yaa10(&2)]αMSH(4-10)NH2 [{&1(CH2)m-4,1-[1,2,3]triazolyl-(CH2)n&2}] (V, VI, VII, VIII, and X)a.
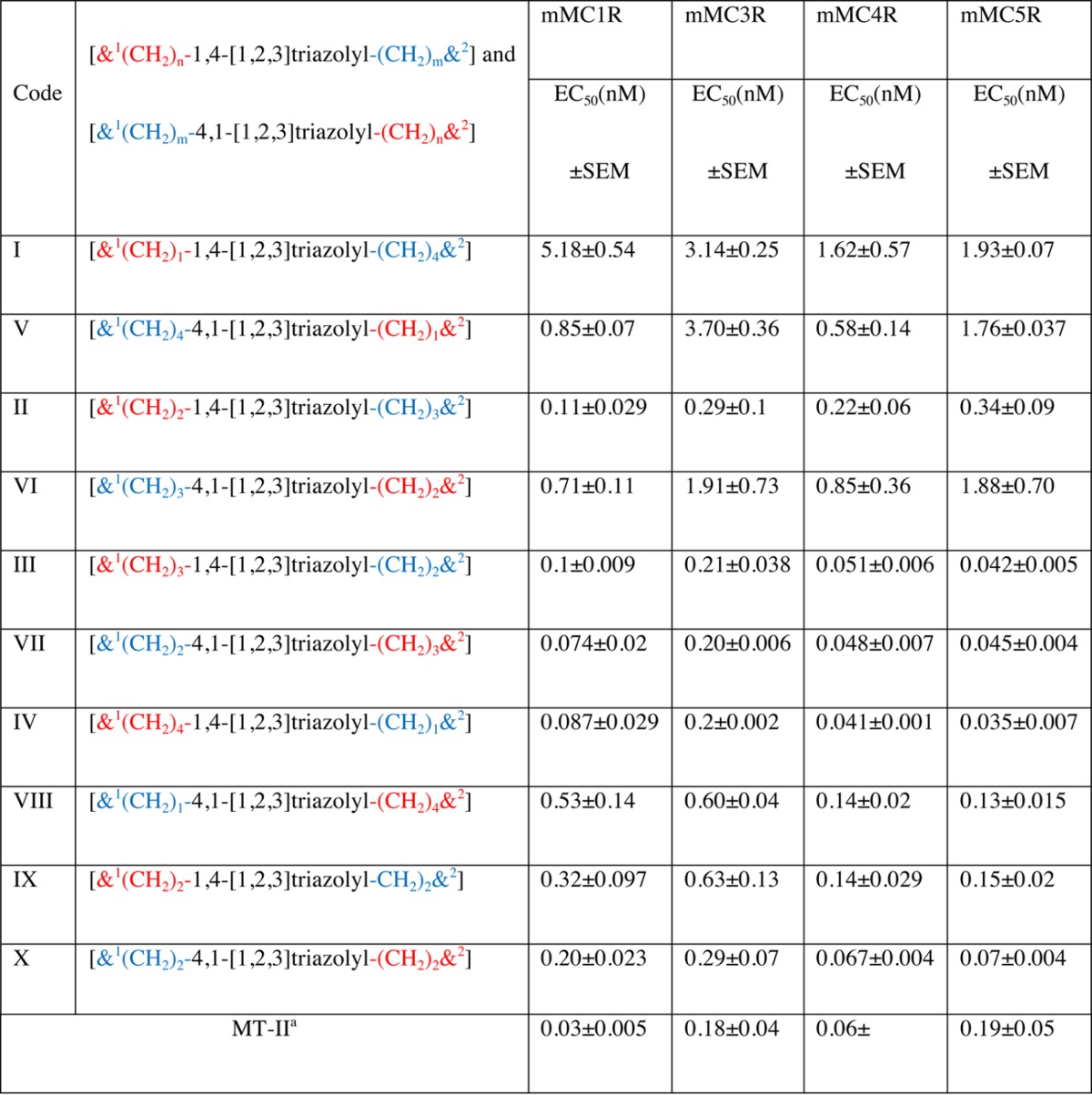
EC50 of MT-II is reported as reference.
Table 2. Functional Potency (EC50) of Linear Peptide Precursors Nα-Ac[Nle4,Yaa5,d-Phe7,Xaa10]αMSH(4–10)NH2 (I′, II′, III′, IV′, and IX′) and Nα-Ac[Nle4,Xaa5,d-Phe7,Yaa10]αMSH(4-10)NH2 (V′, VI′, VII′, VIII′, and X′)a.
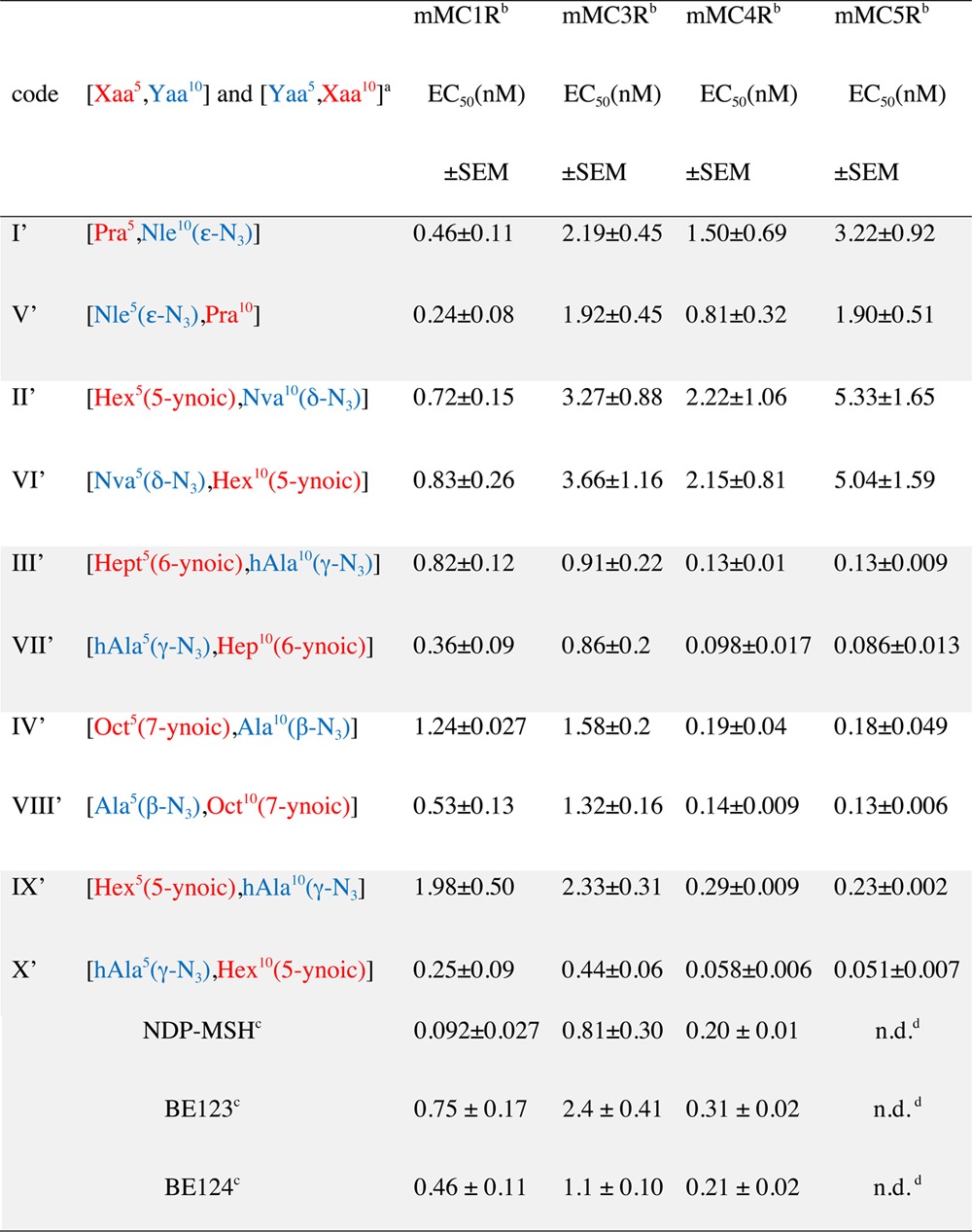
EC50 of NDP-MSH, BE123, and BE124 are reported as references. Notations: Pra, l-propargylglycine; Nva, l-norvaline; hAla, l-homoalanine; Hex, l-α-amino hexanoic acid; Hept, l-α-amino heptanoic acid; Oct, l-α-amino octanoic acid.
Agonist potencies (EC50) of the lactam containing peptide (MT-II) in mMC1R, mMC3R, mMC4R, and mMC5R are 0.03, 018, 0.06, and 0.19 nM, respectively.
The detailed sequences are given in Table 1.
n.d. means not determined.
Synthetic Strategy
In the current study, we explored the potential of the i-to-i+5 side chain-to-side chain 1,4- and 4,1-disubstituted [1,2,3]triazolyl-containing bridges to stabilize β-turn structure, which has been identified as the MT-II bioactive conformation, responsible for its protracted, nonselective, and superagonist activity. To this end, we designed a series of [1,2,3]triazolyl-containing cyclo-heptapeptides, derived from the model lactam peptide, MT-II Nα-Ac[Nle4,c(Asp5,d-Phe7,Lys10)]αMSH(4–10)NH2, that vary in the size of the bridge and the location and orientation of the [1,2,3]triazolyl moiety within this bridge (Schemes 1 and 2).
In this context, Nα-Fmoc-ω-azido-α-amino acids (1–4) and Nα-Fmoc-ω-ynoic-α-amino acids (5–8) with different length of the side chain were introduced in positions i and i+5 to replace Asp5 and Lys10 residues in the sequence of MT-II (see I′–X′, Schemes 1 and 2).21,22 The Nα-Fmoc-ω-azido-α-amino acids (1–4) were synthesized by diazo-transfer reaction starting from the corresponding Nα-protected α,ω-diamino acids (see Supporting Information).22 Except for the commercially available Nα-Fmoc-S-Pra-OH (8), the Nα-Fmoc-ω-alkynyl-α-amino acids (5–7) were synthesized by alkylation of a Ni(II) complex of the Schiff base formed between glycine and (S)-2-(N-benzylprolyl)aminobenzophenone, as a chiral inducer, with alk-ω-ynyl bromides (see Supporting Information).22 Solid-phase peptide synthesis (SPPS) generated a series of linear peptide precursors (I′–X′) in which Lys10 and Asp5 were replaced with ω-azido- and ω-yl-α-amino acid residues with side chains containing 1–4 methylenes. The stepwise solid-phase assembly of the linear precursors I′–X′ was performed following Fmoc/tBu strategy on Rink-amide type resin.23 The chemical inertness of the ω-azido and ω-alkynyl functions toward the entire range of coupling and deprotection reactions and the various nucleophiles and electrophiles present during the multistep synthesis24−26 saves the need for elaborate orthogonal protection schemes. The incorporation of the building blocks Nα-Fmoc-Xaa(ω-N3)-OH and Nα-Fmoc-Yaa(ω-yl)-OH during the SPPS was found to be markedly advantageous to the postpeptide assembly side chain modification that introduces the ω-azido and ω-ynoic functions needed for the CuI-catalyzed intramolecular azide–alkyne cycloaddition.21,22 Solution-phase intramolecular CuAAC converted the linear precursors I′–X′ into the 1,4- and 4,1-disubstituted-[1,2,3]triazolyl-containing cyclopeptides (I–X), presenting different permutation in terms of the size of the [1,2,3]triazolyl-containing bridges and the orientation and position of the [1,2,3]triazolyl moiety within the bridge. Depending on the positions i or i+5 of the azido and alkynyl amino acids in the peptide sequences, we generated two classes of cyclopeptides (Schemes 1 and 2). The intramolecular CuAAC of the linear precursors (I′–X′) was carried out as outlined in Scheme 3.
Scheme 3. General Procedure of CuAAC of the Linear Precursors (I′–X′).
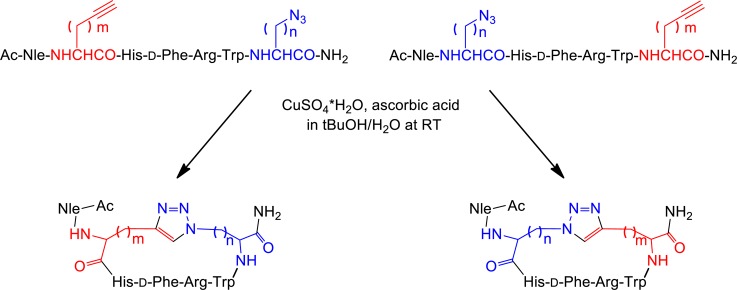
The first class employed linear precursors that had ω-yl- and ω-azido-α-amino acid residues in the respective positions i and i+5 (I′–IV′ and IX′) and yielded upon CuAAC the cyclopeptides presenting the [1,2,3]triazolyl moiety in the 4,1-orientation (I–IV and IX). The second class employed linear precursors that had ω-azido- and ω-yl-α-amino acid residues in the respective positions i and i+5 (V′–VIII′ and X′) and yielded upon CuAAC the cyclopeptides presenting the [1,2,3]triazolyl moiety in the 1,4-orientation (V–VIII and X). The click reaction conditions were identical to those reported by us previously and used a 10-fold molar excess of CuSO4·5H2O and ascorbic acid in tBuOH/H2O (1:2, v/v).21 Under these conditions, there was no formation of oligomeric products resulting from intermolecular click reactions, thus suggesting formation of a CuI/acetylide/azide complex that has high preference for intramolecular cyclizations. Moreover, solution-phase CuAAC avoids dimerizations and macrocyclizations, which are often observed in on-resin CuAAC, resulting in crude products that are easy to purify. All copper salts were eliminated by solid-phase extraction of the crude material with water and elution of the cyclopeptides with 10–30% of ACN in water. The clicked cyclopeptides were further purified by RP-HPLC. The linear analogue of MT-II, BE124 (Ac-Nle-Asp-His-d-Phe-Arg-Trp-Lys-NH2) and the its uncharged variant BE123, Ac-[Asn5,Lys10(Nε-Ac)]BE124, were synthesized by Fmoc/tBu SPPS strategy and used as reference compounds in the biological assays (Table 1 and Table 2).
Table 1. Sequences of MT-II, the Classical NDP-MSH, and the MT-II Linear Analogues BE123 and BE124.
| Reference Peptides |
|---|
| MT-II: Ac-Nle-c[Asp-His-d-Phe-Arg-Trp-Lys]-NH2 |
| NDP-MSH: Ac-Ser-Tyr-Ser-Nle-Glu-His-d-Phe-Arg-Trp-Gly-Lys-Pro-Val-NH2 |
| BE124: Ac-Nle-Asp-His-d-Phe-Arg-Trp-Lys-NH2 |
| BE123: Ac-Nle-Asn-His-d-Phe-Arg-Trp-Lys(NεAc)-NH2 |
Results and Discussion
Biological Activity
The potency of MT-II and of the linear peptide NDP-MSH (Table 1), the superpotent and long acting agonist,27 the reference peptides BE124 and BE123 (Table 1), and the precursors (I′–X′), as well as the [1,2,3]triazolyl-containing cyclopeptides (I–X) (Schemes 1 and 2), was pharmacologically evaluated for functional potency (EC50) and efficacy using a reporter gene based bioassay for cAMP in HEK-293 cells stably expressing the murine (mouse) melanocortin receptor subtypes mMC1R, mMC3R, mMC4R, and mMC5R (Tables 2–3).
Table 2 reports the EC50 values in the adenylyl cyclase based bioassays of the linear peptides: the linear peptide precursors I′–X′, the classical NDP-MSH, and the MT-II analogues, BE123 and BE124. Peptide BE123, in which the charges on Asp and Lys present in BE124 are eliminated, reproduces closely the effect of side chain-to-side chain cyclization present in MT-II. As such, short of the conformational rigidification, BE123 is a linear version of MT-II. Table 3 summarizes the functional agonist potencies of the 1,4- and 4,1-disubstituted-[1,2,3]triazolyl-containing cyclopeptides I–X compared to MT-II. In this study, we used HEK-293 cells stably expressing the murine melanocortin receptors subtypes mMC1R, mMC3R, mMC4R, and mMC5R to assess the agonist potency and subtype selectivity profiles of these MT-II mimetic peptides.28,29
Among the linear peptides, NDP-MSH is the most potent mMC1R agonist, showing an EC50 value that is 5- and 9-fold lower than the respective values for BE124 and BE123 (Table 2). All three linear peptides are good agonists toward the mMCR4R, with very similar subnanomolar EC50s (0.20–0.31 nM), and are 4–9-fold lower than their EC50s toward mMC3R and mMC5R. Relative to the linear peptides, the cyclic MT-II is the most potent agonist toward all four receptor subtypes. It is about 2- and 4-fold more potent than NDP-MSH on the mMC1R and mMC4R, respectively. Moreover, MT-II discriminates nicely between mMC1R or mMC4R and mMC3R or mMC5R. Interestingly, both BE124 and its potential charge-bearing side chains blocked variant BE123 are almost equipotent in all four receptor subtypes. Apparently, the application of the conformational constraint in the form of i-to-i+5 side chain-to-side chain lactam bridge formation as in MT-II and not the neutralization of potential charge-bearing groups as in BE123 play a critical role in potentiation and enhancement of functional selectivity toward certain receptor subtypes.
The potencies of the linear peptide precursors I′–X′ (Table 2, Figure 1) span a range of 2 orders of magnitude between the most potent one X′ (EC50 ≈ 0.050 nM in mMC4R and mMC5R) and the least potent one II′ (EC50 = 5.33 nM in mMC5R). Analogue X′ is the most potent of all four receptor subtypes, with 5–8-fold higher potency on mMC4R and mMC5R than on mMC1R and mMC3R. The selectivity for receptor subtype of a single analogue is in the range of 5–10-fold. For example, IX′ is 10-fold more potent in mMC5R than in mMC3R (EC50s 0.23 and 2.33 nM, respectively).
Figure 1.

Logarithmic graphical representation of functional agonist activity (EC50 in nM) of linear peptide precursors (I′–X′).
The melanocortin receptor subtype related potencies of the two short linear MT-II-related peptides BE123 and BE124 is very similar to that of the linear precursors I′–X′ regardless of the nature of side-chain modifications in positions 5 and 10 (α-MSH-based numbering). Moreover, in general, most of the linear short α-MSH(4–10)-derived peptides are less potent than either MT-II or its cyclic heterodetic “clicked” MT-II-derived peptides I−X.
Neither MT-II nor the 1,4- and 4,1-disubstituted-[1,2,3]triazolyl-containing cyclopeptides I–X show very high selectivity toward a specific receptor subtype, and there is no consistent trend for receptor subtype selectivity in the [1,2,3]triazolyl-contaning analogue series (Table 3, Figure 2). For example, while MT-II is 3–6-fold more potent toward mMC1R and mMC4R (cf. EC50s 0.03 and 0.06 nM on mMC1R and mMC4R, respectively, with 0.18 and 0.19 nM on mMC3R and mMC5R, respectively), the maximal selectivity observed for the least potent analogues I and V toward the different receptor subtypes is 2–3-fold and 3–6-fold, respectively (EC50s 1.62, 3.14, and 5.18 nM of analogue I in mMC4R, mMC3R, and mMC1R, respectively, and 0.58, 1.76, and 3.70 nM of analogue V in mMC4R, mMC5R, and mMC3R, repectively). The most potent [1,2,3]triazolyl-contaning analogues in this series are III, IV, VII, and X. They all have preference to the mMC4R and mMC5R (EC50s 0.051, 0,041, 0.048, and 0.067 nM, respectively, on the former receptor subtype, and 0.042, 0.035, 0.045, and 0.07 nM, respectively, on the latter one) and analogues IV and VII are active to a lesser extent on mMC1R (EC50s 0.087 and 0.074 nM, respectively).
Figure 2.
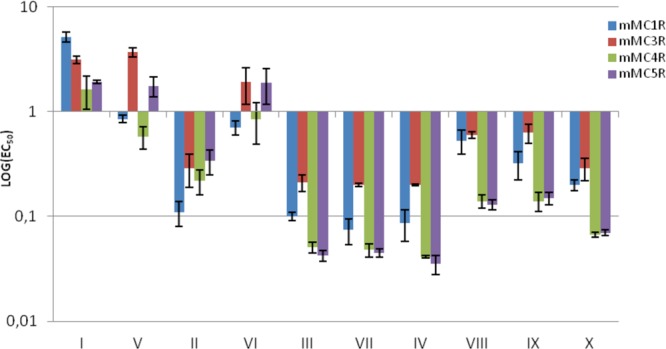
Logarithmic graphical representation of agonist potency (EC50 in nM) of the [1,2,3]triazolyl-containing cyclopeptides (I–X).
[1,2,3]Triazolyl-contaning analogues I–VIII share a common location of the bridgeheads (positions 5 and 10) and an identical size of the bridges (five methylenes in total and a 1,4-disubstituted [1,2,3]triazolyl moiety). As such, they are loosely reproducing the features of the lactam bridge in MT-II (five methylenes in total and one amide bond). However, these heterodetic cyclopeptides differ in the location of the [1,2,3]triazolyl moiety within the bridge, so one or two methylenes are removed from the N-proximal bridgehead (analogues I and VIII and II and VII, respectively) or one or two methylenes from the C-proximal bridgehead (analogues IV and V and III and VI, respectively). The location of the [1,2,3]triazolyl moiety within the bridge in analogues I and VIII is identical with the location of the isosteric amide within the lactam bridge in MT-II. Evidently, I and VIII are less potent than MT-II in mMC1R, mMC3R, and mMC4R (Table 3). Moreover, I, which incorporates a 4,1-[1,2,3]triazolyl moiety, is much less potent than VIII, which incorporates a 1,4-[1,2,3]triazolyl moiety. Interestingly, analogue VII, which incorporates the 1,4-[1,2,3]triazolyl, is more potent than analogue II, which incorporates the 4,1-[1,2,3]triazolyl moiety despite the [1,2,3]triazolyl moiety at the same position in the bridge. In both pairs I and VIII and II and VII, where the position of the [1,2,3]triazolyl ring is closer to the N-proximal bridgehead, the more potent analogues VII and VIII are those that incorporate the 1,4-[1,2,3]triazolyl moiety. On the contrary, in both pairs IV and V and III and VI, where the position of the [1,2,3]triazolyl ring is closer to the C-proximal bridgehead, the more potent analogues III and IV are those that incorporate the 4,1-[triazolyl] moiety. Analogues IX and X have a shorter bridge than analogues I–VIII (four methylenes in total and a 1,4-disubstituted [1,2,3]triazolyl moiety), and the [1,2,3]triazolyl moiety is flanked by two methylenes on each side in either the 4,1- or 1,4-orientation. Moreover, analogues IX and X differ in the orientation of the [1,2,3]triazolyl moiety, which is positioned equal distance from the bridgeheads, being 4,1 in the former and 1,4 in the latter. While both IX and X are more potent at mMC4R and mMC5R than on mMC1R and mMC3R, the activities of analogue IX and X differ by the latter being more potent at the two former receptor subtypes, thus being more selective than analogue IX. The potency profiles of analogues IX and X (EC50s 0.32, 0.63, 0.14, and 0.15 nM and 0.20, 0.29, 0.067, and 0.07 nM at mMC1R, mMC3R, mMC4R, and mMC5R, respectively) follow very closely that of the corresponding analogues VIII and III (EC50s 0.53, 0.60, 0.14, and 0.13 nM and 0.1, 0.21, 0.051, and 0.042 nM at mMC1R, mMC3R, mMC4R, and mMC5R, respectively).
Taken together, these results suggest that close proximity of the [1,2,3]triazolyl moiety to the N-proximal bridgehead, especially in the 4,1-orientation, is disruptive for activity at all four receptor subtypes (analogues I and VIII, respectively). Interestingly, close proximity of the [1,2,3]triazolyl moiety to the C-proximal bridgehead as in analogues V and IV, presenting the [1,2,3]triazolyl in the respective 1,4- and 4,1-orientations, results in an opposite effect in which it is very disruptive for the former and very accommodating for the latter for all four receptor subtypes. Of note is the higher activity of the linear precursors I′ and V′ on the mMC1R as compared to the activity of the corresponding [1,2,3]triazolyl-containing cyclopeptides I and V on the same receptor subtype (EC50s 0.46 and 0.24 nM vs 5.18 and 0.85 nM). This may be the result of the serious perturbation imposed by the 4,1-[1,2,3]triazolyl moiety when it is adjacent to the N-proximal bridgehead and to a lesser extent, regardless of the orientation of the [1,2,3]triazolyl ring, when it is adjacent to the C-proximal bridgehead.
The comparison of the activity profile of the linear peptide precursors and their corresponding [1,2,3]triazolyl-containing cyclopeptides indicates the latter, in general, to be slightly more potent. Clearly, the linear precursors are already very potent indicating that the conformational rigidification in the cyclopeptide is not critical for their high potency. Moreover, only in very few cases the linear precursor peptides are more potent than their corresponding [1,2,3]triazolyl-containing cyclopeptides (cf. I′ and V′ with I and V in mMC1R, respectively).
Conformational Analysis
To gain insight on the relationship among the specific features of the [1,2,3]triazolyl-containing bridge, the potency and receptor subtype selectivity, and the conformation, we selected to study in detail the conformations of three representative heterodetic [1,2,3]triazolyl-containing cyclopeptides, I, IV, and V. The selection includes the least potent analogue I, one of the most potent analogues IV, and analogue V of intermediate potency. In addition, while analogues I and IV incorporate the 4,1-[1,2,3]triazolyl moiety in proximity to the N-proximal- and C-proximal bridgeheads, respectively, both analogues IV and V incorporate the [1,2,3]triazolyl moiety at the C-proximal bridgehead but at opposite orientations, 4,1- and 1,4-, respectively. We anticipated that the opposing location of the [1,2,3]triazolyl ring in the bridge will affect the conformation to the extent that will explain the large difference in activity between analogues I and IV. On the other hand, we speculate that the small difference in biological activity between analogues IV and V, which share the same C-proximal bridgehead location of the [1,2,3]triazolyl ring, may result in similar conformations in spite of the opposite orientations of the [1,2,3]triazolyl ring within the bridge.
To allow comparison with the reported “bioactive” NMR solution structure(s) of the reference homodetic cyclopeptide MT-II, the NMR spectra of the heterodetic cyclopeptides I, IV, and V were acquired in DMSO-d6.30 Moreover, this polar, basic, and aprotic solvent was shown to support intramolecular hydrogen bonds and prevent intermolecular aggregations.31,32 To exclude potential aggregation, we recorded the 1D-1H NMR spectra of the clicked-cyclopeptides at a concentration range spanning 1–0.1 mM. Importantly, at a concentration of 0.1 mM, there was no indication for aggregation. Nevertheless, at 0.1 mM, cyclopeptide I presented double signal pattern spectra indicating the presence of two distinct populations of slow exchanging conformations. Chemical shift assignments of the proton spectra were achieved via the standard systematic application of DQF-COSY,33 TOCSY,34 and NOESY35 experiments, using the SPARKY36 software package according to Wüthrich’s procedure.37
Figure 3 reports sequential and medium range NOEs collected in NOESY spectra of cyclopeptides I, IV, and V in DMSO-d6, confirming the preliminary analysis of 1D-1H NMR spectrum. Two NOEs bar diagrams are reported for [1,2,3]triazolyl-containing cyclopeptide I, corresponding to the observed double signal pattern in the respective NOESY spectrum (Figure 3A,B). The conformer IA is characterized by a more complete sequential and medium range NOE pattern, while conformer IB shows less abundant and, whenever observable, weak, sequential, and medium range connectivities. This observation is consistent with the presence of the two predominant low energy conformational ensembles IA and IB, undergoing slow dynamic interchange. We suggest that the markedly low potency of cyclopeptide I compared to cyclopeptides IV and V in all melanocortin receptor subtypes (EC50s 5.18, 3.14, 1.62, and 1.93 nM for mMC1R, mMC3, mMC4R, and mMC5R, respectively) can be attributed to this conformational heterogeneity. NOE bar diagrams relative to cyclopeptides IV and V (parts C and D of Figure 3, respectively), confirm the presence of numerous and diagnostic sequential and medium range i,i+2 and i,i+3 NOE data in the corresponding NOESY spectra, although a more regular and complete NOE patterns is observed for cyclopeptide V than for cyclopeptide IV. NMR-derived models of cyclopetides IA, IV, and V were calculated using simulated annealing procedures imposing NOE-based interprotonic distances as constraints. Figure 4 shows the NMR-derived bundles of 20 low energy conformations of cyclopeptides IA, IV, and V overlapping with the best calculated structure of MT-II. The NMR structure bundles of cyclopeptides IA and V show high structural agreement with RMSD ∼ 0.50 Å. In agreement with the NOE bar diagrams reported in Figure 3, the structure bundle of cyclopeptide IV is characterized by slightly higher RMSD value (0.80 Å), indicating that cyclopeptide IV is more flexible than cyclopeptides IA and V.
Figure 3.
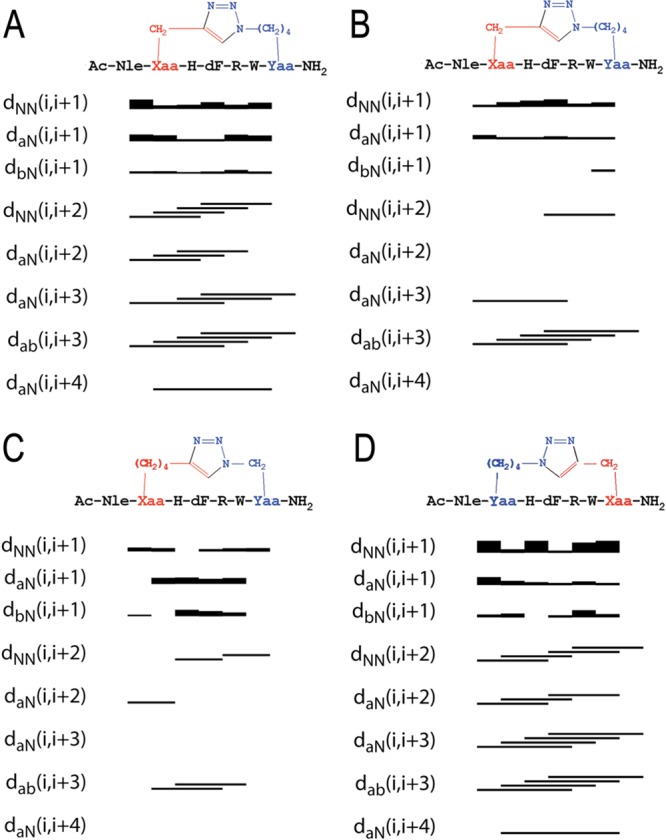
Sequential and medium-range NOEs for [1,2,3]triazolyl-containing cyclopeptides IA (A), IB (B), IV (C), and V (D). (A) and (B) are derived from to the NOE connectivities of the most (IA) and the lesser (IB) abundant conformers of I. Data were obtained from a 600 MHz NOESY experiments with a mixing time of 200 ms and collected in DMSO-d6 at 300 K.
Figure 4.

(a) Overlay of NMR-derived calculated structure bundles of IA, the most abundant conformational ensemble (left), IV (middle), and V (right), all wireframe structures, compared with the best calculated structure of MT-II (ribbon structure). (b) Side chain orientation of cyclopeptides IA, IV, and V. Residue side chains are colored as follow: l-His, light gray; d-Phe, pink; l-Arg, red; l-Trp, blue.
Quantitative analysis of φ and ψ (Supporting Information, Table 1S) dihedral angles of low-energy structures of cyclopeptides IA, IV, and V, using the PROMOTIF program (PROMOTIF, a program to identify and analyze structural motifs in proteins)38 highlights the preponderance of a type I β-turn conformation centered on residues His6-Trp9. In agreement with the preliminary analysis shown in the NOESY connectivity bar diagram (Figure 3), cyclopeptide IB appears to be disordered, presenting many irregular secondary structures including some transient α-helical segments.
The comparison of the 20 lowest-energy NMR structures of cyclopeptides IA, IV, and V (Figure 4) suggests good overlap of the backbone atoms and a common orientation of His and d-Phe side chains. Cyclopeptides IV and V show also a common orientation of the indole ring of Trp.
As shown in Figure 4a the Trp side chain of cyclopeptide IA points to an opposite direction as compared to cyclopeptide IV and V. Moreover, the three cyclopeptides differ in the orientation of the Arg side chain. In cyclopeptides IV and V, as documented by the relative NOE effects, Arg side chains point to [1,2,3]triazolyl ring and d-Phe moiety, respectively (Figure 4a). Consistent with the absence of NOEs suggesting proximity to other side chains, Arg side chain in cyclopeptide IA, displays significantly higher flexibility than in analogues IV and V (Figure 4a,b).
Stabilization of a type II β-turn because of d-Phe and the close proximity of His, d-Phe, and Trp side chains on one of the homodetic cyclopeptide surface and Arg side chain on the opposite surface in MT-II can explain the high potency and affinity to the melanocortin receptors.39,40 It is known that a correct spatial orientation of the hydrophobic aromatic residues and the basic, “arginine-like” moiety, are important for an effective interaction of MT-II and MT-II analogues at the different MCR receptors.30
In summary, conformational analysis of the three selected heterodetic [1,2,3]triazolyl-containing MT-II analogues suggests that (i) the conformational polymorphism of cyclopeptide I contributes to its overall low potency in all receptor subtypes as compared to the more conformationally defined cyclopeptides IV and V, and (ii) the presence, location, and orientation of the [1,2,3]triazolyl ring in the bridge does affect the spatial location of the side chains in general and of the Arg side chain in particular. It appears that biological activity is derived from indirect contribution to the spatial orientations of side chains more then by the direct interaction of the bridge with the target receptor.
Conclusions
In the present paper, we report for the first time a systematic structure–activity conformation relationship study in which a i-to-i+5 side chain-to-side chain CuI-catalyzed azido-to-alkyne 1,3-dipolar cycloaddition (CuAAC) was performed in an attempt to reproduce a type I β-turn, which is considered to be a critical feature in the putative bioactive conformation of MT-II. We demonstrate that (1) i-to-i+5 side chain-to-side chain CuAAC yields potent and interesting melanocortin receptor agonists; (2) side chain-to-side chain conformational stabilization of type I β-turn by i-to-i+5 side chain-to-side chain [1,2,3]triazolyl-containing bridges impacts the in vitro potency and selectivity of these new stapled peptides at the different MCR receptor subtypes. Relative to MT-II, we frequently observe enhancement of potency at the mMC5R and loss of potency at mMC1R. (3) Similar to the general enhancement of potency as a result of homodetic side chain-to-side chain cyclization (cf. BE124 and BE123 with MT-II, Table 2), we also observe enhancement of potency upon transforming the linear precursors I′–X′ to the heterodetic [1,2,3]triazolyl-containing cyclopeptides. Interestingly, similar observation was reported by Cho et al. in i-to-i+5 disulfide-bridged cyclic MSH peptides.41 (4) Regardless of the nature of i-to-i+5 side chain-to-side chain cyclizations, their contribution to the stability of the critical type I β-turn contributes to the enhancement of their agonisit potency at all MCRs. Our observations concur with those that propose side chains orientation of residues within the cyclic segment and those that are flanking it to play a predominant role in determining receptor subtype selectivity. As such, the i-to-i+5 side chain-to-side chain CuAAC offers obvious advantages over the more traditional side chain-to-side chain cyclizations. These heterodetic [1,2,3]triazolyl-containing MT-II mimetics can be constructed by taking advantage of the chemical orthogonality of the azido and alkynyl functions, which reduces synthetic complexity. Moreover, our approach offers access to a vast structural diversity presented by the size of the bridge, the location, and the orientation of the [1,2,3]triazolyl moiety within the ring. In the future, we will seek to reduce the size of the [1,2,3]triazolyl-containing bridge to achieve greater structural rigidification and diversify the nature of the residues flanking the cyclic segment in order to achieve greater receptor subtype selectivity.
Experimental Section
Materials and Methods
The ω-alkynyl-alcohols were purchased from Alfa Aesar; Fmoc-l-Trp-(Boc)-OH, Fmoc-d-Phe-OH, and Fmoc-l-Pra-OH amino acids, Fmoc-Rink amide resin, and HOBt were purchased from Iris Biotech GmbH (Marktredwitz, Germany); Fmoc-l-Arg(Pbf)-OH and Fmoc-l-His(Trt)-OH amino acids were purchased from CBL (Patras, Greece); Fmoc-l-Nle was purchased from NeoMPS (Strasbourg, France); TBTU from Advanced Biotech Italy (Milan, Italy). Peptide-synthesis grade N,N-dimethylformamide (DMF) was purchased from Scharlau (Barcelona, Spain); acetonitrile from Carlo Erba (Milan, Italy); dichloromethane (DCM), trifluoroacetic acid (TFA), piperidine, acetic anhydride (Ac2O), and N-methyl morpholine (NMM) were purchased from Sigma-Aldrich (Milan, Italy). The scavengers for cleavage of peptides from resin, 1,2-ethanedithiol (EDT), thioanisole, and phenol (PhOH), were purchased from Acros Organics (Geel, Belgium), Jansenn Chimica (Beerse, Belgium), and Carlo Erba (Milan, Italy).
The purity of the final linear precursors (I′–X′) and the [1,2,3]triazolyl-containing MT-II analogues (I–X) was established by analytical RP-HPLC and exceeded 95%, their structural integrity was established by ESI-MS.
Synthesis of Nα-Fmoc-Xaa(ω-N3)-OH and Nα-Fmoc-Yaa(ω-yl)-OH
Nα-Fmoc-ω-azido-α-amino- and Nα-Fmoc-ω-ynoic-α-amino acids (1–4 and 5–7, respectively), with (CH2)n/m n/m = 1–4 in the side chain, were synthesized as described before.21,22 In particular, Nα-Fmoc-ω-azido-α-amino acids were obtained by diazo-transfer from correspondent Nα-Fmoc-ω-amino-α-amino acids; Nα-Fmoc-ω-ynoic-α-amino acids were obtained by alkylation of a Ni(II) complex of the Schiff base derived from glycine and the chiral inducer (S)-2-(N-benzylprolyl)aminobenzophenone with alk-ω-ynyl bromides.
Synthesis of Nα-Ac[Nle4,Yaa5,d-Phe7,Xaa10]αMSH(4–10)NH2 (I′, II′, III′, IV′, IX′) and Nα-Ac-[Nle4,Xaa5,d-Phe7,Yaa10]αMSH(4–10)NH2 (VI′, V′, VII′, VIII′, X′)
Peptides I′–X′ were synthesized on a manual batch synthesizer (PLS 4 × 4, Advanced ChemTech) employing Fmoc/tBu chemistry. The syntheses were performed on Rink-amide NovaSyn TGR resin (0.14 mmol/g, 300 mg). The Fmoc-Rink-amide resin was swelled with DMF (1 mL/100 mg of resin) for 20 min before use. Stepwise peptide assembly was performed by repeating for each added amino acid the following deprotection-coupling cycle. (1) Swelling: DMF (1 mL/100 mg of resin) for 5 min. (2) Fmoc-deprotection: resin was washed twice with 20% (v/v) piperidine in DMF (1 mL/100 mg of resin, one wash for 5 min, followed by another wash for 20 min). (3) Resin washing: DMF (3–5 min). (4) Coupling: scale employed TBTU/HOBt/NMM (2.5:2.5:3.5 equiv) as the coupling system and 2.5 equiv of the Fmoc protected amino acids, except for the unnatural amino acids Nα-Fmoc-Xaa(ω-N3)-OH and Nα-Fmoc-Yaa(ω-yl)-OH, for which 1.5 equiv were used. The coupling was carried out in DMF (1 mL/100 mg of resin) for 50 min. (5) Resin washings: DMF (3–5 min) and DCM (1–5 min). Each coupling was monitored by Kaiser test42 and was negative, therefore recouplings were not needed. Acetylation of the amino terminus was carried out in the presence of Ac2O/NMM in DCM (20 equiv, 1.6 mL of DCM). The reaction was monitored by Kaiser test.
Deprotection, Cleavage, and Purification of Peptides: General Procedure
Peptide cleavage from the resin and simultaneous deprotection of the amino acid side chains were carried out with a mixture of TFA/anisole/1,2-ethanedithiol/phenol/H2O (94:1:1:1:1 v/v/v/v/v, 1 mL/100 mg of resin-bound peptide). The cleavage was carried out for 3 h with vigorous shaking at room temperature. Resin was filtered and washed with TFA. The filtrate was concentrated under N2 stream, addition of cold diethyl ether resulted in a precipitate that was separated by centrifugation, dissolved in H2O, and lyophilized on an Edwards apparatus, model Modulyo. Lyophilized crude peptides were prepurified by solid-phase extraction with a RP-18 LiChroprep silica column from Merck (Darmstadt, Germany) using H2O/ACN as eluents. The final purification of the peptides was performed by semipreparative RP-HPLC on a Phenomenex Jupiter C-18 (250 mm × 4.6 mm) column at 28 °C using a Waters instrument (separation module 2695, detector diode array 2996) working at 4 mL/min. The solvent systems used were: A (0.1% TFA in H2O) and B (0.1% TFA in CH3CN). Final purity of all peptides was ≥95%. Peptides were characterized by RP-HPLC ESI-MS. HPLC system was an Alliance Chromatography (Waters) with a Phenomenex Kinetex C-18 column 2.6 μm (100 mm × 3.0 mm) working at 0.6 mL/min, with UV detection at 215 nm, coupled to a single quadrupole ESI-MS (Micromass ZQ). The solvent systems used were: A (0.1% TFA in H2O) and B (0.1% TFA in CH3CN).
Synthesis of Nα-Ac[Nle4,Yaa5(&1),d-Phe7,Xaa10(&2)]αMSH(4–10)NH2 [{&1(CH2)n-1,4-[1,2,3]triazolyl-(CH2)m&2}] (I, II, III, IV, IX) and Nα-Ac[Nle4,Xaa5(&1),d-Phe7,Yaa10(&2)]αMSH(4–10)NH2 [{&1(CH2)m-1,4-[1,2,3]triazolyl-(CH2)n&2}] (V, VI, VII, VIII, X)
Purified linear peptide precursors I′–X′ (>95% purity) were subjected to intramolecular CuI-catalyzed side-chain-to-side-chain azide–alkyne 1,3-dipolar Huisgen cycloaddition (CuAAC). To a solution of the purified linear peptide (4.77 μmol) in H2O/tBuOH (5 mL, 2:1 v/v) were added CuSO4·5H2O (40 μmol) and ascorbic acid (40.3 μmol). The mixture was stirred overnight at room temperature, concentrated, and lyophilized. Complete and clean conversion of all linear precursors into the desired [1,2,3]triazolyl-containing cyclopeptides I–X (Schemes 1 and 2) was observed. The cyclization was monitored by RP-HPLC ESI-MS. The crude [1,2,3]triazolyl-containing cyclopeptides were purified (>97% purity) by solid-phase extraction (SPE) on RP (C18) column, with CH3CN in H2O as eluents. The copper salts were removed by elution with H2O, whereas the desired cyclopeptides were eluted with CH3CN/H2O mixture. Peptides were purified and characterized by semipreparative RP-HPLC and RP-HPLC ESI-MS, respectively.
Solid Phase Synthesis of MT-II: Nα-Ac[Nle4-c(Asp5-d-Phe7-Lys10)]αMSH(4–10)NH2
The fully protected resin-bound peptide was synthesized using a Rink-amide NovaSyn TGR resin (0.63 mmol/g, 300 mg) on a manual batch synthesizer (PLS 4 × 4, Advanced ChemTech) applying the Fmoc/tBu SPPS procedure, as previously reported. The coupling of Nα-Fmoc-amino acids was performed using TBTU/HOBt/NMM (2.5 equiv:2.5 equiv:3.5 equiv) and 2.5 equiv of each Fmoc protected amino acids. The coupling was carried out in DMF (1 mL/100 mg of resin) for 40 min. The formation of the lactam bridges between side chains of Lys10 and Asp5 was performed on the resin-bound peptide, following an orthogonal protocol of deprotection. In particular, Lys10 and Asp5 were protected on side chain respectively with 1-[(4,4-dimethyl-2,6-dioxocyclohex-1-ylidine)ethyl] group (Dde) and with β-4-{N-[1-(4,4-dimethyl-2,6-dioxocyclohexylidene)-3-methylbutyl]-amino} benzyl ester (ODmab), stable under the conditions of peptide elongation. After acetylation of the N-terminal, Dde and ODmab were removed by two consecutive steps, using hydrazine 20% in DMF (1 mL), first for 5 min, followed by an additional step with freshly prepared deprotection mixture for 15 min. After deprotection of Lys10 and Asp5, the formation of intramolecular lactam bridge was performed using TBTU/HOBt/NMM (2.5:2.5:3.5 equiv) as coupling system in DMF for 40 min. The cyclization reaction was monitored by Kaiser test and by RP-HPLC ESI-MS analysis of MW-assisted mini-cleavage resin-bound fragment. Mini-cleavage was carried out with TFA/TIS/water solution (95:2.5:2.5 v/v/v), using a DiscoverTMS-class single-mode MW reactor equipped with an Explorer-48 autosampler (CEM). The reaction was performed at 45 °C, using 15 W for 15 min. The reaction mixture was then filtered, and the crude peptide was precipitated from the cleavage mixture by addition of ice-cold diethyl ether followed by cooling for 5 min at −20 °C. The product was collected by centrifugation and directly subjected to RP-HPLC ESI-MS analysis.
Biological Activity
cAMP based functional bioassay: HEK-293 cells stably expressing the mouse melanocortin receptors were transiently transfected with 4 μg of CRE/β-galactosidase reporter gene as previously described.28,29,43,44 Briefly, 5000–15000 post transfection cells were plated into collagen treated 96-well plates (Nunc) and incubated overnight.
Then 48 h post-transfection, the cells were treated with 100 μL of peptide (10–6–10–12 M) or forskolin (10–4 M) as a control in assay medium (DMEM containing 0.1 mg/mL BSA and 0.1 mM isobutylmethylxanthine) for 6 h. The assay media was aspirated, and 50 μL of lysis buffer (250 mM Tris-HCl pH = 8.0 and 0.1% Triton X-100) was added. The plates were stored at −80 °C overnight. The plates containing the cell lysates were thawed the following day. Aliquots of 10 μL were taken from each well and transferred to another 96-well plate for relative protein determination. To the cell lysate plates, 40 μL of phosphate-buffered saline with 0.5% BSA was added to each well. Subsequently, 150 μL of substrate buffer (60 mM sodium phosphate, 1 mM MgCl2, 10 mM KCl, 5 mM β-mercaptoethanol, 2 mg/mL ONPG) was added to each well, and the plates were incubated at 37 °C. The sample absorbance, OD405, was measured using a 96-well plate reader (Molecular Devices). The relative protein concentration was determined by adding 200 μL of 1:5 dilution Bio Rad G250 protein dye:water to the 10 μL cell lysate sample taken previously, and the OD595 was measured on a 96-well plate reader (Molecular Devices). Data points were normalized to the relative protein content. EC50 values represent the mean of three or more independent experiments. EC50 estimates, and their associated standard errors of the mean, were determined by fitting the data to a nonlinear least-squares analysis using the PRISM program (v4.0, GraphPad Inc.).
NMR Conformational Analysis
Samples for NMR were prepared by dissolving 1.2 mg of the lactam- and heterodetic [1,2,3]triazolyl-containing cycloheptapeptides in 0.5 mL of aqueous phosphate buffer (pH 6.6, 100 mM). Samples were lyophilized and dissolved in a mixture of DMSO/water (0.5 mL, 80:20, v/v). NMR spectra were recorded on a Bruker DRX-600 spectrometer. To exclude potential aggregation, one-dimensional (1D) proton spectra of the cyclopeptides at a concentration range spanning 0.1–1 mM were recorded. At a peptide concentration of 0.1 mM, the peptides did not display any noticeable effects of aggregation. 1D NMR spectra were recorded in the Fourier mode with quadrature detection. The water signal was suppressed by a low-power selective irradiation in the homogated mode. DQF-COSY, TOCSY, and NOESY experiments were run in the phase-sensitive mode using quadrature detection in ω1 by time-proportional phase incrementation of the initial pulse. Data block sizes comprised of 2048 addresses in t2 and 512 equidistant t1 values. Before Fourier transformation, the time domain data matrices were multiplied by shifted sin2 functions in both dimensions. A mixing time of 70 ms was used for the TOCSY experiments. NOESY experiments were run at 300 K with mixing times in the range of 100–250 ms. The qualitative and quantitative analyses of DQF-COSY, TOCSY, and NOESY spectra were obtained using the SPARKY interactive program package. Complete proton resonance assignments were achieved following Wüthrich’s procedure. Sequential and medium range Nuclear Overhauser Effects (NOEs)-derived distances were used to generate 3D-models of the heterodetic [1,2,3]triazolyl-containing cyclo-heptapeptides. The final PDB files were analyzed and validated using PROMOTIF software.
NMR Structure Calculation
On the basis of sequential and medium range NOE derived distances, 3D models of lactam- and [1,2,3]triazolyl-containing peptides were generated with a simulated annealing procedure using the DYANA software package. All structures were energy minimized with the SANDER module of the AMBER 5 program, using for 1000 steps the steepest descent method and for 4000 steps the conjugate gradient method. A nonbonded cutoff of 12 Å and a distance-dependent dielectric term (e = 4 × r) were used. The minimization protocol included three steps in which NOE derived distances were used as constraints with a force constant, respectively, of 1000, 100, and 10 kcal/molÅ. The final PDB files were analyzed and validated using PROCHECK software.
Acknowledgments
We thank Dr. Debora D’Addona and Dr. Susanna Mazzoni for their technical support in preparing the α-alkynyl α-amino acids. This work was partially supported by the “Programme de cotutelle internationale de thèse de doctorat soutenu par la Région Ile-de-France”, for the Ph.D. of C.T., by Agence Nationale de la Recherche “Programme Chaire d’Excellence” ANR-09-CEXC-013-01 to A.M.P., and by the Fondazione Ente Cassa di Risparmio di Firenze (Italy) to PeptLab@UniFi. The biological part has been supported by NIH grant R01DK091906 (C.H.L.).
Glossary
Abbreviations Used
- ACN
acetonitrile
- CuAAC
CuI-catalyzed azido-to-alkyne 1,3-dipolar cycloaddition
- DCM
dichloromethane
- DMF
N,N-dimethylformamide
- EDT
1,2-ethanedithiol
- ESI-MS
electron-spray ionization mass spectrometry
- GPCR
G protein-coupled receptors
- HOBT
N-hydroxybenzotriazole
- MCR
melanocortin GPCR receptors
- MSH
melanocortin stimulating hormones
- MT-II
melanotan II
- NDP-α-MSH
norleucine-4,d-Phe-7 α-MSH(1–13)
- NMM
N-methyl morpholine
- POMC
pro-opiomelanocortin prohormone
- Pra
N-propargyl glycine
- PTHrP
parathyroid hormone-related protein
- SAR
structure–activity relationships
- TBTU
1-[bis(dimethylamino)methylene]-1H-benzotriazolium 3-oxide tetrafluoroborate
- TFA
trifluoroacetic acid
- UPLC
ultraperformance liquid chromatography
Supporting Information Available
Product characterization and NMR data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Chhajlani V.; Wikberg J. E. Molecular cloning and expression of the human melanocyte stimulating hormone receptor cDNA. FEBS Lett. 1992, 309, 417–420. [DOI] [PubMed] [Google Scholar]
- Gantz I.; Miwa H.; Konda Y.; Shimoto Y.; Tashiro T.; Watson S. J.; Del Valle J.; Yamada T. Molecular cloning, expression, and gene localization of a fourth melanocortin receptor. J. Biol. Chem. 1993, 268, 15174–15179. [PubMed] [Google Scholar]
- Wessells H.; Hruby V. J.; Hackett J.; Han G.; Balse-Srinivasan P.; Vanderah T. W. Ac-Nle-c[Asp-His-DPhe-Arg-Trp-Lys]-NH2 induces penile erection via brain and spinal melanocortin receptors. Neuroscience 2003, 118, 755–762. [DOI] [PubMed] [Google Scholar]
- Fan W.; Boston B. A.; Kesterson R. A.; Hruby V. J.; Cone R. D. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature 1997, 385, 165–168. [DOI] [PubMed] [Google Scholar]
- Huszar D.; Lynch C. A.; Fairchild-Huntress V.; Dunmore J. H.; Fang Q.; Berkemeir L. R.; Gu W.; Kesterson R. A.; Boston B. A.; Cone R. D.; Smith F. J.; Campfield L. A.; Burn P.; Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 1997, 88, 131–141. [DOI] [PubMed] [Google Scholar]
- Mogil J. S.; Wilson S. G.; Chesler E. J.; Rankin A. L.; Nemmani V. S.; Lariviere W. R.; Groce M. K.; Wallace M. R.; Kaplan L.; Staud R.; Ness T. J.; Glover T. L.; Stankova M.; Mayorov A. V.; Hruby V. J.; Grisel J. E.; Fillingim R. B. The melanocortin-1 receptor gene mediates female-specific mechanisms of analgesia in mice and humans. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 4867–4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A. S.; Marsh D. J.; Trumbauer M. E.; Frazier E. G.; Guan X. M.; Yu H.; Rosenblum C. I.; Vongs A.; Feng Y.; Cao L.; Metzger J. M.; Strack A. M.; Camacho R. E.; Mellin T. N.; Nunes C. N.; Min W.; Fisher J.; Gopal-Truter S.; Mac Intyre D. E.; Chen H. Y.; Van der Ploeg L. H. Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nature Genet. 2000, 26, 97–102. [DOI] [PubMed] [Google Scholar]
- Butler A. A.; Kesterson R. A.; Khong K.; Cullen M. J.; Pelleymounter M. A.; Dekoning J.; Baetscher M.; Cone R. D. A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology 2000, 141, 3518–3521. [DOI] [PubMed] [Google Scholar]
- Hruby V. J.; Wilkes B. C.; Cody W. L.; Sawyer T. K.; Hadley M. E. Melanotropins: structural, conformational and biological considerations in the development of superpotent and superprolonged analogs. Pept. Protein Rev. 1984, 3, 1–64. [Google Scholar]
- Mountjoy K. G.; Robbins L. S.; Mortrud M. T.; Cone R. D. The cloning of a family of genes that encode the melanocortin receptors. Science 1992, 257, 1248–1251. [DOI] [PubMed] [Google Scholar]
- Holder J. R.; Haskell-Luevano C. Melanocortin ligands: 30 years of structure–activity relationship (SAR) studies. Med. Res. Rev. 2004, 24, 325–356. [DOI] [PubMed] [Google Scholar]
- Lam D. D.; Farooqi I. S.; Hesiler L. K. Melanocortin receptors as targets in the treatment of obesity. Curr. Top. Med. Chem. 2007, 7, 1085–1097. [PubMed] [Google Scholar]
- Haskell-Luevano C.; Nichiforovich G.; Sharma S. D.; Yang Y. K.; Dickinson C.; Hruby V. J.; Gantz I. Biological and conformational examination of stereochemical modifications using the template melanotropin peptide, Ac-Nle-c[Asp-His-Phe-Arg-Trp-Ala-Lys]-NH2, on human melanocortin receptors. J. Med. Chem. 1997, 40, 1738–1748. [DOI] [PubMed] [Google Scholar]
- Al-Obeidi F. A.; Hadley M. E.; Pettitt M. B.; Hruby V. J. Design of a new class of superpotent cyclic α-melanotropins based on quenched dynamic simulations. J. Am. Chem. Soc. 1989, 111, 3413–3416. [Google Scholar]
- Al-Obeidi F. A.; Castrucci A. L.; Hadley M. E. Potent and prolonged acting cyclic lactam analogues of alpha-melanotropin: design based on molecular dynamics. J. Med. Chem. 1989, 32, 2555–2561. [DOI] [PubMed] [Google Scholar]
- Ying J.; Kövér K. E.; Gu X.; Han G.; Trivedi D. B.; Kavarana M. J.; Hruby V. J. Solution structures of cyclic melanocortin agonists and antagonists by NMR. Biopolymers 2003, 71, 696–716. [DOI] [PubMed] [Google Scholar]
- Henchey L. K.; Jochim A. L.; Arora P. S. Contemporary strategies for the stabilization of peptides in the alpha-helical conformation. Curr. Opin. Chem. Biol. 2008, 12, 692–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. W.; Verdine G. L. Stereochemical effects of all-hydrocarbon tethers in i,i+4 stapled peptides. Bioorg. Med. Chem. Lett. 2009, 19, 2533–2536. [DOI] [PubMed] [Google Scholar]
- Cantel S.; Le Chavelier Isaad A.; Scrima M.; Levy J. J.; Di Marchi R. D.; Rovero P.; Halperin J. A.; D’ Ursi A. M.; Papini A. M.; Chorev M. Synthesis and conformational analysis of a cyclic peptide obtained via i to i+4 intramolecular side-chain to side-chain azide–alkyne 1,3-dipolar cycloaddition. J. Org. Chem. 2008, 73, 5663–5674. [DOI] [PubMed] [Google Scholar]
- Spengler J.; Jimenez J.-C.; Burger K.; Giralt E.; Albericio F. Abbreviated nomenclature for cyclic and branched homo- and hetero-detic peptides. J. Peptide Res. 2005, 65, 550–555. [DOI] [PubMed] [Google Scholar]
- Le Chevalier Isaad A.; Papini A. M.; Chorev M.; Rovero P. Side chain-to-side chain cyclization by click reaction. J. Pept. Sci. 2009, 15, 451–454. [DOI] [PubMed] [Google Scholar]
- Le Chevalier Isaad A.; Barbetti F.; Rovero P.; D’Ursi A. M.; Chelli M.; Chorev M.; Papini A. M. Nα-Fmoc-Protected ω-azido- and ω-alkynyl-l-amino acids as building blocks for the synthesis of “clickable” peptides. Eur. J. Org. Chem. 2008, 31, 5308–5314. [Google Scholar]
- Merrifield R. B. Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar]
- Kolb H. C.; Finn M. G.; Sharpless K. B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem., Int. Ed. Engl. 2001, 40, 2004–2021. [DOI] [PubMed] [Google Scholar]
- Link A. J.; Tirrell D. A. Cell surface labeling of Escherichia coli via copper(I)-catalyzed [3 + 2] cycloaddition. J. Am. Chem. Soc. 2003, 125, 11164–11165. [DOI] [PubMed] [Google Scholar]
- Bodine K. D.; Gin D. Y.; Gin M. S. Synthesis of readily modifiable cyclodextrin analogues via cyclodimerization of an alkynyl–azido trisaccharide. J. Am. Chem. Soc. 2004, 126, 1638–1639. [DOI] [PubMed] [Google Scholar]
- Sawyer T. K.; Sanfilippo P. J.; Hruby V. J.; Engel M. H.; Heward C. B.; Burnett J. B.; Hadley M. E. 4-Norleucine, 7-d-phenylalanine-alpha-melanocyte-stimulating hormone: a highly potent alpha-melanotropin with ultralong biological activity. Proc. Natl. Acad. Sci. U. S. A. 1980, 77, 5754–5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haskell-Luevano C.; Cone R. D.; Monck E. K.; Wan Y. P. Structure activity studies of the melanocortin-4 receptor by in vitro mutagenesis: identification of agouti-related protein (AGRP), melanocortin agonist and synthetic peptide antagonist interaction determinants. Biochemistry 2001, 40, 6164–6179. [DOI] [PubMed] [Google Scholar]
- Chen W.; Shields T. S.; Stork P. J. S.; Cone R. D. A colorimetric assay for measuring activation of Gs- and Gq-coupled signaling pathways. Anal. Biochem. 1995, 226, 349–354. [DOI] [PubMed] [Google Scholar]
- Sun H.; Greeley D. N.; Chu X. J.; Cheung A.; Danho W.; Swistok J.; Wang Y.; Zhao C.; Chen L.; Fry D. C. A predictive pharmacophore model of human melanocortin-4 receptor as derived from the solution structures of cyclic peptides. Bioorg. Med. Chem. 2004, 12, 2671–2677. [DOI] [PubMed] [Google Scholar]
- Mierke D. F.; Kessler H. Molecular dynamics with dimethyl-sulfoxide as a solvent—conformation of a cyclic hexapeptide. J. Am. Chem. Soc. 1991, 113, 9466–9470. [Google Scholar]
- Chatterjee J.; Mierke D.; Kessler H. N-Methylated cyclic pentaalanine peptides as template structures. J. Am. Chem. Soc. 2006, 128, 15164–15172. [DOI] [PubMed] [Google Scholar]
- Piantini U.; Sorensen O. W.; Ernst R. R. Multiple quantum filters for elucidating NMR coupling networks. J. Am. Chem. Soc. 1982, 104, 6800–6801. [Google Scholar]
- Bax A.; Davis D. G. Practical aspects of two-dimensional transverse NOE spectroscopy. J. Magn. Reson. 1985, 63, 207–213. [Google Scholar]
- Jeener J.; Meyer B. H.; Bachman P.; Ernst R. R. Investigation of exchange processes by two-dimensional NMR spectroscopy. J. Chem. Phys. 1979, 71, 4546–4553. [Google Scholar]
- Goddard T. D.; Kneller D. G.; SPARKY 3 NMR Software; University of California: San Francisco, 2001.
- Wüthrich K.NMR of Proteins and Nucleic Acids; John Wiley & Sons: New York, 1986. [Google Scholar]
- Hutchinson E. G.; Thornton J. M. PROMOTIF—a program to identify and analyze structural motifs in proteins. Protein Sci. 1996, 5, 212–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugg E. E.; Castrucci A. M.; Hadley M. E.; van Binst G.; Hruby V. J. Cyclic lactam analogues of Ac-[Nle4]alpha-MSH4–11-NH2. Biochemistry 1988, 27, 8181–8188. [DOI] [PubMed] [Google Scholar]
- Al-Obeidi F.; O’ Connor S. D.; Job C.; Hruby V. J.; Pettitt B. M. NMR and quenched molecular dynamics studies of superpotent linear and cyclic alpha-melanotropins. J. Pept. Res. 1998, 51, 420–431. [DOI] [PubMed] [Google Scholar]
- Cho M. K.; Lee C. J.; Lee C. H.; Li S. Z.; Lim S. K.; Baik J. H.; Lee W. Structure and function of the potent cyclic and linear melanocortin analogues. J. Struct. Biol. 2005, 150, 300–308. [DOI] [PubMed] [Google Scholar]
- Kaiser E.; Colescott R.; Bossinger C.; Cook P. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal. Biochem. 1970, 34, 595–598. [DOI] [PubMed] [Google Scholar]
- Xiang Z.; Litherland S. A.; Sorensen N. B.; Proneth B.; Wood M. S.; Shaw A. M.; Millard W. J.; Haskell-Luevano C. Pharmacological characterization of 40 human melanocortin-4 receptor polymorphisms with the endogenous proopiomelanocortin-derived agonists and the agouti-related protein (AGRP) antagonist. Biochemistry 2006, 45, 7277–7288. [DOI] [PubMed] [Google Scholar]
- Xiang Z.; Pogozheva I. D.; Sorenson N. B.; Wilczynski A. M.; Holder J. R.; Litherland S. A.; Millard W. J.; Mosberg H. I.; Haskell-Luevano C. Peptide and small molecules rescue the functional activity and agonist potency of dysfunctional human melanocortin-4 receptor polymorphisms. Biochemistry 2007, 46, 8273–8287. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.