

Background: Sodium-calcium exchanger 1 (NCX1) and calpain are up-regulated in heart failure (HF). Molecular mechanisms and functional consequences of NCX1 cleavage by calpain are not known.
Results: Calpain anchors to two NCX1 regions and cleaves at methionine-369, leading to inactivation.
Conclusion: NCX1 inhibition by calpain might improve cardiac function.
Significance: Calpain might play a pivotal role in NCX1 regulation during HF.
Keywords: Animal Model, Calpain, Computer Modeling, Electrophysiology, Heart Failure, Ion Channel, Peptide Array, Protein-Protein Interaction, Aortic Stenosis, Sodium-Calcium Exchanger
Abstract
Cardiac sodium (Na+)-calcium (Ca2+) exchanger 1 (NCX1) is central to the maintenance of normal Ca2+ homeostasis and contraction. Studies indicate that the Ca2+-activated protease calpain cleaves NCX1. We hypothesized that calpain is an important regulator of NCX1 in response to pressure overload and aimed to identify molecular mechanisms and functional consequences of calpain binding and cleavage of NCX1 in the heart. NCX1 full-length protein and a 75-kDa NCX1 fragment along with calpain were up-regulated in aortic stenosis patients and rats with heart failure. Patients with coronary artery disease and sham-operated rats were used as controls. Calpain co-localized, co-fractionated, and co-immunoprecipitated with NCX1 in rat cardiomyocytes and left ventricle lysate. Immunoprecipitations, pull-down experiments, and extensive use of peptide arrays indicated that calpain domain III anchored to the first Ca2+ binding domain in NCX1, whereas the calpain catalytic region bound to the catenin-like domain in NCX1. The use of bioinformatics, mutational analyses, a substrate competitor peptide, and a specific NCX1-Met369 antibody identified a novel calpain cleavage site at Met369. Engineering NCX1-Met369 into a tobacco etch virus protease cleavage site revealed that specific cleavage at Met369 inhibited NCX1 activity (both forward and reverse mode). Finally, a short peptide fragment containing the NCX1-Met369 cleavage site was modeled into the narrow active cleft of human calpain. Inhibition of NCX1 activity, such as we have observed here following calpain-induced NCX1 cleavage, might be beneficial in pathophysiological conditions where increased NCX1 activity contributes to cardiac dysfunction.
Introduction
The cardiac Na+/Ca2+ exchanger 1 (NCX1)3 is a central component in maintaining normal [Ca2+]i homeostasis and cardiac function. NCX1 is a bidirectional transporter that mediates the exchange of three Na+ for one Ca2+ across the plasma membrane in either forward mode (Ca2+ efflux) or reverse mode (Ca2+ influx) (1, 2). The direction of ion transport depends on the membrane potential and the intracellular and extracellular concentrations of Ca2+ and Na+. Under physiological conditions, NCX1 functions predominantly as a Ca2+ extrusion mechanism, and the contribution to decline of the Ca2+ transient varies from 9 to 30%, dependent on species (3). In pathological settings, the reverse mode of NCX1 function is often augmented (4, 5).
Increased NCX1 expression and/or activity have been linked to disrupted Ca2+ homeostasis during hypertrophy, ischemia/reperfusion, arrhythmia, and heart failure (HF) (4, 5). Interestingly, in animal models of myocardial infarction, increased NCX1 activity was accompanied by only a modest increase in the NCX1 protein level, indicating that other regulators of the exchanger are also involved (6). Mammalian NCX1 consists of nine transmembrane segments (TMs). The large intracellular loop between TM5 and TM6 consists of ∼500 amino acids, containing two Ca2+ binding regulatory domains, CBD1 and CBD2 (7).
Ca2+ also activates a variety of Ca2+-dependent signaling molecules, including the ubiquitously expressed, non-lysosomal cysteine protease calpain (8). Calpain is a cytoplasmic heterodimer composed of a catalytic subunit (80 kDa) and a regulatory subunit (30 kDa). There are two major catalytic isoforms, calpain-1 and calpain-2, which are activated by micromolar and nearly millimolar Ca2+ concentrations, respectively. Active calpain cleaves its substrate with a limited specific proteolysis, suggesting that it is a regulatory protease. Calpain is implicated in various pathological conditions associated with Ca2+ overload (9–12); however, little is known of the precise molecular mechanisms and biological consequences of calpain-dependent cleavage of proteins. Interestingly, the ubiquitously expressed NCX1 has been shown to be cleaved into a proteolytic fragment of ∼75-kDa in various tissues (9, 13, 14).
In the present study, we hypothesized that calpain is an important regulator of NCX1 in response to pressure overload. We aimed to identify the molecular mechanisms and functional consequences of calpain binding and cleavage of NCX1 in normal heart and HF.
EXPERIMENTAL PROCEDURES
Human Left Ventricular (LV) Biopsies
The human myocardial biopsy protocol conformed to the Declaration of Helsinki and was approved by the Regional Committee for Research Ethics in Eastern Norway (Project 2010/2226). Informed written consent was obtained from all patients. LV apical myocardial biopsies (5–10 mg) were obtained immediately before cross-clamping the aorta in eight patients undergoing elective aortic valve replacement for severe aortic stenosis (AS). All patients exhibited preserved ejection fraction (>50%) and no significant coronary artery stenosis. Myocardial biopsies were also obtained from eight patients undergoing elective coronary artery bypass graft (CABG) surgery, which served as controls. This patient group exhibited ejection fraction >50%, stable angina pectoris, and no evidence of peri-operative ischemia, previous myocardial infarction, or significant valvular disease. If left ventriculography was normal, echocardiography was not performed in control patients, in accordance with hospital guidelines. The included patients were receiving the following cardiovascular medications (aortic valve replacement/control): salicylates (2/7), warfarin (2/1), angiotensin-converting enzyme inhibitors (4/3), Ca2+ antagonists (2/1), cholesterol-lowering medication (3/8), diuretics (2/0), and β-blockers (2/3). Biopsies were taken from normal appearing and contracting regions of the myocardium using a 16G MaxCore disposable biopsy instrument (Bard, Tempe, AZ), snap-frozen in liquid nitrogen, and stored at −70 °C.
Animal Model
Animal handling and experiments were approved by the Norwegian Animal Research Committee (FOTS ID 3820) and conformed to the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health (NIH Publication 85-23, revised 1996). Male Wistar rats (Møllergaard Breeding and Research Center, Skensved, Denmark) weighing ∼170 g were subjected to aortic banding as described previously (15, 16). In short, anesthesia was induced in an anesthesia chamber containing a mixture of 67% N2O, 28% O2, and 4% isoflurane. Ventilation was performed by subsequent endotracheal intubation with a respirator (Zoovent, Triumph Technical Services, Milton Keynes, UK), and anesthesia was maintained by administration of a mixture of 69% N2O, 29% O2, and 2% isoflurane. The chest was opened in the left, second intercostal space, and the ascending aorta was carefully dissected. A significant stenosis was induced by a tight banding of the ascending aorta with a 3.0 silk suture. For sham-operated animals, the silk suture around the ascending aorta was not tightened. Buprenorphine was administered for postoperative analgesia. After 6 weeks, echocardiography was performed with a Vevo 2100 (Fujifilm VisualSonics, Canada), and short and long axis images of the LV and atrium were obtained. Flow through the mitral and aortic valve was measured. Classification of aortic banding animals into hypertrophy (ABHT) or congestive HF (ABHF) groupings was determined by echocardiographic, hemodynamic, and post-mortem analyses. Criteria for inclusion in the ABHT group were increased posterior wall diameter (>1.9 mm), increased LV weight (>0.75 g), and preserved lung weight (<2.0 g). The criteria for inclusion in the ABHF group were the same as for ABHT, except with increased lung weight (>2.5 g) and left atrial diameter >5.0 mm (16). The S group served as a control. All LV samples were snap-frozen in liquid nitrogen and stored at −70°C until analyses.
Primary Cultures of Neonatal Rat Cardiomyocytes
Cultures of rat neonatal cardiomyocytes were prepared from the LV of 1–3-day-old Wistar rats. The LV was excised, minced, and enzymatically digested with collagenase solution. The cell suspension was incubated in uncoated culture flasks in a humidified incubator with 5% CO2 at 37 °C for 20 min. After 20 min, the non-adherent cells were isolated as cardiomyocytes and seeded onto 6-well culture plates (precoated with 0.2% gelatin and 0.1% fibronectin (both from Sigma-Aldrich)) at a density of 3.75 × 105 ml−1 in plating medium consisting of DMEM (D1152, Sigma-Aldrich), M-199 (M2520, Sigma-Aldrich), penicillin/streptomycin (P0781, Sigma-Aldrich), horse serum (14-403E, BioWhittaker, Walkersville, MD), and fetal bovine serum (FBS) (14-701F, BioWhittaker). The cells were incubated in a humidified incubator with 5% CO2 at 37 °C for 24 h before protein fractionation.
Adult Cardiomyocyte Isolation
Rats were anesthetized in a chamber filled with 95% room air and 5% isoflurane (Abbott Scandinavia Ab, Solna, Sweden). Cervical dislocation was performed after abolished pain reflexes were verified. Hearts were then quickly excised and placed in cold 0.15 m NaCl solution with heparin (Heparin LEO, 5000 IE/ml; Orifarm AS, Oslo, Norway). The aorta was then cannulated and retrogradely perfused with a cell isolation buffer containing 130 mm NaCl, 25 mm Hepes, 22 mm d-glucose, 5.4 mm KCl, 0.5 mm MgCl2, 0.4 mm NaH2PO4, (pH 7.4) (all from Sigma) to wash out blood. The heart was thereafter perfused with cell isolation buffer containing 200 units ml−1 collagenase type II (Worthington) and 0.1 mm Ca2+. After a 20-min perfusion, the heart was cut down, and the atria and right ventricle were removed. The LV was minced and gently shaken at 37 °C for 3–4 min in the same solution used in the perfusion but with the addition of 1% BSA and 0.02 units/ml deoxyribonuclease I (Worthington). The digested ventricular tissue was then filtered (200-μm nylon mesh), and cardiomyocytes were sedimented. The cardiomyocyte pellet was resuspended in cell isolation buffer with 1% BSA and 0.1 mm Ca2+ solution. Isolated cardiomyocytes were stored at room temperature until use.
Antibodies
Anti-NCX1 (custom-made, epitope: GQPVFRKVHARDHPIPST), anti-Met369-NCX1 (custom-made, epitope: TRLMTGAGNILKRH), and anti-His were obtained from Genscript Corp. (Piscataway, NJ). Anti-calpain (H-240) (sc-30064), anti-PKC-α (C-20) (sc-208), anti-EGFR (1005) (sc-03), and anti-GAPDH (V-18) (sc-20357) were from Santa Cruz Biotechnology, Inc. Anti-calpain-1 (ab39170 and ab89778) were from Abcam (Cambridge, MA), and anti-calpain-1 domain III (9A4H8D3) (208728) was from Calbiochem. Anti-GFP (Living Colors A.v. Monoclonal antibody (JL-8), 632381) was from Clontech (Mountain View, CA). Anti-FLAG (F1804), anti-vinculin (V9131), and anti-biotin-HRP (A-0185) were from Sigma-Aldrich. Anti-calsequestrin (PA1–913) was from Thermo Scientific. Anti-mouse IgG HRP (NA931V), anti-rabbit IgG HRP (NA934V) affinity-purified polyclonal antibody (both from GE Healthcare), and anti-goat IgG HRP (HAF109, R&D Systems, Minneapolis, MI) were used as secondary antibody.
Fractionation
Rat hearts and cardiomyocyte cultures were fractionated according to the manufacturer (2145, Compartment protein extraction kit, Millipore).
Protein Extracts
Human and rat lysates were made in parallel to ensure identical treatment of the samples. Protein lysates from human biopsies were prepared using ice-cold PBS-based lysis buffer containing 1% Triton X-100 (X100-500ML, Sigma-Aldrich), 0.1% Tween 20 (161-0781, Bio-Rad), and protease and phosphatase inhibitors (Complete EDTA-free and PhosStop tablets, Roche Applied Science). Biopsies were homogenized twice for 30 s with a Polytron 1200 homogenizer, left on ice for 30 min, and centrifuged at 20,000 × g for 10 min at 4 °C. Supernatants were stored at −70 °C. Frozen LVs from rats were pulverized in a mortar with liquid nitrogen before transfer to lysis buffer (20 mm Hepes, pH 7.5, 150 mm NaCl, 1 mm EDTA, 0.5% Triton) supplemented with 1 mm PMSF (93482, Sigma-Aldrich) and a Complete Mini EDTA-free tablet (Roche Applied Science). Protease inhibitors were omitted for rat LV lysate treated with active calpain. Tissue samples were homogenized three times for 1 min on ice with a Polytron 1200 homogenizer and centrifuged at 100,000 × g for 60 min at 4 °C. Supernatants were collected and stored at −70 °C. Protein concentrations were determined by the Micro BCA protein assay kit (Pierce).
DNA Constructs
Cloning and mutations of NCX1 and calpain-1 constructs were performed by Genscript. The Mammalian Gene Collection mouse clone BC079673 was used for NCX1 constructs. WT NCX1 (full-length) was cloned into pEGFP-N1 with and without a stop codon before GFP (Clontech) or into the first reading frame of pAdTrack-cytomegalovirus (CMV) shuttle vector (plasmid 16405, Addgene, Cambridge, MA). Alanine mutants (Ala327–336 and Ala364–373) were mutated into NCX1/pEGFP-N1. Deletion mutants of the cytoplasmic loop of NCX1 (NCX1(243–787), NCX1(243–705), NCX1(243–532), NCX1(243–460), and NCX1(243–402)) were cloned into pEGFP-C2 (Clontech). WT rat calpain-1 catalytic subunit (BC061880) was cloned into pCEP4 (Invitrogen), as were the C-terminal FLAG-tagged calpain-1(1–220), calpain-1(213–365), calpain-1(357–525), and calpain-1(519–713). The fidelity of the cloning procedure and mutagenesis were verified by sequence analysis (Genscript). The empty vectors pcDNA3.1 and EGFP-N1 were obtained from Invitrogen and Clontech, respectively. Tobacco etch virus (TEV) protease in pCS2MT was kindly provided by Prof. Pati (17).
HEK293 and Transient Transfection
HEK293 cells were cultured in DMEM (41965-039, Invitrogen) supplemented with 10% FBS (14-701F, BioWhittaker), 1% non-essential amino acids (10370-021, Invitrogen), 100 units/ml penicillin, and 0.1 mg/ml streptomycin (penicillin/streptomycin, P4333, Sigma-Aldrich) and maintained in a 37 °C, 5% CO2 humidified incubator. The HEK293 cells were transfected with DNA using Lipofectamine 2000 as instructed by the manufacturer (11668-019, Invitrogen). After 24 h, the cells were either subjected to cell lysis or transferred to coverslips for patch clamp experiments. Cell lysis buffer contained 20 mm HEPES (pH 7.5), 150 mm NaCl, 1 mm EDTA, and 0.5% Triton X-100 with complete protease inhibitor mixture tablets (Roche Applied Science). Protease inhibitors were omitted for HEK293 lysate treated with active calpain. The glass coverslips were precoated with poly-l-lysine (P4707, Sigma-Aldrich), and the transfected cells on the coverslips were incubated for 24 h prior analysis. GFP was used as a positive control for the transfection.
Peptide Synthesis
Peptides were synthesized to >80% purity by Genscript: anti-NCX1 blocking peptide (amino acids 655–672), CGQPVFRKVHARDHPIPST; anti-Met369-NCX1 blocking peptide (amino acids 366–379), TRLMTGAGNILKRHC; Biotin-calpain-1 (amino acids 29–48), GRHENAIKYLGQDYENLRAR; Biotin-calpain-1 (amino acids 277–296), TDAKQVTYQGQRVNLIRMRN; Biotin-calpain-1 (amino acids 425–444), QKHRRRERRFGRDMETIGFA; Biotin-calpain-1 (amino acids 453–472), AGQPVHLKRDFFLANASRAQ; Biotin-calpain-1 (amino acids 469–488), SRAQSEHFINLREVSNRIRL; Biotin-calpain-2 (amino acids 267–286), TGAEEVESSGSLQKLIRIRN; Biotin-NCX1 (amino acids 251–270)-XIP region, RRLLFYKYVYKRYRAGKQRG (18); Biotin-NCX1 (amino acids 354–373), QQKSRAFYRIQATRLMTGAG.
Spot Membrane Synthesis
The intracellular loop (amino acids 243–799) of rat NCX1 protein (EDM02743) and full-length rat calpain-1 (BC061880) were synthesized as 20-mer peptides with 3-amino acid offsets on cellulose membranes using a Multipep automated peptide synthesizer (INTAVIS Bioanalytical Instruments AG, Köln, Germany) as described (19).
Calpain and NCX1 Overlay
The peptide array membranes were blocked in 1% casein in TBST (Tris-buffered saline with 1% Tween) overnight at 4 °C. The calpain and NCX1 overlay were conducted by incubating the membranes with 1 μg/ml recombinant His-TF-NCX1cyt (in 1% casein) or 1 μg/ml recombinant calpain-1 (human erythrocytes, 208713, Calbiochem) (in 1× calpain buffer: 10 mm EGTA, 0.1% Triton, 20 mm HEPES (pH 7.5), and 20 mm CaCl2). Thereafter, the membranes were washed five times in TBST for 5 min before binding was detected by immunoblotting.
Pull-down Assay
Monoclonal anti-biotin-conjugated beads (A-1559, Sigma-Aldrich) were incubated with 8 μm peptides in 100 μl of PBS for 2 h with rotation. The beads were then washed three times with PBS before incubation with 1 μg of recombinant calpain-1 (Calbiochem) for 2 h at 4 °C. The protein complexes were subsequently washed three times in lysis buffer and boiled for 5 min at 96 °C in 2× SDS loading buffer before SDS-PAGE analysis.
Immunoprecipitation
Immunoprecipitations were performed using 2 μg of the appropriate antibody. The immunocomplexes were collected by protein A/G-agarose beads (sc-2003, Santa Cruz Biotechnology), washed three times in immunoprecipitation buffer, and boiled in SDS loading buffer before SDS-PAGE analysis. Equal amounts of rabbit IgG (sc-2027) and mouse IgG (sc-2025, Santa Cruz Biotechnology) were used as negative controls.
Proximity Ligation Assay and Image Acquisition
Isolated adult rat cardiomyocytes were plated on laminin (Sigma-Aldrich)-coated glass coverslips and left to adhere for 1 h. The cells were fixed in 4% paraformaldehyde (158127, Sigma-Aldrich), permeabilized with 0.3% Triton X-100 (X100, Sigma-Aldrich), and incubated with anti-NCX1 (custom-made, Genscript) and anti-calpain-1 (ab89778, Abcam) overnight at 4 °C. Staining with a custom-made NCX1 blocking peptide (Genscript) or staining in the absence of primary antibodies served as a negative control. The proximity ligation assay (PLA) was performed using the Duolink kit according to the manufacturer's protocol (Sigma-Aldrich). In brief, species-specific secondary antibodies, called PLA probes, were added to the cells. These PLA probes, each having a unique short DNA strand attached to it, bound to the primary antibodies. When the PLA probes were in close proximity, <40 nm, the DNA strands hybridized when enzyme ligase solution was added. The signal was amplified via rolling circle amplification using an amplification solution consisting of polymerase, nucleotides, and fluorescence-labeled oligonucleotides. The output signal was visible as distinct fluorescent green spots because we used the green Duolink® In Situ detection reagent (excitation 488 nm, emission 510 nm). The cells were then incubated with 600-nm SYTOX Orange (S-11368; Molecular Probes, Inc., Eugene, OR) (excitation 543 nm, emission 650 nm), a nucleic acid stain, for 10 min at room temperature and rinsed three times for 5 min with PBS. Imaging experiments were performed at 25 °C. The water-based In Situ Mounting Medium (refractive index 1.44), provided in the kit was used to mount the glass coverslip to the glass slide. The cells were visualized with an inverted LSM 710 confocal microscope (Zeiss GmbH, Jena, Germany) equipped with a LD C-Apochromat ×40 objective (numerical aperture 1.1). Sequential optical scans were acquired using the Zeiss ZEN imaging software.
Calpain-1 and AcTEV Cleavage in Vitro
Rat heart extract or HEK293 lysates were diluted (1:1) in 2× calpain buffer (1× calpain buffer: 10 mm EGTA, 0.1% Triton, 20 mm HEPES (pH 7.5), and 20 mm CaCl2). Subsequently, 1 μg of calpain-1 (Calbiochem) was added, and proteolysis was performed at 37 °C for 1 h. To inhibit calpain, lysates were incubated with 5 μm calpastatin (Millipore) at 37 °C for 30 min before adding active calpain. The reaction was stopped by adding 4× SDS loading buffer. Cleavage was analyzed by SDS-PAGE and immunoblotting. AcTEV cleavage was conducted according to the instructions provided by the manufacturer (12575-015, Invitrogen).
Ca2+ Treatment of Cardiomyocytes
To induce activation of endogenous calpain, 4 mm CaCl2 was added to the neonatal cardiomyocyte culture. To inhibit calpain, 10 μm calpeptin (Calbiochem) was added prior to the addition of CaCl2. After a 24-h incubation, the membrane proteins were isolated using the Compartment protein extraction kit (Millipore).
Immunoblotting
Lysates and immunoprecipitates were analyzed on 4–15 or 15% SDS-PAGE Criterion (Bio-Rad) and blotted onto PVDF membranes (RPN 303F, GE Healthcare). The PVDF membranes and peptide arrays were blocked in 5% nonfat dry milk or 1% casein in TBST for 60 min at room temperature, incubated overnight at 4 °C with primary antibodies, washed 3–5 times for 5 min in TBST, and incubated with an HRP-conjugated secondary antibody. Blots were developed by using ECL Plus or Prime (RPN 2132 or RPN 2232, GE HealthCare). The chemiluminescence signals were detected by Las 1000 or Las-4000 (Fujifilm, Tokyo, Japan).
Bioinformatics
The CaMPDB (Calpain for Modulatory Proteolysis Database) with PSSM and SVM (RBF Kernel) prediction models were used to identify putative calpain cleavage sites in the cytoplasmic region of NCX1 (20).
Patch Clamp Experiments
Whole cell patch clamp experiments were conducted with either pEGFP-N1- or pAdTrack-CMV-transfected HEK293 cells using an Axoclamp 2B amplifier (Axon Instruments) and low resistance pipettes (1–2.5 megaohms). The recordings were performed in an extracellular solution containing 140 mm NaCl, 5 mm CsCl, 1.2 mm MgSO4, 1.2 mm NaH2PO4, 5 mm CaCl2, 10 mm HEPES, 10 mm glucose, pH 7.4 (CsOH), and osmolality 290 mosm. K+, Ca2+, Cl−, and Na+-K+ ATPase currents were blocked by inclusion of cesium, nifedipine (20 μm), niflumic acid (30 μm), and ouabain (1 mm), respectively, in the solution. Patch pipettes were filled with a solution containing 100 mm cesium glutamate, 1 mm MgCl2, 10 mm HEPES, 2.5 mm Na2-ATP, 10 mm EGTA, CsCl2, pH 7.2, and osmolality 270 mosm. The NCX1 reversal potential under these conditions was −43 mV at 37 °C. Cells were voltage-clamped at −43 mV for 4–5 min to allow sufficient intracellular dialysis. Following a prepulse protocol, NCX1 current was elicited by a descending voltage ramp from 120 to −100 mV. The NCX1 current was calculated as the difference current prior to and following the application of 5 mm Ni2+.
Structural Modeling
Atomic coordinates for rat calpain in complex with leupeptin (PDB code 1TL9 (21)) and human calpain in complex with a peptidomimetic inhibitor (PDB code 1ZCM (22)) were used to build models of the 366TRLM369 and 366TRLMT370 binding to calpain. The peptide models were refined using the Rosetta FlexPepDock server (23). For the leupeptin-based model, the rat protein was replaced by human calpain before submitting the input structure to FlexPepDock. Values of the top scoring complexes calculated by FlexPepDock were analyzed and visualized by PyMOL (Schrödinger LLC, New York).
Densitometric Analysis
Densitometric analysis was performed using Image Gauge version 4.0, ImageQuant TL (Amersham Biosciences), or ImageJ (National Institutes of Health).
Statistics
All data were expressed as mean ± S.E. Comparisons between two groups were analyzed using unpaired Student's t test or Mann-Whitney test (Prism version 5.04 (GraphPad Software)). A p value of <0.05 was considered statistically significant.
RESULTS
Increased Levels of NCX1 and a 75-kDa NCX1 Fragment after Chronic Pressure Overload
NCX1 was analyzed in LV lysates isolated from patients with AS and rats 6 weeks after aortic banding by immunoblotting. Patient and animal clinical characteristics are listed in Tables 1 and 2, respectively. Full-length NCX1 and a proteolytic 75-kDa NCX1 fragment were increased 2.0- and 3.7-fold in AS patients with myocardial hypertrophy, respectively, compared with controls (CAD; coronary artery disease without LV remodeling and biopsied in non-ischemic myocardium) (Fig. 1A). Similarly, rats with hypertrophic, non-failing (ABHT) hearts showed a somewhat increased level of both full-length NCX1 and 75-kDa NCX1 fragment, whereas both levels were significantly increased in failing (ABHF) hearts compared with sham-operated controls (Fig. 1B). The increase in NCX1 fragmentation was paralleled by increased proteolysis of PKCα, a known substrate for calpain (24), implying increased calpain activity in ABHT and ABHF hearts. Consistent with increased NCX1 and PKCα fragmentation, calpain was increased 1.8-fold in ABHF versus sham hearts (Fig. 1C). The addition of active calpain into rat LV lysate resulted in an increased NCX1 fragment (Fig. 1D, top, lane 2) of a size similar to that of endogenous cleaved NCX1 in ABHT and ABHF hearts (Fig. 1B). A somewhat different pattern of cleavage products might be due to in vitro calpain cleavage. No NCX1 fragmentation was observed when the calpain inhibitor, calpastatin, was added. Moreover, treating cardiomyocytes with Ca2+ to induce activation of endogenous calpain resulted in an increased 75-kDa NCX1 fragment (Fig. 1E, lanes 2 and 3). The 75-kDa NCX1 fragment was not increased when the calpain inhibitor, calpeptin, was added (lane 4).
TABLE 1.
Clinical characteristics of aortic stenosis (AS) and control patients (CAD)
Patient medications are detailed under “Experimental Procedures.” NA, not available; LVIDd/s, LV internal diameter in diastole and systole, respectively; IVSd, interventricular septal thickness in diastole; LVPWd, LV posterior wall thickness in diastole.
| AS | CAD | |
|---|---|---|
| No. of patients (no. of females) | 8 (4) | 8 (1) |
| Years of age at myocardial tissue biopsy | 74.88 ± 2.53 | 68.34 ± 2.65 |
| Ejection fraction | All >50% | All >50% |
| Aortic valve area (cm2) | 0.66 ± 0.05 | NA |
| Maximum aortic gradient (mm Hg) | 85.38 ± 7.91 | NA |
| Mean aortic gradient (mm Hg) | 54.63 ± 4.50 | NA |
| LVIDd (cm) | 4.83 ± 0.25 | NA |
| LVIDs (cm) | 2.88 ± 0.29 | NA |
| IVSd (cm) | 1.16 ± 0.07 | NA |
| LVPWd (cm) | 1.09 ± 0.03 | NA |
TABLE 2.
Clinical characteristics of rats with hypertrophic, non-failing (ABHT) and failing (ABHF) hearts compared with sham-operated controls
LVDd/s, LV diameter in diastole and systole; FS, fractional shortening in LVD; PWd, posterior wall thickness in diastole; LAD, left atrial diameter; LVOT, LV outlet tract; CO, cardiac output.
| Sham | ABHT | ABHF | |
|---|---|---|---|
| Number (n) | 14 | 10 | 10 |
| Body weight (g) | 357 ± 6 | 332 ± 7a | 338 ± 13 |
| Heart weight (g) | 0.96 ± 0.03 | 1.53 ± 0.05a | 2.7 ± 0.13a,b |
| LV weight (g) | 0.56 ± 0.03 | 0.97 ± 0.02a | 1.3 ± 0.05a,b |
| LV/body weight ratio (mg/g) | 1.58 ± 0.08 | 2.92 ± 0.09a | 4.0 ± 0.22a,b |
| Lung weight (g) | 1.11 ± 0.06 | 1.55 ± 0.04a | 4.0 ± 0.14a,b |
| LVDd (mm) | 6.2 ± 0.1 | 5.4 ± 0.1a | 7.5 ± 0.2a,b |
| LVDs (mm) | 3.0 ± 0.2 | 2.2 ± 0.1a | 4.5 ± 0.3a,b |
| FS (%) | 57 ± 2 | 59 ± 2 | 40 ± 3a,b |
| PWd (mm) | 1.6 ± 0.04 | 2.3 ± 0.1a | 2.4 ± 0.1a |
| LAD (mm) | 3.4 ± 0.1 | 4.2 ± 0.1a | 6.1 ± 0.3a,b |
| Peak mitral flow (mm/s) | 859 ± 30 | 925 ± 47 | 1076 ± 42a,b |
| Mitral deceleration (mm/s) | 2714 ± 145 | 3314 ± 242a | 4940 ± 418a |
| Peak LVOT flow (mm/s) | 1087 ± 62 | 917 ± 39a | 824 ± 90 |
| Heart rate | 430 ± 6 | 416 ± 12 | 383 ± 8a |
| CO in LVOT (ml/min) | 129 ± 6 | 115 ± 7 | 103 ± 12 |
| Maximal velocity (mm/s) | 73 ± 3 | 51 ± 3a | 38 ± 4a,b |
| Minimal velocity (mm/s) | 78 ± 3 | 56 ± 4a | 51 ± 3a |
a p < 0.05 versus sham.
b p < 0.05 versus ABHT.
FIGURE 1.

Levels of NCX1, 75-kDa NCX1, and calpain after chronic pressure overload. NCX1 and calpain were analyzed in LV lysates from AS patients (A) and ABHT and ABHF rats (B and C) using anti-NCX1 and anti-calpain. A 40-kDa proteolytic PKCα fragment was used as a control for calpain activity (24). Bar graphs show calpain and NCX1 levels semiquantified by densitometry analysis. Differences were tested using unpaired Student's t test (*, p < 0.05; **, p < 0.01; ***, p < 0.001) (n = 5–8). Error bars, S.E. D, NCX1 fragmentation in LV lysate after treatment with recombinant active calpain (n = 3). Calpastatin, a calpain inhibitor, was used as a control for specificity. GAPDH and vinculin were used as loading controls (A–D, bottom panels). E, NCX1 fragmentation in membrane fractions isolated from neonatal cardiomyocytes treated with and without Ca2+ in combination with the calpain inhibitor, calpeptin. Non-treated cardiomyocytes were used as a control (lane 1). Calsequestrin was used as a loading control (bottom).
Association of Calpain-NCX1 in Cardiac Membranes
Although calpain is predominantly a cytoplasmic protease (25), it was also observed together with NCX1 in membrane fractions from rat LV (Fig. 2A, left, weak signal) and cardiomyocytes (right, strong signal). To test whether calpain was recruited to membrane fractions after chronic pressure overload, subcellular fractionation of LV from sham, ABHT, and ABHF hearts was performed. Immunoblotting showed that calpain levels were increased by 2.3- and 1.8-fold in ABHT and ABHF membrane fractions, respectively (Fig. 2B and 2C, middle). Consistent with findings in LV lysates (Fig. 1, A and B), NCX1 levels tended to be elevated in ABHT membrane fractions and were significantly increased in ABHF (Fig. 2, B and C, top panels). Although calpain expression also was increased in membrane fractions after pressure overload (Fig. 2, B and C), immunoprecipitation showed that less calpain precipitated with NCX1 in ABHF compared with sham (Fig. 2D). Taken together, our data indicate that calpain associates with NCX1 in membrane fractions in normal and hypertrophic hearts but might dissociate after cleavage of NCX1 in HF.
FIGURE 2.

NCX1 and calpain levels in isolated cardiac membrane fractions. A, NCX1 and calpain were analyzed in cytoplasmic and membrane fractions isolated from rat LV and neonatal cardiomyocytes using anti-NCX1 and anti-calpain. GAPDH and EGFR were used as controls for cytoplasmic and membrane fractions, respectively. NCX1 and calpain were analyzed in membrane fractions isolated from ABHT (B) and ABHF (C) versus sham using anti-NCX1 and anti-calpain. D, lysates from sham, ABHT, and ABHF were subjected to immunoprecipitation (IP) using anti-NCX1. The presence of endogenous NCX1 and calpain in immunoprecipitates was analyzed by immunoblotting. B–D, differences were tested using unpaired Student's t test (*, p < 0.05; **, p < 0.001) (n = 4–6). Error bars, S.E.
Calpain Interacts through its Catalytic Domain IIb and Domain III to NCX1
Several experiments were performed to analyze whether calpain interacted directly with NCX1. Calpain and NCX1 were observed to colocalize within a range of ∼40 nm in intact cardiomyocytes (Fig. 3A, left) using specific NCX1 (13) and calpain antibodies (Fig. 3B) in a proximity ligation assay. No signal was observed when primary antibodies were omitted (Fig. 3A, middle) or anti-NCX1 was preincubating with an anti-NCX1 blocking peptide (Fig. 3A, right). Immunoprecipitation of calpain from cardiomyocyte membrane fractions revealed co-precipitation of NCX1 (Fig. 3C). Finally, immunoprecipitation of recombinant His-TF-NCX1cyt (Fig. 3D, top) using His antibodies showed that active calpain precipitated only in the presence of His-TF-NCX1cyt (bottom), suggesting NCX1 and calpain to be direct binding partners.
FIGURE 3.

NCX1 binding in calpain. A, confocal images of in situ proximity ligation assay. NCX1-calpain-1 association analyzed in adult cardiomyocytes using anti-NCX1 and anti-calpain (left; interaction indicated by green spots; see “Experimental Procedures” for details). Incubation without primary antibodies (middle) and preincubating anti-NCX1 with blocking peptide (right) were used as negative controls. Nuclei were stained with SYTOX orange. B, epitope was identified by overlaying an array of immobilized overlapping 20-mer calpain catalytic subunit peptides with anti-calpain (sc-30064, top). Amino acids 201–232 are relevant for anti-calpain binding and are within the 230-amino acid fragment used as antigen by the manufacturer (Santa Cruz Biotechnology, Inc.). Immunoblotting without anti-calpain was used as a negative control (bottom). C, membrane fraction was subjected to immunoprecipitation using anti-calpain or control rabbit IgG (n = 3). Lysates and immunoprecipitates were analyzed for the presence of endogenous NCX1 and calpain by immunoblotting. D, schematic illustration of recombinant His-TF-NCX1cyt (top). Shown is immunoprecipitation (IP) of recombinant calpain-1 with His-TF-NCX1cyt using anti-His (bottom). Lysates and precipitates were analyzed for the presence of calpain and NCX1 by immunoblotting. E, NCX1 binding identified by overlaying His-TF-NCX1cyt on membranes containing 20-mer overlapping calpain-1 peptides and immunoblotting with anti-NCX1 (n = 3). Incubation without His-TF-NCX1cyt was used as a negative control (bottom). Underlined amino acids indicate sequences used in the pull-down experiment in F. F, pull-down assays with biotinylated calpain peptides against His-TF-NCX1cyt were performed using anti-biotin-coupled agarose beads. NCX1 binding was analyzed by immunoblotting with anti-NCX1. Biotin-NCX1(251–270) containing the self-dimerization region XIP (18) or beads only was used as positive or negative control, respectively (n = 4).
To identify NCX1 binding in calpain, calpain-1 was synthesized as 20-mer overlapping peptides on a membrane and incubated with His-TF-NCX1cyt, followed by NCX1 immunoblotting. Strong NCX1 binding was observed in five regions of calpain-1 (Fig. 3E, top). To confirm overlay data, pull-down experiments with biotinylated peptides covering the five putative NCX1 binding sites in calpain (amino acids 29–48, 277–396, 425–444, 453–472, and 469–488) (underlined sequences in Fig. 3E) together with recombinant His-TF-NCX1cyt were performed with anti-biotin-agarose beads. Only calpain(277–296) and calpain(425–444), residing in catalytic domain IIb and domain III, respectively, co-precipitated His-TF-NCX1cyt (Fig. 3F). Thus, the other sequences were revealed as false positives. Interestingly, NCX1 bound to catalytic domain IIb, containing His272 and Asn296 (Fig. 3E, boldface type), which together with Cys115 forms the catalytic triad in the active site in presence of Ca2+ (26). The NCX1-XIP region containing the self-dimerization domain (18) was used as a positive control for the assay. Beads without any peptide were used as a negative control.
The two NCX1-calpain interaction sites were also confirmed by binding studies in HEK293 cells. Five constructs were expressed in HEK293: calpain-1(FL)-FLAG, calpain-1(1–220)-FLAG, calpain-1(213–365)-FLAG, calpain-1(357–525)-FLAG, and calpain-1(519–731)-FLAG (Fig. 4A). Fishing with His-TF-NCX1cyt in HEK293 lysates containing each variant revealed that NCX1 bound specifically to calpain-1(213–365) and calpain-1(357–525), containing catalytic domain IIb and domain III, respectively (Fig. 4B, stars). NCX1 bound only very weakly to these calpain-1 domains compared with calpain-1(FL)-FLAG (top left), suggesting that both regions were required to obtain strong binding. No His-TF-NCX1cyt binding was observed to calpain-1(1–220)-FLAG or calpain-1(519–731)-FLAG, not even after longer time exposures.
FIGURE 4.

Binding analysis in HEK293 cells and protein sequence alignments. A, schematic illustration of calpain-FLAG variants expressed in HEK293 cells. The calpain catalytic subunit consists of four domains: the N terminus (domain I), the catalytic domain (domain IIa and IIb), domain III, and domain IV (based on the calpain-2 crystal structure (51)). Cys115, His272, and Asn296 in domain IIa and IIb form the active catalytic site in the presence of Ca2+ (26). B, pull-down assays with recombinant His-TF-NCX1cyt against calpain-1-FLAG, calpain(1–220)-FLAG, calpain(213–365)-FLAG, calpain(357–525)-FLAG, and calpain(519–731)-FLAG were performed using His antibodies. Inputs and immunoprecipitates were analyzed with anti-NCX1 and anti-FLAG (n = 2–6). Mouse IgG was used as a negative control. Shown is alignment of NCX1 binding sites I and II in human, rat, and mouse calpain-1 catalytic domain IIb (C) and domain III (D), respectively. E, alignment of the NCX1 binding site in rat calpain-1 versus calpain-2 catalytic domain. His272 and Asn296 in the catalytic cleft are indicated with arrows. Black boxes, functional similar amino acids in C–E (DNA Star, Madison, WI). F, immunoprecipitation of His-TF-NCX1cyt and biotin-calpain-2(267–286) was performed with anti-NCX1. Input and immunoprecipitates were analyzed with anti-NCX1 and anti-biotin-HRP. Immunoprecipitation without His-TF-NCX1cyt was used as negative control. IP, immunoprecipitation.
Sequence alignments show that NCX1 binding site I in catalytic domain IIb (Fig. 4C) and site II in domain III (Fig. 4D) were highly conserved across human, rat, and mouse calpain-1. Although there was less sequence similarity between catalytic domain IIb of calpain-1 and calpain-2 (Fig. 4E), His-TF-NCX1cyt also bound calpain-2 (amino acids 267–286) (Fig. 4F), suggesting NCX1 to be both a calpain-1 and calpain-2 substrate. Indeed, the substrate specificities have been shown to be almost indistinguishable (12).
Identification of a Novel Calpain Binding and Cleavage Site at Met369 in NCX1
By using the CaMPDB prediction database (20), two high score calpain cleavage sites were identified at Lys331 and Met369 in the catenin-like domain (CLD) of NCX1 (Fig. 5A). Cleavage of either site could theoretically result in an NCX1 fragment of ∼75 kDa. The two sites were each mutated to 10 alanines in NCX1-GFP (Fig. 5A, underlined) because this mutation strategy has been shown to inhibit calpain cleavage (24). NCX1(Ala327–336)-GFP (Fig. 5B), but not NCX1(Ala364–373)-GFP (Fig. 5C) was cleaved by calpain, suggesting Met369 to be a novel calpain cleavage site. The apparent higher molecular mass of cleaved fragment (95 kDa) was due to GFP tag. Moreover, NCX1 (Ala364–373)-GFP precipitated less calpain than NCX1-GFP (Fig. 5D), suggesting that an intact calpain cleavage site was required to obtain a strong calpain-NCX1 interaction. In an in vitro calpain cleavage assay, the presence of a substrate competitor peptide, NCX1(354–373), containing Met369, reduced cleavage of NCX1-GFP (Fig. 5E). Finally, to confirm Met369 as the cleavage site, an antibody against the Met369 cleavage site was generated. Anti-Met369-NCX1 detected NCX1 only when epitope TRLMTGAGNILKRH was intact (Fig. 5F). In contrast to the regular anti-NCX1, which detects both FL and 75-kDa NCX1 (Figs. 1–4 and 5G, top), anti-Met369-NCX1 detected only FL NCX1 (Fig. 5G, middle), suggesting that the 75-kDa fragment originates from cleavage at Met369. Preincubation of anti-Met369-NCX1 with a blocking peptide gave no signals from FL NCX1 or 75-kDa NCX1 (Fig. 5G, bottom).
FIGURE 5.

Identification of novel calpain cleavage at NCX1-Met369. A, bioinformatics using CaMPDB identified Lys331 and Met369 as putative calpain cleavage sites (in boldface type). Underlined amino acids were subjected to alanine substitutions. NCX1-GFP, NCX1(Ala327–336)-GFP (B), and NCX1(Ala364–375)-GFP (C) were treated with active calpain-1. Cleavage was analyzed by immunoblotting with anti-NCX1. D, immunoprecipitation of NCX1-GFP and NCX1(Ala364–373) with recombinant calpain-1 using anti-NCX1. Lysates and immunoprecipitates were analyzed with anti-NCX1 and anti-calpain (n = 3). E, cleavage of NCX1-GFP analyzed in the presence of a peptide substrate, biotin-NCX1(354–373). GAPDH was used as loading control in B, C, and E. F, epitope mapping of anti-Met369-NCX1. Antibody epitope (TRLMTGAGNILKRH) and N-terminal deletions were synthesized on membrane and analyzed using anti-Met369-NCX1. Intact epitope was a requirement for a strong signal. G, levels of FL NCX1 and the 75-kDa NCX1 fragment in ABHF versus sham were analyzed by regular anti-NCX1 (top, GQPVFRKVHARDHPIPST epitope, positive control), anti-Met369-NCX1 (middle, TRLMTGAGNILKRH epitope), or anti-Met369-NCX1 preincubated with blocking peptide (TRLMTGAGNILKRH, negative control). Shown is sequence alignment of the calpain cleavage site in human, rat, and mouse NCX1 (H) and rat NCX1–3 (I). Black boxes, functional similar amino acids (DNA Star, Madison, WI).
Conclusively, our data indicate a novel calpain cleavage site at Met369 in NCX1-CLD. The sequence spanning Met369 is identical in human, rat, and mouse NCX1 (Fig. 5H) and highly conserved in NCX1–3 isoforms (Fig. 5I).
Cleavage at Met369 Reduces NCX1 Activity
To investigate the biological role of specific cleavage of NCX1 at Met369, we engineered cleavage without calpain activation. This was accomplished by replacing amino acids surrounding the calpain cleavage site with a TEV protease site (Fig. 6A), which cleaves amino acid sequence ENLYFQ(G/S) after Q with high specificity (27). We transiently expressed NCX1(TEV), NCX1(Ala364–373), or NCX1(WT) into HEK293 cells. In contrast to NCX1(WT) (without GFP), which was cleaved to a fragment of 75 kDa, neither NCX1(TEV) nor NCX1(Ala364–373) was cleaved by active recombinant calpain-1 (data not shown). However, transient expression of NCX1(TEV) with TEV protease into HEK293 resulted in generation of the 75-kDa proteolytic NCX1 fragment in membrane fraction (Fig. 6B, denoted with stars).
FIGURE 6.

Biological function of cleavage at NCX1-Met369. A, substitution of amino acids surrounding NCX1-Met369 into the TEV recognition site (marked in boldface type). Arrows, respective cleavage sites. B, NCX1 in membrane fractions isolated from HEK293 transfected with NCX1 or NCX1(TEV) in combination with or without TEV protease or control plasmids. C and D, whole cell patch clamp recordings from transfected HEK293. Different current traces were evoked using a voltage ramp from 120 to −100 mV. Typical current traces are shown in C. D, corresponding I-V curve from −100 to 100 mV of NCX1(TEV), TEV-cleaved NCX1(TEV), and GFP (n = 5).
Total NCX1 current before and after TEV protease cleavage was analyzed by using whole cell patch clamp techniques. NCX1 and NCX1(TEV) in combination with TEV protease were transiently expressed into HEK293. NCX1 current was isolated by measuring Ni2+-sensitive current elicited during a slow voltage ramp from 120 to −100 mV (Fig. 6C). Plotted current-voltage relationships show reduced current in both forward and reverse mode in cleaved NCX1(TEV) (Fig. 6D). Non-cleaved NCX1(TEV) showed the same current profile as NCX1(WT) (data not shown). Taken together, our data strongly suggest that cleavage of NCX1 at Met369 inhibits NCX1 activity.
Structural Modeling of the NCX1-Met369 Peptide into the Calpain Active Cleft
Upon Ca2+ binding, calpain domains IIa and IIb move toward each other to form an active narrow cleft that substrate fits into (28). Peptide docking models of 366TRLM369 or 366TRLMT370 peptides from NCX1 onto human calpain are shown in Fig. 7. Docking of 366TRLMT370, using leupeptin as a template (21), showed that Met369 fitted well in the cleft between domain IIa and IIb in calpain (Fig. 7A). In this model, the scissile peptide bond was not positioned close to catalytic Cys115. However, docking the shorter 366TRLM369 peptide, using a peptidomimetics inhibitor as a template (22), gave a model where the peptide bond was positioned close to Cys115 (Fig. 7B). In this latter model, the NCX1-Met369 residue was still observed to bind in the cleft but was not positioned as close to surface of calpain. This model, which was based on a covalently linked inhibitor, did not give any indication as to where the C-terminal part of the NCX1 peptide would be located. However, inspection of several top ranked docking models suggested two alternative positions for the Arg367 residue (Fig. 7B). Both peptide docking models have the Leu368 residue positioned in the same place as the corresponding Leu residues in leupeptin and peptidomimetics inhibitor templates (21, 22).
FIGURE 7.
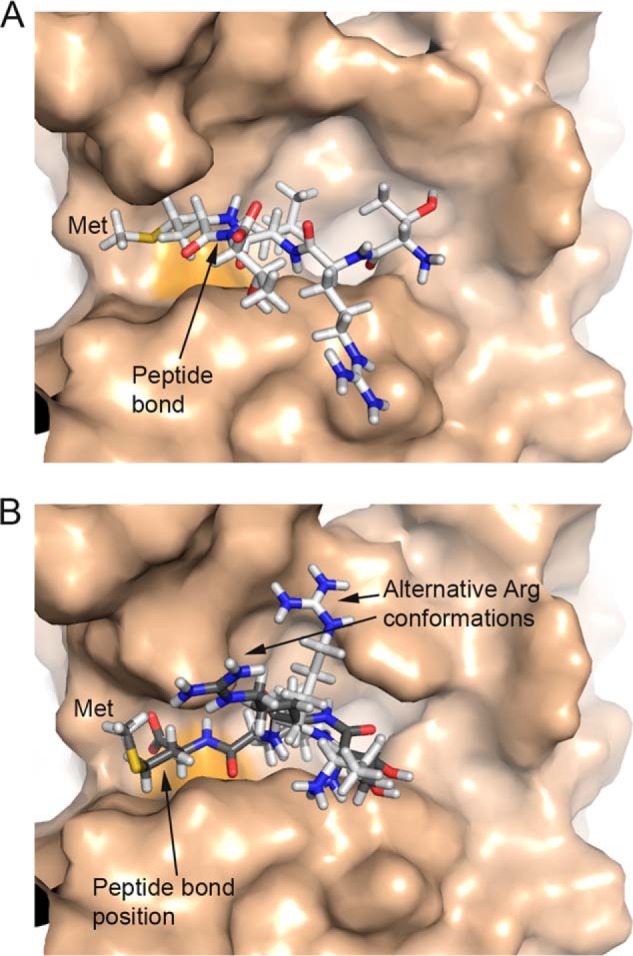
Peptide docking models of NCX1-TRLM(T) peptide into human calpain. A, ball-and-stick model of 366TRLMT370 peptide binding to calpain. The scissile peptide bond (arrow) is distant from active site Cys115 (orange surface). Met369 fits snuggly into a cleft formed by two loops in calpain. B, ball-and-stick models of two different conformations (white and gray) of 366TRLM369 peptide binding to calpain. The scissile peptide bond (arrow) is close to active site Cys115 (orange surface). Met369 fits into the cleft between two loops (domain IIa and IIb) in calpain.
Identification of a Second Calpain Binding Site in the NCX1-CBD1
As indicated in Figs. 3F and 4B, calpain domain III also bound to NCX1. To identify domain III binding in NCX1, NCX1-GFP and GFP-NCX1 variants were designed and expressed in HEK293: NCX1(FL)-GFP, GFP-NCX1(243–787), GFP-NCX1(243–705), GFP-NCX1(243–532), and GFP-NCX1(243–402) (Fig. 8A). Fishing with active calpain-1 revealed that all GFP-NCX1 variants precipitated, except GFP-NCX1(243–402) (Fig. 8B). This finding suggested the second calpain binding site to be within amino acids 402–460 in NCX1. This observation was confirmed by overlaying calpain-1 onto NCX1cyt spot-synthesized as 20-mer overlapping peptides on membranes. Immunoblotting with anti-calpain identified calpain binding to amino acids 345–373 and 411–439 in NCX1 (Fig. 8C). Importantly, amino acids 345–373 contain the novel calpain binding and cleavage site, Met369, identified above. The second calpain binding site resided within the first regulatory calcium binding region of NCX1(CBD1).
FIGURE 8.

Calpain binding in NCX1. A, schematic illustrations of NCX1 variants expressed in HEK293 cells. Shown are the CLD as well as CBD1 and CBD2 (7, 42). B, immunoprecipitation (IP) of calpain-1 with NCX1 variants using anti-calpain. Input lysates and precipitates were analyzed with immunoblotting. C, identification of calpain binding by overlaying calpain-1 on 20-mer overlapping NCX1 peptides synthesized on membrane (n = 3). Binding was analyzed using anti-calpain. Incubation without calpain was used as a negative control (bottom). Shown is an alignment of calpain binding site II in human, rat, and mouse NCX1 (D) and rat NCX1–3 (E). Black boxes, functionally similar amino acids (DNA Star).
Sequence alignments show that the second calpain binding site was almost identical in human, rat, and mouse NCX1 (Fig. 8D) and was similar between rat NCX1–3 isoforms (Fig. 8E). A model of calpain binding and cleavage of NCX1 is summarized in Fig. 9.
FIGURE 9.
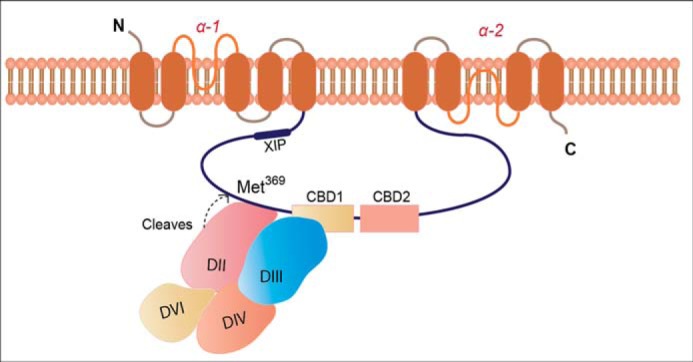
Schematic illustration of calpain binding and cleavage of NCX1. Calpain binds to NCX1-CLD (amino acids 345–373) and NCX1-CBD1 (amino acids 411–439) through its catalytic domain (amino acids 269–304 in domain II) and domain III (amino acids 413–448), respectively. When calpain is activated, it cleaves NCX1-Met369, leading to inhibition of NCX1. After cleavage, calpain dissociates from NCX1. Recent evidence suggests that mammalian NCX1 may have 10 TM domains similar to NCX_Mj from archaebacterial Methanococcus jannaschii (52).
DISCUSSION
In an ongoing effort to understand regulation of cardiac NCX1 activity, we sought to identify molecular mechanisms and consequences of calpain binding and cleavage of NCX1 in normal, hypertrophic non-failing, and failing hearts.
Calpain Activation and Cleavage of Cardiac NCX1
We observed increasing levels of a 75-kDa proteolytic NCX1 fragment along with FL NCX1 in LV lysates isolated from aortic-banded rats with hypertrophic, non-failing heart (ABHT) and HF (ABHF). Similar results were observed in LV biopsies from aortic stenosis (AS) patients with hypertrophic remodeling. The observed increase in NCX1 fragmentation was paralleled by an increase in calpain levels, an observation consistent with previous reports on involvement of calpains in various heart diseases (29). Moreover, calpain interacted directly with NCX1 in membrane fractions, but the interaction was reduced in ABHF compared with sham, suggesting that calpain dissociated from NCX1 after cleavage. A low level of the 75-kDa proteolytic NCX1 fragment was also observed in sham-operated rats, consistent with a low level of proteolytic calpain activity in non-stressed myocardium (30).
The precise activation mechanism of calpains is not known. Calpain is believed to predominately present as an inactive enzyme in the cytosol, where it binds to the endogenous inhibitor calpastatin. When Ca2+ rises, calpain translocates to membrane, where it associates with phospholipids. Calpain-1 activation reportedly requires 3–50 μm [Ca2+]i, whereas calpain-2 requires 400–800 μm [Ca2+]i (8). Because bulk cytosolic [Ca2+]i is only ∼0.1 μm in resting cells, it is believed that calpain activation occurs at epicenters of Ca2+ influx and/or release and then quickly inactivates as Ca2+ diffuses away or is removed by various pumps (8). In cardiomyocytes, NCX1 localizes to surface sarcolemma and T-tubules at dyadic junctions with the sarcoplasmic reticulum (SR). Opening of L-type Ca2+ channels during action potential triggers a large release of Ca2+ from SR via clusters of ryanodine receptors, which has been estimated to result in peak dyadic Ca2+ concentrations between 10 and 15 μm (31). Thus, systolic dyadic [Ca2+] appears to be sufficient for at least calpain-1 activation and cleavage of neighboring NCX1. However, in situ experiments indicate that increasing the mean cytosolic [Ca2+] to as little as 451 nm Ca2+ can activate calpain in cardiac myocytes (32). Thus, there may be additional activators of calpain present in vivo (e.g. phospholipids (12)) that lower the Ca2+ concentration required to activate calpain.
A Novel Inhibitory Calpain Cleavage Site at NCX1-Met369
Our screening strategy utilizing peptide arrays combined with bioinformatics is an effective approach to identify novel binding and cleavage sites in proteins. Calpain-1 catalytic domain was observed to bind to amino acids 345–373 in the disordered CLD, resulting in cleavage at NCX1-Met369. In support of our assertion that NCX1-Met369 is a novel calpain cleavage site, we observed that 1) CaMPDB prediction database (20) predicted Met369 to be a high score cleavage site; 2) cleavage of Met369 resulted in a 75-kDa NCX1 fragment, which is of a size similar to that reported for proteolytic NCX1 digestion in LV lysate (13), brain (9), isolated caveolae vesicles, and endoplasmic reticulum (ER) from bovine pulmonary artery smooth muscle (14, 33); 3) calpain bound directly to the NCX1-Met369-containing region using peptide arrays; 4) mutation of the region spanning Met369 into alanines or TEV protease cleavage site inhibited calpain cleavage of NCX1, thus suggesting that Met369 is the sole or primary cleavage site; 5) an excess of a soluble substrate peptide containing the region spanning Met369 blocked cleavage of NCX1; and 6) antibody with an epitope covering the Met369-spanning sequence was not able to detect the endogenous 75-kDa NCX1 fragment.
Engineering Met369 into a TEV recognition sequence enabled us to explore the biological effect of NCX1-Met369 cleavage without calpain activation. Cleavage of NCX1-Met369 led to a reduction in NCX1 activity (both forward and reverse mode) compared with full-length NCX1 when expressed in HEK293. Contrary to our findings, calpain treatment was reported to lead to constitutive activation of cardiac NCX1 when expressed in Xenopus oocytes (34). However, the authors reported that the results could not be replicated when Ca2+ binding sites in CBD1 or CBD2 were mutated. Thus, in light of these data, we suggest that calpain is able to activate full-length NCX1 through an indirect mechanism. For example, PKCα, which phosphorylates and activates NCX1 (35), is reported to be constitutively activated by calpain cleavage, leading to hyperphosphorylation of its substrates (24).
Calpain Cleavage of Other Ion Channels
Other Ca2+ transporters are also suggested to be substrates for calpain cleavage. These include the plasma membrane Ca2+-ATPase, which, like NCX1, extrudes Ca2+ (36, 37), SR Ca2+-handling proteins like RYR2 (11, 38) and SERCA2 (11, 39), mitochondrial inner membrane NCLX (10), ER NCX1 (33), and brain NCX3 (9). However, there is, in general, a paucity of data concerning the precise mechanisms and sites at which calpain cleaves these proteins. Functional consequences are also poorly understood, although it is generally suggested that the activity of Ca2+ transporters is inhibited following cleavage. Plasma membrane Ca2+-ATPase has been shown to be sequentially cleaved, resulting in an initial functional activation as the C-terminal autoinhibitory domain was cleaved (36), followed by a secondary cleavage resulting in functional inhibition (40). Moreover, although cleavage of NCLX (110 kDa) and ER NCX1 led to Ca2+ overload in mitochondria (10) and inhibited Ca2+ uptake into ER (33), no mechanisms or cleavage sites were identified. Cleavage of NCX3 after brain ischemia contributed to glutamate-mediated neuronal cytotoxicity (9). A cluster of three calpain cleavage sites, within a region without any homology to NCX1, was identified in NCX3 (9). A fourth cleavage site, NCX3-Lys377, was reported to be inaccessible in all three NCX isoforms in vivo (9). Consistent with their observations, no calpain binding was observed at NCX1-Lys377 in our study using peptide arrays.
Calpain Domain III Binds to the First Ca2+ Binding Region in NCX1(CBD1)
We identified that calpain domain III bound to amino acids 411–439 within NCX1-CBD1. Domain III, which is also referred to as the C2-like domain, contains a β-sandwich tertiary structure, similar to although with a different topology compared with known C2 domains found in enzymes that transiently bind to the membrane. Domain III is suggested to be involved in membrane translocation of calpain (41). However, to our knowledge, NCX1 is the first protein described to bind to domain III, suggesting an anchoring role of this domain in calpain.
NCX1-CBD1 is known to be a high affinity Ca2+ binding site and constitutes a crucial site for Ca2+ sensing leading to NCX1 activation. CBD1-Glu385, residing in the domain III binding site, has been shown to be important for coordinating Ca2+ binding (42). Thus, calpain anchoring might impair Ca2+ binding and consequently NCX1 function. It has been recently suggested that Ca2+ binding to CBD1 constrains the conformational freedom of CBD1-CBD2, rigidifying the intracellular loop and thus propagating the signal to ion transport sites in membrane (5). Because the calpain binding site in CBD1 was less conserved in NCX2 and NCX3, the presented calpain binding and cleavage mechanisms might be unique to NCX1.
Is Calpain Cleavage of Cardiac NCX1 a Compensatory Mechanism?
Inhibition of NCX1 might be beneficial in pathophysiological conditions or etiologies where increased NCX1 activity contributes to cardiac dysfunction. NCX and SERCA compete for the same pool of Ca2+, and increased NCX1 activity during conditions such as HF is believed to attenuate SR Ca2+ load and reduce the magnitude of Ca2+ transients (43). Indeed, NCX1 blockade has been observed to augment SR content and Ca2+ transients (44), and similar compensatory effects might be expected to result following calpain cleavage of NCX1. However, the specific phenotype of HF should be considered because there has been a recent appreciation that more than 50% of HF patients exhibit normal systolic function but impaired diastolic function (45). Reduced NCX1 activity can promote slowed removal of cytosolic Ca2+, leading to slowed cardiomyocyte relaxation during HF, and this deficit may become particularly significant when SERCA levels are also reduced (46). Although it is as yet unclear whether calpain-mediated cleavage may promote such dysfunction, reduced NCX1 activity is also known to result from elevated intracellular [Na+]i in failing cells due to reduced expression of the Na+-K+ ATPase (47), augmentation of late Na+ current (48), and intracellular acidosis (49). Thus, there is general agreement that reduced NCX1 activity, such as we have observed following calpain-induced NCX1 cleavage, is expected to be compensatory for systolic function in HF but detrimental for diastolic function.
Inhibition of NCX1 activity may also have potential beneficial effects in the setting of arrhythmia. Spontaneous SR Ca2+ release occurs more frequently during conditions such as HF, and resulting extrusion of released Ca2+ by NCX1 results in delayed after-depolarizations. Reducing NCX1 activity can reduce the magnitude of these depolarizing currents, which will reduce the likelihood that they will trigger action potentials and ectopic beats (50). Reducing NCX1 current also shortens action potential, which makes the occurrence of early after-depolarizations less likely (50). Finally, inhibition of NCX1 current during ischemia and reperfusion may also be compensatory, where NCX1-mediated Ca2+ entry is a key contributor to cellular damage.
Acknowledgments
We thank Bjørg Austbø, Marita Martinsen, Ståle Nygård, and the animal facility staff at Oslo University Hospital for excellent technical assistance. Moreover, we thank the surgical teams at Oslo University Hospital for providing human biopsies and the patients who donated the biopsies.
This work was supported by the Research Council of Norway, Norwegian National Health Association, Stiftelsen Kristian Gerhard Jebsen, Anders Jahre's Fund for the promotion of Science, the South-Eastern Norway Regional Health Authority, and the Simon Fougner Hartmanns Family Fund, Denmark.
- NCX
- Na+/Ca2+ exchanger
- HF
- heart failure
- LV
- left ventricle
- FL
- full-length
- TM
- transmembrane segment
- CBD
- Ca2+ binding regulatory domain
- CLD
- catenin-like domain
- XIP
- exchanger inhibitory peptide
- AS
- aortic stenosis
- ABHT
- aortic banding hypertrophy
- ABHF
- aortic banding heart failure
- HEK
- human embryonic kidney
- TEV
- tobacco etch virus
- CaMPDB
- Calpain for Modulatory Proteolysis Database
- ER
- endoplasmic reticulum
- SERCA
- sarcoplasmic endoplasmic reticulum Ca2+-ATPase
- SR
- sarcoplasmic reticulum
- PLA
- proximity ligation assay.
REFERENCES
- 1. Blaustein M. P., Lederer W. J. (1999) Sodium/calcium exchange: its physiological implications. Physiol. Rev. 79, 763–854 [DOI] [PubMed] [Google Scholar]
- 2. Kang T. M., Hilgemann D. W. (2004) Multiple transport modes of the cardiac Na+/Ca2+ exchanger. Nature 427, 544–548 [DOI] [PubMed] [Google Scholar]
- 3. Bers D. M. (2001) Excitation-Contraction Coupling and Cardiac Contractile Force, 2nd Ed., pp. 147, 152,, 253, Kluwer Academic, Dordrecht, Netherlands [Google Scholar]
- 4. Khananshvili D. (2013) The SLC8 gene family of sodium-calcium exchangers (NCX): structure, function, and regulation in health and disease. Mol. Aspects Med. 34, 220–235 [DOI] [PubMed] [Google Scholar]
- 5. Khananshvili D. (2014) Sodium-calcium exchangers (NCX): molecular hallmarks underlying the tissue-specific and systemic functions. Pflugers Arch. 466, 43–60 [DOI] [PubMed] [Google Scholar]
- 6. Wasserstrom J. A., Holt E., Sjaastad I., Lunde P. K., Odegaard A., Sejersted O. M. (2000) Altered E-C coupling in rat ventricular myocytes from failing hearts 6 wk after MI. Am. J. Physiol. Heart Circ. Physiol. 279, H798–H807 [DOI] [PubMed] [Google Scholar]
- 7. Hilge M., Aelen J., Vuister G. W. (2006) Ca2+ regulation in the Na+/Ca2+ exchanger involves two markedly different Ca2+ sensors. Mol. Cell 22, 15–25 [DOI] [PubMed] [Google Scholar]
- 8. Campbell R. L., Davies P. L. (2012) Structure-function relationships in calpains. Biochem. J. 447, 335–351 [DOI] [PubMed] [Google Scholar]
- 9. Bano D., Young K. W., Guerin C. J., Lefeuvre R., Rothwell N. J., Naldini L., Rizzuto R., Carafoli E., Nicotera P. (2005) Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell 120, 275–285 [DOI] [PubMed] [Google Scholar]
- 10. Kar P., Chakraborti T., Samanta K., Chakraborti S. (2009) μ-Calpain-mediated cleavage of the Na+/Ca2+ exchanger in isolated mitochondria under A23187 induced Ca2+ stimulation. Arch. Biochem. Biophys. 482, 66–76 [DOI] [PubMed] [Google Scholar]
- 11. Singh R. B., Chohan P. K., Dhalla N. S., Netticadan T. (2004) The sarcoplasmic reticulum proteins are targets for calpain action in the ischemic-reperfused heart. J. Mol. Cell Cardiol. 37, 101–110 [DOI] [PubMed] [Google Scholar]
- 12. Sorimachi H., Ono Y. (2012) Regulation and physiological roles of the calpain system in muscular disorders. Cardiovasc. Res. 96, 11–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wanichawan P., Louch W. E., Hortemo K. H., Austbø B., Lunde P. K., Scott J. D., Sejersted O. M., Carlson C. R. (2011) Full-length cardiac Na+/Ca2+ exchanger 1 protein is not phosphorylated by protein kinase A. Am. J. Physiol. Cell Physiol. 300, C989–C997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shaikh S., Samanta K., Kar P., Roy S., Chakraborti T., Chakraborti S. (2010) m-Calpain-mediated cleavage of Na+/Ca2+ exchanger-1 in caveolae vesicles isolated from pulmonary artery smooth muscle. Mol. Cell Biochem. 341, 167–180 [DOI] [PubMed] [Google Scholar]
- 15. Brattelid T., Qvigstad E., Birkeland J. A., Swift F., Bekkevold S. V., Krobert K. A., Sejersted O. M., Skomedal T., Osnes J. B., Levy F. O., Sjaastad I. (2007) Serotonin responsiveness through 5-HT2A and 5-HT4 receptors is differentially regulated in hypertrophic and failing rat cardiac ventricle. J. Mol. Cell Cardiol. 43, 767–779 [DOI] [PubMed] [Google Scholar]
- 16. Lunde I. G., Aronsen J. M., Kvaløy H., Qvigstad E., Sjaastad I., Tønnessen T., Christensen G., Grønning-Wang L. M., Carlson C. R. (2012) Cardiac O-GlcNAc signaling is increased in hypertrophy and heart failure. Physiol. Genomics 44, 162–172 [DOI] [PubMed] [Google Scholar]
- 17. Panigrahi A. K., Zhang N., Mao Q., Pati D. (2011) Calpain-1 cleaves Rad21 to promote sister chromatid separation. Mol. Cell. Biol. 31, 4335–4347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li Z., Nicoll D. A., Collins A., Hilgemann D. W., Filoteo A. G., Penniston J. T., Weiss J. N., Tomich J. M., Philipson K. D. (1991) Identification of a peptide inhibitor of the cardiac sarcolemmal Na+-Ca2+ exchanger. J. Biol. Chem. 266, 1014–1020 [PubMed] [Google Scholar]
- 19. Frank R., Overwin H. (1996) SPOT synthesis: epitope analysis with arrays of synthetic peptides prepared on cellulose membranes. Methods Mol. Biol. 66, 149–169 [DOI] [PubMed] [Google Scholar]
- 20. DuVerle D. A., Ono Y., Sorimachi H., Mamitsuka H. (2011) Calpain cleavage prediction using multiple kernel learning. PLoS One 6, e19035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moldoveanu T., Campbell R. L., Cuerrier D., Davies P. L. (2004) Crystal structures of calpain-E64 and -leupeptin inhibitor complexes reveal mobile loops gating the active site. J. Mol. Biol. 343, 1313–1326 [DOI] [PubMed] [Google Scholar]
- 22. Li Q., Hanzlik R. P., Weaver R. F., Schönbrunn E. (2006) Molecular mode of action of a covalently inhibiting peptidomimetic on the human calpain protease core. Biochemistry 45, 701–708 [DOI] [PubMed] [Google Scholar]
- 23. London N., Raveh B., Cohen E., Fathi G., Schueler-Furman O. (2011) Rosetta FlexPepDock web server: high resolution modeling of peptide-protein interactions. Nucleic Acids Res. 39, W249–W253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kang M. Y., Zhang Y., Matkovich S. J., Diwan A., Chishti A. H., Dorn G. W. (2010) Receptor-independent cardiac protein kinase Cα activation by calpain-mediated truncation of regulatory domains. Circ. Res. 107, 903–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Inserte J., Hernando V., Garcia-Dorado D. (2012) Contribution of calpains to myocardial ischaemia/reperfusion injury. Cardiovasc. Res. 96, 23–31 [DOI] [PubMed] [Google Scholar]
- 26. Moldoveanu T., Hosfield C. M., Lim D., Elce J. S., Jia Z., Davies P. L. (2002) A Ca2+ switch aligns the active site of calpain. Cell 108, 649–660 [DOI] [PubMed] [Google Scholar]
- 27. Parks T. D., Leuther K. K., Howard E. D., Johnston S. A., Dougherty W. G. (1994) Release of proteins and peptides from fusion proteins using a recombinant plant virus proteinase. Anal. Biochem. 216, 413–417 [DOI] [PubMed] [Google Scholar]
- 28. Strobl S., Fernandez-Catalan C., Braun M., Huber R., Masumoto H., Nakagawa K., Irie A., Sorimachi H., Bourenkow G., Bartunik H., Suzuki K., Bode W. (2000) The crystal structure of calcium-free human m-calpain suggests an electrostatic switch mechanism for activation by calcium. Proc. Natl. Acad. Sci. U.S.A. 97, 588–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Letavernier E., Zafrani L., Perez J., Letavernier B., Haymann J. P., Baud L. (2012) The role of calpains in myocardial remodelling and heart failure. Cardiovasc. Res. 96, 38–45 [DOI] [PubMed] [Google Scholar]
- 30. Galvez A. S., Diwan A., Odley A. M., Hahn H. S., Osinska H., Melendez J. G., Robbins J., Lynch R. A., Marreez Y., Dorn G. W. (2007) Cardiomyocyte degeneration with calpain deficiency reveals a critical role in protein homeostasis. Circ. Res. 100, 1071–1078 [DOI] [PubMed] [Google Scholar]
- 31. Acsai K., Antoons G., Livshitz L., Rudy Y., Sipido K. R. (2011) Microdomain [Ca2+] near ryanodine receptors as reported by L-type Ca2+ and Na+/Ca2+ exchange currents. J. Physiol. 589, 2569–2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Matsumura Y., Saeki E., Otsu K., Morita T., Takeda H., Kuzuya T., Hori M., Kusuoka H. (2001) Intracellular calcium level required for calpain activation in a single myocardial cell. J. Mol. Cell Cardiol. 33, 1133–1142 [DOI] [PubMed] [Google Scholar]
- 33. Samanta K., Kar P., Chakraborti T., Chakraborti S. (2010) Calcium-dependent cleavage of the Na+/Ca2+ exchanger by m-calpain in isolated endoplasmic reticulum. J. Biochem. 147, 225–235 [DOI] [PubMed] [Google Scholar]
- 34. Hnatowich M., Le H. D., DeMoissac D., Ranson K., Yurkov V., Gilchrist J. S., Omelchenko A., Hryshko L. V. (2012) μ-Calpain-mediated deregulation of cardiac, brain, and kidney NCX1 splice variants. Cell Calcium 51, 164–170 [DOI] [PubMed] [Google Scholar]
- 35. Iwamoto T., Pan Y., Wakabayashi S., Imagawa T., Yamanaka H. I., Shigekawa M. (1996) Phosphorylation-dependent regulation of cardiac Na+/Ca2+ exchanger via protein kinase C. J. Biol. Chem. 271, 13609–13615 [DOI] [PubMed] [Google Scholar]
- 36. James P., Vorherr T., Krebs J., Morelli A., Castello G., McCormick D. J., Penniston J. T., De Flora A., Carafoli E. (1989) Modulation of erythrocyte Ca2+-ATPase by selective calpain cleavage of the calmodulin-binding domain. J. Biol. Chem. 264, 8289–8296 [PubMed] [Google Scholar]
- 37. Salamino F., Sparatore B., Melloni E., Michetti M., Viotti P. L., Pontremoli S., Carafoli E. (1994) The plasma membrane calcium pump is the preferred calpain substrate within the erythrocyte. Cell Calcium 15, 28–35 [DOI] [PubMed] [Google Scholar]
- 38. Pedrozo Z., Sánchez G., Torrealba N., Valenzuela R., Fernández C., Hidalgo C., Lavandero S., Donoso P. (2010) Calpains and proteasomes mediate degradation of ryanodine receptors in a model of cardiac ischemic reperfusion. Biochim. Biophys. Acta 1802, 356–362 [DOI] [PubMed] [Google Scholar]
- 39. French J. P., Quindry J. C., Falk D. J., Staib J. L., Lee Y., Wang K. K., Powers S. K. (2006) Ischemia-reperfusion-induced calpain activation and SERCA2a degradation are attenuated by exercise training and calpain inhibition. Am. J. Physiol. Heart Circ. Physiol 290, H128–H136 [DOI] [PubMed] [Google Scholar]
- 40. Brown C. S., Dean W. L. (2007) Regulation of plasma membrane Ca2+-ATPase in human platelets by calpain. Platelets 18, 207–211 [DOI] [PubMed] [Google Scholar]
- 41. Tompa P., Emori Y., Sorimachi H., Suzuki K., Friedrich P. (2001) Domain III of calpain is a Ca2+-regulated phospholipid-binding domain. Biochem. Biophys. Res. Commun. 280, 1333–1339 [DOI] [PubMed] [Google Scholar]
- 42. Hilge M., Aelen J., Foarce A., Perrakis A., Vuister G. W. (2009) Ca2+ regulation in the Na+/Ca2+ exchanger features a dual electrostatic switch mechanism. Proc. Natl. Acad. Sci. U.S.A. 106, 14333–14338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Despa S., Bers D. M. (2013) Na+ transport in the normal and failing heart: remember the balance. J. Mol. Cell Cardiol. 61, 2–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ozdemir S., Bito V., Holemans P., Vinet L., Mercadier J. J., Varro A., Sipido K. R. (2008) Pharmacological inhibition of Na/Ca exchange results in increased cellular Ca2+ load attributable to the predominance of forward mode block. Circ. Res. 102, 1398–1405 [DOI] [PubMed] [Google Scholar]
- 45. Paulus W. J., Tschöpe C., Sanderson J. E., Rusconi C., Flachskampf F. A., Rademakers F. E., Marino P., Smiseth O. A., De Keulenaer G., Leite-Moreira A. F., Borbély A., Edes I., Handoko M. L., Heymans S., Pezzali N., Pieske B., Dickstein K., Fraser A. G., Brutsaert D. L. (2007) How to diagnose diastolic heart failure: a consensus statement on the diagnosis of heart failure with normal left ventricular ejection fraction by the Heart Failure and Echocardiography Associations of the European Society of Cardiology. Eur. Heart J. 28, 2539–2550 [DOI] [PubMed] [Google Scholar]
- 46. Louch W. E., Hougen K., Mørk H. K., Swift F., Aronsen J. M., Sjaastad I., Reims H. M., Roald B., Andersson K. B., Christensen G., Sejersted O. M. (2010) Sodium accumulation promotes diastolic dysfunction in end-stage heart failure following Serca2 knockout. J. Physiol. 588, 465–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Swift F., Birkeland J. A., Tovsrud N., Enger U. H., Aronsen J. M., Louch W. E., Sjaastad I., Sejersted O. M. (2008) Altered Na+/Ca2+-exchanger activity due to downregulation of Na+/K+-ATPase α2-isoform in heart failure. Cardiovasc. Res. 78, 71–78 [DOI] [PubMed] [Google Scholar]
- 48. Sossalla S., Wagner S., Rasenack E. C., Ruff H., Weber S. L., Schöndube F. A., Tirilomis T., Tenderich G., Hasenfuss G., Belardinelli L., Maier L. S. (2008) Ranolazine improves diastolic dysfunction in isolated myocardium from failing human hearts: role of late sodium current and intracellular ion accumulation. J. Mol. Cell Cardiol. 45, 32–43 [DOI] [PubMed] [Google Scholar]
- 49. Li L., Louch W. E., Niederer S. A., Aronsen J. M., Christensen G., Sejersted O. M., Smith N. P. (2012) Sodium accumulation in SERCA knockout-induced heart failure. Biophys. J. 102, 2039–2048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ter Keurs H. E., Boyden P. A. (2007) Calcium and arrhythmogenesis. Physiol. Rev. 87, 457–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Goll D. E., Thompson V. F., Li H., Wei W., Cong J. (2003) The calpain system. Physiol. Rev. 83, 731–801 [DOI] [PubMed] [Google Scholar]
- 52. Liao J., Li H., Zeng W., Sauer D. B., Belmares R., Jiang Y. (2012) Structural insight into the ion-exchange mechanism of the sodium/calcium exchanger. Science 335, 686–690 [DOI] [PubMed] [Google Scholar]