

Abstract
In large-scale proteomic experiments, multiple peptide precursors are often cofragmented simultaneously in the same mixture tandem mass (MS/MS) spectrum. These spectra tend to elude current computational tools because of the ubiquitous assumption that each spectrum is generated from only one peptide. Therefore, tools that consider multiple peptide matches to each MS/MS spectrum can potentially improve the relatively low spectrum identification rate often observed in proteomics experiments. More importantly, data independent acquisition protocols promoting the cofragmentation of multiple precursors are emerging as alternative methods that can greatly improve the throughput of peptide identifications but their success also depends on the availability of algorithms to identify multiple peptides from each MS/MS spectrum. Here we address a fundamental question in the identification of mixture MS/MS spectra: determining the statistical significance of multiple peptides matched to a given MS/MS spectrum. We propose the MixGF generating function model to rigorously compute the statistical significance of peptide identifications for mixture spectra and show that this approach improves the sensitivity of current mixture spectra database search tools by a ≈30–390%. Analysis of multiple data sets with MixGF reveals that in complex biological samples the number of identified mixture spectra can be as high as 20% of all the identified spectra and the number of unique peptides identified only in mixture spectra can be up to 35.4% of those identified in single-peptide spectra.
The advancement of technology and instrumentation has made tandem mass (MS/MS)1 spectrometry the leading high-throughput method to analyze proteins (1, 2, 3). In typical experiments, tens of thousands to millions of MS/MS spectra are generated and enable researchers to probe various aspects of the proteome on a large scale. Part of this success hinges on the availability of computational methods that can analyze the large amount of data generated from these experiments. The classical question in computational proteomics asks: given an MS/MS spectrum, what is the peptide that generated the spectrum? However, it is increasingly being recognized that this assumption that each MS/MS spectrum comes from only one peptide is often not valid. Several recent analyses show that as many as 50% of the MS/MS spectra collected in typical proteomics experiments come from more than one peptide precursor (4, 5). The presence of multiple peptides in mixture spectra can decrease their identification rate to as low as one half of that for MS/MS spectra generated from only one peptide (6, 7, 8). In addition, there have been numerous developments in data independent acquisition (DIA) technologies where multiple peptide precursors are intentionally selected to cofragment in each MS/MS spectrum (9, 10, 11, 12, 13, 14, 15). These emerging technologies can address some of the enduring disadvantages of traditional data-dependent acquisition (DDA) methods (e.g. low reproducibility (16)) and potentially increase the throughput of peptide identification 5–10 fold (4, 17). However, despite the growing importance of mixture spectra in various contexts, there are still only a few computational tools that can analyze mixture spectra from more than one peptide (18, 19, 20, 21, 8, 22). Our recent analysis indicated that current database search methods for mixture spectra still have relatively low sensitivity compared with their single-peptide counterpart and the main bottleneck is their limited ability to separate true matches from false positive matches (8). Traditionally problem of peptide identification from MS/MS spectra involves two sub-problems: 1) define a Peptide-Spectrum-Match (PSM) scoring function that assigns each MS/MS spectrum to the peptide sequence that most likely generated the spectrum; and 2) given a set of top-scoring PSMs, select a subset that corresponds to statistical significance PSMs. Here we focus on the second problem, which is still an ongoing research question even for the case of single-peptide spectra (23, 24, 25, 26). Intuitively the second problem is difficult because one needs to consider spectra across the whole data set (instead of comparing different peptide candidates against one spectrum as in the first problem) and PSM scoring functions are often not well-calibrated across different spectra (i.e. a PSM score of 50 may be good for one spectrum but poor for a different spectrum). Ideally, a scoring function will give high scores to all true PSMs and low scores to false PSMs regardless of the peptide or spectrum being considered. However, in practice, some spectra may receive higher scores than others simply because they have more peaks or their precursor mass results in more peptide candidates being considered from the sequence database (27, 28). Therefore, a scoring function that accounts for spectrum or peptide-specific effects can make the scores more comparable and thus help assess the confidence of identifications across different spectra. The MS-GF solution to this problem is to compute the per-spectrum statistical significance of each top-scoring PSM, which can be defined as the probability that a random peptide (out of all possible peptide within parent mass tolerance) will match to the spectrum with a score at least as high as that of the top-scoring PSM. This measures how good the current best match is in relation to all possible peptides matching to the same spectrum, normalizing any spectrum effect from the scoring function. Intuitively, our proposed MixGF approach extends the MS-GF approach to now calculate the statistical significance of the top pair of peptides matched from the database to a given mixture spectrum M (i.e. the significance of the top peptide–peptide spectrum match (PPSM)). As such, MixGF determines the probability that a random pair of peptides (out of all possible peptides within parent mass tolerance) will match a given mixture spectrum with a score at least as high as that of the top-scoring PPSM.
Despite the theoretical attractiveness of computing statistical significance, it is generally prohibitive for any database search methods to score all possible peptides against a spectrum. Therefore, earlier works in this direction focus on approximating this probability by assuming the score distribution of all PSMs follows certain analytical form such as the normal, Poisson or hypergeometric distributions (29, 30, 31). In practice, because score distributions are highly data-dependent and spectrum-specific, these model assumptions do not always hold. Other approaches tried to learn the score distribution empirically from the data (29, 27). However, one is most interested in the region of the score distribution where only a small fraction of false positives are allowed (typically at 1% FDR). This usually corresponds to the extreme tail of the distribution where p values are on the order of 10−9 or lower and thus there is typically lack of sufficient data points to accurately model the tail of the score distribution (32). More recently, Kim et al. (24) and Alves et al. (33), in parallel, proposed a generating function approach to compute the exact score distribution of random peptide matches for any spectra without explicitly matching all peptides to a spectrum. Because it is an exact computation, no assumption is made about the form of score distribution and the tail of the distribution can be computed very accurately. As a result, this approach substantially improved the ability to separate true matches from false positive ones and lead to a significant increase in sensitivity of peptide identification over state-of-the-art database search tools in single-peptide spectra (24).
For mixture spectra, it is expected that the scores for the top-scoring match will be even less comparable across different spectra because now more than one peptide and different numbers of peptides can be matched to each spectrum at the same time. We extend the generating function approach (24) to rigorously compute the statistical significance of multiple-Peptide-Spectrum Matches (mPSMs) and demonstrate its utility toward addressing the peptide identification problem in mixture spectra. In particular, we show how to extend the generating approach for mixture from two peptides. We focus on this relatively simple case of mixture spectra because it accounts for a large fraction of mixture spectra presented in traditional DDA workflows (5). This allows us to test and develop algorithmic concepts using readily-available DDA data because data with more complex mixture spectra such as those from DIA workflows (11) is still not widely available in public repositories.
MATERIALS AND METHODS
Spectral Probability for a Mixture Spectrum
For single-peptide spectra, the statistical significance of a particular peptide P matched to a spectrum S with score T is determined by the probability that a random peptide R (out of all possible peptides) when matched to S has a score greater or equal to T: Pr(Score(R, S)≥T) where Score(R, S) is a scoring function for a peptide-spectrum-match. From here on, we will refer to this as the Single-peptide probability in order to distinguish it from the other definitions introduced below. Analogously, to compute the statistical significance of a particular peptide pair (P, Q) matched to a mixture spectrum (M) with a score of T, we are interested in two statistical questions: 1) Joint probability ≡ Pr(Score(R1,R2,M) ≥T): the probability that a random peptide pair (R1, R2) (out of all possible peptide pairs) when matched to M yields a score greater or equal to T and 2) Conditional probability ≡ Pr(Score(R1,R2,M) ≥T | R1 = P): given a peptide P, the probability that a random peptide R2 (out of all possible peptides) together with P when matched to M yields a score greater or equal to T. Intuitively a peptide–peptide spectrum match (PPSM) can fall into three categories: (1) Correct-match: both peptides are correct matches; (2) Half-correct match: one peptide is correct and the other peptide is an incorrect match; and 3) Incorrect-match: both peptides are incorrect matches. We are interested in separating the correct matches from incorrect and half-correct matches. The definitions above address this question in two steps. The joint probability assesses the chance that two random peptides have the same or higher score than a given match. When this probability is very low, this means that at least one peptide is a statistically significant match to the spectrum (i.e. it is a correct or half-correct match). Once we assume that at least one peptide is a true match, the conditional probability assesses whether the second peptide is also a statistically significant match (i.e. correct matches). In summary, one is looking for PPSMs with both low joint probability and conditional probability.
Scoring Function for Mixture Spectrum
Before describing the computation of the different probability measures, first we review the basics of our scoring function for a peptide-spectrum-match. We represent a tandem mass (MS/MS) spectrum with parent mass N as a real-valued vector: V = ν1 … νN with N elements, where νi is the sum of intensity of all the peaks with mass between i − 0.5 and i + 0.5 and parent mass is defined as the sum of the masses of all amino acids in the peptide that generated the spectrum. A prefix residue mass (PRM) spectrum is a transformation of an MS/MS spectrum into a scored version S = s1 … sN using a probabilistic model as described before (34). In brief, at every mass position i of the PRM spectrum is a score si that represents the log-likelihood that the peptide from which the spectrum was generated contains a prefix mass i (35). Given a peptide P, its prefix masses are defined by the amino acid masses for each peptide prefix. For a peptide P of length n with prefix masses p1 … pn, we define its parent mass as pn and the score of matching peptide P to a spectrum is the sum of all the scores at its theoretical prefix masses in the PRM spectrum:
Note that the probabilistic model used to generate the PRM spectrum depends on the precursor charge state of the MS/MS spectrum (34), thus when matching P to S, the precursor charge state for S is determined such that the parent mass of P is equal to that of S within the specified mass error tolerance.
We define a mixture spectrum as a spectrum from two different peptides. When interpreting an MS/MS spectrum as a mixture spectrum M, we construct two PRM spectra, MH and ML, each generated using the corresponding scoring models for high and low-abundance peptides present in a mixture spectrum. As shown in MixDB (8), different scoring models are needed for high and low-abundance peptides because they exhibit substantially different fragmentation statistics in mixture spectra. Intuitively, this is because the low-abundance peptides will generate less intense peaks in the mixture spectrum and, in general, it also has less number of detectable peaks above noise level. For example, the median peak intensity rank (ranked by decreasing peak intensity) for a y-ion from high-abundance peptides is 19, whereas the median peak rank for a y-ion from low-abundance peptide drops to 35. Without loss of generality, when matching a mixture spectrum (M) against a pair of peptides (P, Q) we assume that the first peptide (P) is the high-abundance peptide. Thus, the score of a pair of peptides (P, Q) against a mixture spectrum M will be the sum of scoring P with MH and scoring Q with ML:
To avoid double counting, when a prefix mass of P is the same as a prefix mass of Q, only the bin with the higher score is considered and the other peptide gets a score of zero for that particular mass position:
Computing Spectral Probabilities
In order to compute the probabilities mentioned above we need to know the score distribution for all possible peptides and peptide pairs. The original MS-GF (24) approach uses dynamic programming to efficiently compute the single-peptide probability without explicitly considering the scores for all peptides. Here we extend this generating function approach to compute the distributions for the joint and conditional probabilities. Let JM be a three-dimensional dynamic programming matrix where each element JM(p, q, T) stands for the joint probability that a pair of peptides P, Q with parent mass p and q match to M with score higher than or equal to T. This means P matches to MH up to the p-th bin and Q matches to ML up to q-th bin. The following recurrence can then be used to compute the joint probability:
 |
In the equation above a, a1, and a2 denote amino acids; mass(a) denotes the mass of an amino acid; prob(a) denotes the probability that a particular amino acid occurs in a peptide and recall that MH and ML are the PRM spectra defined in the previous section. When considering all possible peptide sequences this probability is uniform and has a value of for each of the 20 standard amino acids. To better reflect the amino acid composition observed in real protein sequences we can also define this probability by computing the frequency of each amino acid in the protein sequence database against which the spectra are searched. To start the computation of the recurrence, we initialize Jm(0, 0, 0) = 1 and JM(p,q,s) for all entries where p or q is smaller than the smallest mass of an amino acid or s is less than zero.
The computation of the conditional probability is similar to that of single-peptide probability, except that it is conditioned on the first peptide being accepted as a match. Specifically, for a peptide pair (P, Q) matched to a spectrum M with score T, we define that peptide P and Q contribute Tp and TQ to the total score, respectively. Assuming that peptide P was matched to M, we define a two-dimensional dynamic programming matrix CM where each element CM(q,T|P) represents the conditional probability that a peptide with parent mass q together with P match M with a score greater than or equal to T. To compute this probability, we first modify ML by setting all the bins corresponding to a prefix mass of P to zero if MH has a higher score at the same location. Then Conditional probability can be computed using the following recursion:
We initialize the recurrence with the base case: CM(0,TP|P). The base case starts at score TP rather than zero because the first peptide P already contributes TP to the total score.
We note that even though the joint probability assesses whether at least one peptide is a significant match to the spectrum, it does not determine which peptide is the significant match in the case when only one peptide is a significant match. More importantly, when calculating the conditional probability one assumes that the first peptide is a true match but it is unclear which peptide is the first peptide from the joint probability assessment. In order to resolve this ambiguity, for a candidate peptide pair (P, Q) matched to a spectrum M, we compute their respective single-peptide probabilities and the peptide with lower (i.e. statistically more significant) single-peptide probability is designated as the first peptide. The dynamic programming method described above assumes that peptide fragment ions have integer masses. However, this is not appropriate for data sets with high mass accuracy in the MS/MS spectra. The details of how to extend this method for high mass accuracy data are described in the Supplementary Material. The current implementation of mixgf considers the set of all unmodified peptides or peptide pairs when computing the conditional and joint probability, however as shown in unpublished work MSGF+ (36) it is possible to extend this approach to take variable modifications into consideration.
Approximating Joint Probability
The dynamic programming approach described above enables the computation of the Joint probability without explicitly computing the scores for all peptide pairs. However, the computational complexity still scales exponentially with the number of peptides that possibly generated the observed spectrum (e.g. quadratic for two peptides), making it difficult to generalize to cases with more than two peptides. Thus, it is desirable to find a way to efficiently approximate this probability. To derive this approximation we borrow an intuition from the definition of conditional probability where the joint probability of two random events (R1, R2), is equal to the probability of one event times the conditional probability of the second event given the first event:
Analogously we can decompose the joint probability question into two simpler questions: (1) what is the probability Pr(Score(R1,M) ≥TP) of finding a random peptide R1 that matches to M with a score equal or better than Tp = Score(P, M)? and (2) once we find a first peptide P, what is the probability Pr(Score(R1,R2,M) ≥T|R1 = P) of finding a random peptide R1 that together with P scores equal or higher than T when matched to M? Note that the first question is just the single-peptide probability and the second question is the conditional probability. Therefore, we can define the following approximation:
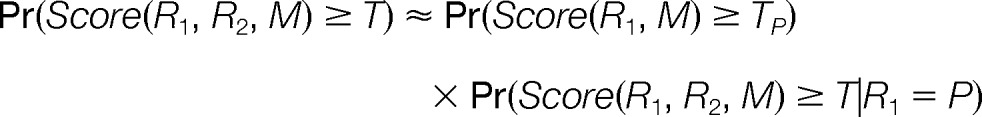 |
From here on, we refer to this approximation as the Product probability. This formulation is not exactly equivalent to the definition of joint probability because it fixes R1 = P in the conditional probability term (where P is the first peptide in the PPSM) and thus does not explicitly consider the dependences between all possible pairs of peptides that can be matched to the mixture spectrum. However, both single-peptide probability and conditional probability can be computed efficiently in linear time and we show in the next section that this approximation is sufficiently accurate for our main use of the joint probability – to separate correct from incorrect matches to mixture spectra.
Classification of Matches
Because a typical proteomics data set contains both single-peptide and mixture spectra we consider three possible outcomes when searching a given query spectrum M: (1) No-match: M does not match any peptide in the database; (2) Single-peptide match: M matches one peptide in the database; and (3) Mixture match: M matches a pair of peptides in the database. Every query spectrum is initially assumed to be a putative mixture spectrum and is assigned to its top-scoring PPSM. Then a two-step procedure is used to separate true mixture matches from false mixture matches. At the first stage, all PPSMs with joint probability less than a threshold are accepted. Then PPSMs with conditional probability less than a second threshold are accepted as Mixture-matches. The probability thresholds are determined in a way such that it enforces a selected false discovery rate (FDR, see next section). Next, all the remaining spectra that do not pass either probability threshold are reconsidered as single-peptide spectra. Each PPSM is converted into a PSM by considering the first peptide as the match to the spectrum. Single-peptide probabilities are computed for all PSMs and a probability threshold is determined to enforce a selected FDR for single-peptide spectra. A graphical illustration of this classification procedure is provided in the Fig. 1.
Fig. 1.
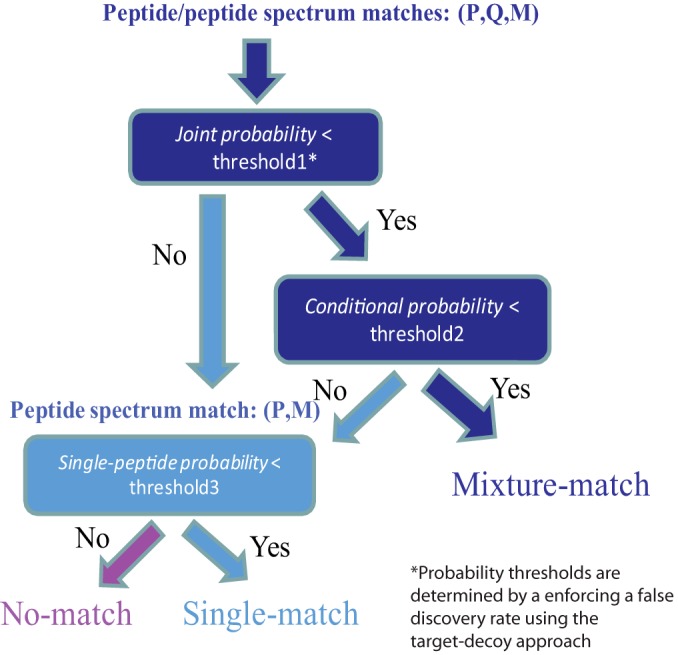
Classification of matches. All query spectra are first assumed to be putative mixture spectra and their top-scoring PPSMs are considered. PPSMs with joint and conditional probabilities passing a particular threshold are classified as Mixture-match - spectra that match to two peptides in the database. The probability thresholds for joint and conditional probability are determined by enforcing a chosen FDR using a target/decoy approach (37). Next, spectra that do not pass either probability threshold are treated as single-peptide spectra by considering the first peptide in each PPSM as the peptide match to the spectrum. Then single-peptide probabilities are calculated and those with probability passing a FDR-imposed threshold are considered as Single-matches - spectra that match to only one peptide in the database.
Estimation of False Discovery Rates
In the classification steps of PPSMs, the probability thresholds are determined to enforce a certain FDR. For the joint probability we are interested in the FDR that an incorrect mixture match is accepted either as half-correct or correct match:
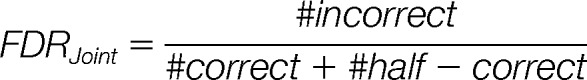 |
For the conditional probability we only want to accept correct matches so we are interested in the FDR where either half-correct or incorrect matches are accepted as correct mixture matches:
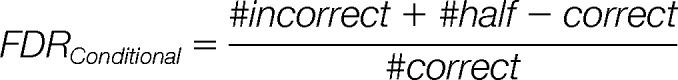 |
Each of the above FDRs is estimated by extending the Target-Decoy Approach (TDA) for single-peptide spectra (37). However, the assumptions used in TDA first need to be generalized to the case of PPSMs and their validity also needs to be tested (the detailed derivation of the TDA approach for PPSMs is given in the Supplementary Material). In brief, for a set of PPSMs if we define TT to be the number of PPSMs where both peptide matches are from the target database; TD or DT to be the number of cases where one peptide is from the target and the other peptide is from the decoy database and DD to be the cases where both peptides are from the decoy database, the two FDRs mentioned above can be computed using the following formulae:
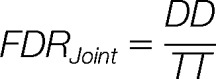 |
and
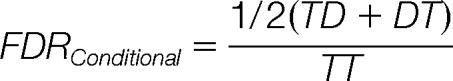 |
Finally, for the single-peptide probability the FDR is estimated using the standard TDA approach:
 |
where T is the number of PSMs from the target database and D is the number PSMs from the decoy database.
In summary two types of matches can be returned by MixGF: Single-match and Mixture-match. FDRSingle enforces the FDR for Single-match, whereas FDRJoint and FDRConditional enforce the FDR for Mixture-match. All three FDR operates on PSMs level (a Mixture-match is essentially treated as two PSMs, see Supplementary Material). Therefore to enforce a global FDR of 1% for all matches returned by MixGF, all three FDR thresholds were set to 1%.
Data sets and Data Processing
The performance of MixGF was first evaluated on a set of simulated mixture spectra (21). In brief, mixture spectra were created by linearly combining two single-peptide spectra with predefined mixture coefficients α – a parameter that reflects the relative abundance of the two peptides in the mixture spectrum. In addition, MixGF was tested on three data sets (38, 39, 40) representing typical experimental setups in proteomics studies. In brief, the Yeast data set (38) is from a tryptic digest of Saccharomyces cerevisiae that was analyzed on an LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific) and MS/MS spectra were acquired using a data-dependent scanning mode in which each full MS scan (m/z 300–2000) was acquired on the Orbitrap at resolution 60,000, followed by eight MS/MS scans collected on the LTQ. Two Human data sets were also analyzed. The Human-L data set (39) (which stands for human data set with low mass accuracy MS/MS) is from a tryptic digest of HEK293 cell lysate that was fractioned using Strong Cation Exchange (SCX) and each fraction was analyzed by an LTQ Orbitrap XL ETD mass spectrometer (Thermo Fisher Scientific) in data dependent mode. MS full scans were acquired from m/z 350–1500 with a resolution of 60,000. The two most intense ions were fragmented in the linear ion trap using CID and ETD. The Human-H data set (40) (human data set with high mass accuracy) is from a tryptic digest of Human LOVO cell lysates. The sample was analyzed using an LTQ-Orbitrap XL mass spectrometer operated in data-dependent mode with MS full scans acquired at resolution 30,000 and MS/MS (CID and ETD) for the three most abundant peptides recorded at resolution 7500. For the Human data sets, only CID spectra were considered in the present study. All database searches were performed with precursor m/z tolerance of 3.0 Th to allow for the possible identification of co-eluting peptides in mixture spectra (the instrument does not record any parent mass for the least abundant peptide). Only tryptic peptides were considered and only precursor charge of two and three are considered; no variable modifications and only carbamidomethylation on cystein (C+57.021) was considered as a fixed modification. The MS/MS fragment mass tolerance was set to 0.5 Da for the low mass accuracy data and 0.05 Da for the high mass accuracy human data. For ProbIDtree, we separated all the spectra into two sets depending on whether ProbIDtree identified only one or multiple peptides match to the spectrum, then an FDR was enforced using the standard TDA method (37) for each subset. As shown before (8) the rationale for separate FDR determination is that an FDR calculation combining both mixture and single-peptide spectra leads to underestimation of FDR for mixture spectra. M-SPLIT searches were done with 3.0Da parent mass tolerance against the yeast (ver. 05/04/2009) and human (ver. 01/14/2010) spectral libraries downloaded from National Institute of Standards and Technology (NIST) (41). The yeast spectral library contain 86,861 unique peptide ions (i.e. same peptides with different charge states are counted as different ions), whereas the human spectral library contains 343,301 unique peptide ions. The protein sequence databases used were the SGD yeast protein database (ver.5/8/2009) and the Human protein database (downloaded from NCBI refseq, ver.10/29/2010).
RESULTS
Separating True and False Mixture Spectrum Matches
As described above, our main goal of computing the statistical significance of PPSMs is to separate correct mixture matches from half-correct and incorrect matches. To test MixGF's ability in these tasks, we built a set of simulated mixture spectra by linearly combining pairs of single-peptide spectra same as before (21). Because we know a priori the peptides that generated each simulated mixture spectrum, we can extract the top-scoring correct, half-correct, and incorrect matches returned by MixDB and compute their joint and conditional probabilities. As shown in Fig. 2A, joint probability performs very well when separating correct matches from incorrect matches but there is considerable overlap between the joint probability of correct-matches and that of half-correct matches (see Fig. 2B). Further investigation of cases in the overlap region shows that for correct-matches usually both peptides contribute moderate scores to the final combined score but for the half-correct matches the correct peptide often contributes a very high score and thus even when paired with an incorrect match, the resulting combined high score still yields a low joint probability. Intuitively in order to separate half-correct matches from correct matches we need to look for cases that have high combined score as well as both peptides contributing significantly to the total score. The concept of conditional probability defined above aims to address exactly this question—is the score of the peptide pair (P, Q) significantly higher than that of the single peptide P? As illustrated in Fig. 2C, conditional probability is indeed better at separating correct matches from half-correct matches. Therefore, a two-step procedure is used to separate correct matches from false matches: at the first stage of MixGF, joint probability is used to filter out incorrect matches and then conditional probability is used to filter out half-correct matches.
Fig. 2.
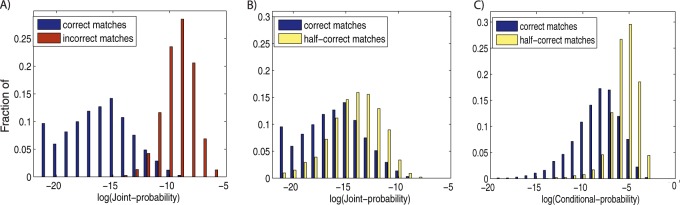
Separating true matches from false matches. Mixture spectra were simulated by a linear combination of two single-peptide spectra. Correct matches are cases where both peptides in a PPSM are correct, incorrect matches are cases where both peptides are incorrect, and half-correct matches are cases where one peptide is correct and one peptide is incorrect. The distribution of joint probability and conditional probability for correct matches (blue bars), incorrect matches (red bars), and half-correct matches (yellow bars) are shown. As shown in A, the distributions of joint probability are well-separated between correct and incorrect matches. However, there is considerable overlap between the joint probability distribution of correct matches and half-correct matches (see B). On the other hand, conditional probability is a better approach for separating correct matches from half-correct matches as shown in C).
Approximating Joint by the Product of Conditional Probability
To test whether the approximation of joint probability is accurate, The joint and product probability were computed for each spectrum in the simulated mixture spectra data set. As shown in Fig. 3, in most cases the joint probability is accurately approximated by product probability as most data points clustered tightly along the main diagonal. For correct matches, the product probability is sometimes lower than the true joint probability. This can be attributed to the fact that the approximation does not explicitly consider all pairs of peptides – because P is fixed, there are less opportunities for false positive matches to achieve high scores and thus the resulting spectral probability can be smaller in such cases. However, the range of probabilities where this underestimation occurs is well below the range where incorrect matches tend to occur. Therefore, for the purpose of separating correct matches from incorrect matches, using the approximation is nearly equivalent to computing the exact joint probability. As shown in Fig. 3B, correct matches and incorrect matches remain very well-separated using either the product or joint probability. In addition, the product probability can be computed much more efficiently than the joint probability. In practice the average run time for joint probability is 205.7 s per PPSM, whereas the average time it takes to compute the product probability is only 0.12 s on a Windows 7 machine with an Intel Xeon(R) E5430 CPU, resulting in a ≈1700 times speedup. This makes MixGF computation time in similar scale as searching for the top-scoring PPSMs by MixDB which takes 0.49 s per spectrum on the Yeast data set and 1.08 s on the Human data sets with a 3.0Th precursor mass tolerance.
Fig. 3.
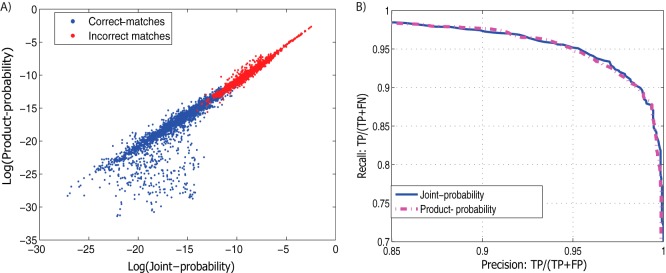
Approximation of joint probability. Because it is computationally expensive to compute the exact joint probability for a mixture spectrum (scales exponentially with the number of peptides), we approximates it using the product of single-peptide and conditional probabilities, which can be computed in linear time. As shown in A, for most cases the product probability accurately approximate the joint probability (most points cluster tightly around the main diagonal). For correct-matches, there are some cases falling below the main diagonal showing that the approximated probability is sometimes lower than the true joint probability for correct matches. However, as shown in the Precision/Recall curve in B, for the practical purpose of distinguishing correct-matches and incorrect-matches, the two distributions remain very well separated using either the exact joint probability or its approximation.
Joint and Product Probability Improve the Detection of Mixture Spectra
For mixture spectra, we expect joint probability to perform better at separating correct matches from incorrect matches by explicitly considering two peptides. Intuitively we expect that single-peptide probabilities for correct peptide matches to mixture spectra to be higher (i.e. worse) than those for correct matches to single-peptide spectra. This is because the presence of a second peptide in mixture spectra will allow more peptides to match to the spectrum with high score. However, for false matches the single-peptide probability distribution remains comparable for both single-peptide and mixture spectra because they are random matches in either case. Therefore, the distribution of single-peptide probabilities between correct and incorrect matches should be less well-separated for mixture spectra than for single-peptide spectra. To show this we used the simulated mixture spectra where the first peptide is mixed with a second peptide at 100%, 50%, 30% of the first peptide's total intensity and then computed the single-peptide probability, joint probability and product probability for the correct matches as well as the top-scoring incorrect matches. The performance of each probability function in separating correct from incorrect matches is shown in Table I. As expected, when the second peptide is at relatively low abundance (i.e. 30%), the FDR-controlled performance of single-peptide probability is nearly identical to that of joint probability because the mixture spectra are more similar to single-peptide spectra than at higher second-peptide abundances. However, as we increase the relative abundance of the second peptide, joint probability performs considerably better at separating correct matches from incorrect matches. Thus, we expect that as mixture spectra with more peptides become more common in experiments, joint probability and its product probability approximation will substantially improve our ability to identify mixture spectra.
Table I. Sensitivity for accepting correct Peptide–Peptide Spectrum Matches: A set of simulated mixture spectra were constructed with mixture coefficients α = 1.0,0.5,0.3. Single-peptide probability, joint-probability and product probability were computed for the correct mixture matches as well as the top-scoring incorrect mixture matches returned by MixDB. Then each probability was used to separate correct from incorrect mixture matches. The false discovery rate is computed as the ratio of the number of accepted incorrect mixture matches over the total number accepted matches. The sensitivity of accepting correct matches at different FDR levels is shown.
| Mixture coefficient | Probability | False discovery rate |
||
|---|---|---|---|---|
| 1% | 2% | 5% | ||
| α = 1.0 | Single-peptide probability | 71.9 | 74.7 | 81.8 |
| Joint probability | 93.4 | 94.7 | 96.6 | |
| Product probability | 93.6 | 94.0 | 96.7 | |
| α = 0.5 | Single-peptide probability | 85.7 | 87.5 | 92.0 |
| Joint probability | 93.5 | 94.3 | 96.0 | |
| Product probability | 92.6 | 94.1 | 96.1 | |
| α = 0.3 | Single-peptide probability | 89.4 | 90.8 | 92.8 |
| Joint probability | 90.2 | 91.8 | 93.8 | |
| Product probability | 90.4 | 91.7 | 93.8 | |
Identification of Mixture Spectra in Complex Biological Samples
To illustrate MixGF's ability to identify mixture spectra in realistic scenarios, we evaluated its performance on one yeast (38) and two human data sets (39, 40) that represent typical proteomics analysis of whole cell lysates. We compared the performance of MixGF with two current database search methods for identification of mixture spectra: MixDB and ProbIDtree. As shown in Table II, MixGF is able to outperform MixDB and ProbIDtree by identifying 30–76% and 160–390% more mixture spectra, respectively. To estimate their sensitivity, the various database search methods were also benchmarked against M-SPLIT, a spectral library search method able to identify mixture spectra. In general, spectral library search methods have shown to be more sensitive than database search methods (42, 21) because of their smaller search space and the known peptide fragmentation contained in the spectral library (43). Thus, the number of mixture spectra identified by M-SPLIT can be used as an estimate of the upper bound of mixture spectra that can be identified in each sample by database search methods. As shown in Table II, whereas MixDB and ProbIDtree only identify an average of 50 and 21% of the mixture spectra identified by M-SPLIT (respectively), MixGF is closing the gap between database search and spectral-library search methods by identifying up to 84% of the mixture spectra identified by library search (in the Human-H data set). Comparing search results across different data sets reflects their varying sample complexity. In the yeast data set, the number of mixture spectra identified by MixGF is only 4.71% of the single-peptide spectra. However, in the human data set the number of identified mixture spectra increased to 17.8–23.7% of the identified single-peptide spectra. This shows that as the complexity of the sample increases, more peptides with similar precursors will co-elute at same retention time and thus mixture spectra indeed constitute a significant fraction of all MS/MS spectra in the data. Such observations have been reported in studies showing that multiple precursors often coincide within the same MS/MS precursor isolation window for as many as 50% of all MS/MS spectra (5, 4). But while it remains unclear what fraction of these cofragmented peptides is identifiable, here we show that a large number of co-eluting peptides can be identified. It is worth noting that because mixture spectra contain two peptides per spectrum, they potentially contain more information in each spectrum. For example in the yeast data set even though the number of mixture spectra identified by MixGF is only 4.71% of the single-peptide spectra, the number of unique peptides identified in mixture spectra is 22.1% of the total number of peptides identified in single-peptide spectra. For the human data sets the number of unique peptides identified in mixture spectra is a strikingly 50.3–65.24% of the number of peptides identified in single-peptide spectra. Of course, while many of the peptides identified in mixture spectra were also identified in single-peptide spectra, the combined result still leads to a gain of 23.4–34.5% more unique peptides identified than single-peptide spectra. The comparison between the results in Human-L and Human-H data set also reveals that the high mass accuracy available in the Human-H data enables more mixture spectra to be identified. The total numbers of spectra identified in both human data sets are similar but the fraction of all identified spectra that are mixture spectra increased by 33.2% in the Human-H data set. Analyzing the data with a regular search engine, MSGFDB (39), shows that it was able to identify a similar number of spectra as MixGF, indicating that a large fraction of mixture spectra were also identified as single-peptide matches by MSGFDB. However, MixGF's ability to identify two peptides per mixture spectrum leads to an improvement of 13.3–15.4% in the total number of unique peptide identifications compared with a conventional analysis tool.
Table II. Identification of mixture spectra in complex biological sample: MixGF, MixDB, ProbIDtree, MSGFDB and M-SPLIT were tested on three datasets that are from tryptic digest of yeast and human cell lysate respectively. Numbers of spectra and unique peptides identified by each tool at 1% FDR are summarized. “Single” indicates spectra from which only one peptide is identified and “Mixture” indicates spectra from which more than one peptides are identified. *Note that M-SPLIT is a spectral library search tool. Since spectral library search method is generally considered to be more sensitive, its result is included in the comparison as an estimate of the upper bound of the number mixture spectra that can be identified by database search tools in each dataset. **The search of the Human-H dataset using ProbIDtree did not finish.
| Data set | Method | Identified spectra |
Identified peptides |
||||
|---|---|---|---|---|---|---|---|
| Single | Mixture | Total | Single | Mixture | Total | ||
| Yeast | ProbIDtree | 21807 | 504 | 22311 | 4826 | 495 | 4936 |
| MixDB | 25033 | 748 | 25778 | 5702 | 895 | 5924 | |
| MixGF | 28022 | 1320 | 29342 | 6315 | 1398 | 6637 | |
| MSGFDB | 26657 | n/a | 26657 | 5752 | n/a | 5752 | |
| M-SPLIT* | 28417 | 2053 | 30470 | 5997 | 2033 | 6684 | |
| Human-L | ProbIDtree | 28614 | 1433 | 30036 | 8479 | 1675 | 9153 |
| MixDB | 38855 | 5420 | 44275 | 13021 | 5735 | 15298 | |
| MixGF | 39701 | 7052 | 46783 | 13027 | 6982 | 16080 | |
| MSGFDB | 46137 | n/a | 46137 | 14027 | n/a | 14027 | |
| M-SPLIT* | 49585 | 8425 | 58023 | 16504 | 8300 | 19826 | |
| Human-H | ProbIDtree** | – | – | – | – | – | – |
| MixDB | 34790 | 5395 | 40185 | 10317 | 4325 | 12350 | |
| MixGF | 35760 | 8462 | 44222 | 10280 | 6707 | 13824 | |
| MSGFDB | 46674 | n/a | 46674 | 12202 | n/a | 12202 | |
| M-SPLIT* | 45680 | 10935 | 56615 | 12447 | 7988 | 16363 | |
DISCUSSION
It is increasingly being recognized that MS/MS spectra from more than one peptide are common and important in proteomics data (4, 5) but processing them will require the development of accurate computational tools to identify multiple peptides in each MS/MS spectrum. Two fundamental questions need to be addressed in the pursuit of this goal: (1) to separate correct multiple-peptide-spectrum matches (mPSMs) from false positive matches, and (2) to estimate the false positive or false discovery rate (FDR) in a set of mPSMs. Here we addressed these questions by computing the statistical significance of mPSMs for the special case of two peptides per spectrum. Given an MS/MS spectrum, a database search tool can always return a top-scoring peptide or multiple peptides matched to the query spectrum and by random chance, it is always possible for false peptide matches to also obtain high scores. This is especially true in the case of mixture spectra because the explosion in the search space dramatically increases the occurrence of high-scoring false matches. Thus, it is crucial to be able to compute the statistical significance of mPSMs. Here we show that for the two-peptide case, it is possible to rigorously compute the statistical significance using the generating function approach and show that the joint and conditional probabilities can be used to separate true PPSMs from false positive matches. We further show that the computationally-expensive joint probability can be accurately approximated in our range of interest using a product of probabilities that can be computed efficiently in linear time, resulting in a ≈1700 times speedup and thus potentially allowing MixGF to scale to an arbitrary number of peptides. In order to estimate the false discovery rate (FDR) for mixture spectra, we extended the traditional target-decoy approach (TDA) to perform the database search using a concatenated target-decoy sequence database allowing for the estimation half-correct matches where one peptide in the PPSM is correct and one peptide is incorrect. This is important because these matches constitute a large fraction of false positive matches in mixture spectra and, as shown in Fig. 2, these are more difficult to separate from correct matches than cases when both peptides are incorrect.
Benchmarking MixGF performance on three data sets showed that MixGF identified 30.1–390% more mixture spectra than MixDB and ProbIDtree, respectively. In addition, we found that the number of unique peptides identified in mixture spectra can be up to 65% of the number of peptides identified in single-peptide spectra with up to 34.5% of all unique peptides being identified only in mixture spectra. Though, many of the peptides in mixture spectra were also identified by MSGFDB as single-peptide matches, making MixGF overall gain in peptide identification to be 13.3–15.4% compared with MSGFDB. These observations highlight the importance of relaxing the one-peptide-one-spectrum assumption when designing the next generation of computational tools for identifying MS/MS spectra as instruments advances and higher complexity samples are being analyzed in less time in proteomic experiments. This also illustrates the potential of emerging data acquisition protocols (9, 10, 11, 12, 13, 14, 15, 44) where multiple peptides are intentionally cofragmented in each MS/MS spectrum. Finally, even though the proposed MixGF approach focuses on mixture spectra from two peptides, the same approach should be extensible to more than two peptides. Focusing on the case of pairs allowed for a comprehensive assessment of the various performance aspects of the proposed approach and the analysis of several data sets showed that properly interpreting mixture spectra from two peptides already greatly improves the number of spectra and peptides identified in current experimental setups. Therefore solving the problem for the case of two peptides provides a foundation toward addressing the more general scenario of mixture spectra from any number of peptides.
Supplementary Material
Acknowledgments
We thank the National Institute of Science and Technology (NIST), ProteomeCommons, and Vanderbilt University for the public availability of the mass spectrometry data and Dr. Fedor Kryuchkov and Frank Kjeldsen for the availability of the Human-H data.
Footnotes
Author contributions: J.W., P.E.B., and N.B. designed research; J.W. performed research; J.W. and N.B. analyzed data; J.W., P.E.B., and N.B. wrote the paper.
* This work was supported in part by the National Institutes of Health grant GM078596 (JW and PEB) and 8 P41 GM103485-05 (JW and NB) from the National Center for Research Resources.
 This article contains supplemental Fig. S1 and Table S1.
This article contains supplemental Fig. S1 and Table S1.
1 The abbreviations used are:
- MS/MS
- tandem mass spectrometry
- M-SPLIT
- mixture-spectrum partitioning using libraries of identified tandem mass spectra
- PSM
- peptide spectrum match
- PPSM
- peptide/peptide spectrum match
- FDR
- false discovery rate
- PRM
- prefix residue mass
- mPSM
- multipeptide spectrum match
- DDA
- data dependent acquisition
- DIA
- data independent acquisition
- TDA
- target decoy approach.
REFERENCES
- 1. Washburn M. P., Wolters D., Yates J. R. (2001) Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 19, 242–247 [DOI] [PubMed] [Google Scholar]
- 2. Brunner E., Ahrens C. H., Mohanty S., Baetschmann H., Loevenich S., Potthast F., Deutsch E. W., Panse C., de Lichtenberg U., Rinner O., Lee H., Pedorli P. G. A., Malmstrom J., Koehler K., Schrimpf S., Krijgsveld J., Kregenow F., Heck A. J. R., Hafen E., Schlapbach R., Aebersold R. (2007) A high-quality catalog of the Drosophila melanogaster proteome. Nat. Biotechnol. 25, 576–583 [DOI] [PubMed] [Google Scholar]
- 3. Aebersold R., Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 4. Michalski A., Cox J., Mann M. (2011) More than 100,000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC MS/MS. J. Proteome Res. 10, 1785–1793 [DOI] [PubMed] [Google Scholar]
- 5. Luethy R., Kessner D. E., Katz J. E., MacLean B., Grothe R., Kani K., Faca V., Pitteri S., Hanash S., Agus D. B., Mallick P. (2008) Precursor-ion mass re-estimation improves peptide identification on hybrid instruments. J. Proteome Res. 7, 4031–4039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alves G., Ogurtsov A. Y., Kwok S., Wu W. W., Wang G., Shen R. F., Yu Y. K. (2008) Detection of co-eluted peptides using database search methods. Biol. Direct 3, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Houel S., Abernathy A., Renganathan K., Meyer-Arendt M., Ahn N. A., Old W. M. (2010) Quantifying the impact of chimera ms/ms spectra on peptide identification in large-scale proteomics studies. J. Proteome Res. 9, 4152–4160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang J., Bourne P. E., Bandeira N. (2011) Peptide identification by database search of mixture tandem mass spectra. Mol. Cell. Proteomics 10 10.12, M111-010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Masselon C., Paša-Tolić L., Lee S. W., Li L., Anderson G. A., Harkewicz R., Smith R. D. (2003) Identification of tryptic peptides from large databases using multiplexed tandem mass spectrometry: simulations and experimental results. Proteomics 3, 1279–1286 [DOI] [PubMed] [Google Scholar]
- 10. Venable J. D., Dong M. Q., Wohlschlegel J., Dillin A., Yates J. R., III (2004) Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat. Methods, 1, 39–45 [DOI] [PubMed] [Google Scholar]
- 11. Plumb R. S., Johnson K. A., Rainville P., Smith B. W., Wilson I. D., Castro-Perez J. M., Nicholson J. K. (2006) UPLC/MSE; a new approach for generating molecular fragment information for biomarker structure elucidation. Rapid Commun. Mass Sp. 20, 1989–1994 [DOI] [PubMed] [Google Scholar]
- 12. Chakraborty A. B., Berger S. J., Gebler J. C. (2007) Use of an integrated ms–multiplexed ms/ms data acquisition strategy for high-coverage peptide mapping studies. Rapid Commun. Mass Sp. 21, 730–744 [DOI] [PubMed] [Google Scholar]
- 13. Panchaud A., Scherl A., Shaffer S. A., von Haller P. D., Kulasekara H. D., Miller S. I., Goodlett D. R. (2009) Precursor acquisition independent from ion count: how to dive deeper into the proteomics ocean. Anal. Chem. 81, 6481–6488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Geiger T., Cox J., Mann M. (2010) Proteomics on an orbitrap benchtop mass spectrometer using all-ion fragmentation. Mol. Cell. Proteomics 9, 2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gillet L. C., Navarro P., Tate S., Röst H., Selevsek N., Reiter L., Bonner R., Aebersold R. (2012) Targeted data extraction of the ms/ms spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics 11 0111-016717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tabb D. L., Vega-Monototo L., Rudnick P., Mulayathy A., Ham A. J. L., Bunk M. D., Kilpatrick L.E., Billheimer D. D., Blackman R. K., Cardasis H. L., Carr S. A., Clauser K. R., Jaffee J. D., Kowalski K. A., Neubert T. A., Regnier F. E., Schilling B., Tegeler T. J., Wang M., Wang P., Whiteaker J. R., Zimmerman L. J., Fisher S. J., Gibson B. W., Kinsinger C. R., Mesri M., Rodriquez H., Stein S. E., Tempst P., Paulovich A. G., Liebler C., Spiegelman C. (2009) “Repeatability and Reproducibility in Proteomic Identifications by Liquid Chromatography–Tandem Mass Spectrometry.” Journal of Proteome Research 9, 761–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Blackburn K., Mbeunkui F., Mitra S. K., Mentzel T., Goshe M. B. (2010) Improving protein and proteome coverage through data-independent multiplexed peptide fragmentation. J. Proteome Res. 9, 3621–3637 [DOI] [PubMed] [Google Scholar]
- 18. Zhang N., Li X., Ye M., Pan S., Schwikowski B., Aebersold R. (2005) ProbIDtree: an automated software program capable of identifying multiple peptides from a single collision-induced dissociation spectrum collected by a tandem mass spectrometer. Proteomics 5, 4096–4106 [DOI] [PubMed] [Google Scholar]
- 19. Li G. Z., Vissers J. P. C., Silva J. C., Golick D., Gorenstein M. V., Geromanos S. J. (2009) Database searching and accounting of multiplexed precursor and product ion spectra from the data independent analysis of simple and complex peptide mixtures. Proteomics 9, 1696–1719 [DOI] [PubMed] [Google Scholar]
- 20. Bern M., Finney G., Hoopmann M. R., Merrihew G., Toth M. J., MacCoss M. J. (2009) Deconvolution of mixture spectra from ion-trap data-independent-acquisition tandem mass spectrometry. Anal. Chem. 82, 833–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang J., Perez-Santiago J., Katz J. E., Mallick P., Bandeira N. (2010) Peptide identification from mixture tandem mass spectra. Mol. Cell. Proteomics 9, 1476–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M. (2011) Andromeda: a peptide search engine integrated into the maxquant environment. J, Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 23. Kall L., Canterbury J. D., Weston J., Noble W. S., MacCoss M. S. (2007) Semi-supervised learning for peptide identification from shotgun proteomics data sets. Nat. Methods 4, 923–925 [DOI] [PubMed] [Google Scholar]
- 24. Kim S., Gupta N., Pevzner P. A. (2008) Spectral probabilities and generating functions of tandem mass spectra: a strike against decoy databases. J. Proteome Res. 7, 3354–3363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Choi H., Nesvizhskii A. I. (2007) Semisupervised model-based validation of peptide identifications in mass spectrometry-based proteomics. J. Proteome Res. 7, 254–265 [DOI] [PubMed] [Google Scholar]
- 26. Nesvizhskii A. I. (2010) A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J. Proteomics 73, 2092–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Klammer A. A., Park C. Y., Noble W. S. (2009) Statistical calibration of the sequest xcorr function. J. Proteome Res. 8, 2106–2113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Granholm V., Käll L. (2011) Quality assessments of peptide–spectrum matches in shotgun proteomics. Proteomics 11, 1086–1093 [DOI] [PubMed] [Google Scholar]
- 29. Sadygov R. G., Yates J. R. (2003) A hypergeometric probability model for protein identification and validation using tandem mass spectral data and protein sequence databases. Anal. Chem. 75, 3792–3798 [DOI] [PubMed] [Google Scholar]
- 30. Geer L. Y., Markey S. P., Kowalak J. A., Wagner L., Xu M., Maynard D. M., Yang X., Shi W., Bryant S. H. (2004) Open mass spectrometry search algorithm. J. Proteome Res. 3, 958–964 [DOI] [PubMed] [Google Scholar]
- 31. Fenyö D., Beavis R. C. (2003) A method for assessing the statistical significance of mass spectrometry-based protein identifications using general scoring schemes. Anal. Chem. 75, 768–774 [DOI] [PubMed] [Google Scholar]
- 32. Spirin V., Shpunt A., Seebacher J., Gentzel M., Shevchenko A., Gygi S., Sunyaev S. (2011) Assigning spectrum-specific p-values to protein identifications by mass spectrometry. Bioinformatics 27, 1128–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alves G., Yu Y-K. (2008) Statistical characterization of a 1d random potential problem–with applications in score statistics of ms-based peptide sequencing. Physica A 387, 6538–6544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim S., Gupta N., Bandeira N., Pevzner P. A. (2009) Spectral dictionaries: Integrating de novo peptide sequencing with database search of tandem mass spectra. Mol. Cell. Proteomics 8, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dancík V., Addona T. A., Clauser K. R., Vath J. E., Pevzner P. A. (1999) De novo peptide sequencing via tandem mass spectrometry. J. Comput. Biol. 6, 327–342 [DOI] [PubMed] [Google Scholar]
- 36. Kim S., Pevzner P. A. Universal database search tool for mass spectrometry. submitted for publication. [Google Scholar]
- 37. Elias J. E., Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 38. Li J., Zimmerman L. J., Park B. H., Tabb D. L., Liebler D. C., Zhang B. (2009) Network-assisted protein identification and data interpretation in shotgun proteomics. Mol. Syst. Biol. 5, 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim S., Mischerikow N., Bandeira N., Navarro J. D., Wich L., Mohammed S., Heck A. J. R., Pevzner P. (2010) The generating function of CID, ETD, and CID/ETD pairs of tandem mass spectra: applications to database search. Mol. Cell. Proteomics 9, 2840–2852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kryuchkov F., Verano-Braga T., Hansen T. A., Sprenger R. R., Kjeldsen K. (2013) Deconvolution of mixture spectra and increased throughput of peptide identification by utilization of intensified complementary ions formed in tandem mass spectrometry. J. Proteome Res. 12, 3362–3371 [DOI] [PubMed] [Google Scholar]
- 41. Eds. Stein S. E., Rudnick P. A. NIST Peptide Tandem Mass Spectra LIbraries. Yeast Peptide Mass Spectral Reference Data, ion trap, 2009, National Institute of Standards and Technology, Gaithersburg, MD, 20899 [Google Scholar]
- 42. Lam H., Deutsch E. W., Eddes J. S., Eng J. K., King N., Stein S. E., Aebersold R. (2007) Development and validation of a spectral library searching method for peptide identification from ms/ms. Proteomics 7, 655–667 [DOI] [PubMed] [Google Scholar]
- 43. Zhang X., Li Y., Shao W., Lam H. (2011) Understanding the improved sensitivity of spectral library searching over sequence database searching in proteomics data analysis. Proteomics 11, 1075–1085 [DOI] [PubMed] [Google Scholar]
- 44. Weisbrod C. R., Eng J. K., Hoopmann M. R., Baker T., Bruce J. E. (2012) Accurate peptide fragment mass analysis: multiplexed peptide identification and quantification. J. Proteome Res. 11, 1621–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.