

Abstract
Aim
Further to its pivotal role in haemostasis, factor Xa (FXa) promotes effects on the vascular wall. The purpose of the study was to evaluate if FXa modifies the expression level of energy metabolism and oxidative stress-related proteins in femoral arteries obtained from type 2 diabetic patients with end-stage vasculopathy.
Methods
Femoral arteries were obtained from 12 type 2 diabetic patients who underwent leg amputation. Segments from the femoral arteries were incubated in vitro alone and in the presence of 25 nmol l−1 FXa and 25 nmol l−1 FXa + 50 nmol l−1 rivaroxaban.
Results
In the femoral arteries, FXa increased triosephosphate isomerase and glyceraldehyde-3-phosphate dehydrogenase isotype 1 expression but decreased pyruvate dehydrogenase expression. These facts were accompanied by an increased content of acetyl-CoA. Aconitase activity was reduced in FXa-incubated femoral arteries as compared with control. Moreover, FXa increased the protein expression level of oxidative stress-related proteins which was accompanied by an increased malonyldialdehyde arterial content. The FXa inhibitor, rivaroxaban, failed to prevent the reduced expression of pyruvate dehydrogenase induced by FXa but reduced acetyl-CoA content and reverted the decreased aconitase activity observed with FXa alone. Rivaroxaban + FXa but not FXa alone increased the expression level of carnitine palmitoyltransferase I and II, two mitochondrial long chain fatty acid transporters. Rivaroxaban also prevented the increased expression of oxidative stress-related proteins induced by FXa alone.
Conclusions
In femoral isolated arteries from type 2 diabetic patients with end-stage vasculopathy, FXa promoted disruption of the aerobic mitochondrial metabolism. Rivaroxaban prevented such effects and even seemed to favour long chain fatty acid transport into mitochondria.
Keywords: energy metabolism, factor Xa, femoral arteries, oxidative stress, protein expression
What is Already Known about this Subject
Factor Xa (FXa) modifies the vascular wall which suggests that FXa is a modulator of vascular function.
Patients with type 2 diabetes mellitus have an increased risk of cardiovascular events associated with thrombotic and pro-coagulant conditions.
In diabetes, abnormalities in vascular energy metabolism contribute to vascular dysfunction and higher risk of cardiovascular events
What this Study Adds
FXa stimulates the glycolytic pathway in diabetic arteries but not to pyruvate anaerobic catabolism.
There is no involvement of long chain fatty acid beta oxidation as alternative source to increase acetyl-CoA in FXa-incubated arteries.
FXa changes the expression level of glucose oxidation-related proteins suggesting disruption of mitochondrial metabolism.
Introduction
During thrombosis, the vascular wall is exposed to clotting factors, including the procoagulant protease factor Xa (FXa). FXa is a critical convergence point for the intrinsic and extrinsic coagulation pathways. FXa is produced from factor X and catalyzes the conversion of pro-thrombin into thrombin during blood coagulation 1.
Further to its key function as a clotting enzyme, FXa also produces effects on the vascular wall suggesting the involvement of FXa as a modulator of vascular function. In this regard, and as an example, systemic administration of FXa to anaesthetized rats caused a hypotensive response associated with the release of nitric oxide from rat isolated aortic rings 2,3. Moreover, in cultured endothelial cells FXa reduced the intracellular calcium transients and stimulated cell proliferation and pro-inflammatory cytokine production 4–6.
Patients with type 2 diabetes mellitus have an increased risk of cardiovascular events associated with thrombotic and pro-coagulant conditions 7. In this regard, data have shown that diabetic patients with vascular complications are a diabetic subgroup of patients with increased risk of coagulation disorders and fibrinolysis 8. In this regard, the vascular wall of diabetic patients with end-stage vasculopathy has a higher risk to increase platelet activation. Interestingly, it has been demonstrated that platelets from both experimental diabetic animals and diabetic patients are an important source for FXa generation, which may contribute to increase the local concentration of FXa, enhancing the exposition of the diabetic vascular wall to FXa during thrombosis 9.
Vascular function is a complex processes that it is dependent on several molecular mechanisms and among them vascular energetic metabolism has a relevant role 10. The arterial wall is supplied with oxygen and nutrients and aerobic glycolysis is considered characteristic of its physiological metabolism 11. In this regard, it is known that in diabetic patients, abnormalities in vascular energy metabolism contribute to vascular dysfunction and, therefore, to a higher risk of cardiovascular events 12.
Oxidative stress is also closely associated with disturbed coagulation 13. Moreover, a close relationship between energy metabolism and oxidative stress has been also postulated. In this regard, mitochondria are not only key in aerobic metabolism but they are also the major site of reactive oxygen species (ROS) production. Several reports have associated mitochondrial dysfunction with type 2 diabetes mellitus 14–16. However, to our knowledge, oxidative actions of FXa have only been previously been reported in human smooth muscle cells from saphenous veins but not yet explored in the arterial wall of diabetic patients 17.
Therefore, trying to resemble a coagulating situation, where probably FXa concentration should be locally increased, our aim was to determine if exogenous addition of FXa to femoral arteries from patients with type 2 diabetes mellitus and end-stage vasculopathy could modify the expression level of proteins associated with glycolytic metabolism and/or with oxidative stress.
Methods
Collection of femoral arteries and in vitro incubations
Femoral arterial segments were obtained from 12 patients diagnosed of type 2 diabetes mellitus undergoing leg amputations by thrombosis. Type 2 diabetes was defined according to the Clinical Guidelines Task Force from the International Diabetes Federation 18. Patients were excluded if they were under anticoagulant and/or antiplatelet treatment 10 days before surgery or the cause of leg amputation was related to infectious disease i.e. osteomyelitis and phlegmon. The investigation conformed to the principles outlined in the Declaration of Helsinki. All subjects gave fully informed consent and the Institutional Ethics Committee approved the study.
The femoral artery was carefully isolated, washed with serum saline and cut into portions (aproximately 5 mm each). Each portion was incubated in RPMI medium containing 1% fetal calf serum, 5 mmol l−1 glutamine, 0.01 mmol l−1 L-arginine, 2 × 10−5μg l−1 streptomycin and 2 × 10−5 U l−1 penicillin. All the procedures were performed under sterile conditions.
From each femoral artery segments were incubated in triplicate as follows: (a) none (control), (b) 25 nmol l−1 FXa and (c) 25 nmol l−1 FXa + rivaroxaban (Bay 59-7939) (50 nmol l−1), an specific reversible inhibitor of FXa activity 19. Rivaroxaban was added to the incubation medium 5 min before FXa.
The FXa concentration used was chosen according to previous in vitro experiments that demonstrated stimulation of pro-collagen production and induction of fibroblasts proliferation for this FXa concentration 19. Rivaroxaban was purchased from Bayer Health Care AG and the chosen concentration was based on previous studies that demonstrated that this concentration fully inhibited FXa activity 20. Furthermore, the rivaroxaban concentration used in the present experiments is clinically relevant 21,22. In vitro incubations were followed up for 18 h and after that, the femoral arterial segments were collected and stored at −80°C until the molecular determinations were performed.
Two-dimensional electrophoresis (2-DE) and mass spectrometry (MS) analysis
Femoral arterial samples were homogenized with an Ultra-Turrax T8 IKA-Werke in a buffer containing 8 mol l−1 urea, 2% CHAPS w: v, 40 mmol l−1 dithiothreitol, 0.2% Bio-LyteTH ampholyte (Bio-Rad) and 0.01% w : v bromophenol blue. After incubation in a wheel revolving for 12 h at 4°C, the homogenate tissues were centrifuged at 10 000 g for 10 min and the supernatant stored at −80°C until further analysis. Protein determination was performed using bicinchoninic acid reagent (Pierce). Before analysis, samples were submitted to cleanup for isoelectric focusing and 2-DE electrophoresis using a commercial kit, ReadyPrep 2-D Cleanup Kit (Bio-Rad Laboratories, USA) following the manufacturer's instruction. Total protein (250 μg) was then loaded onto each gel on immobilized gradient IPG strips (pH 3–10) and isoelectric focusing was performed using a Protean IEF cell system (Bio-Rad Laboratories, USA) as reported 23. In the second dimension, the proteins from femoral arteries were resolved on 10% SDS-PAGE gels using a Protean II XL system (Bio-Rad Laboratories, USA) as reported 24. After electrophoresis, the gels were fixed and silver stained, following the manufacturer's recommendations using a Silver Stain Plus Kit (Bio-Rad laboratories). The gels were then scanned using a UMAX POWERLOOK III Scanner operated by the software Magic Scan V 4.5 and the image analysis was performed using Quantity One 4.2.3 (Bio-Rad Laboratories, USA). Each spot intensity volume was processed by background subtraction.
For MS, spots were extracted from silver-stained 2-DE gels and digested with sequence-grade modified trypsin (Promega, Madison, WI, USA) as reported 25. Samples were analyzed using a 4700 Proteomic Analyzer (Applied Biosystem, Old Connecticut Path, Framingham, MA, USA) operated in reflector positive mode. The data generated from spectra were used to identify proteins by peptide mass finger printing, using the MASCOT program and comparing with the Mascot database 1.9 (http://www.matrixscience.com). As mentioned in previous papers 24,25, MS identifications were accepted based on a tripartite evaluation that takes into account significant molecular weight search (worse) scores, spectrum annotation and observed vs. expected migration on 2-DE gels.
Western blot analysis
Protein expression levels of triosephosphate isomerase, pyruvate dehydrogenase, lactate dehydrogenase, carnitine palmitoyltransferase (CPT) I and II (CPT-I and CPT-II) and the gp91-phox, p47-phox and p67-phox subunits of NADPH oxidase were analyzed by Western blot. In brief, the homogenized vascular segments were solubilized in Laemmli buffer containing 2-mercaptoethanol. The obtained proteins were separated on denaturing SDS-PAGE 15% (w: v) polyacrylamide gels. Equal amounts of proteins (20 μg/lane), estimated by bicinchoninic acid reagent (Pierce), were loaded. Proteins were blotted onto nitrocellulose (Immobilion-P; Millipore, USA) and the blots were blocked overnight at 4°C with 5% (w: v) non-fat dry milk. Membranes were then incubated with different antibodies against each of the above-mentioned proteins. For this purpose, triosephosphate isomerase was determined using a polyclonal antibody (TIM (FL-249); sc-30145, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA, dilution 1:500). Lactate dehydrogenase was determined using a monoclonal antibody (sc-133123 Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA, dilution 1:100). Pyruvate dehydrogenase was determined by using a polyclonal antibody (PDK1 sc-7140 dilution 1:1000, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA). CPT-I and CPT-II and gp91-phox NADPH were determined using polyclonal antibodies at dilution 1:1000 purchased from Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA (sc-20670, sc20526 and sc-5827, respectively). The cytosolic NADPH oxidase gp47-phox and gp67-phox subunits were determined using polyclonal antibodies at dilution 1:1500 purchased from Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA (sc-14015 and sc-7663, respectively). The same samples were also incubated with a monoclonal anti-β-actin antibody as loading control. After overnight incubation at 4°C, nitrocellulose membranes were washed twice and incubated with a peroxidase-conjugated anti-goat IgG antibody (for pyruvate dehydrogenase, CPT-II, and the gp91-phox and gp67-phox NADPH oxidase isotypes), a peroxidase-conjugated anti-rabbit IgG antibody (for triosephosphate isomerase, CPT-I and p47-phox NADPH oxidase) and with an anti-mouse IgG antibody (for lactate dehydrogenase and β-actin), and developed with enhancing chemoluminiscence reagents (ECL; GE Healthcare, Little Chalfont Buckinghamshire, UK).
Determination of lactate, acetyl-coenzyme A (CoA) and malonyldialdehyde (MDA) content and aconitase activity in the femoral arteries
The content of lactate in the human aortic samples was determined using a lactate assay kit (K607-100; BioVision Incoporated CA, USA) following the manufacturer's instructions.
In the femoral arteries, the content of acetyl-CoA was determined using a double-antibody sandwhich enzyme-linked immunosorbent assay (ELISA) kit (Shanghai Qayee Biotechnology Co., Shanghai, China). The MDA content in the femoral arteries was also determined by an enzyme immunoassay technique using a commercial kit (Cloud-Clone Corporation, USA) following the manufacturer's instructions.
Aconitase activity was determined using a commercial colorimetric assay kit (BioVision Incorporated, CA, USA) following the manufacturer's instructions.
To determine the lactate, acetyl-CoA and MDA content and the aconitase activity in the femoral arteries, 80 μg of each homogenized femoral artery was used.
Statistical analysis
Results are expressed as mean ± SEM. Wilcoxon's test was used to determine statistical significances. To control the potential antioxidant effects of statin treatment multivariate linear regression analyses was performed. The dependent variable was the level of MDA content and the level of expression of the oxidative-related proteins in the femoral arteries, the fixed factor was the sample type (control, FXa- and FXa + rivaroxaban-incubated arteries) and statin treatment was used as a covariant. A P value <0.05 was considered statistically significant.
Results
Effect of FXa in the level of expression of glycolytic-related proteins in the femoral arterial wall
Table 1 shows the clinical features of the included patients. All the included patients had diabetes mellitus and they had not undergone any antiplatelet and/or anticoagulant treatments from at least 10 days before the femoral artery was obtained. At inclusion, a number of patients were under statin treatment.
Table 1.
Clinical features of the included type 2 diabetes mellitus patients
| Parameters | Patients (n = 12) |
|---|---|
| Age (years) | 73.8 ± 2.65 |
| Male/Female | 11/1 |
| HbA1c (%) | 7.90 ± 1.62 |
| Risk factors (%) | |
| Hypertension | 9/12 (75%) |
| Dyslipidaemia | 4/12 (33%) |
| Diabetes mellitus | 12/12 (100%) |
| Previous history | |
| Congestive cardiac failure | 3/12 (25.0%) |
| Acute myocardial infarction | 3/12 (25.0%) |
| Cerebral infarction | 3/12 (25.0%) |
| Pharmacological treatment | |
| Statins | 7/12 (58.3%) |
| Oral anti-diabetic drugs + diet | 4/12 (33.3%) |
| Insulin + diet | 6/12 (50.0%) |
| Diet alone | 2/12 (16.6%) |
Results are represented as mean ± SEM.
In the proteomic analysis the studied spots were those expressed in at least 80% of the 2-DE gels within each experiment group. MS analysis was only performed in spots in which statistical differences between control and FXa-incubated arteries were observed. Table 2 shows the MS features of such spots identified in the femoral arteries.
Table 2.
Proteins identified using mass spectrometry after 2-DE electrophoresis sorted according with the metabolic pathways

In FXa-incubated femoral arteries the protein expression of two identified triosephosphate isomerase isoforms was increased as compared with those arteries incubated under control conditions (Table 3). This greater expression observed in the FXa-incubated femoral arteries was further confirmed by Western blot experiments (Figure 1A, B). Moreover, the protein expression of glyceraldehyde-3-phosphate dehydrogenase isoform 1 was also higher in FXa-incubated femoral arteries than in controls (Table 3).
Table 3.
Level of expression energy metabolism-related proteins in the femoral artery
| Protein | Control (AU) | FXa (AU) | FXa + rivaroxaban (AU) | |
|---|---|---|---|---|
| Energetic metabolism | Fructose 1,6-bisphosphate aldolase | 18.32 ± 7.6 | 19.65 ± 6.43 | 12.00 ± 4.80 |
| Triosephosphate isomerase | ||||
| Isoform 1 | 7.80 ± 2.46 | 13.34 ± 3.68* | 6.23 ± 1.79** | |
| Isoform 2 | 8.20 ± 3.12 | 29.75 ± 6.60* | 13.55 ± 5.82 | |
| Glyceraldehyde-3-phosphate dehydrogenase | ||||
| Isoform 1 | 8.91 ± 3.12 | 19.28 ± 5.35* | 7.19 ± 2.71** | |
| Isoform 2 | 28.4 ± 9.13 | 19.23 ± 6.27 | 26.46 ± 13.98 | |
| Isoform 3 | 39.08 ± 16.05 | 33.45 ± 14.79 | 20.10 ± 13.26 | |
| Phosphoglycerate kinase | ||||
| Isoform 1 | 16.20 ± 4.64 | 16.00 ± 6.31 | 18.50 ± 4.24 | |
| Isoform 2 | 10.88 ± 4.04 | 9.60 ± 3.23 | 16.52 ± 4.68 | |
| Phosphoglycerate mutase | 12.87 ± 4.21 | 14.94 ± 9.87 | 13.73 ± 3.41 | |
| Alpha-enolase | 13.78 ± 3.48 | 13.06 ± 4.84 | 8.27 ± 1.90 | |
| Gamma enolase | 15.48 ± 4.24 | 16.57 ± 4.15 | 13.20 ± 3.74 | |
| Pyruvate kinase | 15.90 ± 4.05 | 24.27 ± 5.56 | 15.07 ± 2.09 | |
| Lactate dehydrogenase | 17.20 ± 5.59 | 16.14 ± 3.77 | 15.84 ± 4.83 | |
| Aconytate hydratase | 12.13 ± 3.46 | 15.99 ± 3.11 | 15.35 ± 1.96 | |
| Mitochondrial precursor acyl CoA dehydrogenase | ||||
| Isoform 1 | 6.40 ± 1.91 | 9.79 ± 3.54 | 5.79 ± 1.46 | |
| Isoform 2 | 5.22 ± 1.46 | 5.39 ± 1.42 | 4.54 ± 0.84 | |
| Creatine kinase M chain | 13.39 ± 4.37 | 33.96 ± 7.96* | 15.16 ± 3.45** |
P < 0.05 with respect to control.
P < 0.05 with respect to FXa alone. The results are represented as mean ± SEM. AU, arbitrary units.
Figure 1.
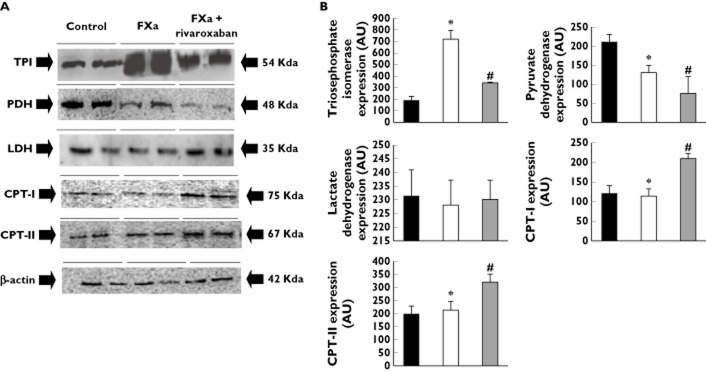
(A) Representative Western blots of triosephosphate isomerase (TPI), lactate dehydrogenase (LDH), pyruvate dehydrogenase (PDH) and carnitine palmitoyltransferase (CPT)-I and CPT-II. The expression of β-actin was used as loading protein control. (B) Bar graphs showing the densitometric analysis in arbitrary units (AU) of the entire Western blots. Densitometric values are represented as mean ± SEM. *P < 0.05 with respect to control. #P < 0.05 with respect to FXa alone.  , control;
, control;  , FXa;
, FXa;  , FXa+ rivaroxaban
, FXa+ rivaroxaban
The proteomic analysis also revealed that lactate dehydrogenase expression was similar in FXa-incubated femoral arterial segments and in controls (Table 3). This was also confirmed by Western blot analysis (Figure 1A, B). In addition, lactate content in the FXa-incubated femoral arteries was also similar to control (lactate content in nmol mg−1 protein: control 17.04 ± 1.42; FXa 15.92 ± 2.31; NS).
Pyruvate dehydrogenase expression (the enzyme involved in the conversion of pyruvate into acetyl-CoA) determined by Western blot was significantly reduced in FXa-incubated arteries with respect to control (Figure 1A, B). However, acetyl-CoA content in the FXa-incubated femoral arteries was greater than that observed in control conditions (acetyl-CoA content in pmol mg−1 protein: control: 2.02 ± 0.39, FXa-incubated arteries: 5.20 ± 0.67 P < 0.05).
Two isoforms of the mitochondrial precursors of acyl-CoA dehydrogenase, an enzyme involved in long chain fatty acids beta-oxidation, were also identified in the femoral arterial wall, although the protein expression level of none of them was changed by FXa (Table 3). The protein expression level of CPT-I and CPT-II was also determined by Western blotting and they were not significantly different between FXa-incubated and control arteries (Figure 1A, B).
In the tricarboxylic acid cycle, citrate is converted by aconitase into isocitrate. In the femoral arteries, FXa significantly reduced aconitase activity as compared with control (Figure 2).
Figure 2.
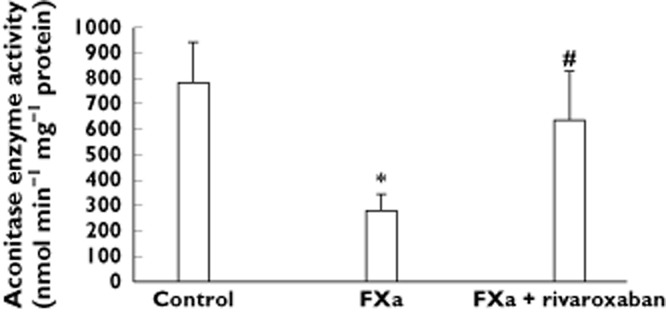
Bar graphs show aconitase activity in diabetic femoral arteries incubated in presence and absence of FXa and rivaroxaban. Results are represented as mean ± SEM. *P < 0.05 with respect to control. #P < 0.05 with respect to FXa alone
The proteomic analysis also showed a higher protein expression level of creatine kinase M chain in FXa-incubated femoral arteries than in control (Table 3). The level of expression of other identified energy metabolism-related proteins was not different between control and FXa-incubated arteries (Table 3).
Effects of rivaroxaban on the modifications induced by FXa on the glycolytic metabolism
Addition of rivaroxaban to FXa-incubated femoral arteries reduced the expression level of triosaphosphate isomerase isoforms 1 and 2 with respect to those arteries incubated with FXa alone (Table 3 and Figure 1A, B). The protein expression level of glyceraldehyde-3-phosphate dehydrogenase isoform 1 was also decreased in the femoral arteries incubated with rivaroxaban + FXa as compared with FXa alone and it was not statistically different with respect to control (Table 3).
In the diabetic femoral arterial wall, addition of rivaroxaban to FXa did not modify either lactate dehydrogenase expression or lactate content as compared with control and with FXa-incubated arteries (lactate content in rivaroxaban + FXa-incubated femoral arteries: 18.52 ± 02.89 nmol mg−1 protein).
In rivaroxaban + FXa-incubated femoral arteries, pyruvate dehydrogenase expression remained reduced with respect to control and this reduction tended to be more marked than that observed with FXa alone (Figure 1A, B). Acetyl-CoA content in the rivaroxaban + FXa-incubated femoral arteries was similar to control and significantly reduced with respect to FXa-incubated arteries (acetyl-CoA content: rivaroxaban + FXa 1.39 ± 0.28 pmol mg−1 arterial protein; P < 0.05 with respect to FXa alone).
Protein expression level of CPT-I and CPT-II was significantly increased in FXa + rivaroxaban-incubated arteries as compared with both FXa-incubated and control arteries (Figure 1A, B). In addition, aconitase activity was increased in FXa + rivaroxaban-incubated arteries with respect to those incubated with FXa alone (Figure 2). Finally, the expression of creatine kinase M chain was reduced in the femoral arteries incubated with Rivaroxaban + FXa as compared with those incubated with FXa alone and it was similar to that found in control arteries (Table 3).
Effects of FXa and rivaroxaban on the expression of proteins related to oxidative stress
As Table 4 shows, FXa apparently increased the protein expression level of glutathione-S-transferase and two aldehyde dehydrogenase isoforms as compared with control. The protein expression level of the mitochondrial gp91-phox NADPH oxidase isotype and the cytosolic gp47-phox and gp67-phox NADPH oxidase isotypes was also significantly higher in FXa-incubated femoral arteries than in control (Figure 3). It was accompanied by an increased content of MDA in the FXa-incubated arteries with respect to control (Figure 4). The differences in MDA content and the protein expression level of the cytosolic and mitochondrial gp-phox NADPH isotypes between FXa-incubated and control arteries remained significantly increased after adjustment by statin treatment, a drug with antioxidant properties, suggesting that the effects of FXa on these parameters were independent of the use of statins (Figures 3 and 4). However, neither the protein expression level of S–gluthatione transferase nor the two aldehyde dehydrogenase isoforms was different between control and FXa-incubated arteries when results were adjusted for statins in the linear regression model (Table 4).
Table 4.
Expression level of oxidative stress-related proteins in the femoral artery
| Protein | Control (AU) | FXa (AU) | FXa + rivaroxaban (AU) | |
|---|---|---|---|---|
| Oxidative stress | Glutathione-S-transferase | 56.44 ± 11.57 | 98.09 ± 8.06 | 43.49 ± 7.53 |
| Aldehyde dehydrogenase | ||||
| Isoform 1 | 12.29 ± 3.88 | 27.66 ± 7.99 | 15.42 ± 3.33 | |
| Isoform 2 | 17.14 ± 6.20 | 31.25 ± 6.99 | 15.24 ± 4.02 |
The results are represented as mean ± SEM. Statin treatment was used as covariant in the model to give adjusted P values. AU, arbitrary units.
Figure 3.
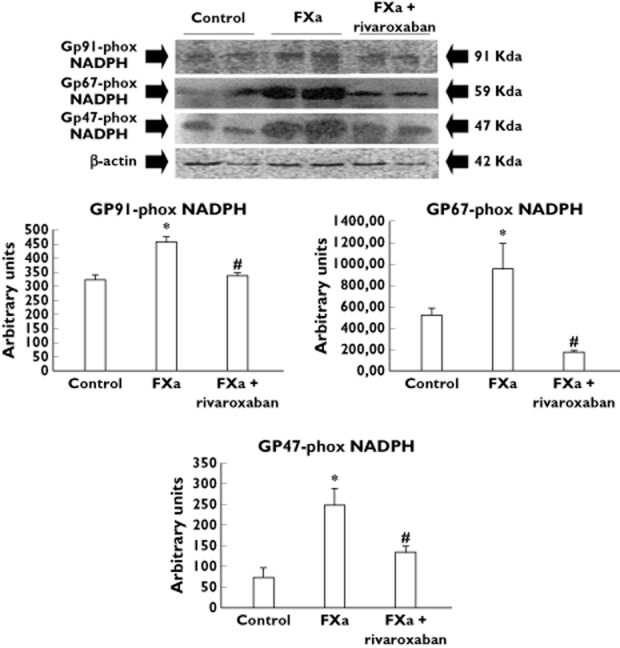
Representative Western blots of the mitochondrial gp91-phox NADPH oxidase isotype and the cytosolic gp47-phox and gp67-phox NADPH oxidase isotypes. Bar graphs show the densitometric analysis in arbitrary units (AU) of all the Western blots. The expression of β-actin was used as loading protein control. Densitometric values are represented as mean ± SEM. *P < 0.05 with respect to control. #P < 0.05 with respect to FXa alone. Statin treatment was used as covariant into the model to give adjusted P values
Figure 4.
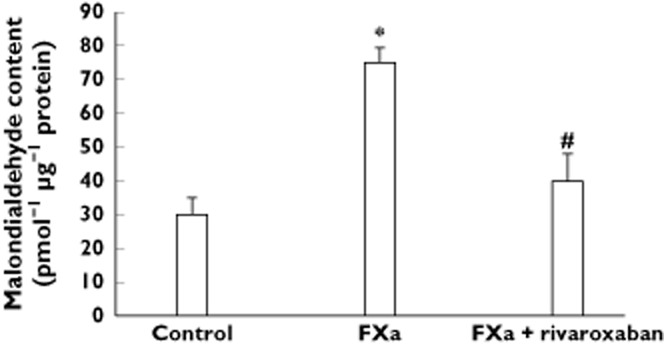
Bar graphs show malondialdehyde content in diabetic femoral arteries incubated in presence and absence of FXa and rivaroxaban. Results are represented as mean ± SEM. *P < 0.05 with respect to control. #P < 0.05 with respect to FXa alone. Statin treatment was used as covariant into the model to give adjusted P values
Addition of rivaroxaban to FXa reduced the increased protein expression level of either gp91-, gp47- and gp67-phox NADPH oxidase isotypes induced by FXa alone even when the results were adjusted by statin treatment (Figure 3). MDA content in the femoral arteries was also reduced in FXa + rivaroxaban-incubated femoral arteries as compared with those incubated with FXa alone and this difference remained significant after used statin treatment as covariant (Figure 4).
Discussion
The present study shows for the first time the effects of FXa on the expression of proteins related to energetic metabolism in human arteries and identified its impact upon levels of proteins involved in energy metabolism. The results showed that in femoral arteries from type 2 diabetic patients with end-stage vasculopathy, FXa modified the expression level of proteins involved in glycolytic metabolism. It was accompanied by a significant increased expression of proteins associated with oxidative stress. Moreover, in the diabetic femoral arteries rivaroxaban, a specific inhibitor of FXa activity, reverted most of the effects elicited by FXa on the energy metabolism and oxidative stress and seemed to favour long chain fatty acid transport into the mitochondria.
Glucose metabolism is an important determinant of vascular reactivity 26,27. The present study showed that in the diabetic femoral arterial wall, the level of expression of two triosephosphate isomerase isoforms, a glycolitic key-step enzyme, was significantly increased by FXa as compared with control. FXa also increased the level of expression of an isoform of another important glycolytic enzyme, glyceraldehyde-3-phosphate dehydrogenase. Initially, these findings might suggest that FXa stimulated the glycolytic pathway in diabetic arteries.
In the arterial wall, the reported stimulation of different molecular mechanisms elicited by FXa, i.e. cell proliferation and synthesis of inflammatory mediators, may require an increased energy demand 28,29. Therefore, despite of the well known abnormal glucose transport in human type 2 diabetes, the possible greater energy demand related to FXa may promote the increase in triosephosphate isomerase expression and glyceraldehyde-3-phosphate dehydrogenase, two of the main enzymes that regulate the glycolytic pathway. However, the expression level of pyruvate dehydrogenase, an enzyme included in the pyruvate dehydrogenase complex that mediates pyruvate oxidation to yield acetyl-CoA, was reduced in FXa-incubated arteries. This pyruvate dehydrogenase downexpression in FXa-incubated femoral arteries may direct pyruvate into lactate promoting an anaerobic state. However, the anaerobic catabolism of pyruvate seems to be unaffected by FXa since in the femoral arteries neither lactate dehydrogenase expression nor lactate content were changed by FXa.
Apparently, a paradoxical result may be the observation that the acetyl-CoA content increased in FXa-incubated arteries with respect to control since pyruvate dehydrogenase has been downexpressed by FXa. Long chain fatty acid beta-oxidation is an alternative metabolic pathway to glucose oxidation and also yields acetyl-CoA that then is catabolized by the tricarboxylic acid cycle. In this regard, circulating levels of free fatty acids have been reported elevated in diabetes because of their excess liberation from adipose tissue and diminished uptake by skeletal muscle 30,31. However, the expression level of two enzymes, CPTI and CPTII, involved in the mitochondrial fatty acid transport is a rate-limiting step in long chain fatty acid oxidation. In addition, the protein expression levels of CPT-I and CPT-II were not modified by FXa. Moreover, protein expression level of two isotypes of the mitochondrial precursors of acyl-CoA dehydrogenase, enzyme involved in fatty acid beta-oxidation, was not changed by FXa. Taken together, these findings seem to diminish the involvement of long chain fatty acid β-oxidation as an alternative source to increase acetyl-CoA in FXa-incubated arteries.
Another possible cause to increase acetyl-CoA accumulation in FXa-incubated diabetic femoral arteries could be the disruption of the energy metabolism downstream to acetyl-CoA. Acetyl-CoA is metabolized throughout the tricarboxylic acid cycle in the mitochondria. In this regard, a reduced tricarboxylic acid cycle flux has also been shown in type 2 diabetic patients 32. Therefore, in the diabetic femoral wall FXa may increase the dysfunction of the tricarboxylic acid cycle promoting acetyl-CoA accumulation. Accordingly, in FXa-incubated femoral arteries aconitase activity, an enzyme that converts citrate to isocitrate in the tricarboxylic acid cycle, was significantly reduced. Moreover, creatine kinase M chain expression was increased by FXa. Creatine kinase M chain maintains elevated ATP levels when other ATP generating sources are not able to support ATP requirements 33, further supporting that probably FXa-incubated arteries may need alternative sources than either glycolysis and long chain fatty acid β-oxidation to provide ATP to the vascular wall.
Oxidative stress is closely associated not only with energy metabolism and mitochondrial dysfunctionality but also with disturbed coagulation. As an example, in haemodialysis patients NADPH oxidase activity was correlated with coagulation priming 34. Although evidence have suggested that generation of reactive oxygen species may play an important role in the pathogenesis and complication of diabetes 35,36, less is known about the role of FXa on oxidative stress in the vascular wall. In this regard, previous work has demonstrated that FXa increased oxidative stress in human vascular smooth muscle cells from saphenous vein 17. We here showed that in diabetic femoral arteries, FXa increased the protein expression level of gp 47-phox, 67-phox and gp91-phox NADPH oxidase isotypes, catalytic subunits of cytosolic and mitochondrial NADPH oxidases, supporting pro-oxidative features for FXa. It was further supported by the fact that in the femoral arteries FXa increased the content of MDA, a sign for lipid peroxidation since it is produced during the attack of free radicals to membrane lipoproteins and polyunsaturated fatty acids 37. Accordingly, increased circulating MDA levels were previously reported in diabetic patients 38. Therefore, as speculation, FXa may exacerbate oxidative stress in the vascular wall of diabetic patients and may participate in the vascular damage associated with type 2 diabetes mellitus.
The effect of FXa on the expression of these pro-oxidant enzymes and MDA content in the arteries was not modified after adjustment by statin treatment suggesting that these pro-oxidant effects of FXa also occurred in the presence of statins. However, statin treatment affected the apparent modification elicited by FXa on other oxidative-related proteins, such as gluthatione-S-transferase, an antioxidant protein 39, and cytosolic aldehyde dehydrogenase, an enzyme that oxidizes aldehyde to carboxylic acid. Taken together, these findings may suggest that the presence of statins probably reduced the impact of FXa on the expression of specific oxidative stress-related proteins but not on others.
Effects of rivaroxaban on the changes induced by FXa
Although rivaroxaban prevented most of the effects of FXa on the energetic metabolism, it failed to modify FXa-induced pyruvate dehydrogenase down expression. Since rivaroxaban specifically inhibits FXa activity, this seems difficult to explain. However, Bono et al. have previously demonstrated that the catalytic inactivation of FXa suppressed FXa-induced increase in cytosolic free Ca2+ but failed to reduce ligand binding of FXa to affect cell protease receptor-1 (EPR-1) 40. EPR-1 behaves as a cofactor for FXa to catalyze prothrombin activation in the absence of FXa. Interestingly, it has been reported that EPR-1 is expressed in endothelial cells 41,42. Therefore, further experiments are needed to characterize if in the diabetic femoral arteries EPR-1 is coupled to down expression of pyruvate dehydrogenase mediated by FXa. The fact that addition of rivaroxaban to FXa increased the expression level of two carnitine palmitoyltransferases, proteins that transport fatty acids into the mitochondria, may suggest that in the diabetic arterial wall rivaroxaban facilitates the activity of alternative metabolic pathways other than glucose oxidation, such as fatty acid β-oxidation. The common end product of both fatty acid β-oxidation and glucose oxidation, acetyl-CoA, was reduced in rivaroxaban + FXa-incubated arteries suggesting that tricarboxylic acid cycle activity was probably improved by rivaroxaban. It was further supported by the fact that aconitase activity was markedly increased in rivaroxaban + FXa-incubated femoral arteries as compared with those incubated with FXa alone. Taken together, the increased aconitase activity and the increased expression level of the two mitochondrial fatty acid transporters, CPT-I and CPT-II, support improvement of mitochondrial energetic metabolism by rivaroxaban.
In diabetic femoral arteries, rivaroxaban also prevented the upexpression of oxidative stress-related proteins and prevented the increased arterial MDA content induced by FXa. It is also in accordance with the apparent better mitochondrial functionality observed in the experiments with rivaroxaban + FXa with respect to those with FXa alone.
Comments and study limitations
At present, some controversial results exist in the available sparse published data about whether FXa is increased or not in diabetic patients 43,44. However, it was postulated that diabetic patients showing vascular complications could be a subgroup of diabetic patients with increased risk of coagulation and fibrinolytic disorders 8. Moreover, platelets from both experimental diabetic animals and diabetic patients showed higher FXa generation, which additionally may contribute to greater exposition of the diabetic vascular wall to FXa during thrombosis 9. Based on this knowledge, we here only tried to test in vascular wall obtained from diabetic patients with end-stage vasculopathy whether an increased exposure to FXa may modify the protein expression level of energetic metabolism and oxidative-stress-related proteins. Moreover, we must to point out that the arterial samples we used were obtained during leg amputation by thrombosis from diabetic patients with end-stage vasculopathy. It is important to point out that there was not any other practical way to obtain such a type of tissue. Therefore, the present findings should be only considered for the type of arterial samples tested here.
An observation extracted from the present findings was that rivaroxaban might exert additional effects non-related to FXa, since FXa by itself did not modify the expression of CPT-I and CPT-II in the diabetic femoral arteries but it was increased in presence of rivaroxaban 45. In this regard, the existence of rivaroxaban-derived metabolites has been reported. Therefore, we cannot discard that the observed effect of rivaroxaban on CPT-I and CPT-II expression could due to rivaroxaban by itself but also by its identified metabolites. Although it seems to be an interesting observation, at present it is out of the scope of the present study and further experiments are needed to clarify it.
A limitation of the present study is the difficulty to determine the clinical implications of the effects of FXa on the energy metabolism and oxidative stress in the femoral diabetic arteries. The fact that FXa may exert vascular effects further to participate in coagulation should increase the interest for the pharmacological perspectives of the FXa inhibitors. In this regard, it is plausible that incorporation of specific FXa inhibitors, such as rivaroxaban, to the convential treatment of higher risk cardiovascular patients, such as type 2 diabetic patients, may improve prevention of acute vascular events where vascular functionality plays a pivotal role. In this regard, rivaroxaban reduced ischaemic events including mortality across a broad-acute coronary syndrome population in the ATLAS-ACS trial 46.
In conclusion, in femoral isolated arteries from type 2 diabetic patients with end-stage vasculopathy, FXa promoted changes in the expression level of proteins related to glucose oxidation suggesting disruption of mitochondrial metabolism and it was also accompanied by an increased expression of oxidative stress-related proteins. The better knowledge of the molecular mechanisms triggered by FXa may open new clinical approaches for the use of FXa inhibitors.
Funding
This work was supported by Fondo de Investigaciones de la Seguridad Social [Redes temáticas de Investigación Cooperativa (RETICs) RD12/0042/0040], Fondo Europeo de Desarrollo Regional (Fondos FEDER) and Bayer Pharmaceuticals.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare no support from any organization for the submitted work, no financial relationships with any organization that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work.
Javier Modrego is a staff member of RETICs. José J. Zamorano León is a staff member of Comunidad de Madrid (S2010/BMD-2374). We thank Begoña Larrea for secretarial assistance.
References
- Perzborn E, Roehrig S, Straub A, Kubitza D, Mueck W, Laux V. Rivaroxaban: a new oral factor Xa inhibitor. Arterioscler Thromb Vasc Biol. 2010;30:376–381. doi: 10.1161/ATVBAHA.110.202978. [DOI] [PubMed] [Google Scholar]
- Papapetropoulos A, Piccardoni P, Cirino G, Bucci M, Sorrentino R, Cicala C, Johnson K, Zachariou V, Sessa WC, Altieri DC. Hypotension and inflammatory cytokine gene expression triggered by factor Xa-nitric oxide signaling. Proc Natl Acad Sci U S A. 1998;95:4738–4742. doi: 10.1073/pnas.95.8.4738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer P, Mares AM, Dol F, Bono F, Herbert JM. Coagulation factor Xa induces endothelium-dependent relaxations in rat aorta. Circ Res. 1997;81:824–828. doi: 10.1161/01.res.81.5.824. [DOI] [PubMed] [Google Scholar]
- Senden NH, Jeunhomme TM, Heemskerk JW, Wagenvoord R, van't Veer C, Hemker HC, Buurman WA. Factor Xa induces cytokine production and expression of adhesion molecules by human umbilical vein endothelial cells. J Immunol. 1998;161:4318–4324. [PubMed] [Google Scholar]
- Koo BH, Kim DS. Factor Xa induces mitogenesis of vascular smooth muscle cells via autocrine production of epiregulin. J Biol Chem. 2003;278:52578–52586. doi: 10.1074/jbc.M310007200. [DOI] [PubMed] [Google Scholar]
- Busch G, Seitz I, Steppich B, Hess S, Eckl R, Schömig A, Ott I. Coagulation factor Xa stimulates interleukin-8 release in endothelial cells and mononuclear leukocytes: implications in acute myocardial infarction. Arterioscler Thromb Vasc Biol. 2005;25:461–466. doi: 10.1161/01.ATV.0000151279.35780.2d. [DOI] [PubMed] [Google Scholar]
- Sowers JR, Lester MA. Diabetes and cardiovascular disease. Diabetes Care. 1999;22(Suppl. 3):C14–20. [PubMed] [Google Scholar]
- Asakawa H, Tokunaga K, Kawakami F. Elevation of fibrinogen and thrombin–antithrombin III complex levels of type 2 diabetes mellitus patients with retinopathy and nephropathy. J Diabetes Complications. 2000;14:121–126. doi: 10.1016/s1056-8727(00)00075-1. [DOI] [PubMed] [Google Scholar]
- Lupu C, Calb M, Ionescu M, Lupu F. Enhanced prothrombin and intrinsic factor X activation on blood platelets from diabetic patients. Thromb Haemost. 1993;70:579–583. [PubMed] [Google Scholar]
- Olive JL, Ballard KD, Miller JJ, Milliner BA. Metabolic rate and vascular function are reduced in women with a family history of type 2 diabetes mellitus. Metabolism. 2008;57:831–837. doi: 10.1016/j.metabol.2008.01.028. [DOI] [PubMed] [Google Scholar]
- Morrison AD, Berwick L, Orci L, Winegrad AI. Morphology and metabolism of an aortic intima-media preparation in which an intact endothelium is preserved. J Clin Invest. 1976;57:650–660. doi: 10.1172/JCI108321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison AD, Clements RS, Jr, Winegrad AI. Effects of elevated glucose concentrations on the metabolism of the aortic wall. J Clin Invest. 1972;51:3114–3123. doi: 10.1172/JCI107138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlak K, Borawski J, Naumnik B, Mysliwiec M. Relationship between oxidative stress and extrinsic coagulation pathway in haemodialyzed patients. Thromb Res. 2003;109:247–251. doi: 10.1016/s0049-3848(03)00241-x. [DOI] [PubMed] [Google Scholar]
- Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–2950. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- Green K, Brand MD, Murphy MP. Prevention of mitochondrial damage as a therapeutic strategy in diabetes. Diabetes. 2004;58:S110–118. doi: 10.2337/diabetes.53.2007.s110. [DOI] [PubMed] [Google Scholar]
- Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobi K, Rauch BH, Dangwal S, Freidel K, Doller A, Eberhardt W, Fischer JW, Schrör K, Rosenkranz AC. Redox regulation of human protease-activated receptor-2 by activated factor X. Free Radic Biol Med. 2011;51:1758–1764. doi: 10.1016/j.freeradbiomed.2011.08.003. [DOI] [PubMed] [Google Scholar]
- IDF Clinical Guidelines Task Force. Global Guideline for Type 2 Diabetes: recommendations for standard, comprehensive, and minimal care. Diabet Med. 2006;23:579–593. doi: 10.1111/j.1464-5491.2006.01918.x. [DOI] [PubMed] [Google Scholar]
- Blanc-Brude OP, Archer F, Leoni P, Derian C, Bolsover S, Laurent GJ, Chambers RC. Factor Xa stimulates fibroblast procollagen production, proliferation, and calcium signaling via PAR1 activation. Exp Cell Res. 2005;304:16–27. doi: 10.1016/j.yexcr.2004.10.021. [DOI] [PubMed] [Google Scholar]
- Perzborn E, Strassburger J, Wilmen A, Pohlmann J, Roehrig S, Schlemmer KH, Straub A. In vitro and in vivo studies of the novel antithrombotic agent BAY 59-7939-an oral, direct Factor Xa inhibitor. J Thromb Haemost. 2005;3:514–521. doi: 10.1111/j.1538-7836.2005.01166.x. [DOI] [PubMed] [Google Scholar]
- Haas S. Rivaroxaban – an oral, direct Factor Xa inhibitor: lessons from a broad clinical study programme. Eur J Haematol. 2009;82:339–349. doi: 10.1111/j.1600-0609.2009.01230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubitza D, Becka M, Mueck W, Zuehlsdorf M. Rivaroxaban (BAY 59-7939) – an oral, direct Factor Xa inhibitor – has no clinically relevant interaction with naproxen. Br J Clin Pharmacol. 2006;63:469–476. doi: 10.1111/j.1365-2125.2006.02776.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateos-Cáceres PJ, García-Méndez A, López Farré A, Macaya C, Núñez A, Gómez J, Alonso-Orgaz S, Carrasco C, Burgos ME, de Andrés R, Granizo JJ, Farré J, Rico LA. Proteomic analysis of plasma from patients during an acute coronary syndrome. J Am Coll Cardiol. 2004;44:1578–1583. doi: 10.1016/j.jacc.2004.06.073. [DOI] [PubMed] [Google Scholar]
- Modrego J, Maroto L, Tamargo J, Azcona L, Mateos-Cáceres P, Segura A, Moreno-Herrero R, Pérez-Castellanos N, Delpón E, Pérez-Villacastín J, Rodríguez E, Macaya C, López-Farré AJ. Comparative expression of proteins in left and right atrial appendages from patients with mitral valve disease at sinus rhythm and atrial fibrillation. J Cardiovasc Electrophysiol. 2010;21:859–868. doi: 10.1111/j.1540-8167.2010.01718.x. [DOI] [PubMed] [Google Scholar]
- Modrego J, López-Farré AJ, Martínez-López I, Muela M, Macaya C, Serrano J, Moñux G. Expression of cytoskeleton and energetic metabolism-related proteins at human abdominal aortic aneurysm sites. J Vasc Surg. 2012;55:1124–1133. doi: 10.1016/j.jvs.2011.10.033. [DOI] [PubMed] [Google Scholar]
- Hall JL, Matter CM, Wang X, Gibbons GH. Hyperglycemia inhibits vascular smooth muscle cell apoptosis through a protein kinase C-dependent pathway. Circ Res. 2000;87:574–580. doi: 10.1161/01.res.87.7.574. [DOI] [PubMed] [Google Scholar]
- Cline GW, Petersen KF, Krssak M, Shen J, Hundal RS, Trajanoski Z, Inzucchi S, Dresner A, Rothman DL, Shulman GI. Impaired glucose transport as a cause of decreased insulin-stimulated muscle glycogen synthesis in type 2 diabetes. N Engl J Med. 1999;341:240–246. doi: 10.1056/NEJM199907223410404. [DOI] [PubMed] [Google Scholar]
- Steinhoff M, Vergnolle N, Young SH, Tognetto M, Amadesi S, Ennes HS, Trevisani M, Hollenberg MD, Wallace JL, Caughey GH, Mitchell SE, Williams LM, Geppetti P, Mayer EA, Bunnett NW. Agonists of proteinase-activated receptor 2 induce inflammation by a neurogenic mechanism. Nat Med. 2000;6:151–158. doi: 10.1038/72247. [DOI] [PubMed] [Google Scholar]
- Bono F, Lamarche I, Herbert JM. Induction of vascular smooth muscle cell growth by selective activation of the proteinase activated receptor-2 (PAR-2) Biochem Biophys Res Commun. 1997;24:762–764. doi: 10.1006/bbrc.1997.7847. [DOI] [PubMed] [Google Scholar]
- Kelley DE, Simoneau JA. Impaired free fatty acid utilization by skeletal muscle in non-insulin-dependent diabetes mellitus. J Clin Invest. 1994;94:2349–2356. doi: 10.1172/JCI117600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boden G. Free fatty acids, insulin resistance, and type 2 diabetes mellitus. Proc Assoc Am Physicians. 1999;111:241–248. doi: 10.1046/j.1525-1381.1999.99220.x. [DOI] [PubMed] [Google Scholar]
- Gaster M, Nehlin JO, Minet AD. Impaired TCA cycle flux in mitochondria in skeletal muscle from type 2 diabetic subjects: marker or maker of the diabetic phenotype? Arch Physiol Biochem. 2012;118:156–189. doi: 10.3109/13813455.2012.656653. [DOI] [PubMed] [Google Scholar]
- Wyss M, Kaddurah-Daouk R. Creatine and creatinine metabolism. Physiol Rev. 2000;80:1107–1213. doi: 10.1152/physrev.2000.80.3.1107. [DOI] [PubMed] [Google Scholar]
- Cariello M, Simone S, Loverre A, Gigante M, Incampo F, Pietanza S, Colucci M, Schena FP, Gesualdo L, Grandaliano G, Pertosa G. Coagulation activation is associated with nicotinamide adenine dinucleotide phosphate oxidase-dependent reactive oxygen species generation in hemodialysis patients. Antioxid Redox Signal. 2012;16:428–439. doi: 10.1089/ars.2011.4062. [DOI] [PubMed] [Google Scholar]
- Chang YC, Chuang LM. The role of oxidative stress in the pathogenesis of type 2 diabetes: from molecular mechanisms to clinical implication. Am J Transl Res. 2010;2:316–331. [PMC free article] [PubMed] [Google Scholar]
- Niedowicz DM, Daleke DL. The role of oxidative stress in diabetic complications. Cell Biochem Biophys. 2005;43:289–330. doi: 10.1385/CBB:43:2:289. [DOI] [PubMed] [Google Scholar]
- Yang RL, Shi YH, Hao G, Li W, Le GW. Increasing oxidative stress with progressive hyperlipidemia in human: relation between malondialdehyde and atherogenic index. Clin Biochem Nutr. 2008;43:154–158. doi: 10.3164/jcbn.2008044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamuna Rani A, Mythili SV. Study on total antioxidant status in relation to oxidative stress in type 2 diabetes mellitus. J Clin Diagn Res. 2014;8:108–110. doi: 10.7860/JCDR/2014/7603.4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaziri ND, Lin CY, Farmand F, Sindhu RK. Superoxide dismutase, catalase, glutathione peroxidase and NADPH oxidase in lead-induced hypertension. Kidney Int. 2003;63:186–194. doi: 10.1046/j.1523-1755.2003.00711.x. [DOI] [PubMed] [Google Scholar]
- Bono F, Schaeffer P, Hérault JP, Michaux C, Nestor AL, Guillemot JC, Herbert JM. Factor Xa activates endothelial cells by a receptor cascade between EPR-1 and PAR-2. Arterioscler Thromb Vasc Biol. 2000;20:e107–112. doi: 10.1161/01.atv.20.11.e107. [DOI] [PubMed] [Google Scholar]
- Ambrosini G, Altieri DC. Molecular dissection of effector cell protease receptor-1 recognition of factor Xa. J Biol Chem. 1996;271:1243–1248. doi: 10.1074/jbc.271.2.1243. [DOI] [PubMed] [Google Scholar]
- Bono F, Herault JP, Avril C, Schaeffer P, Lormeau JC, Herbert JM. Human umbilical vein endothelial cells express high affinity receptor for factor Xa. J Cell Physiol. 1997;172:36–43. doi: 10.1002/(SICI)1097-4652(199707)172:1<36::AID-JCP4>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Myrup B, Rossing P, Jensen T, Gram J, Kluft C, Jespersen J. Procoagulant activity and intimal dysfunction in IDDM. Diabetologia. 1995;38:73–78. doi: 10.1007/BF02369355. [DOI] [PubMed] [Google Scholar]
- Lefebvre PJ. The role of hyperglycaemia-induced alterations of antithrombin III and factor X activation in the thrombin hyperactivity of diabetes mellitus. Diabetic Med. 1990;7:343–348. doi: 10.1111/j.1464-5491.1990.tb01402.x. [DOI] [PubMed] [Google Scholar]
- Weinz C, Schwarz T, Kubitza D, Mueck W, Lang D. Metabolism and excretion of Rivaroxaban, an oral, direct factor Xa inhibitor, in rats, dogs, and humans. Drug Metab Dispos. 2009;37:1056–1064. doi: 10.1124/dmd.108.025569. [DOI] [PubMed] [Google Scholar]
- Gibson CM, Chakrabarti AK, Mega J, Bode C, Bassand JP, Verheugt FW, Bhatt DL, Goto S, Cohen M, Mohanavelu S, Burton P, Stone G, Braunwald E ATLAS-ACS 2 TIMI 51 Investigators. Reduction of stent thrombosis in patients with acute coronary syndromes treated with rivaroxaban in ATLAS-ACS 2 TIMI 51. J Am Coll Cardiol. 2013;62:286–290. doi: 10.1016/j.jacc.2013.03.041. [DOI] [PubMed] [Google Scholar]