

Abstract
Background
Synthesis and uptake of pyoverdine, the primary siderophore of the opportunistic pathogen Pseudomonas aeruginosa, is dependent on two extra-cytoplasmic function (ECF) sigma factors, FpvI and PvdS. FpvI and PvdS are required for expression of the ferri-pyoverdine receptor gene fpvA and of pyoverdine synthesis genes respectively. In the absence of pyoverdine the anti-sigma factor FpvR that spans the cytoplasmic membrane inhibits the activities of both FpvI and PvdS, despite the two sigma factors having low sequence identity.
Results
To investigate the interactions of FpvR with FpvI and PvdS, we first used a tandem affinity purification system to demonstrate binding of PvdS by the cytoplasmic region of FpvR in P. aeruginosa at physiological levels. The cytoplasmic region of FpvR bound to and inhibited both FpvI and PvdS when the proteins were co-expressed in Escherichia coli. Each sigma factor was then subjected to error prone PCR and site-directed mutagenesis to identify mutations that increased sigma factor activity in the presence of FpvR. In FpvI, the amino acid changes clustered around conserved region four of the protein and are likely to disrupt interactions with FpvR. Deletion of five amino acids from the C-terminal end of FpvI also disrupted interactions with FpvR. Mutations in PvdS were present in conserved regions two and four. Most of these mutations as well as deletion of thirteen amino acids from the C-terminal end of PvdS increased sigma factor activity independent of whether FpvR was present, suggesting that they increase either the stability of PvdS or its affinity for core RNA polymerase.
Conclusions
These data show that FpvR binds to PvdS in both P. aeruginosa and E. coli, inhibiting its activity. FpvR also binds to and inhibits FpvI and binding of FpvI is likely to involve conserved region four of the sigma factor protein.
Electronic supplementary material
The online version of this article (doi:10.1186/s12866-014-0287-2) contains supplementary material, which is available to authorized users.
Keywords: Siderophore, Gene expression, ECF sigma factor, Anti-sigma factor, Pyoverdine, Pseudomonas, Cell surface signaling, TonB-dependent signaling, Tandem affinity purification
Background
Pseudomonas aeruginosa is a widespread opportunistic pathogen recognized for its role in morbidity and mortality in cystic fibrosis and burns patients [1]. Like other bacteria P. aeruginosa has a requirement to take up iron, which is an essential co-factor in a number of proteins. P. aeruginosa achieves this via active uptake of iron-chelating siderophores, with pyoverdine being the primary siderophore secreted by this bacterium [2]. Once pyoverdine has bound iron, the cell-surface receptor FpvA binds and transports ferri-pyoverdine into the cell (reviewed in [3]). Expression of pyoverdine synthesis genes and the fpvA gene is directed by the alternative sigma factors PvdS and FpvI respectively [4-7], and PvdS is also required for maximal expression of two secreted virulence factors, exotoxin A and PrpL protease [8,9]. In the absence of pyoverdine the activities of PvdS and FpvI are inhibited by an anti-sigma factor, FpvR, which spans the cytoplasmic membrane [6,10,11]. In a positive feedback loop, interaction of ferri-pyoverdine with FpvA results in proteolytic degradation of FpvR, a process that requires the energy-transducing protein TonB as well as interaction between periplasmic domains of the Fpv proteins [12-15]. The sigma factors are then free to recruit core RNA polymerase, facilitating promoter recognition with consequent up-regulation of the pyoverdine synthesis genes and the genes encoding FpvA, exotoxin A and PrpL protease. Signal transduction systems of this sort (cell-surface signaling) are widespread in Gram negative bacteria and control the expression of a large number of genes encoding ferri-siderophore receptor proteins, in a wide range of species [11,16,17]. However the ferri-pyoverdine system is the only cell-surface signaling pathway known in which a single anti-sigma factor (FpvR) inhibits two different sigma factors (PvdS and FpvI). In some cell-surface signaling pathways, such as the Fec (ferric citrate) pathway in Escherichia coli [18] and the Fox (desferrioxamine) and Fiu (ferrichrome) pathways in P. aeruginosa [19] the anti-sigma factors are also required for sigma factor function and so are considered to be sigma factor regulators. However, there is no evidence that FpvR is required for activity of PvdS or FpvI.
FpvI and PvdS belong to the class IV or extra-cytoplasmic function (ECF) sigma factors, alternative sigma factors that control a wide range of functions in bacteria and are the largest and most diverse group of sigma factors known (reviewed in [20,21]). Class IV sigma factors are relatively small and share only two of the four conserved functional regions present in other sigma factors, region two and region four, these being connected by a flexible non-conserved linker. Region two plays a specific role in −10 promoter recognition and in DNA melting, and region four recognizes the −35 promoter region [20]. In a previous study, alanine scanning mutagenesis was used to elicit more detail on the functions of these regions in PvdS [22]. Mutations in region 2.1 and 2.2 reduced binding to core RNA polymerase and mutations in regions 2.3, 2.4 and 4.2 impaired DNA binding without affecting binding to core RNA polymerase. It is very likely that the corresponding regions in FpvI have equivalent functions.
Binding of sigma factors by anti-sigma factor proteins provides an effective mechanism of post-translational control of protein activity in cell-surface signaling and many other bacterial systems. However, there are very few cases in which sigma/anti-sigma factor interactions have been characterized at the molecular level and none of these closely parallel the FpvR/FpvI/PvdS system. One well-studied example is the stress response sigma factor σE in complex with the N-terminal (residues 1–66) region of its anti-sigma factor RseA from E. coli [23]. The region RseAN1–66 slots between regions two and four of σE, making extensive interactions that sterically prevent σE from recruiting core RNA polymerase. The cytoplasmic domain of the anti-sigma factor ChrR from Rhodobacter sphaeroides has a similar structure to RseAN1–66 [24]. This observation in combination with bioinformatic analysis has led to the proposal that there is a common structure for the cytoplasmic domain of class IV anti-sigma factors [24], despite low sequence identity. However, there is considerable variability amongst ECF sigma factors for the regions bound by the cognate anti-sigma factor, which can be both region two and four [23-25], region two alone [26] or region four alone [27].
Although both are inhibited by FpvR, FpvI and PvdS have low sequence identity with each other (34.7%) [6]. Previously, bacterial 2-hybrid analysis demonstrated an interaction between FpvI and FpvR, and PvdS and FpvR, when each pair was expressed in E. coli [28]. It was also shown that the cytoplasmic N-terminal 67 amino acids of FpvR comprised the minimum region required for interaction with PvdS and FpvI [28]. Mutations were identified in fpvI that interfered with the interaction of FpvR and FpvI although the effects of most of these mutations were attributed to reduced amounts of FpvI protein, with only one mutation specifically affecting binding of FpvI by FpvR. The effect of mutations in PvdS on interactions with FpvR was not investigated.
The overall aim of the research described here was to investigate the interactions of FpvR with PvdS and FpvI in vivo, and to identify amino acid residues in each of the sigma factors that contribute to those interactions.
Results
Co-purification of FpvR and PvdS from P. aeruginosa
We validated binding of PvdS by FpvR in P. aeruginosa by purifying the cytoplasmic portion of FpvR and determining whether PvdS was co-purified. P. aeruginosa (PAO1) was engineered to express the cytoplasmic portion and predicted sigma factor inhibitory region of FpvR (residues 1–89) [6,28] fused to a C-terminal tandem affinity purification (TAP) tag [29]. The FpvR1–89 -TAP fusion, expressed from the fpvR promoter, was either integrated into the bacterial chromosome using mini-CTX or was expressed from plasmid pUCP23. Chromosomal integration was used to demonstrate that FpvR1–89 and PvdS interact when expressed in physiological amounts. Higher plasmid-based expression was expected to titrate out any regulatory factors that may have limited FpvR1–89 expression, ensuring sufficient FpvR1–89 was present for visualisation and co-purification with PvdS. As expected, expression of FpvR from the chromosomally-integrated construct was repressed by the presence of iron in the King’s B medium (Additional file 1: Figure S1). FpvR1–89 fused to calmodulin binding protein (CBP) was purified using the TAP protocol. The purification resulted in a 15 kDa protein, the predicted size for FpvR1–89–CBP, which could be detected using antibodies against either CBP or FpvR1–89 (Figure 1). Fractions that contained purified FpvR1–89–CBP also contained co-purified PvdS. PvdS was not present in fractions obtained using the purification protocol with bacteria that did not contain the FpvR1–89–TAP construct, confirming that purification of PvdS was dependent on the presence of FpvR1–89–TAP. FpvI was not detected following FpvR1–89–TAP purification using a polyclonal FpvI antibody and a suitable monoclonal antibody was not available.
Figure 1.
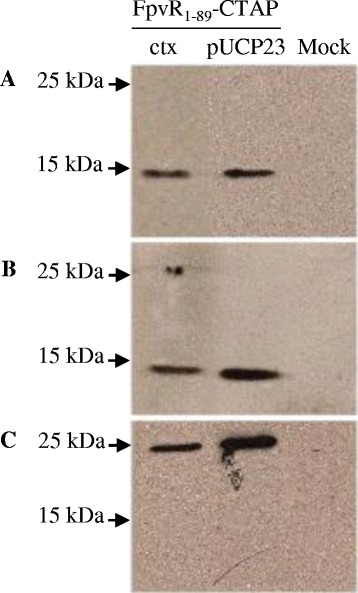
Co-purification of PvdS with FpvR 1–89 –TAP from P. aeruginosa . Soluble protein was prepared from P. aeruginosa PAO1 fpvR expressing plasmid-borne (pUCP23) or chromosomally-integrated (ctx) FpvR1–89 fused to a C-terminal TAP tag. Protein was purified using the TAP protocol and the purified protein analyzed by Western blotting for FpvR1–89-CBP or PvdS. (A) anti-CBP; (B) anti-FpvR; (C) anti-PvdS. A mock purification was carried out with P. aeruginosa PAO1 fpvR carrying pUCP23 without the fpvR 1–89-TAP fusion as a negative control for the TAP tag purification procedure. The positions of molecular weight markers are shown.
The cytoplasmic region of FpvR inhibits sigma factor activity in E. coli
To investigate the inhibition of FpvI and PvdS by FpvR1–89, a system was established for detecting the activity of FpvI or PvdS when co-expressed with FpvR1–89 in E. coli. The pETDuet vector allows a 1:1 molar ratio of expression from two multiple cloning sites (MCS). DNA encoding fpvR1–89 was inserted into MCS2 and either fpvI or pvdS was inserted into MCS1, giving plasmids pETDuetfpvI_fpvR1–89 and pETDuetpvdS_fpvR1–89 respectively (Table 1). Reporter plasmids (Table 1) carrying fpvA or pvdE promoters fused upstream of lacZ were used to detect FpvI or PvdS activity respectively [6,30]. In the absence of FpvR1–89, FpvI induced fpvA promoter activity and PvdS induced pvdE promoter activity (Figure 2). The activity of FpvI and PvdS was strongly repressed in the presence of FpvR1–89. These results demonstrated the inhibitory function of the cytoplasmic region of FpvR on both FpvI and PvdS activity in E. coli.
Table 1.
Bacterial strains and plasmids used in this study
| Strains | Genotype/phenotype | Reference |
|---|---|---|
| P. aeruginosa | ||
| PAO1 | Wild-type | [31] |
| PAO1 fpvR | PAO1 with fpvR (PA2388) deletion | [10] |
| PAO1 fpvR 1–89-TAP | PAO1 containing mini CTX2:: fpvR 1–89-TAP | This study |
| PAO1fpvR; pUCP23::fpvR 1–89-TAP | PAO1fpvR containing pUCP23::fpvR 1–89-TAP; GmR, CbR | This study |
| E. coli | ||
| S17-1 | hsd R hsdM + recA thi pro [integrated RP4-2-Tc::Mu, Km::Tn7]; Sm R Tp R | [32] |
| JM83 | F- ara Δ (lac-proAB) rpsL [Φ80, lacZΔM15] thi | [33] |
| MC1061 | ΔlacX74 Hsr− Hsm+ rpsL | [34] |
| MC1061 (DE3) | MC1061 lysogenized with λDE3 [lacI lacUV5-T7 gene 1] | This study |
| Plasmids | ||
| pUCP23 | lacI q lacZ(α-fragment) aacC1 ColE1 ori; RO1600 ori; Cb/AmpR, GmR | [35] |
| pUCP23::fpvR 1-89TAP | 519 bp PCR fragment containing the fpvR promoter and 5′end of fpvR, joined to a C-terminal TAP-tag and cloned into pUCP23 | This study |
| Mini-CTX2 | Self-proficient integration vector; TcR | [36] |
| Mini-CTX2::fpvR 1-89TAP | 519 bp PCR fragment containing the fpvR promoter and 5′end of fpvR, joined to a C-terminal TAP-tag and cloned into Mini-CTX2 | This study |
| pFLP2 | pRO1600 ori, sacB, flp recombinase; ApR/CbR | [37] |
| pMP190::PfpvA_lacZ | fpvA promoter cloned upstream of lacZ in pMP190; CmR | [6] |
| pMP190::PpvdE_lacZ | pvdE promoter cloned upstream of lacZ in pMP190; CmR | [30,38] |
| pETDuet | Dual expression vector; ApR | [39] |
| pETDuet::fpvR 1–89 | fpvR 1–89 cloned into MCS2 of pETDuet | This study |
| pETDuet::fpvI | fpvI cloned into MCS1 of pETDuet | This study |
| pETDuet::pvdS | pvdS cloned into MCS1 of pETDuet | This study |
| pETDuet::fpvI_fpvR 1–89 | fpvI cloned into MCS1 and fpvR 1–89 cloned into MCS2 of pETDuet | This study |
| PETDuet::pvdS_fpvR 1–89 | pvdS cloned into MCS1 and fpvR 1–89 cloned into MCS2 of pETDuet | This study |
Figure 2.
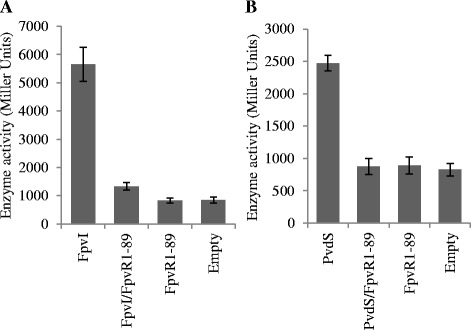
The activity of PvdS and FpvI in the presence and absence of FpvR 1–89 . β-galactosidase assays were carried out with E. coli MC1061 (DE3) containing either (A) pMP190::PfpvA_lacZ or (B) pMP190::PpvdE_lacZ, along with pETDuet expressing FpvR1–89 and either (A) FpvI or (B) PvdS. An empty pETDuet control is also shown to control for background expression of lacZ from pMP190::PfpvA_lacZ or pMP190::PpvdE_lacZ. Averages were obtained from three biological replicates. Error bars are ±1 SD.
Co-purification of FpvR1–89 and FpvI or PvdS from E. coli
The pETDuet co-expression system was used to investigate the interaction of FpvI and PvdS with FpvR1–89, via purification of hexahistidine-tagged FpvI or PvdS. FpvR1–89 co-purified with His6-FpvI and with His6-PvdS (Figure 3A and B). FpvR1–89 was not purified when His6-FpvI or His6-PvdS were absent (Additional file 1: Figure S2). In a reciprocal experiment, untagged FpvI and PvdS co-purified with His6-FpvR1–67 that contains only the 67 N-terminal residues of FpvR (Additional file 1: Figure S3). These findings demonstrated that the cytoplasmic region of FpvR forms stable complexes with either FpvI or PvdS when co-expressed in E. coli.
Figure 3.
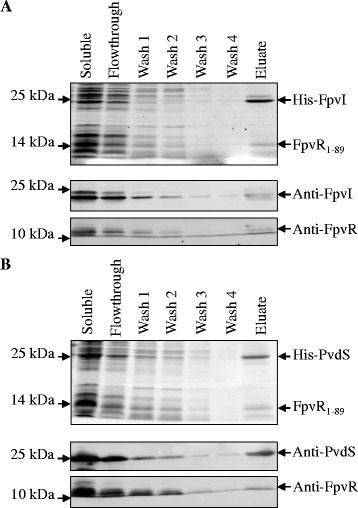
Co-purification of FpvR 1–89 with either His 6 -FpvI or His 6 -PvdS from E. coli . Soluble protein was obtained from E. coli MC1061 (DE3) co-expressing either His6-FpvI and FpvR1-89 or His6-PvdS and FpvR1–89. Protein was purified by nickel affinity chromatography via the His6-tags on PvdS and FpvI and analyzed by SDS-PAGE (top panels) and Western blotting (lower panels) using anti-FpvI, anti-PvdS or anti-FpvR antibodies as shown. (A) Co-purification of FpvR1–89 with His6-FpvI; (B) co-purification of FpvR1-89 with His6-PvdS. The positions of molecular weight markers are shown.
Identification of mutations that increase sigma factor activity in the presence of FpvR1-89
The above data demonstrated binding and inhibition of FpvI and PvdS by FpvR1–89. To further investigate interactions of each sigma factor with FpvR1–89 we generated, selected and characterized mutations that increased the activity of FpvI and PvdS in the presence of FpvR1-89. Error-prone PCR was used to introduce random mutations into fpvI or pvdS and libraries of mutated genes were cloned into MCS1 of pETDuet co-expressing fpvR1–89. The mutagenesis method used minimizes bias in mutations [40] and sequence analysis of 9 independent clones (a total of 25 mutations) of mutagenized pvdS did not suggest any mutational bias (Additional file 1: Table S2). The resulting plasmid libraries were transformed into E. coli containing either PfpvA::lacZ or PpvdE::lacZ, to screen for gain-of-function mutations in fpvI and pvdS respectively. Mutants exhibiting increased activity in the presence of FpvR1–89 were identified on a qualitative basis as colonies showing increased β-galactosidase activity on agar plates supplemented with BCIG, as described in Methods.
Approximately 6% of FpvI mutant colonies and 0.5% of PvdS mutant colonies had increased sigma factor activity in this screen. Mutant fpvI and pvdS genes were sequenced and their lacZ activities quantified. The properties of each mutant confirmed to have increased sigma factor activity are summarized in Additional file 1: Table S3 and S4. The FpvI mutants contained one to five amino acid changes with an average of two, and PvdS mutants contained one to three amino acid changes with an average of two. The activities of the FpvI and PvdS mutants were generally significantly higher than those of WT FpvI or PvdS (Figure 4; Additional file 1: Table S3 and S4). One exception was mutant PvdS S65G/Y136N/Q176* that had clearly increased activity relative to WT PvdS on screening medium but not in quantitative assays.
Figure 4.
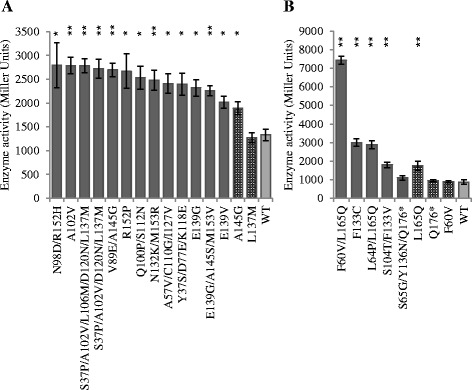
The activity of FpvI and PvdS mutants in the presence of FpvR 1–89 . β-galactosidase assays were carried out with E. coli MC1061 (DE3) containing (A) pMP190::PfpvA_lacZ or (B) pMP190::PpvdE_lacZ, along with pETDuet expressing FpvR1–89 and (A) mutant FpvI or (B) mutant PvdS. Dark grey bars: FpvI and PvdS mutants generated by error prone PCR. Patterned bars: FpvI and PvdS mutants engineered by site directed mutagenesis. Light grey bars: WT FpvI or PvdS. Mutants that were further investigated in Figure 5 are indicated in bold. Data were obtained from three biological replicates and error bars are ±1 SD. Statistically significant difference to WT according to Student's T-test is indicated: *p <0.05, **p <0.01.
Many of the mutant genes contained multiple mutations but all of the mutant genes contained at least one mutation in or around conserved region four. Two single mutations were engineered into fpvI and three into pvdS by site-directed mutagenesis in order to investigate the effects of the individual mutations on interactions of the sigma factors with FpvR1–89. These mutations were chosen because the corresponding amino acid residues were altered in more than one mutant, they were located in region four, or they apparently enhanced the activity of a change in region four. Of these mutants, FpvI A145G and PvdS L165Q had higher activity than WT, although the activity of the latter was not as high as in F60V/L165Q or L64P/L165Q double mutants. PvdS F60V alone showed no difference in activity from WT indicating that F60V only had an effect on activity in the presence of L165Q.
Activity of FpvI and PvdS mutants in the absence of FpvR1–89
Increased activity of FpvI or PvdS mutants in the presence of FpvR1–89 could be due to reduced affinity of the mutant proteins for FpvR1–89 or to an intrinsic increase in sigma factor activity, which could arise from several factors including increased protein stability, improved affinity for core RNA polymerase, or stronger promoter DNA binding. To distinguish between reduced affinity for FpvR1–89 and intrinsically increased activity of the sigma factors, five FpvI and four PvdS mutants were compared with WT for activity in the absence of FpvR1–89. The activities of the FpvI mutants (Figure 5A) were not higher than WT FpvI; indeed, for four of these mutants the activity was lower than WT FpvI, which may indicate impaired function or destabilized protein folding as a result of the mutations. These data suggest that the increased activity of the FpvI mutants in the presence of FpvR1–89 (Figure 4A) is because in each case the mutations reduce the ability of FpvR1–89 to bind FpvI. In contrast, three of the PvdS mutants had significantly higher activity than WT PvdS in the absence of FpvR1–89 (Figure 5B). The increased activity of these mutants in the presence of FpvR1–89 (Figure 4B) may therefore be due at least in part to improved sigma factor function or protein stability.
Figure 5.
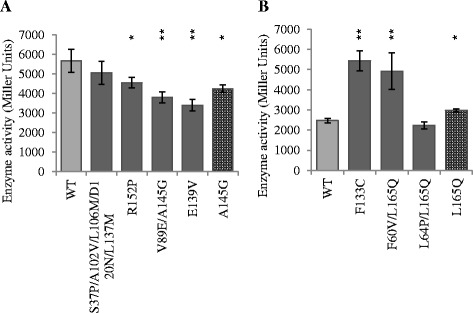
The activity of FpvI and PvdS mutants in the absence of FpvR 1–89 . β-galactosidase assays were carried out with E. coli MC1061 (DE3) containing (A) pMP190::PfpvA_lacZ or (B) pMP190::PpvdE_lacZ, along with pETDuet expressing (A) mutant FpvI or (B) mutant PvdS. Dark grey bars: FpvI and PvdS mutants generated by error prone PCR. Patterned bars: FpvI and PvdS mutants engineered by site directed mutagenesis. Light grey bars: WT FpvI or PvdS. Data were obtained from three biological replicates and error bars are ±1 SD. Statistically significant difference to WT according to Student's T-test is indicated: *p <0.05, **p <0.01.
The mutations identified in FpvI cluster in region four
The identified mutations were mapped onto the conserved regions of FpvI and PvdS [22]. The mutations clustered in region four of FpvI (Figure 6), including single mutations that increased the activity of FpvI in the presence of FpvR1–89, suggesting that this region is important for binding of FpvI by FpvR. No obvious clustering of mutations was observed for PvdS, however the sample size was small.
Figure 6.

The location of mutations in FpvI and PvdS that increased sigma factor activity in the presence of FpvR 1–89 . The approximate location of mutations are shown on the ECF sigma factor functional regions two (2.1-2.4) and four (4.1 and 4.2) of (A) FpvI and (B) PvdS according to the following classifications: ▲mutation(s) that gave higher activity in the presence, not absence of FpvR1–89; ■ mutation(s) that gave higher activity whether or not FpvR1–89 was present; ● mutation(s) that gave higher activity in the presence of FpvR1–89 and were not tested in the absence of FpvR1–89. Each mutant is represented on a different line.
Investigating the effects of FpvI and PvdS C-terminal deletions on activity
The truncation of PvdS by 12 amino acids due to the mutation Q176* had no effect on activity in quantitative assays (Figure 4B). To further investigate the role of the amino acids beyond region four in sigma factor activity, five amino acids were removed from the C-terminal end of FpvI (giving construct FpvI1–154) and 13 amino acids were removed from the C-terminal end of PvdS (PvdS1–174) (Figure 7A). FpvI1–154 had higher activity than WT FpvI in the presence of FpvR1-89, and lower activity than WT FpvI in the absence of FpvR1–89 (Figure 7B). This indicated that the amino acids beyond the C-terminal of region four are involved in binding to FpvR1–89. PvdS1–174 had higher activity than WT in both the presence and absence of FpvR1–89 (Figure 7C) suggesting that the removal of the C-terminal 13 amino acids improved protein stability or sigma factor function.
Figure 7.
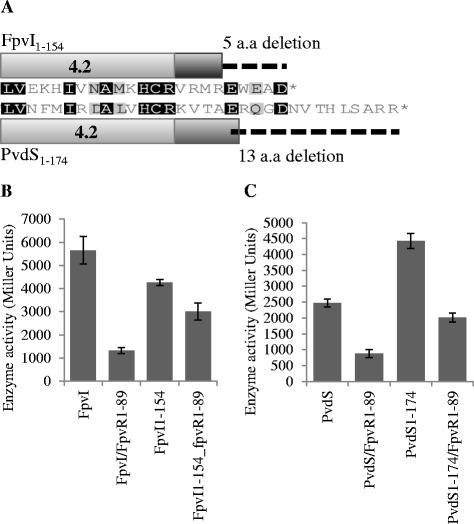
The activity of FpvI and PvdS C-terminal deletion mutants. (A) An alignment of FpvI showing the 5 amino acid C-terminal deletion and PvdS showing the 13 amino acid C-terminal deletion. (B and C) β-galactosidase assays were carried out with E. coli MC1061 (DE3) containing pMP190::PfpvA_lacZ or pMP190::PpvdE_lacZ, along with pETDuet expressing FpvR1–89 and C-terminal deletion mutants of (B) FpvI or (C) PvdS. Values are compared to WT FpvI and PvdS and were obtained from three biological replicates. Error bars are ±1 SD.
Discussion
Interactions between ECF sigma and anti-sigma factor proteins have been experimentally demonstrated for a small number of systems [23-27,41]. FpvR is unusual amongst anti-sigma factors in that it inhibits the activities of two different sigma factors, FpvI and PvdS [6,10]. Genetic evidence has indicated that FpvR interacts directly with both FpvI and PvdS [28]. Using the TAP-tag approach we have now demonstrated that the cytoplasmic region of FpvR does indeed bind to PvdS in P. aeruginosa, with binding being stable enough to permit co-purification of PvdS with the FpvR1–89-TAP construct. A key aspect of the TAP-tag methodology is that proteins are expressed at physiological levels, avoiding any artefacts that may result from the use of overexpression constructs and providing confidence that FpvR naturally binds PvdS. Binding of FpvR1–89-TAP to PvdS in conjunction with extensive genetic evidence for FpvR/FpvI interactions ([6,7,28], this study) allow us to conclude that FpvR also binds to FpvI, inhibiting its activity. However, we were unable to detect co-purification of FpvI with FpvR1–89-TAP (data not shown). This may be because the amount of FpvI in P. aeruginosa, which has not been measured, is much lower than the amount of PvdS (500–700 molecules per cell [42,43]), or because FpvR1–89 has lower affinity for FpvI than for PvdS, or due to limitations of the polyclonal FpvI antibody.
Co-expression of FpvR1–89 with either PvdS or FpvI in E. coli enabled purification of FpvR1-89–sigma factor complexes, providing further evidence that the cytoplasmic region of FpvR can bind to both FpvI and PvdS. The use of reporter gene constructs showed that binding inhibited sigma factor activity. The pETDuet vector used for these experiments gives approximately equal expression of co-expressed genes (in this case fpvR1–89 with either fpvI or pvdS). The strong inhibition of sigma factor activity seen in reporter gene assays (Figure 2), as well as the approximately equi-molar amounts of FpvR1–89 and FpvI/PvdS obtained following co-purification (Figure 3), suggest that FpvR has a high affinity for each of its partner sigma factor proteins despite their very different sequences. Comparisons of the small number of ECF sigma factor structures available indicate that all have a similar structure involving two alpha helix bundles, corresponding to regions two and four, connected by a disordered region [23-26,41]. It is likely that FpvI and PvdS have the same overall structure with sigma factor-specific features that are recognized by FpvR.
A bacterial 2-hybrid system was used previously to identify mutations that interfere with FpvR-sigma factor binding [28] although mutagenesis was not carried out on PvdS and was only performed on the C-terminal section of FpvI (FpvI95–159). A disadvantage of this system is that loss of protein-protein interaction can occur through mutations that have a non-specific effect on protein structure or amount and indeed the majority of mutations identified in that study resulted in reduced amounts of the mutant protein. One amino acid change (L103P) was identified that reduced affinity of FpvI for FpvR in the bacterial 2-hybrid system, while not affecting the amount of FpvI protein. We reasoned that selection for gain-of-function mutations using full-length FpvI and PvdS would identify further residues involved in sigma-FpvR interactions, while excluding mutations that caused reduced amounts of sigma factor or disrupted the overall protein structure. We therefore used the pETDuet co-expression system, in conjunction with error-prone PCR and reporter gene assays, to identify gain-of-function mutations that increased the activity of full length FpvI and PvdS in the presence of FpvR1–89. A number of mutations (for example, FpvI E139V/G; FpvI N132K/M153R; PvdS F133C/V and PvdS L165Q) were obtained in independent screens, from different pools of mutagenized genes. This parallel evolution suggests that these mutations were particularly effective in increasing sigma factor activity in the presence of FpvR1–89. Additional mutant genes containing single mutations were engineered by site-directed mutagenesis. Single mutations in FpvI (residues A102V, E139V/G, R152P and A145G) increased the activity of FpvI in the presence of FpvR1–89, implicating these residues in FpvR – FpvI interactions. None of the mutant FpvI variants tested had higher activity than WT in the absence of FpvR1–89 (Figure 5) showing that most if not all of the FpvI mutations specifically affect interactions with FpvR.
Three of the four PvdS mutants tested had higher activity than WT PvdS in the absence as well as the presence of FpvR1–89. This suggests that the effect of these mutations was at least partially due to increased sigma factor function or protein stability. In the absence of FpvR, PvdS expressed in E. coli forms inclusion bodies [44] and it may be that these mutations improve the solubility of PvdS or alternatively its affinity for core RNA polymerase or promoter DNA. PvdS L64P/L165Q was the only PvdS mutant to show increased activity in the presence of FpvR1–89 and similar activity to WT PvdS in the absence of FpvR1-89, suggesting that this combination of mutations disrupted interactions with FpvR1–89.
Screening for increased activity of FpvI and PvdS in the presence of FpvR1–89 gave different outcomes for the two sigma factors. FpvI mutants were obtained at a high frequency (approximately 6% of colonies) and the mutants tested had similar or lower activity than WT FpvI in the absence of FpvR1–89. PvdS mutations were obtained at a lower frequency (approximately 0.5% of colonies) and three of the four tested had higher activity than WT PvdS in the absence of FpvR1–89. One explanation for these differences is that mutations which disrupt interactions of PvdS with FpvR1–89 also reduce the activity of PvdS and so would not be identified in our gain-of-function screen. Alternatively, interaction of FpvI with FpvR1–89 may involve a relatively small number of amino acid residue interactions and disrupting one of these significantly reduces the affinity of FpvI for FpvR, whereas PvdS-FpvR1–89 interactions may involve a larger number of weaker interactions and disrupting only one of these may not give a detectable reduction in inhibition by FpvR1–89.
All the FpvI mutants that showed enhanced activity in the presence of FpvR1–89 had mutations in or around region four (Figure 6). This provides clear evidence for the role of this region in FpvR binding, consistent with earlier findings [28]. The PvdS mutants that showed enhanced activity in the presence of FpvR also had mutations in region four. However, fewer mutants were identified and the majority of those tested had enhanced sigma factor function in the absence of FpvR1–89 making the role of region four in binding to FpvR less clear-cut. Region four in each of FpvI and PvdS is predicted to be involved in promoter recognition at the −35 site [20,22,45] and structural data for E. coli housekeeping sigma 70 shows all regions of sigma 70 to make contact with core RNA polymerase [45]. Therefore, FpvR binding to region four might sterically interfere with both promoter recognition and recruitment of core RNA polymerase. There is considerable variability amongst ECF sigma factors in the region bound by the cognate anti-sigma factor which can be both region two and four [23-25], region two alone [26] or region four alone [27]. FpvR binding and sequestering any of these regions would likely disrupt recruitment of core RNA polymerase.
Although region four of FpvI is clearly critical for interactions with FpvR1–89, our results also suggest some involvement of region two in either improving sigma factor function or in interactions with FpvR1–89. For example, changes at residue S37 in region 2.2 of FpvI arose in three separate screens in combination with additional mutations. The PvdS double mutants F60V/L165Q and L64P/L165Q showed markedly higher activity than the PvdS L165Q mutant in the presence of FpvR1–89. For PvdS F60V/L165Q the effect appeared to be synergistic because the individual mutants showed no difference in activity to WT PvdS. These results, together with the fact that no improved mutants were identified containing only a mutation in region two, suggest that changes in region two stabilized the protein or improved sigma factor function.
One mutation identified in our initial screen was PvdS Q176*, although this mutation did not increase activity of PvdS in quantitative assays. This suggested that the 12 amino acids at the C-terminus of PvdS, which occur after region four, are not required for sigma factor function. An engineered mutant where the last 13 amino acids of PvdS were deleted showed higher activity than WT while apparently retaining interaction with FpvR1–89. It is possible that the residues at the C-terminal end of region four act as a signal targeting PvdS for degradation [46]. Removal of these residues could improve stability of the protein. Alternatively it could enhance interactions with core RNA polymerase if the flexibility of these residues decreases the strength of interaction. Deletion of five residues at the C-terminal of FpvI resulted in increased sigma factor activity in the presence of FpvR1–89 suggesting that this region also contributes to binding by FpvR1–89.
Conclusions
The cytoplasmic domain of FpvR forms stable interactions with PvdS in both P. aeruginosa and E. coli, and with FpvI in E. coli, despite the relatively low sequence identity of the sigma factors. Region four of FpvI as well as the C-terminus of this protein are of primary importance in binding to FpvR and we have identified four amino acid residues in FpvI that are likely to play a key role in its interaction with FpvR. It is likely that FpvI and PvdS share the same general tertiary structure that, in combination with specific residue interactions, is recognized by FpvR although we were unable to identify any single mutations that clearly reduced the affinity of PvdS for FpvR1–89. Our data are consistent with a model whereby FpvR inhibits FpvI and PvdS by occluding their binding to core RNA polymerase.
Methods
General methods
Bacterial strains used in this study are listed in Table 1. Bacteria were routinely grown in LB medium or on LB agar at 37°C. P. aeruginosa was grown using King’s B medium [47] for preparation of protein extracts. Media were supplemented with ampicillin (50 μg · mL−1), chloramphenicol (30 μg · mL−1), carbenicillin (300 μg · mL−1), tetracycline (25 μg · mL−1) or gentamicin (300 μg · mL−1) as required. E. coli MC1061 (DE3) was derived by lysogenizing E. coli MC1061 with λDE3 prophage (Novagen) using the manufacturer’s protocol.
Genetic manipulations
DNA constructs were made using PCR, with the PCR primers (Additional file 1: Table S1) containing introduced restriction sites to enable cloning. A PCR fragment comprising the fpvR promoter and the 5′ end of fpvR, encoding the predicted cytoplasmic part of FpvR (residues 1–89; designated fpvR1–89) was amplified from P. aeruginosa PAO1 genomic DNA using primers miniPfpvRXbaIfor and fpvRNEcorRIrev. This fragment was ligated (via the introduced EcoRI site) to an EcoRI-digested fragment encoding a C-terminal TAP tag (C-TAP) that had been amplified with primers CTAPEcoRIfor and CTAPBamHIrev from plasmid pCTAPi [48]. The resulting fpvR1–89-TAP fragment was cloned into pUCP23 [35] and miniCTX2 [36] using the introduced XbaI and BamHI restriction sites. Plasmids were introduced into P. aeruginosa by transformation or by conjugation from E. coli S17-1, and miniCTX2 vector sequences were then excised from the integrated miniCTX2 construct, as described previously [36]. pETDuet constructs were made by amplifying fpvR1–89, pvdS and fpvI from P. aeruginosa PAO1 genomic DNA using appropriate primers (Additional file 1: Table S1) and then cloning the resulting PCR fragments into pETDuet-1 [39]. The fidelity of all constructs was verified by DNA sequencing.
Protein purification
Proteins were purified from P. aeruginosa using the tandem affinity tag (TAP tag) method [29], with all purification steps carried out at 4°C. Following 24 h incubation at 37°C, bacteria were collected from 1.2 L of culture by centrifugation (6160 × g, 15 min), resuspended in chilled lysis buffer (10 mM Tris-Cl [pH 8.0], 75 mM NaCl, 0.05% (v/v) Tween 20, 20% (v/v) glycerol, 25 mM KCl, 2 mM EDTA [pH 8.0], 0.5 mM DTT, 0.5 mM PMSF) (10 mL) and lysed by sonication (Sonics Vibra Cell). The lysed cells were centrifuged (16,000 × g, 30 min) and the supernatant was applied to an IgG sepharose column (Amersham Biosciences/GE Healthcare) (0.5 mL) that had been equilibrated with lysis buffer. The column was washed with approximately 20 volumes of lysis buffer then equilibrated with AcTEV cleavage buffer (Invitrogen). The column-bound protein was treated with 100 U of AcTEV protease (Invitrogen) (12 h). The released protein was collected, mixed with an equal volume (1 mL) of calmodulin binding buffer (10 mM Tris-Cl [pH 8.0], 75 mM NaCl, 2 mM CaCl2, 0.05% (v/v) Tween 20, 10 mM β-mercaptoethanol, 0.5 mM PMSF) containing Complete™ protease inhibitor Mini tablet (Roche) (1 tablet/100 ml of buffer). It was then applied to a calmodulin sepharose column (Amersham Biosciences/GE Healthcare), washed twice with calmodulin binding buffer and then washed using fresh calmodulin binding buffer in which the concentration of Tween 20 had been amended to 0.02% (w/v). Elution buffer (10 mM Tris-Cl [pH 8.0], 100 mM NH4HCO3, 10 mM EGTA, 10 mM β-mercaptoethanol) (1.2 mL applied in 6 aliquots) was then added and the protein collected.
Proteins expressed as hexahistidine fusions in pETDuet were purified from E. coli by nickel-affinity chromatography. Cultures grown for 16 h were used to inoculate fresh media to OD600 = 0.1. Expression was induced at OD600 = 0.6 using a final IPTG concentration of 14 μg · mL−1 at 18°C for 16 h. The cells were collected by centrifugation and then resuspended in binding buffer (50 mM sodium phosphate [pH 8.0], 300 mM NaCl). Cell lysis was carried out using sonication as described above. The soluble fractions were obtained by centrifugation and applied to nickel-affinity chromatography resin (BioRad) equilibrated in binding buffer. The resin was washed four times in wash buffer (50 mM sodium phosphate [pH 8.0], 300 mM NaCl, 30 mM imidazole) prior to elution in elution buffer (50 mM sodium phosphate [pH 8.0], 300 mM NaCl, 250 mM imidazole).
Proteins were analyzed by SDS-PAGE and Western blotting as described previously [14,43] using monoclonal antibodies against CBP (Upstate, Millipore), PvdS [49], the N-terminal (cytoplasmic) portion of FpvR [14], and a polyclonal antibody against FpvI (generated in-house), applied to the membrane sequentially.
β-galactosidase reporter gene assays
Bacteria were inoculated in duplicate into 150 μL overnight cultures in LB amended with appropriate antibiotics and 0.4% glucose in wells of a 96-well flat bottom microtiter plate and grown for 16 h, 200 rpm at 37°C. Wells of a fresh microtiter plate, each containing 200 μL LB, antibiotics and 0.2% glucose, were inoculated with 15 μL of the overnight culture and incubated for 4 h, 200 rpm at 30°C. Portions (100 μL) of the resulting micro-cultures were inoculated into wells of a microtiter plate containing induction media (100 μL LB, antibiotics, 0.05 mM IPTG, 0.2% glucose) and incubated for 1 h, 200 rpm at 30°C. Absorbance was measured at OD600 using an Enspire plate reader (Perkin Elmer). For the assay, 20 μL of each induced culture were added to sodium phosphate buffer (84 μL, 40 mM, pH 7.0) containing ZOB buffer (46 μL) [50] and immediately incubated in the plate reader at 37°C without shaking. Absorbance readings at OD420 were taken at time zero and then every 2 minutes for 30 minutes. Absorbance readings for LB control wells were subtracted from the data. The enzyme activity for each well was calculated using the Miller equation [51]. Each assay was carried out in triplicate.
Mutagenic PCR
A mutagenic PCR protocol was developed using Red Hot Taq polymerase (ABgene) that lacks 3′-5′ exonuclease activity, and error prone buffer conditions based on those described in [40]. The error prone PCR protocol used has been shown to minimize the intrinsic nucleotide substitution bias of Taq polymerase and the occurrence of mutation hotspots. Each reaction contained 2 μL 10 × mutagenic PCR buffer (70 mM MgCl2, 500 mM KCl, 100 mM Tris–HCl, [pH 8.2]), 2 μL 10 × dNTP (2 mM dGTP, 2 mM dATP, 15 mM dCTP, 10 mM dTTP), 1 μL 6 pmol · μL−1 forward primer, 1 μL 6 pmol · μL−1 reverse primer, 11 μL ddH2O, 2 μL 5 mM MnCl2, 0.4 μL Red Hot Taq polymerase, and 0.6 μL 5 ng · μL−1 template DNA. The reactions were incubated for 4 minutes at 94°C, followed by 30 cycles of 30 seconds at 94°C, 30 seconds at 55°C and 2 minutes at 72°C. For mutagenesis of pvdS, mutagenic PCR was carried out using primers pvdSBamHIfor and pvdSSalIrev (Additional file 1: Table S1), with pETDuet::pvdS as template. For mutagenesis of fpvI, mutagenic PCR was carried out using primers fpvIBamHIfor and fpvISacIstoprev (Additional file 1: Table S1) with pETDuet::fpvI as template. Libraries of mutated PCR products were cloned into pGEM-T Easy (Promega) and then sub-cloned into pETDuet_fpvR1–89 for screening. Three separate pvdS and fpvI libraries were made. Libraries were grown on LB agar containing 24 μg · mL−1 IPTG and 120 μg · mL−1 BCIG. Mutants that had enhanced reporter plasmid activity were identified as deeper blue colonies and were analyzed by DNA sequencing and β-galactosidase assay.
Overlap PCR was used to generate single mutations in fpvI and pvdS genes. The genes were amplified from PAO1 genomic DNA using Phusion High-Fidelity polymerase (Thermo Scientific) in two fragments using primers (Additional file 1: Table S1) that created a 25 bp overlap. Overlap PCR was performed using Biomix Red master mix (Bioline) with the two purified template fragments at equi-molar concentration totaling 100 ng in a volume of 40 μL. The reactions were initially incubated in the absence of primers for 2 minutes at 95°C, followed by 15 cycles of 30 seconds at 95°C, 30 seconds at 55°C, 30 seconds/kb at 72°C. The full length primers were then added in 20 μL of Biomix Red, adjusted for the final reaction volume of 60 μL. The reactions were incubated for an additional 15 cycles of 30 seconds at 95°C, 30 seconds at 67°C, 30 seconds/kb at 72°C, followed by 5 minutes at 72°C.
Acknowledgements
This research was supported in part by the Marsden Fund administered by the Royal Society of New Zealand (contracts UOO0502 and VUW0901) and by a University of Otago Research Grant. Rebecca Edgar was the recipient of a Prestigious PhD scholarship from the University of Otago. We are grateful to Dulantha Ulluwishewa and Matthew McNeil for their assistance in establishing the mutagenic PCR protocol and mutation screen and to Catherine Day for her advice during establishment of the TAP purification protocol.
Abbreviations
- CBP
Calmodulin binding protein
- ECF
Extracytoplasmic function
- MCS
Multiple cloning site
- TAP-tag
Tandem affinity purification tag
- WT
Wild-type
Additional file
Supplemental tables S1-S4 and figures S1-S3 associated with this manuscript.
Footnotes
Authors’ contributions
RE carried out protein co-purifications from E. coli and reporter gene assays, generated mutants by site directed mutagenesis, analyzed data and co-wrote the manuscript; XX carried out mutagenic PCR, identified and sequenced clones containing mutant fpvI or pvdS with increased activity in reporter gene assays, and created the C-terminal deletion mutants; MS engineered and carried out all experiments with TAP-tag constructs in P. aeruginosa and analyzed data; AK and LM generated and tested all constructs for co-purification of proteins and mutagenesis in E. coli; DA contributed to experimental design, interpreted data and co-wrote the manuscript; IL conceived and coordinated the study, contributed to experimental design, interpreted data and co-wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Rebecca J Edgar, Email: edgre961@student.otago.ac.nz.
Xin Xu, Email: xinxu9@gmail.com.
Matt Shirley, Email: barshirley1996@gmail.com.
Anna F Konings, Email: afkonings@gmail.com.
Lois W Martin, Email: lois.martin@otago.ac.nz.
David F Ackerley, Email: david.ackerley@vuw.ac.nz.
Iain L Lamont, Email: iain.lamont@otago.ac.nz.
References
- 1.Driscoll JA, Brody SL, Kollef MH. The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs. 2007;67(3):351–368. doi: 10.2165/00003495-200767030-00003. [DOI] [PubMed] [Google Scholar]
- 2.Ratledge C, Dover LG. Iron metabolism in pathogenic bacteria. Annu Rev Microbiol. 2000;54:881–941. doi: 10.1146/annurev.micro.54.1.881. [DOI] [PubMed] [Google Scholar]
- 3.Schalk IJ. Metal trafficking via siderophores in Gram-negative bacteria: specificities and characteristics of the pyoverdine pathway. J Inorg Biochem. 2008;102(5–6):1159–1169. doi: 10.1016/j.jinorgbio.2007.11.017. [DOI] [PubMed] [Google Scholar]
- 4.Cunliffe HE, Merriman TR, Lamont IL. Cloning and characterization of pvdS, a gene required for pyoverdine synthesis in Pseudomonas aeruginosa: PvdS is probably an alternative sigma factor. J Bacteriol. 1995;177:2744–2750. doi: 10.1128/jb.177.10.2744-2750.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miyazaki H, Kato H, Nakazawa T, Tsuda M. A positive regulatory gene, pvdS, for expression of pyoverdin biosynthetic genes in Pseudomonas aeruginosa PAO. Mol Gen Genet. 1995;248:17–24. doi: 10.1007/BF02456609. [DOI] [PubMed] [Google Scholar]
- 6.Beare PA, For RJ, Martin LW, Lamont IL. Siderophore-mediated cell signalling in Pseudomonas aeruginosa: divergent pathways regulate virulence factor production and siderophore receptor synthesis. Mol Microbiol. 2003;47(1):195–207. doi: 10.1046/j.1365-2958.2003.03288.x. [DOI] [PubMed] [Google Scholar]
- 7.Redly GA, Poole K. Pyoverdine-mediated regulation of FpvA synthesis in Pseudomonas aeruginosa: Involvement of a probable extracytoplasmic-function sigma factor, FpvI. J Bacteriol. 2003;185(4):1261–1265. doi: 10.1128/JB.185.4.1261-1265.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ochsner UA, Johnson Z, Lamont IL, Cunliffe HE, Vasil ML. Exotoxin A production in Pseudomonas aeruginosa requires the iron-regulated pvdS gene encoding an alternative sigma factor. Mol Microbiol. 1996;21(5):1019–1028. doi: 10.1046/j.1365-2958.1996.481425.x. [DOI] [PubMed] [Google Scholar]
- 9.Wilderman PJ, Vasil AI, Johnson Z, Wilson MJ, Cunliffe HE, Lamont IL, Vasil MI. Characterization of an endoprotease (PrpL) encoded by a PvdS-regulated gene in Pseudomonas aeruginosa. Infect Immun. 2001;69(9):5385–5394. doi: 10.1128/IAI.69.9.5385-5394.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lamont IL, Beare PA, Ochsner U, Vasil AI, Vasil ML. Siderophore-mediated signaling regulates virulence factor production in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2002;99(10):7072–7077. doi: 10.1073/pnas.092016999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Llamas MA, Imperi F, Visca P, Lamont IL. Cell-surface signaling in Pseudomonas: stress responses, iron transport and pathogenicity. FEMS Microbiol Rev. 2014;38(4):569–597. doi: 10.1111/1574-6976.12078. [DOI] [PubMed] [Google Scholar]
- 12.James HE, Beare PA, Martin LW, Lamont IL. Mutational analysis of a bifunctional ferrisiderophore receptor and signal-transducing protein from Pseudomonas aeruginosa. J Bacteriol. 2005;187(13):4514–4520. doi: 10.1128/JB.187.13.4514-4520.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen J, Meldrum A, Poole K. FpvA receptor involvement in pyoverdine biosynthesis in Pseudomonas aeruginosa. J Bacteriol. 2002;184(12):3268–3275. doi: 10.1128/JB.184.12.3268-3275.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Draper RC, Martin LW, Beare PA, Lamont IL. Differential proteolysis of sigma regulators controls cell-surface signalling in Pseudomonas aeruginosa. Mol Microbiol. 2011;82(6):1444–1453. doi: 10.1111/j.1365-2958.2011.07901.x. [DOI] [PubMed] [Google Scholar]
- 15.Shirley M, Lamont IL. Role of TonB1 in pyoverdine-mediated signaling in Pseudomonas aeruginosa. J Bacteriol. 2009;191(18):5634–5640. doi: 10.1128/JB.00742-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Braun V, Mahren S. Transmembrane transcriptional control (surface signalling) of the Escherichia coli Fec type. FEMS Microbiol Rev. 2005;29(4):673–684. doi: 10.1016/j.femsre.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 17.Visca P, Leoni L, Wilson MJ, Lamont IL. Iron transport and regulation, cell signalling and genomics: lessons from Escherichia coli and Pseudomonas. Mol Microbiol. 2002;45(5):1177–1190. doi: 10.1046/j.1365-2958.2002.03088.x. [DOI] [PubMed] [Google Scholar]
- 18.Ochs M, Veitinger S, Kim I, Welz D, Angerer A, Braun V. Regulation of citrate-dependent iron transport of Escherichia coli: fecR is required for transcription activation by FecI. Mol Microbiol. 1995;15(1):119–132. doi: 10.1111/j.1365-2958.1995.tb02226.x. [DOI] [PubMed] [Google Scholar]
- 19.Mettrick KA, Lamont IL. Different roles for anti-sigma factors in siderophore signalling pathways of Pseudomonas aeruginosa. Mol Microbiol. 2009;74(5):1257–1271. doi: 10.1111/j.1365-2958.2009.06932.x. [DOI] [PubMed] [Google Scholar]
- 20.Helmann JD. The extracytoplasmic function (ECF) sigma factors. Adv Microb Physiol. 2002;46:47–110. doi: 10.1016/S0065-2911(02)46002-X. [DOI] [PubMed] [Google Scholar]
- 21.Staron A, Sofia HJ, Dietrich S, Ulrich LE, Liesegang H, Mascher T. The third pillar of bacterial signal transduction: classification of the extracytoplasmic function (ECF) sigma factor protein family. Mol Microbiol. 2009;74(3):557–581. doi: 10.1111/j.1365-2958.2009.06870.x. [DOI] [PubMed] [Google Scholar]
- 22.Wilson MJ, Lamont IL. Mutational analysis of an extracytoplasmic-function sigma factor to investigate its interactions with RNA polymerase and DNA. J Bacteriol. 2006;188(5):1935–1942. doi: 10.1128/JB.188.5.1935-1942.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Campbell EA, Tupy JL, Gruber TM, Wang S, Sharp MM, Gross CA, Darst SA. Crystal structure of Escherichia coli σE with the cytoplasmic domain of its anti-sigma RseA. Mol Cell. 2003;11(4):1067–1078. doi: 10.1016/S1097-2765(03)00148-5. [DOI] [PubMed] [Google Scholar]
- 24.Campbell EA, Greenwell R, Anthony JR, Wang S, Lim L, Das K, Sofia HJ, Donohue TJ, Darst SA. A conserved structural module regulates transcriptional responses to diverse stress signals in bacteria. Mol Cell. 2007;27(5):793–805. doi: 10.1016/j.molcel.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shukla J, Gupta R, Thakur KG, Gokhale R, Gopal B. Structural basis for the redox sensitivity of the Mycobacterium tuberculosis SigK-RskA sigma-anti-sigma complex. Acta Crystallogr D Biol Crystallogr. 2014;70(Pt 4):1026–1036. doi: 10.1107/S1399004714000121. [DOI] [PubMed] [Google Scholar]
- 26.Li W, Stevenson CE, Burton N, Jakimowicz P, Paget MS, Buttner MJ, Lawson DM, Kleanthous C. Identification and structure of the anti-sigma factor-binding domain of the disulphide-stress regulated sigma factor sigma R from Streptomyces coelicolor. J Mol Biol. 2002;323(2):225–236. doi: 10.1016/S0022-2836(02)00948-8. [DOI] [PubMed] [Google Scholar]
- 27.Jaiswal RK, Prabha TS, Manjeera G, Gopal B. Mycobacterium tuberculosis RsdA provides a conformational rationale for selective regulation of sigma-factor activity by proteolysis. Nucleic Acids Res. 2013;41(5):3414–3423. doi: 10.1093/nar/gks1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Redly GA, Poole K. FpvIR control of fpvA ferric pyoverdine receptor gene expression in Pseudomonas aeruginosa: demonstration of an interaction between FpvI and FpvR and identification of mutations in each compromising this interaction. J Bacteriol. 2005;187(16):5648–5657. doi: 10.1128/JB.187.16.5648-5657.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Seraphin B. A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol. 1999;17(10):1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- 30.Wilson MJ, McMorran BJ, Lamont IL. Analysis of promoters recognized by PvdS, an extracytoplasmic-function sigma factor protein from Pseudomonas aeruginosa. J Bacteriol. 2001;183(6):2151–2155. doi: 10.1128/JB.183.6.2151-2155.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FSL, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GK, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock RE, Lory S, et al. Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature. 2000;406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 32.Simon R, Priefer U, Pühler A. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Biotechnology. 1983;1:784–791. doi: 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 33.Yanisch-Perron C, Vieira J, Messing J. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene. 1985;33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- 34.Casabadan MJ, Cohen SN. Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J Mol Biol. 1980;138:179–207. doi: 10.1016/0022-2836(80)90283-1. [DOI] [PubMed] [Google Scholar]
- 35.West SE, Schweizer HP, Dall C, Sample AK, Runyen-Janecky LJ. Construction of improved Escherichia-Pseudomonas shuttle vectors derived from pUC18/19 and sequence of the region required for replication in P. aeruginosa. Gene. 1994;148:81–86. doi: 10.1016/0378-1119(94)90237-2. [DOI] [PubMed] [Google Scholar]
- 36.Hoang TT, Kutchma AJ, Becher A, Schweizer HP. Integration-proficient plasmids for Pseudomonas aeruginosa: site-specific integration and use for engineering of reporter and expression strains. Plasmid. 2000;43(1):59–72. doi: 10.1006/plas.1999.1441. [DOI] [PubMed] [Google Scholar]
- 37.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. A broad-host-range Flp-FRT recombination system for site-specific exision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene. 1998;212:77–86. doi: 10.1016/S0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- 38.Rombel IT, McMorran BJ, Lamont IL. Identification of a DNA sequence motif required for expression of iron-regulated genes in pseudomonads. Mol Gen Genet. 1995;246:519–528. doi: 10.1007/BF00290456. [DOI] [PubMed] [Google Scholar]
- 39.Novy R, Yaeger K, Held D, Mierendorf R. Coexpression of multiple target proteins in E. coli. inNovations. 2002;15:2–6. [Google Scholar]
- 40.Cadwell RC, Joyce GF. Randomization of genes by PCR mutagenesis. PCR Methods Appl. 1992;2(1):28–33. doi: 10.1101/gr.2.1.28. [DOI] [PubMed] [Google Scholar]
- 41.Maillard AP, Girard E, Ziani W, Petit-Hartlein I, Kahn R, Coves J. The crystal structure of the anti-sigma factor CnrY in complex with the sigma factor CnrH shows a new structural class of anti-sigma factors targeting extracytoplasmic function sigma factors. J Mol Biol. 2014;426(12):2313–27. doi: 10.1016/j.jmb.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 42.Tiburzi F, Imperi F, Visca P. Intracellular levels and activity of PvdS, the major iron starvation sigma factor of Pseudomonas aeruginosa. Mol Microbiol. 2008;67(1):213–227. doi: 10.1111/j.1365-2958.2007.06051.x. [DOI] [PubMed] [Google Scholar]
- 43.Spencer MR, Beare PA, Lamont IL. Role of cell surface signaling in proteolysis of an alternative sigma factor in Pseudomonas aeruginosa. J Bacteriol. 2008;190(14):4865–4869. doi: 10.1128/JB.01998-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilson MJ, Lamont IL. Characterization of an ECF sigma factor protein from Pseudomonas aeruginosa. Biochem Biophys Res Commun. 2000;273(2):578–583. doi: 10.1006/bbrc.2000.2996. [DOI] [PubMed] [Google Scholar]
- 45.Murakami KS. X-ray crystal structure of Escherichia coli RNA polymerase sigma70 holoenzyme. J Biol Chem. 2013;288(13):9126–9134. doi: 10.1074/jbc.M112.430900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sauer RT, Baker TA. AAA + proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem. 2011;80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- 47.King EO, Ward MK, Raney DE. Two simple media for the demonstration of pyocyanin and fluorescein. J of Lab Medicine. 1954;44:301–307. [PubMed] [Google Scholar]
- 48.Rohila JS, Chen M, Cerny R, Fromm ME. Improved tandem affinity purification tag and methods for isolation of protein heterocomplexes from plants. Plant J. 2004;38(1):172–181. doi: 10.1111/j.1365-313X.2004.02031.x. [DOI] [PubMed] [Google Scholar]
- 49.Xiong YQ, Vasil ML, Johnson Z, Ochsner UA, Bayer AS. The oxygen- and iron-dependent sigma factor PvdS of Pseudomonas aeruginosa is an important virulence factor in experimental infective endocarditis. J Infect Dis. 2000;181(3):1020–1026. doi: 10.1086/315338. [DOI] [PubMed] [Google Scholar]
- 50.Alksne LE, Burgio P, Hu W, Feld B, Singh MP, Tuckman M, Petersen PJ, Labthavikul P, McGlynn M, Barbieri L, McDonald L, Bradford P, Dushin RG, Rothstein D, Projan SJ. Identification and analysis of bacterial protein secretion inhibitors utilizing a SecA-LacZ reporter fusion system. Antimicrob Agents Chemother. 2000;44(6):1418–1427. doi: 10.1128/AAC.44.6.1418-1427.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller JH, editor. Assay of Beta-Galactosidase. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory; 1972. [Google Scholar]