

Abstract
Mutations in LRRK2 cause a dominantly inherited form of Parkinson’s disease (PD) and are the most common known genetic determinant of PD. Inhibitor-based therapies targeting LRRK2 have emerged as a key therapeutic strategy in PD; thus, understanding the consequences of inhibiting the normal cellular functions of this protein is vital. Despite much interest, the physiological functions of LRRK2 remain unclear. Several recent studies have linked the toxicity caused by overexpression of pathogenic mutant forms of LRRK2 to defects in the endolysosomal and autophagy pathways, raising the question of whether endogenous LRRK2 might play a role in these processes. Here, we report the characterization of multiple novel ethyl methanesulfonate (EMS)-induced nonsense alleles in the Drosophila LRRK2 homolog, lrrk. Using these alleles, we show that lrrk loss-of-function causes striking defects in the endolysosomal and autophagy pathways, including the accumulation of markedly enlarged lysosomes that are laden with undigested contents, consistent with a defect in lysosomal degradation. lrrk loss-of-function also results in the accumulation of autophagosomes, as well as the presence of enlarged early endosomes laden with mono-ubiquitylated cargo proteins, suggesting an additional defect in lysosomal substrate delivery. Interestingly, the lysosomal abnormalities in these lrrk mutants can be suppressed by a constitutively active form of the small GTPase rab9, which promotes retromer-dependent recycling from late endosomes to the Golgi. Collectively, our data provides compelling evidence of a vital role for lrrk in lysosomal function and endolysosomal membrane transport in vivo, and suggests a link between lrrk and retromer-mediated endosomal recycling.
KEY WORDS: LRRK2, Lysosome, Parkinson’s disease, Drosophila, Autophagy, Endosomes, Rab7, Rab9
INTRODUCTION
Parkinson’s disease (PD) is a common and devastating neurodegenerative movement disorder. The leucine-rich repeat kinase 2 (LRRK2) gene is an important therapeutic target for PD because it is the most common known genetic determinant of the disease (Dawson et al., 2010; Kett and Dauer, 2012). LRRK2 mutations were originally identified as the causative factor in dominantly inherited forms of PD linked to the PARK8 locus (Paisán-Ruíz et al., 2004; Zimprich et al., 2004) and, more recently, sequence variation at the LRRK2 locus has been associated with an increased risk of developing sporadic PD in genome-wide association studies (Satake et al., 2009; Simón-Sánchez et al., 2009). LRRK2 encodes a large multi-domain protein characterized by leucine-rich repeats, a GTPase domain and a kinase domain (Bosgraaf and Van Haastert, 2003). The cellular functions of LRRK2 remain unclear because it has been linked to multiple diverse cellular processes, including mitochondrial function (Smith et al., 2005), regulation of transcription (Kanao et al., 2010) and translation (Gehrke et al., 2010; Imai et al., 2008; Martin et al., 2014), Golgi protein sorting (Sakaguchi-Nakashima et al., 2007), apoptosis (Ho et al., 2009), and regulation of the dynamics of actin (Jaleel et al., 2007; Parisiadou et al., 2009) and microtubules (Gandhi et al., 2008; Gillardon, 2009; Kett et al., 2012; Lin et al., 2009).
Understanding the normal cellular functions of LRRK2 is vital because the mechanisms mediating the pathogenicity of mutant forms of LRRK2 are likely to be related to the physiological functions of the wild-type protein. Moreover, although inhibitor-based therapies targeting LRRK2 have emerged as a prime therapeutic target in PD (Lee et al., 2012), the effects of inhibiting endogenous LRRK2 are not clear. Several recent studies have suggested a role for LRRK2 and its homologs in lysosomal function and in membrane trafficking in the endolysosomal and autophagy pathways; however, these interpretations derive largely from studies of overexpression of LRRK2 and its pathological mutant forms. This raises the question of whether endogenous LRRK2 plays a role in endolysosomal processes under physiological conditions.
Despite the diversity of cellular functions to which LRRK2 has been linked, LRRK2 and its homologs have been consistently found to localize to intracellular membranes in the endolysosomal pathway (Alegre-Abarrategui et al., 2009; Berger et al., 2010; Biskup et al., 2006; Hatano et al., 2007; Higashi et al., 2009; Shin et al., 2008). We have previously reported that the protein encoded by the Drosophila homolog of LRRK2, lrrk, localizes specifically to the membranes of late endosomes and lysosomes, and to a lesser extent to early endosomes, in vivo (Dodson et al., 2012). We have found that Lrrk physically binds to the late endosomal protein Rab7, and overexpression of a PD-causing mutant form of lrrk results in Rab7-mediated lysosomal positioning defects (Dodson et al., 2012). Interestingly, mammalian LRRK2 has recently been found to interact physically with Rab7L1 (Beilina et al., 2014; MacLeod et al., 2013), a homolog of Rab7 (Shimizu et al., 1997). Via this interaction, LRRK2 has been linked in overexpression-based studies to retromer-dependent endosome-to-Golgi membrane transport via interactions with Vps35 (MacLeod et al., 2013), and to Rab7L1-dependent lysosomal clearance of Golgi-derived vesicles (Beilina et al., 2014). Moreover, multiple studies have reported that overexpression of either wild-type or pathogenic mutant forms of LRRK2 result in the accumulation of aberrant lysosomal and autophagosomal structures (Alegre-Abarrategui et al., 2009; Bravo-San Pedro et al., 2012; Gómez-Suaga et al., 2012; MacLeod et al., 2006; MacLeod et al., 2013; Orenstein et al., 2013; Plowey et al., 2008; Ramonet et al., 2011).
TRANSLATIONAL IMPACT.
Clinical issue
Parkinson’s disease (PD) is a devastating neurodegenerative movement disorder with no cure. Mutations in the leucine-rich repeat kinase 2 (LRRK2) gene cause a dominantly inherited form of PD. In addition, sequence variation at the LRRK2 locus has been associated with risk for sporadic PD, making LRRK2 the most common genetic determinant of PD. Despite intense research efforts, the normal cellular functions of LRRK2 remain unclear. Understanding the in vivo functions of LRRK2 is crucial because LRRK2 inhibition has emerged as a prime therapeutic strategy for PD.
Results
In this study, multiple nonsense mutations in the Drosophila LRRK2 homolog lrrk were characterized, demonstrating that these novel lrrk mutants show multiple defects in the endolysosomal and autophagy pathways. These include accumulation of markedly enlarged lysosomes containing undigested cellular contents, enlarged early endosomes laden with monoubiquitylated cargo proteins, and autophagosomes. Interestingly, the lysosomal defects in these lrrk mutant flies can be suppressed by enhancing the expression of rab9, a gene encoding a small GTPase with which Lrrk colocalizes and physically binds to at the late endosomal membrane. This suggests that augmenting Rab9 activity, which is involved in retrograde transport from late endosomes to the trans-Golgi network, might bypass a need for lrrk, supporting a role for lrrk in the Rab9-trafficking pathway.
Implications and future directions
These results demonstrate the crucial role played by the Drosophila LRRK2 homolog in lysosomal transport and in cargo trafficking to the lysosome in the endocytic pathway. The interactions between Lrrk and Rab9 suggest that a defect in rab9-dependent endosome-to-Golgi transport might underlie lysosome dysfunction in lrrk mutant flies. The findings suggest that efforts to target LRRK2 activity therapeutically will need to be specifically aimed at mutant LRRK2 alleles. Reducing the activity of endogenous wild-type LRRK2 might in fact result in cellular toxicity due to impaired lysosomal function and endolysosomal membrane transport.
Although these studies suggest a role for LRRK2 in endolysosomal processes, what is lacking is strong evidence for a physiological role for LRRK2 in endolysosomal functions in vivo. The strongest evidence to date comes from analysis of renal epithelial cells in LRRK2 knockout mice, showing changes in the numbers of autophagosomes and autolysosomes, as well as accumulation of lipofuscin granules, which can be suggestive of lysosome dysfunction (Tong et al., 2012). Loss-of-function of Drosophila lrrk has been reported to cause defects in synaptic vesicle endocytosis at the larval neuromuscular junction (NMJ) that is associated with decreased uptake of the tracer FM1-43, which is internalized via endocytosis (Matta et al., 2012). However, this experimental system is limited because the NMJ lacks the resolution for detailed subcellular analysis. In our previous work, we used the best-characterized Drosophila lrrk loss-of-function allele, which results from insertion of a PiggyBac transposable element within a lrrk intron (Imai et al., 2008; Lee et al., 2007; Matta et al., 2012; Wang et al., 2008), and found only mild defects in the endolysosomal pathway under endogenous conditions. Our data suggested that endogenous lrrk does play a role in regulating lysosome positioning, but only in a sensitized genetic system in which a constitutively active form of rab7 is expressed (Dodson et al., 2012).
Because proper functioning of the endolysosomal pathway is crucial for cellular homeostasis, it is puzzling that stronger phenotypes have not been detected in the context of loss-of-function of LRRK2 or its homologs, if indeed it plays a vital role in these processes. Thus, we hypothesized that the published Drosophila lrrk allele might not uncover the full range of phenotypes caused by lrrk loss-of-function. To address this issue, we have generated multiple novel alleles of Drosophila lrrk with in-frame stop codons, which should thus function as true null alleles. We report here striking phenotypes in both the endolysosomal and autophagy pathways in these flies.
RESULTS
Novel lrrk nonsense alleles cause reduced female fertility associated with premature caspase-dependent apoptosis of follicle cells
The best-characterized loss-of-function mutation in Drosophila lrrk, lrrke03680, results from a PiggyBac insertion in a lrrk intron (Fig. 1A) (Imai et al., 2008; Lee et al., 2007; Matta et al., 2012; Wang et al., 2008). Previously, we reported a reduction in lrrk transcript levels caused by the lrrke03680 allele by quantitative RT-PCR (Dodson et al., 2012). However, in a more detailed analysis using multiple primer pairs distributed across the lrrk genomic region, we found heterogeneity in lrrk transcript abundance in lrrke03680 flies (Fig. 1A). For example, primer pairs spanning the exon 6/7 and exon 9/10 splice junctions yielded a transcript abundance of about 25% of control in lrrke03680 homozygous flies, whereas primer pairs spanning the exon 7/8 junction yielded an abundance of 90% of control, and primers spanning the exon 4/5 junction gave a 36% increase in transcript abundance (Fig. 1A). These results were highly reproducible across multiple replicates, and suggest the presence of multiple transcripts that are differentially affected by the PiggyBac insertion, and indeed that some of these transcripts might be upregulated in lrrke03680 flies.
Fig. 1.

Characterization of lrrk NS alleles, which result in reduced female fertility. (A) Quantitative RT-PCR analysis of lrrk alleles, expressed as fold change in transcript abundance relative to wild type. Expected products from primer pairs used in this analysis are depicted in the schematic of the lrrk genomic region above (exons 1–11 depicted as gray rectangles and introns depicted as black lines). There is marked heterogeneity in transcript abundance in lrrke03680 homozygous flies depending on which primer pairs were used, ranging from a transcript abundance of 25% relative to wild type using primers flanking the exon 6/7 and 9/10 splice sites, to a 36% increase in transcript abundance using primers flanking the exon 4/5 splice site. Note that the PiggyBac insertion site in the lrrke03680 line is within the intron between exons 5 and 6. In contrast, regardless of which primer pairs were used, transcript abundance in lrrk NS flies was either not significantly changed relative to wild type, or mildly reduced to 60–80% of control. Note that trans-heterozygous combinations of lrrk NS alleles were used for some of this analysis because background lethal or semi-lethal mutations in the lrrk1 and lrrk2 stocks make obtaining adult homozygous flies difficult. (B) Schematic representation of Lrrk protein domains (LRR, leucine-rich repeats; Roc, GTPase domain; Cor, C-terminal of Roc domain; Kin, kinase domain), showing locations of nonsense mutations in lrrk NS alleles (lrrk1–lrrk4). Note that the lrrk2 allele is generated by a single-nucleotide insertion that results in a frameshift that creates a new stop codon 11 residues downstream. Shown in red is the location of the G1849S mutation within the kinase domain (referred to as lrrkGS), which is analogous to the most common PD-associated mutation in human LRRK2, G2019S. (C) As measured by average number of adult progeny produced per single female of the indicated genotype, trans-heterozygous combinations of lrrk NS alleles show markedly reduced female fertility, which can be rescued by targeted follicle-cell-specific expression of wild-type lrrk (lrrkWT), lrrkGS or human LRRK2 (huLRRK2). n=20 single females per genotype. *P<0.0001.
These results suggested the possibility that the lrrke03680 allele does not represent a true null owing to differentially expressed alternative isoforms. To generate new lrrk nonsense alleles, we used TILLING, a high-throughput method by which point mutations in a gene of interest are identified in a population of chemically mutagenized flies (Till et al., 2003). This yielded four ethyl methanesulfonate (EMS)-induced nonsense mutations in lrrk (these alleles are hereafter collectively referred to as lrrk NS). The truncated proteins encoded by all four of the lrrk NS alleles are predicted to lack the kinase domain, and three of the four would additionally lack the GTPase domain (Fig. 1B). In contrast to the lrrke03680 allele, the lrrk NS alleles consistently and reproducibly yielded transcript abundances that were either not significantly changed compared with wild type, or that were modestly reduced to 60–80% of control (Fig. 1A), suggesting that the transcript abundance of the different lrrk isoforms is less differentially affected by the NS alleles than by the lrrke03680 PiggyBac insertion. Unfortunately, using the only published antibody to Drosophila Lrrk (Imai et al., 2008), we were unable to detect a specific Lrrk signal in wild-type flies, nor in lrrk overexpression flies, under multiple experimental conditions. Thus, we are unable to assess the net effect of these alleles on Lrrk protein abundance.
Because alleles generated by EMS mutagenesis are likely to also carry background mutations, we exclusively focused our studies on trans-heterozygous combinations of these alleles because they would be highly unlikely to carry similar EMS-induced background mutations. All phenotypes described below were highly similar if not identical in all trans-heterozygous combinations of these four nonsense alleles. lrrk NS flies did not show any discernible defects in dopaminergic neuron number or morphology with age (supplementary material Fig. S1), which is consistent with what has previously been reported for lrrke03680 flies (Wang et al., 2008). This is not unexpected given that dominant mutations in LRRK2, rather than recessive loss-of-function mutations, cause PD.
Previously, we and others have reported that lrrke03680 homozygous females show a dramatic reduction in female fertility (Dodson et al., 2012; Imai et al., 2008; Lee et al., 2007), which can be rescued by restoring wild-type lrrk function specifically in follicle cells (Dodson et al., 2012). Follicle cells are a somatic epithelial cell monolayer that surrounds the developing oocyte during oogenesis (Fig. 2A) (King, 1970; Spradling, 1993). Follicle cells have a number of advantages that make them an attractive cell biological system, including their large size, ease of accessibility and squamous morphology that aids in visualization of subcellular structures. As expected, all trans-heterozygous combinations of lrrk NS alleles showed reduced female fertility that could be rescued by follicle-cell-specific expression of wild-type lrrk (Fig. 1C). Expression of lrrkGS, which is analogous to the most common PD-causing mutation in human LRRK2 (G2019S) (Bonifati, 2007; Taylor et al., 2006), also suppressed female infertility in lrrk NS flies (Fig. 1C). This suggests that lrrkGS retains at least some of the functions of wild-type lrrk, consistent with what we have previously reported (Dodson et al., 2012). Moreover, follicle-cell-specific expression of human LRRK2 also restored fertility to lrrk NS flies (Fig. 1C), suggesting that the human and Drosophila proteins are functionally conserved.
Fig. 2.
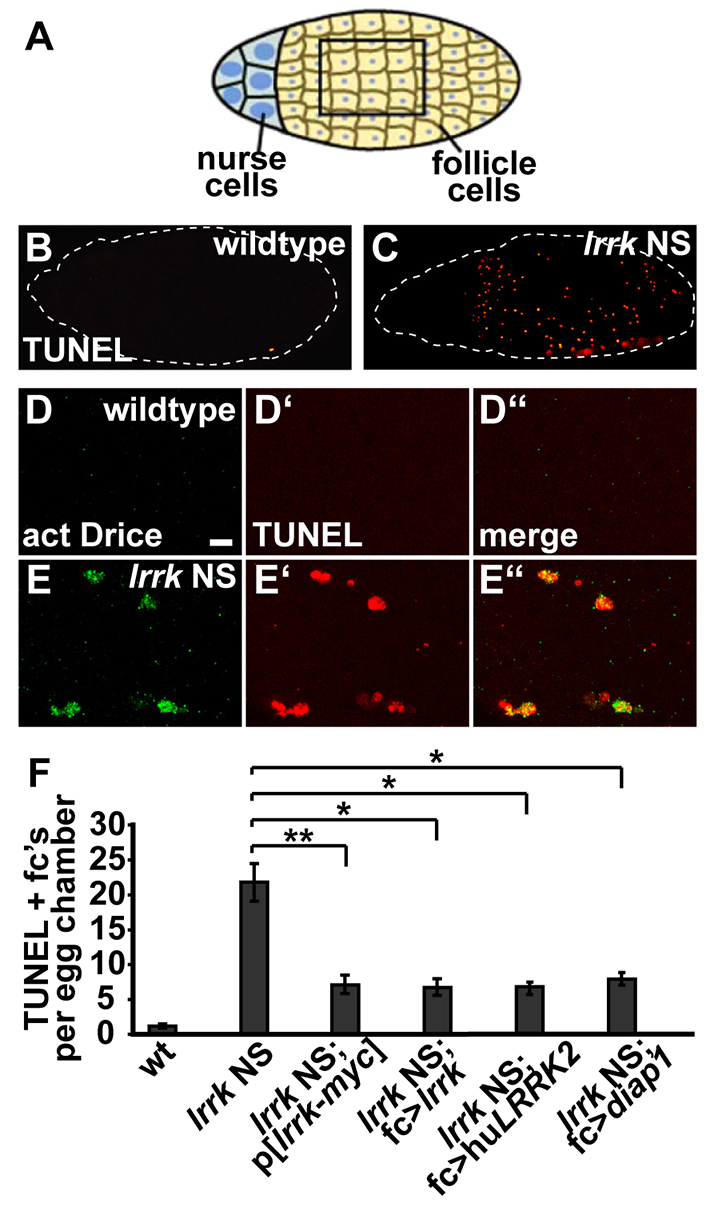
Follicle cells undergo premature caspase-dependent cell death in lrrk NS flies. (A) Schematic depicting a stage-12 egg chamber, with anterior to the left and posterior to the right. Most of the exterior of the egg chamber is composed of an epithelial layer of somatic follicle cells that surrounds the developing oocyte, whereas the anterior of the egg chamber contains the germline nurse cells, which are clonally related to the oocyte. (B–E) Note that, whereas B and C depict whole egg chambers, D and E are higher-magnification views of follicle cells, similar to what is outlined by the box in A. (B,C) Single stage-12 egg chambers from wild-type (B) and lrrk1/4 (C) females labeled by TUNEL to highlight apoptotic nuclei, show a dramatic increase in follicle cell death in lrrk NS egg chambers. The egg chamber is outlined with a dashed line for ease of visualization. Note that the area to the anterior (right) of the lrrk NS egg chamber (C) is devoid of TUNEL-positive signal because this area is occupied predominantly by nurse cells rather than follicle cells. (D,E) Higher-magnification images of follicle cells co-stained for activated caspase-3 (called Drice in Drosophila) (D,E) and TUNEL (D′,E′) demonstrates increased caspase activation in apoptotic follicle cells from stage-12 lrrk1/2 egg chambers (E) relative to wild type (D). Merged images show that, in lrrk NS flies, follicle cells that accumulate activated Drice are also TUNEL-positive (E″), suggesting that caspase activation occurs specifically in cells undergoing apoptosis. (F) Quantification of average number of TUNEL-positive follicle cells per egg chamber of the indicated genotype. Whereas lrrk NS alleles show a dramatic increase in follicle cell apoptosis relative to wild type, this is significantly rescued by a myc-tagged lrrk genomic rescue transgene, and by follicle-cell-specific expression of either Drosophila lrrk or human LRRK2. Apoptosis of lrrk NS follicle cells is also suppressed by expression of the caspase inhibitor diap1, suggesting that this process is caspase-dependent. n=56, 41, 58, 26, 107 and 61 egg chambers each, respectively, for the genotypes in panel F. *P<0.0001. **P=0.002. Scale bar in D: 10 μm.
Egg chamber development in Drosophila is divided into 14 stages, and follicle cells normally undergo programmed cell death at the culmination of egg chamber development (King, 1970; Spradling, 1993). In lrrk NS flies, however, TUNEL staining revealed premature follicle cell death at stages 11–13 (Fig. 2C vs 2B; Fig. 2F). As with all other lrrk NS follicle cell phenotypes reported below, there was marked heterogeneity in the number of TUNEL-positive follicle cells per egg chamber, with some lrrk NS egg chambers severely affected and others appearing completely normal. Thus, quantification has been employed for lrrk NS follicle cell phenotypes where possible, such that the magnitude of the phenotype is averaged over all egg chambers. Premature follicle cell death in lrrk NS flies was rescued by targeted expression of lrrk or human LRRK2 in follicle cells, and by a single copy of a lrrk genomic rescue transgene (Dodson et al., 2012) (Fig. 2F). lrrke03680 homozygous flies did not show premature follicle cell death (supplementary material Fig. S2C), which is consistent with our hypothesis that this allele might not uncover the full range of lrrk loss-of-function phenotypes. Premature follicle cell death in lrrk NS flies was associated with increased staining for activated caspase-3 (called Drice in Drosophila) (Fig. 2E vs 2D), and double labeling with activated caspase-3 and TUNEL confirmed that caspase activation was specific to dying cells (Fig. 2E″ vs 2D″). Moreover, follicle cell death in lrrk NS flies was suppressed by expression of the caspase inhibitor Drosophila inhibitor of apoptosis 1 (diap-1) (Hay et al., 1995) (Fig. 2F). Collectively, these data suggest that premature follicle cell death in lrrk NS flies is caspase-dependent.
lrrk NS flies show massively expanded lysosomal compartments that aberrantly accumulate lipid and autophagic components
As we have previously reported, Lrrk localizes predominantly to the membranes of late endosomes and lysosomes, and to a lesser extent to early endosomes, in vivo (Dodson et al., 2012). We thus first asked whether premature follicle cell death in lrrk NS flies is associated with defects in the late endosomal and lysosomal compartments. Using multiple late endosomal and lysosomal markers, including Rab7 (Fig. 3C vs 3B), Lamp1:GFP (Fig. 3C′ vs 3B′) and the acidophilic dye Lysotracker (Fig. 3E vs 3D), we found that many lrrk NS follicle cells showed markedly enlarged late endosomal/lysosomal structures. There did not seem to be any appreciable alteration in lysosome position in lrrk NS flies: the enlarged lysosomes occurred in both the perinuclear region as well as the cell periphery. Expression of the caspase inhibitor diap1 had no significant effect on lysosome enlargement in lrrk NS flies (supplementary material Fig. S3), suggesting that lysosome enlargement is not a consequence of caspase activation. In contrast to the massively enlarged lysosomal structures observed in lrrk NS flies, lrrke03680 homozygous flies showed a mild increase in the size of the Rab7-positive late endosomal compartment in a subset of cells (supplementary material Fig. S2F) and no discernible change in lysosome morphology by Lysotracker staining (supplementary material Fig. S2I) (Dodson et al., 2012). Interestingly, the massive accumulation of Lamp1:GFP within enlarged lysosomes in lrrk NS flies (Fig. 3C′) suggests that their expansion in size is associated with, and perhaps due to, a defect in the degradative properties of the lysosome, because Lamp1:GFP is normally degraded rapidly upon reaching the lysosome (Pulipparacharuvil et al., 2005).
Fig. 3.

Abnormally expanded late endosomal and lysosomal compartments in lrrk NS flies. (A) Schematic depicting the endosomal and autophagy pathways, and highlighting markers for different pathway compartments. The lysosomal compartment, indicated by the red box, is the focus of this figure. Nu, nucleus. (B,C) Antibody staining for the late endosomal protein Rab7 in follicle cells from stage-12 wild-type (B) and lrrk NS (C) egg chambers shows dramatically enlarged Rab7-positive compartments in lrrk NS. These enlarged Rab7-positive compartments accumulate the lysosomal marker Lamp1:GFP (C′ vs B′). Arrowheads indicate Rab7 staining of the late endosomal membrane. (D–I) Staining of lysosomes with the acidophilic dye Lysotracker in the indicated genotypes shows dramatically enlarged lysosomes in lrrk NS follicle cells (E) relative to wild type (D); this phenotype can be rescued by follicle-cell-specific expression of wild-type lrrk (H). In an otherwise wild-type background, expression of lrrkGS (G), but not wild-type lrrk (F), results in perinuclear clustering of lysosomes. In lrrk NS flies, expression of lrrkGS suppresses the enlarged lysosome phenotype; however, perinuclear clustering of lysosomes is still observed (I). Arrowheads indicate perinuclear lysosome clusters. (J) Quantification of average lysosome size in the indicated genotypes as determined by Lysotracker staining of follicle cells from stage-12 egg chambers. Whereas lrrk NS results in a dramatic increase in average lysosome size, this phenotype is significantly suppressed by follicle-cell-specific expression of wild-type lrrk, lrrkGS or human LRRK2. n=16, 34, 16, 16 and 17 egg chambers each, respectively, for the genotypes in J. *P<0.0001. lrrk NS is lrrk1/2. Follicle cell nuclei are outlined by dashed white lines in B–I. Scale bar: 5 μm.
The enlarged-lysosome phenotype of lrrk NS flies could be rescued by follicle-cell-specific expression of either Drosophila lrrk (Fig. 3H vs 3E; Fig. 3J) or human LRRK2 (Fig. 3J). Previously, we reported that the pathogenic LrrkGS (G2019S) mutation alters the function of Lrrk such that LrrkGS promotes perinuclear lysosome transport, whereas equivalent expression of LrrkWT (wild type) inhibits this process (Dodson et al., 2012). Interestingly, expression of lrrkGS suppressed the enlarged-lysosome phenotype caused by lrrk NS as efficiently as did expression of lrrkWT (Fig. 3E–J). The finding that the suppression of lysosome enlargement in lrrk NS flies was identical between lrrkGS and lrrkWT indicates that LrrkGS retains at least some of the functions of LrrkWT, at least with respect to the Lrrk-dependent processes controlling lysosome size. However, perinuclear lysosome clustering was still observed with expression of lrrkGS (Fig. 3G,I), but not lrrkWT (Fig. 3F,H), regardless of whether these were expressed in the wild-type or lrrk NS background. This is consistent with a model we previously proposed in which LrrkGS retains at least some of the normal functions of the wild-type protein, while also possessing neomorphic effects (Dodson et al., 2012).
On transmission electron microscopy (TEM) analysis, the expanded lysosomes in lrrk NS flies contained prominent large clear inclusions reminiscent of lipid (Fig. 4B), as well as undigested cytosolic contents, including intact mitochondria (Fig. 4C). These findings were confirmed with immunofluorescence analysis, in which the enlarged Lysotracker-positive lysosomes in lrrk NS flies also contained lipid, as labeled by the lipophilic dye BODIPY 493/503 (Fig. 4E), as well as mitochondria, as labeled by GFP fused to a mitochondrial targeting sequence (mitoGFP) (Fig. 4G). BODIPY 493/503 (Fig. 4D)- or mitoGFP (Fig. 4F)-positive signals were never seen within Lysotracker-positive lysosomes in stage-matched wild-type follicle cells. Thus, the aberrantly enlarged lysosomes in lrrk NS cells accumulate both lipid and intact mitochondria, suggesting a primary defect in the degradative properties of lysosomes in lrrk NS flies.
Fig. 4.

lrrk NS flies accumulate cytosolic material, including undigested organelles and lipid inclusions, within lysosomes, and accumulate autophagosomes. (A–C) Transmission electron micrographs (TEMs) from wild-type (A) and lrrk NS mutant (B,C) follicle cells from stage-12 egg chambers. In wild-type follicle cells (A), no enlarged lysosomes are observed (follicle cell nuclei are highlighted in purple). Observed in the cytosol of lrrk NS flies (B,C) are aberrantly enlarged membrane-bound structures (outlined with dashed black line) filled with clear inclusions of lipid (B) and undigested cytosolic contents (C), including intact mitochondria (marked with yellow arrowheads). (D,E) Co-staining with Lysotracker (red) and the lipophilic dye BODIPY 493/503 (green) shows that the aberrantly enlarged lysosomes in follicle cells from lrrk NS flies (E) accumulate lipid inclusions, whereas this was not observed in wild type (D). (F,G) Similarly, the enlarged lysosomes in lrrk NS flies (G) also accumulate numerous mitochondria as labeled with a mitochondrially targeted GFP, whereas mitochondria always appeared to be distinct from lysosomes in wild-type follicle cells (F). (H,I) TEM images from lrrk NS follicle cells demonstrate the presence of double-membrane autophagosomes often containing intact mitochondria. Compare with wild-type follicle cells in A, in which autophagosomes are not observed by TEM analysis. (J,K) Lysotracker staining in follicle cells expressing the autophagosome marker Atg8a:GFP (J′,K′) in wild-type (J) and lrrk NS (K) follicle cells. In wild-type cells, Atg8a:GFP is mostly uniformly distributed throughout the cytosol (J′), indicating the relative absence of autophagosomes. In lrrk NS follicle cells, however, Atg8a:GFP shifts to a punctate distribution (K′), and these puncta occasionally, but not always, colocalize with Lysotracker (K″), indicating the presence of both autophagosomes and autolysosomes in lrrk NS flies. lrrk NS is lrrk1/2. Follicle cell nuclei are outlined by dashed white lines in J–K″. Scale bars: 1 μm in A–C and H,I, and 5 μm in D–G and J–K″.
Autophagy is dysregulated in lrrk NS flies
The lysosome is the final degradative compartment for multiple cellular pathways, including both the endocytic and autophagy pathways. The finding of intact organelles within enlarged lysosomes in lrrk NS flies suggests that at least some of these lysosomal contents are derived from the autophagy pathway. The hallmark of autophagy is the formation of double-membrane structures called autophagosomes, which label specifically with the marker Atg8a:GFP (Fig. 3A), the Drosophila homolog of mammalian LC3 (Geng and Klionsky, 2008). Autophagosomes form in the cytosol, engulf cytosolic components and are then transported to the lysosome for degradation (Feng et al., 2014). In lrrk NS follicle cells, autophagosomes were indeed apparent both by TEM analysis (Fig. 4H,I) and by a marked redistribution of Atg8a:GFP from a diffuse pattern in wild-type cells (Fig. 4J′), indicating a relative absence of autophagosomes, to predominantly punctate pattern in lrrk NS cells (Fig. 4K′). Moreover, autophagosomes were not observed in stage-matched wild-type follicle cells by TEM analysis (Fig. 4A). These puncta of Atg8a:GFP in lrrk NS flies often localized to Lysotracker-positive lysosomes, but were also frequently distinct from lysosomes (Fig. 4K″). Thus, lrrk NS cells show an increase in structures derived from autophagy; this increase could be due to either increased autophagy induction, decreased clearance of autophagosomes and autolysosomes, or both.
To evaluate this further, we studied the consequences of suppressing autophagy in lrrk NS flies. Atg7 is a component of the ubiquitin-like conjugation system required for autophagosome formation (Geng and Klionsky, 2008), and removing the function of atg7 in Drosophila has been shown to inhibit starvation-induced autophagy in the fat body (Juhász et al., 2007). Interestingly, we found that removing atg7 function in lrrk NS flies resulted in a significant increase in follicle cell death as assessed by TUNEL staining (Fig. 5A). This was not associated with any significant enhancement in the lysosome-enlargement phenotype observed in lrrk NS flies (Fig. 5B–E). These atg7/lrrk NS double mutants also displayed a mild rough-eye phenotype (Fig. 5I), which was not observed in either single mutant alone (Fig. 5G,H). The increased toxicity observed in atg7/lrrk double mutants versus lrrk NS flies alone is consistent with autophagy induction as a prosurvival response in lrrk NS flies. That this increased toxicity in the double mutants is not associated with enhancement of the lrrk NS lysosome-enlargement defect suggests that the lysosomal defects in lrrk NS flies are independent of the autophagy pathway.
Fig. 5.
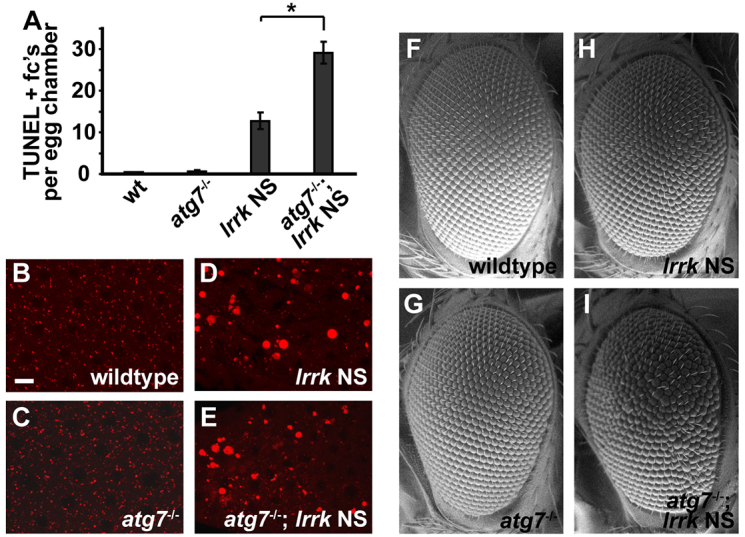
atg7 loss-of-function enhances cell death in lrrk NS flies, but does not enhance lysosome enlargement. (A) Average number of TUNEL-positive follicle cell nuclei in the indicated genotypes shows an increase in follicle cell death in atg7/lrrk NS double mutants relative to lrrk NS alone. atg7 loss-of-function alone did not cause any significant follicle cell death by TUNEL staining. n=61, 63, 62 and 57 egg chambers each, respectively, for the genotypes in A. *P<0.0001. (B–E) Lysotracker staining of the indicated genotypes shows no significant enhancement of lysosome enlargement in atg7/lrrk NS double mutants (E) versus lrrk NS mutants alone (D). Relative to wild type (B), atg7 loss-of-function alone (C) has no significant effect on lysosome morphology. (F–I) Scanning electron micrographs of adult fly eyes of the indicated genotypes shows that atg7/lrrk NS double mutants display a mild rough-eye phenotype (I). In contrast, eye morphology is indistinguishable from wild type (F) in atg7 (G) and lrrk NS (H) single mutants. lrrk NS is lrrk1/1. Scale bar: 5 μm for B–E.
lrrk NS flies display aberrant early endosomes laden with ubiquitylated cargoes
Previously, we reported that Lrrk also physically binds to the early endosomal protein Rab5 (Dodson et al., 2012), which plays a vital role in the maturation of early endosomes to late endosomes and, thus, in endosomal substrate delivery to the lysosome (Huotari and Helenius, 2011). Genetic and physical interactions have also been reported between mammalian LRRK2 and Rab5 in cell-culture models under conditions of LRRK2 overexpression (Heo et al., 2010; Shin et al., 2008). Thus, we investigated whether lrrk also acts to regulate cargo delivery to the lysosome in the endocytic pathway. Interestingly, we found that the early endosomes, marked by either anti-Rab5 (Fig. 6D vs 6C) or anti-hepatocyte-growth-factor-regulated tyrosine kinase substrate (Hrs) antibodies (Fig. 6F vs 6E), were enlarged in lrrk NS follicle cells relative to wild type. In the endocytic pathway, monoubiquitylation serves as a sorting signal for protein cargoes that are bound for the lysosome to be degraded (Piper and Luzio, 2007; Raiborg and Stenmark, 2009). Interestingly, lrrk NS cells showed a marked accumulation of ubiquitylated proteins (as identified by an antibody that recognizes mono- or polyubiquitin conjugated to any target protein, but not free ubiquitin) (Fig. 6H vs 6G; Fig. 6B). Interestingly, this ubiquitin-positive staining often appeared as halos surrounding a non-stained central region (Fig. 6H, inset), suggesting the accumulation of ubiquitylated cargoes on vesicle membranes. Consistent with this hypothesis, the ubiquitylated-protein accumulations in lrrk NS cells colocalized with the expanded anti-Rab5-positive (Fig. 6D″) and anti-Hrs-positive (Fig. 6F″) early endosomes. These structures also accumulated the endocytic tracer dextran (Fig. 6H″), which is taken up from the extracellular space via endocytosis and subsequently trafficked through the endocytic pathway, thus confirming the identity of these structures as endosomes. Moreover, these ubiquitin accumulations consisted primarily of monoubiquitylated proteins, as evidenced by a lack of staining with an antibody that recognizes polyubiquitin chains (Fig. 6J′). This is distinct from what is observed with expression of the dominant-negative proteosome subunit DTS7 (serving as a control), which resulted in the accumulation of predominantly polyubiquitylated proteins recognized by both antibodies (Fig. 6K″). Importantly, the accumulation of monoubiquitylated proteins in lrrk NS flies could be rescued by follicle-cell-specific expression of wild-type lrrk (Fig. 6B). lrrke03680 homozygous flies showed a similar, albeit weaker, accumulation of ubiquitylated proteins (supplementary material Fig. S4C versus S4B), which also colocalized with internalized dextran (supplementary material Fig. S4C″) and Hrs (supplementary material Fig. S4F″). Collectively, these results show that lrrk NS flies accumulate aberrantly enlarged early endosomes that are laden with monoubiquitylated endosomal cargo proteins, suggesting a defect in substrate delivery to the lysosome in the endocytic pathway.
Fig. 6.

lrrk NS follicle cells accumulate enlarged early endosomes laden with ubiquitylated cargo proteins. (A) Schematic depicting the endosomal and autophagy pathways. Highlighted is the early endosome compartment, which is specifically labeled with anti-Rab5 and anti-Hrs antibodies. Nu, nucleus. (B) Quantification of the number of ubiquitin-positive endosomes per microscope field in the indicated genotypes, showing a dramatic increase in the number of endosomes laden with ubiquitylated proteins in lrrk NS flies, which is significantly suppressed by follicle-cell-specific expression of wild-type lrrk. n=18, 15 and 20 egg chambers each, respectively, for the genotypes in B. *P=0.02. (C–F) Antibody staining for the early endosomal proteins Rab5 (C,D) and Hrs (E,F) in wild-type (C,E) and lrrk NS (D,F) stage-12 follicle cells. In lrrk NS cells, the Rab5 (D vs C)- and Hrs (F vs E)-positive compartments are dramatically expanded. Co-staining with an antibody (D′ and F′) that recognizes either monoubiquitin or polyubiquitin chains conjugated to any target protein, but not free ubiquitin, in lrrk NS cells shows that these enlarged Rab5 (D″)- and Hrs (F″)-positive compartments are laden with ubiquitylated proteins. Arrowheads indicate enlarged endosomes labeled by Rab5/Hrs and ubiquitin. (G,H) In wild-type follicle cells (G), there is no significant labeling by the antibody recognizing ubiquitin-protein conjugates, whereas, in lrrk NS follicle cells (H), these large ubiquitin-positive structures are frequently seen. Note that the ubiquitin-protein conjugate staining in lrrk NS cells frequently appears as a halo surrounding a non-stained central region (inset), suggesting accumulation of ubiquitylated proteins on endosomal membranes. This is consistent with the co-labeling of these structures with the early endosomal proteins Rab5 and Hrs as seen in D and F. These accumulations of ubiquitin-protein conjugates in lrrk NS also colocalize with endocytosed dextran (H′ and H″), confirming the identity of these structures as endosomes. Arrowheads indicate enlarged endosomes labeled by dextran and ubiquitin. (I–K) The ubiquitin-protein conjugates that accumulate in lrrk NS flies are predominantly monoubiquitylated. In wild-type follicle cells (I), there is no significant staining with an antibody recognizing ubiquitin-protein conjugates (I), nor with an antibody recognizing polyubiquitin chains (I′). In lrrk NS cells, the accumulations of ubiquitin-protein conjugates (J) do not co-label with the antibody to polyubiquitin chains (J′,J″), suggesting that these accumulations are predominantly monoubiquitylated proteins. As a positive control, the dominant-negative proteasome subunit DTS7 accumulates inclusions that stain positive both for ubiquitin-protein conjugates (K) and for polyubiquitin chains (K′,K″). lrrk NS is lrrk1/2. Follicle cell nuclei are outlined by dashed white lines in G and H. Scale bars: 5 μm in all images.
lrrk NS phenotypes are rescued by expression of constitutively active rab9
Previously, we found, from predominantly overexpression-based studies, that lrrk inhibits rab7-dependent perinuclear lysosome clustering (Dodson et al., 2012). We therefore asked whether the lysosomal-enlargement phenotype seen with lrrk loss-of-function is due to an interaction between lrrk and rab7. However, we found that lrrk loss-of-function does not phenocopy the lysosome clustering caused by expression of a constitutively active form of rab7 (rab7CA) (supplementary material Fig. S5B), as would be expected according to this model. Moreover, rab7CA had no effect on lysosome enlargement in lrrk NS flies (supplementary material Fig. S5E vs S5D,G). In addition, average lysosome size was no different in lrrk NS flies alone compared with lrrk NS flies expressing a dominant-negative version of rab7 (rab7DN) (supplementary material Fig. S5G). These lrrk NS/rab7DN flies did not show the massively enlarged lysosomes often seen in lrrk NS flies (supplementary material Fig. S5F vs S5D), but rather showed a more moderate but consistent lysosome-enlargement phenotype, the net result being no overall change in average lysosome size compared with lrrk NS (supplementary material Fig. S5G). This result is not unexpected given that dominant-negative rab7 results in lysosome dispersal and decreased lysosome fusion (Bucci et al., 2000). Taken together, these data suggest that the lysosome enlargement observed in lrrk NS flies is at least partially Rab7-independent.
Recently, it was reported that LRRK2 forms a complex with VPS35 (MacLeod et al., 2013), which is a core component of the retromer complex, involved in retrograde trafficking of cargoes from the endocytic pathway to the trans-Golgi network (Bonifacino and Rojas, 2006). This pathway is required for the retrograde transport of multiple substrates, including the mannose 6-phosphate receptor (MPR) (Arighi et al., 2004; Hierro et al., 2007; Seaman, 2004), a process that also requires the late endosomal GTPase Rab9 (Barbero et al., 2002; Lombardi et al., 1993; Riederer et al., 1994). Overexpression of LRRK2 G2019S (LrrkGS) has been reported to disrupt the normal trafficking of the MPR; this disruption can be rescued by overexpression of Vps35 (MacLeod et al., 2013). Inhibition of the retromer pathway increases lysosomal degradation of the MPR and thereby results in swelling of lysosomes due to intra-lysosomal accumulation of undigested material, presumably due to defects in trafficking of hydrolases to the lysosome (Arighi et al., 2004). Because this phenotype is highly reminiscent of what we have observed in lrrk NS flies, we hypothesized that augmenting retrograde transport might suppress lrrk NS phenotypes.
As we have previously reported, Drosophila Lrrk colocalizes with Rab9 at the late-endosome membrane (Dodson et al., 2012). Interestingly, we found that Lrrk-myc co-immunoprecipitated with Rab9-YFP, but did not show significant binding with either YFP alone or with Rab11-YFP (Fig. 7A), which served as controls. Expression of a constitutively active form of rab9 (rab9CA) in follicle cells resulted in marked suppression of the lysosome-enlargement phenotype observed in lrrk NS cells (Fig. 7D vs 7C; Fig. 7E). Similarly, rab9CA significantly, although incompletely, suppressed premature follicle cell death in lrrk NS cells (Fig. 7F). One mechanism by which lrrk could be required for rab9-dependent processes is through regulation of the membrane recruitment of Rab9; however, we did not detect any significant difference in the subcellular localization of Rab9 between the wild-type and lrrk-NS-mutant backgrounds (supplementary material Fig. S6). Collectively, these data suggest that augmenting Rab9 function can, at least in part, bypass the need for lrrk.
Fig. 7.

Lrrk physically binds to Rab9, and expression of activated rab9 rescues lrrk NS phenotypes. (A) In lysates from transiently transfected cultured Drosophila S2 cells, Lrrk-Myc co-immunoprecipitates with YFP-tagged Rab5 and more strongly with Rab9, but not with Rab11 or YFP alone. (B–D) Lysotracker staining of stage-12 follicle cells from wild-type flies (B), lrrk NS flies (C) and lrrk NS flies expressing constitutively active rab9 (rab9CA) specifically in follicle cells (D) shows dramatic suppression of the abnormal lysosome morphology seen in lrrk NS flies when rab9 is expressed. (E) Quantification of average lysosome size in Lysotracker-stained follicle cells shows suppression of lysosome enlargement by rab9CA in lrrk NS flies. n=16, 16, 34 and 33 egg chambers each, respectively, for the genotypes in E. **P<0.0001. (F) Similarly, premature follicle cell death in lrrk NS flies, as assessed by TUNEL staining, is suppressed by rab9CA. n=61, 68, 99 and 102 egg chambers each, respectively, for the genotypes in F. *P<0.05. (G–J) Staining of the presynaptic motor neuron at the neuromuscular junction (NMJ) at abdominal segment 4, muscle 4 in late third instar larvae with anti-cysteine string protein (CSP; red), which labels synaptic vesicles. Compared with wild type (G), lrrk NS larvae (H) show an overgrowth phenotype in which numerous small satellite boutons (marked with arrowheads) form adjacent to the larger normal boutons. This overgrowth phenotype is significantly suppressed by expression of either wild-type lrrk (I) or rab9CA (J) using the motor neuron-specific OK6-Gal4 driver. (K) Quantification of the numbers of boutons per NMJ in the indicated genotypes shows significant suppression of lrrk NS synaptic overgrowth with expression of both wild-type lrrk and rab9CA. n=27 NMJs analyzed for each genotype in K. ***P=0.0003, *P<0.05. lrrk NS is lrrk1/2. Scale bars: 5 μm in B–D, 10 μm in G–J.
To further substantiate the finding that overactivation of rab9 can bypass the requirement for lrrk, we examined genetic interactions between lrrk and rab9 in another tissue that lrrk has been shown to act on: the larval NMJ. Previously, it has been reported that lrrke03680 mutants show a defect in NMJ morphology characterized by overgrowth of the synapse between the presynaptic motor neuron and postsynaptic muscle cell, with increased branching and number of boutons (Lee et al., 2010). We observed a similar, albeit stronger, phenotype in lrrk NS flies, where the synaptic overgrowth was mostly observed as small supernumerary satellite boutons surrounding the larger normal boutons (Fig. 7H vs 7G; Fig. 7K). This phenotype could be rescued by motor-neuron-specific expression of wild-type lrrk (Fig. 7I vs 7H; Fig. 7K). As observed in follicle cells, expression of rab9CA significantly suppressed the NMJ morphology defect in lrrk NS flies (Fig. 7J vs 7H; Fig. 7K). Together, these results suggest that lrrk plays a role in retromer-mediated endosomal recycling, and that augmentation of late endosome-to-Golgi retrograde transport can bypass the requirement for lrrk.
DISCUSSION
Here, we provide evidence of a vital physiological role for a LRRK2 homolog in endolysosomal membrane transport and autophagy in vivo, by characterizing novel loss-of-function nonsense alleles in Drosophila lrrk. These lrrk NS alleles cause accumulation of markedly enlarged lysosomes full of undigested lipid and other cytosolic contents, as well as accumulation of autophagosomes and enlarged early endosomes laden with monoubiquitylated cargo proteins. The interactions that we describe between lrrk and rab9 suggest that the role of Lrrk in maintaining proper lysosome function involves the Rab9-dependent late-endosome-to-Golgi pathway.
Whereas the increase in perinuclear lysosome clustering caused by expression of lrrkGS is rab7-dependent (Dodson et al., 2012), the effects of lrrk loss-of-function on lysosome size are rab7-independent, and rather seem to be mediated through the Rab9-retromer pathway. rab9 promotes retromer-dependent recycling of cargoes from late endosomes to the Golgi (Barbero et al., 2002; Lombardi et al., 1993; Riederer et al., 1994). Defects in retromer-dependent cargo trafficking lead to impaired lysosomal degradation and the accumulation of undigested materials within lysosomes (Arighi et al., 2004), a phenotype highly reminiscent of what is seen in lrrk NS flies. Rab9 and Lrrk colocalize at the late endosomal membrane (Dodson et al., 2012), and, as we show in this work, physically bind to one another. Augmentation of rab9 activity rescues lysosomal enlargement and premature follicle cell death in lrrk NS flies. Enhancing rab9 activity also rescues the NMJ morphology defect in lrrk NS flies, which have an NMJ phenotype identical to that reported with loss-of-function of the retromer component Vps35 (Korolchuk et al., 2007). Thus, in multiple tissues, augmentation of rab9 activity bypasses the need for lrrk, suggesting a role for lrrk in the Rab9-retromer pathway.
Defects in the autophagy pathway have previously been reported in the context of lrrk loss-of-function, but the results have been conflicting, including the finding of decreased autophagosome formation with LRRK2 knockdown in immune cells (Schapansky et al., 2014) and biphasic alterations in autophagy in the kidney of LRRK2 knockout mice, with markers of autophagy increased at 7 months of age and reduced at 20 months (Tong et al., 2012). Here, we report increased autophagic structures in lrrk NS flies, including both autophagosomes and autolysosomes. Although there is probably a component of decreased turnover of autophagic structures due to the lysosome dysfunction that occurs in lrrk NS flies, our data also suggest that autophagy induction is increased in lrrk NS flies, possibly as a prosurvival stress response to lysosome dysfunction or ubiquitylated-protein accumulation. This hypothesis is supported by the increased toxicity in lrrk NS flies when atg7 function is removed. Caspase-dependent apoptosis seems to be the end result when the autophagic response is insufficient, and occurs downstream of both lysosome dysfunction and autophagy induction. We cannot, however, exclude the possibility that lrrk loss-of-function increases autophagy activation via a more direct mechanism. Interestingly, rab9 has been implicated in autophagosome formation in mammalian cells in an alternative atg7-independent autophagy pathway that can be induced by certain cellular stressors (Nishida et al., 2009). That some lrrk NS phenotypes are enhanced by atg7 loss-of-function suggests that autophagy induction in the context of lrrk loss-of-function is atg7-dependent rather than atg7-independent. Still, it remains an intriguing possibility that lrrk could directly regulate autophagy induction, perhaps via its interaction with rab9.
Another important question is whether the defects seen in lrrk NS flies at both the early endosomal compartment and the late endosomal/lysosomal compartments reflect distinct roles for Lrrk at multiple endolysosomal transport steps. For example, via its physical binding to the early endosomal protein Rab5, which we and others have reported (Dodson et al., 2012; Shin et al., 2008), LRRK2/Lrrk might exert a distinct Rab9-independent role in the trafficking of early endosomes. Interestingly, loss-of-function of Drosophila lrrk has also been reported to impair synaptic vesicle endocytosis via an interaction with endophilin A, which is involved in the early membrane tubulation steps involved in vesicle budding from the plasma membrane (Matta et al., 2012). Thus, Lrrk might function as a general regulator of multiple distinct membrane transport steps.
Although our work has focused predominantly on follicle cells in the Drosophila ovary owing to the aforementioned benefits as a cell biological model system, we believe that our results have significance to the function of lrrk in neurons for several reasons. Lysosomal defects in lrrk NS flies can be rescued by human LRRK2, suggesting functional conservation of the endolysosomal functions of the two proteins. Moreover, both follicle cell lysosomal defects and abnormal NMJ morphology in lrrk NS flies are rescued by augmenting rab9 function, suggesting similar defects in rab9-dependent membrane transport processes due to lrrk loss-of-function in both neuronal and non-neuronal tissues. Why might follicle cells be particularly sensitive to the effects of lrrk loss-of-function? Ultrastructural analyses have reported an increase in the number of lysosomes in follicle cells from stage-12 egg chambers (Cummings and King, 1970), the stage at which lysosome defects in lrrk NS flies become apparent. Moreover, this stage-specific increase in lysosome abundance plays a role in the degradation of nurse cells, which are germline cells clonally related to the oocyte that undergo programmed cell death at the culmination of oogenesis (King, 1970; Spradling, 1993). Thus, we speculate that an increased requirement for lysosomal degradation in late-stage follicle cells could underlie the sensitivity of these cells to lrrk loss-of-function.
Our work demonstrates a vital physiological role for endogenous lrrk in endolysosomal membrane transport and lysosome function in vivo, and suggests that the toxicity of the pathogenic mutant lrrkGS allele is related to dysregulation of its endolysosomal functions. These findings have important implications for the development of inhibitor-based therapies for PD that target LRRK2, because excessive inhibition might result in endolysosomal dysfunction and thus cellular toxicity. Regulation of lysosomal function and autophagy have emerged as key themes in the pathogenesis of neurodegenerative diseases, from inherited diseases of lysosomal storage to diseases such as PD, involving the accumulation of protein inclusions. Together with a striking amount of recent genetic data linking PD risk to genes involved in endolysosomal functions, including β-glucocerebrosidase (Aharon-Peretz et al., 2004; Gan-Or et al., 2008), ATP13A2 (Ramirez et al., 2006), VPS35 (Vilariño-Güell et al., 2011; Zimprich et al., 2011) and RAB7L1 (Gan-Or et al., 2012), our data establishes dysfunction of the endolysosomal pathway as a central pathogenic mechanism in PD.
MATERIALS AND METHODS
Molecular biology
The UAS-lrrk, UAS-lrrkGS and CaSpeR-lrrk genomic rescue constructs were previously described (Dodson et al., 2012). For UAS-LRRK2, the human LRRK2 cDNA was introduced into the pTHW gateway vector (which contains an N-terminal HA tag as well as the UASt promoter) by recombination. All cloned PCR products were confirmed by DNA sequencing. The Rab-YFP constructs were obtained from Matthew Scott (Stanford University, Palo Alto, CA).
RNA purification and quantitative PCR
RNA was isolated from 1-day-old flies using Trizol, and purified using Phase Lock Gel Tubes to remove DNA. Two independent cDNA synthesis reactions were performed for each genotype using Clontech EcoDry Premix cDNA Kit (Double Primed Oligo-dT and Random Hexamers), after normalizing the amount of input RNA across genotypes. Quantitative PCR was performed in triplicate for each cDNA reaction and the six values for each genotype across the two cDNA reactions averaged. All primer pairs spanned an intron as depicted in Fig. 1A. Values were normalized to the geometric mean of three control genes, rpl32, eIF1a and aTub84B, for each genotype, and then normalized to wild type to calculate a fold change in gene expression.
Drosophila genetics
To generate loss-of-function mutations in lrrk, we used TILLING (Till et al., 2003), which is a method for detecting point mutations in a gene of interest following chemical mutagenesis. EMS mutations were generated in a prior screen (Koundakjian et al., 2004), and were recovered using the Drosophila Tilling Service (Fred Hutchinson Cancer Research Center). lrrk1, lrrk3 and lrrk4 are nonsense alleles resulting from substitution of a stop codon for L389, Q622 and W1221, respectively. lrrk2, which was recovered from a mixed stock, has a single-nucleotide insertion changing L584 to F, and resulting in a frameshift that generates a stop codon 11 codons downstream. lrrke03680 results from the insertion of a PiggyBac transposable element in the intron between exons 5 and 6, and was obtained from the Harvard Drosophila Stock Center. The follicle-cell-specific driver CY2-GAL4 was a gift from Celeste Berg (University of Washington, Seattle, WA). atg7 mutants were obtained from Thomas Neufeld (Juhász et al., 2007). UAS-rab7DN, -rab7CA and -rab9CA are as described (Zhang et al., 2007). UASp-diap1 flies were obtained from Bruce Hay (California Institute of Technology, Pasadena, CA) and Andreas Bergann (University of Massachusetts, Worchester, MA). All other stocks are from the Bloomington Drosophila Stock Center. For experiments involving transgenic flies, multiple lines were generated (Rainbow Transgenic Flies) and tested for each transgene. Drosophila strains were maintained in a 25°C humidified incubator unless otherwise noted.
Immunofluorescence and confocal microscopy
For follicle cell experiments, freshly eclosed females were maintained on wet yeast paste for 24 hours prior to ovary dissection, and individual stage 11–13 egg chambers were hand dissected following fixation. The NMJ at abdominal segment 4, muscle 4 was used for all NMJ experiments. All tissues were fixed in either 3.7% formaldehyde or 4% paraformaldehyde in PBS. PBS + 0.1% Triton X-100 (or 0.2% Tween) + 2% BSA was used for blocking and antibody incubations. The following primary antibodies were used for immunocytochemistry: mouse anti-ubiquitin protein conjugates (Biomol, FK2, 1:300), mouse anti-polyubiquitin chains (Biomol, FK1, 1:25), rabbit anti-Rab5 (Abcam, 1:20), rabbit anti-Rab7 [a gift from Yashodhan Chinchore (Harvard University, Boston, MA) and Patrick Dolph (Dartmouth University, Hanover, NH), 1:100], mouse anti-tyrosine hydroxylase (Immunostar, 1:300), guinea pig anti-Hrs [a gift from Hugo Bellen (Baylor University, Houston, TX), 1:100], rabbit anti-cleaved-caspase-3 (Cell Signaling, 1:300), mouse anti-CSP (DHSB, 1:10) and fluorescein-conjugated anti-HRP (1:250). Secondary antibodies were Alexa Fluor 488 or 568 (Molecular Probes, 1:250). For Lysotracker staining, ovaries were dissected and incubated for 15 minutes in 100 nM Lysotracker red DND-99 (Molecular Probes), washed briefly, then mounted and imaged immediately, all in Schneider’s Drosophila Medium (Gibco). For lipid visualization, the Lysotracker staining solution was supplemented with 2 mg/ml BODIPY 493/503 (Molecular Probes). Images were obtained with a Zeiss LSM5 confocal microscope. All follicle cell images shown are from stage-12 or -13 egg chambers.
Dextran uptake assay
Freshly dissected stage-12 egg chambers were incubated in 0.5 mM Texas-Red–dextran (3000 MW, Invitrogen) for 15 minutes, washed briefly five times, then incubated for 30 minutes prior to fixation in 4% paraformaldehyde. All steps were performed at 25°C in Schneider’s Drosophila Medium (Gibco).
TUNEL assay
Ovaries were fixed in 3.7% formaldehyde for 20 minutes, washed in PBS, and stage 11–13 egg chambers hand dissected. Egg chambers were permeabilized for 30 minutes in 50 mM Tris-Cl + 188 mM NaCl + 0.1% Triton X-100 + 3% BSA, washed in PBS, then incubated for 1 hour at 37°C in 100 μl of TUNEL reaction mix according to the manufacturer’s instructions (Roche In Situ Cell Death Detection kit, TMR-red).
Transmission electron microscopy
All samples were prepared for electron microscopy as described (Deng et al., 2008). 70- to 80-nm sections were stained with uranyl acetate and lead citrate and visualized using a JEOL 100CX transmission electron microscope (UCLA Brain Research Institute EM Facility). At least three individual flies of each genotype were examined for TEM studies.
Scanning electron microscopy
Freshly sacrificed flies were mounted on their side, with one eye upward, on white tape using clear nail polish. All flies were placed on a rotating platform to permit for orientation under vacuum and were imaged as described (Gross et al., 2008).
Female fertility tests
Single 0- to 3-day-old females were placed in a vial supplemented with dry yeast along with three sibling males and maintained at 25°C. After 4 days, the flies were removed and progeny were allowed to develop at 25°C for an additional 14 days, at which time the number of adult progeny per vial was quantified.
S2 cell culture and transfection
S2 cells were cultured in Schneider’s Drosophila Medium (Gibco) + 10% fetal bovine serum (Invitrogen) + 1% penicillin/streptomycin (Invitrogen). For immunoprecipitations, cells were plated on 10-cm dishes at a density of 3.6×106. Transfections were performed 24 hours later using the Qiagen Effectene kit according to the manufacturer’s recommendations. pMT-lrrk-9myc was transfected along with pAC-Gal4 and pUASp-Rab:YFP constructs (obtained from Matthew Scott) as indicated. pMT-lrrk-9myc expression was induced by adding 0.5 mM copper sulfate 24 hours after transfection, and cells were harvested 24 hours later.
Lysate preparation, immunoprecipitation and western blotting
Cells were lysed in 800 ml of RIPA buffer (Upstate) containing protease inhibitor cocktail (Roche), and immunoprecipitations performed using Dynabeads from Invitrogen according to the manufacturer’s instructions. Rabbit anti-GFP (Invitrogen) or mouse anti-Myc antibodies were used for immunoprecipitation. Bound proteins were eluted at 72°C for 10 min in 1× SDS sample buffer containing 5% 2-mercaptoethanol, and were resolved on a 6% or 10% polyacrylamide gel. Proteins were transferred to an Immobilon membrane (Millipore), and labeled with mouse antibodies against Myc (Millipore, 1:10,000), GFP (Roche, 1:5000), or α-tubulin (Sigma, 1:15,000) and goat anti-mouse HRP. Blots were developed using SuperSignal West Pico Chemiluminescent Substrate (Pierce).
Quantifications and statistical analysis
Ubiquitin-positive endosome density was calculated by manually counting the number of ubiquitin-positive puncta (labeled with FK2) per 63× field per egg chamber. Lysosome size was calculated from Lysotracker-stained samples using ImageJ software. All quantifications were performed on randomly selected egg chambers. Error bars represent standard error of the mean (s.e.m.). The two-tailed Student’s t-test was used to determine statistical significance.
Supplementary material
Supplementary material available online at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.017020/-/DC1
Acknowledgments
We are very grateful to P. Dolph, C. Berg, M. Scott, H. Bellen, H. Kramer, B. Hay and A. Bergmann for fly strains, antibodies and DNA constructs; to C. C. Ho and W. Dauer for generating the human LRRK2 construct; to F. Laski for use of his microtome; and to E. Dell’Angelica and B. A. Hay for discussions.
Footnotes
Competing interests
The authors declare no competing financial interests.
Author contributions
M.W.D. and M.G. conceived and designed the experiments. M.W.D., L.K.L., M.L. and M.A.L. performed the experiments. M.W.D., L.K.L., M.L., M.A.L. and M.G. analyzed the experiments. M.W.D. and M.G. wrote the paper.
Funding
This work was supported by the National Institutes of Health (NIH) NRSA predoctoral fellowship and the Medical Scientist Training Program (MSTP) to M.W.D., the UCLA Philip Whitcome Predoctoral Fellowship to L.K.L., as well as grants and funds from NIH (R01, K02), the Alfred P. Sloan Foundation, the Glenn Family Foundation, the Klingenstein Fund (Robert H. Ebert Clinical Scholar), and the McKnight Foundation of Neuroscience to M.G.
References
- Aharon-Peretz J., Rosenbaum H., Gershoni-Baruch R. (2004). Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N. Engl. J. Med. 351, 1972–1977. [DOI] [PubMed] [Google Scholar]
- Alegre-Abarrategui J., Christian H., Lufino M. M., Mutihac R., Venda L. L., Ansorge O., Wade-Martins R. (2009). LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum. Mol. Genet. 18, 4022–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arighi C. N., Hartnell L. M., Aguilar R. C., Haft C. R., Bonifacino J. S. (2004). Role of the mammalian retromer in sorting of the cation-independent mannose 6-phosphate receptor. J. Cell Biol. 165, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbero P., Bittova L., Pfeffer S. R. (2002). Visualization of Rab9-mediated vesicle transport from endosomes to the trans-Golgi in living cells. J. Cell Biol. 156, 511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilina A., Rudenko I. N., Kaganovich A., Civiero L., Chau H., Kalia S. K., Kalia L. V., Lobbestael E., Chia R., Ndukwe K., et al. International Parkinson’s Disease Genomics Consortium; North American Brain Expression Consortium (2014). Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc. Natl. Acad. Sci. USA 111, 2626–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger Z., Smith K. A., Lavoie M. J. (2010). Membrane localization of LRRK2 is associated with increased formation of the highly active LRRK2 dimer and changes in its phosphorylation. Biochemistry 49, 5511–5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biskup S., Moore D. J., Celsi F., Higashi S., West A. B., Andrabi S. A., Kurkinen K., Yu S. W., Savitt J. M., Waldvogel H. J., et al. (2006). Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann. Neurol. 60, 557–569. [DOI] [PubMed] [Google Scholar]
- Bonifacino J. S., Rojas R. (2006). Retrograde transport from endosomes to the trans-Golgi network. Nat. Rev. Mol. Cell Biol. 7, 568–579. [DOI] [PubMed] [Google Scholar]
- Bonifati V. (2007). LRRK2 low-penetrance mutations (Gly2019Ser) and risk alleles (Gly2385Arg)-linking familial and sporadic Parkinson’s disease. Neurochem. Res. 32, 1700–1708. [DOI] [PubMed] [Google Scholar]
- Bosgraaf L., Van Haastert P. J. (2003). Roc, a Ras/GTPase domain in complex proteins. Biochim. Biophys. Acta 1643, 5–10. [DOI] [PubMed] [Google Scholar]
- Bravo-San Pedro J. M., Gómez-Sánchez R., Niso-Santano M., Pizarro-Estrella E., Aiastui-Pujana A., Gorostidi A., Climent V., López de Maturana R., Sanchez-Pernaute R., López de Munain A., et al. (2012). The MAPK1/3 pathway is essential for the deregulation of autophagy observed in G2019S LRRK2 mutant fibroblasts. Autophagy 8, 1537–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucci C., Thomsen P., Nicoziani P., McCarthy J., van Deurs B. (2000). Rab7: a key to lysosome biogenesis. Mol. Biol. Cell 11, 467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings M. R., King R. C. (1970). Ultrastructural changes in nurse and follicle cells during late stages of oogenesis in Drosophila melanogaster. Z. Zellforsch. Mikrosk. Anat. 110, 1–8. [DOI] [PubMed] [Google Scholar]
- Dawson T. M., Ko H. S., Dawson V. L. (2010). Genetic animal models of Parkinson’s disease. Neuron 66, 646–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H., Dodson M. W., Huang H., Guo M. (2008). The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc. Natl. Acad. Sci. USA 105, 14503–14508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson M. W., Zhang T., Jiang C., Chen S., Guo M. (2012). Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum. Mol. Genet. 21, 1350–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y., He D., Yao Z., Klionsky D. J. (2014). The machinery of macroautophagy. Cell Res. 24, 24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan-Or Z., Giladi N., Rozovski U., Shifrin C., Rosner S., Gurevich T., Bar-Shira A., Orr-Urtreger A. (2008). Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 70, 2277–2283. [DOI] [PubMed] [Google Scholar]
- Gan-Or Z., Bar-Shira A., Dahary D., Mirelman A., Kedmi M., Gurevich T., Giladi N., Orr-Urtreger A. (2012). Association of sequence alterations in the putative promoter of RAB7L1 with a reduced parkinson disease risk. Arch. Neurol. 69, 105–110. [DOI] [PubMed] [Google Scholar]
- Gandhi P. N., Wang X., Zhu X., Chen S. G., Wilson-Delfosse A. L. (2008). The Roc domain of leucine-rich repeat kinase 2 is sufficient for interaction with microtubules. J. Neurosci. Res. 86, 1711–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrke S., Imai Y., Sokol N., Lu B. (2010). Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature 466, 637–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng J., Klionsky D. J. (2008). The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 9, 859–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillardon F. (2009). Leucine-rich repeat kinase 2 phosphorylates brain tubulin-beta isoforms and modulates microtubule stability – a point of convergence in parkinsonian neurodegeneration? J. Neurochem. 110, 1514–1522. [DOI] [PubMed] [Google Scholar]
- Gómez-Suaga P., Luzón-Toro B., Churamani D., Zhang L., Bloor-Young D., Patel S., Woodman P. G., Churchill G. C., Hilfiker S. (2012). Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum. Mol. Genet. 21, 511–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross G. G., Feldman R. M. R., Ganguly A., Wang J., Yu H., Guo M. (2008). Role of X11 and ubiquilin as in vivo regulators of the amyloid precursor protein in Drosophila. PLoS ONE 3, e2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatano T., Kubo S., Imai S., Maeda M., Ishikawa K., Mizuno Y., Hattori N. (2007). Leucine-rich repeat kinase 2 associates with lipid rafts. Hum. Mol. Genet. 16, 678–690. [DOI] [PubMed] [Google Scholar]
- Hay B. A., Wassarman D. A., Rubin G. M. (1995). Drosophila homologs of baculovirus inhibitor of apoptosis proteins function to block cell death. Cell 83, 1253–1262. [DOI] [PubMed] [Google Scholar]
- Heo H. Y., Kim K. S., Seol W. (2010). Coordinate regulation of neurite outgrowth by LRRK2 and its interactor, Rab5. Exp Neurobiol 19, 97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hierro A., Rojas A. L., Rojas R., Murthy N., Effantin G., Kajava A. V., Steven A. C., Bonifacino J. S., Hurley J. H. (2007). Functional architecture of the retromer cargo-recognition complex. Nature 449, 1063–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi S., Moore D. J., Yamamoto R., Minegishi M., Sato K., Togo T., Katsuse O., Uchikado H., Furukawa Y., Hino H., et al. (2009). Abnormal localization of leucine-rich repeat kinase 2 to the endosomal-lysosomal compartment in lewy body disease. J. Neuropathol. Exp. Neurol. 68, 994–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho C. C., Rideout H. J., Ribe E., Troy C. M., Dauer W. T. (2009). The Parkinson disease protein leucine-rich repeat kinase 2 transduces death signals via Fas-associated protein with death domain and caspase-8 in a cellular model of neurodegeneration. J. Neurosci. 29, 1011–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huotari J., Helenius A. (2011). Endosome maturation. EMBO J. 30, 3481–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y., Gehrke S., Wang H. Q., Takahashi R., Hasegawa K., Oota E., Lu B. (2008). Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 27, 2432–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaleel M., Nichols R. J., Deak M., Campbell D. G., Gillardon F., Knebel A., Alessi D. R. (2007). LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson’s disease mutants affect kinase activity. Biochem. J. 405, 307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhász G., Erdi B., Sass M., Neufeld T. P. (2007). Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev. 21, 3061–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanao T., Venderova K., Park D. S., Unterman T., Lu B., Imai Y. (2010). Activation of FoxO by LRRK2 induces expression of proapoptotic proteins and alters survival of postmitotic dopaminergic neuron in Drosophila. Hum. Mol. Genet. 19, 3747–3758. [DOI] [PubMed] [Google Scholar]
- Kett L. R., Dauer W. T. (2012). Leucine-rich repeat kinase 2 for beginners: six key questions. Cold Spring Harb Perspect Med 2, a009407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kett L. R., Boassa D., Ho C. C., Rideout H. J., Hu J., Terada M., Ellisman M., Dauer W. T. (2012). LRRK2 Parkinson disease mutations enhance its microtubule association. Hum. Mol. Genet. 21, 890–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King R. C. (1970). Ovarian Development in Drosophila Melanogaster. New York, NY: Academic Press Inc. [Google Scholar]
- Korolchuk V. I., Schütz M. M., Gómez-Llorente C., Rocha J., Lansu N. R., Collins S. M., Wairkar Y. P., Robinson I. M., O’Kane C. J. (2007). Drosophila Vps35 function is necessary for normal endocytic trafficking and actin cytoskeleton organisation. J. Cell Sci. 120, 4367–4376. [DOI] [PubMed] [Google Scholar]
- Koundakjian E. J., Cowan D. M., Hardy R. W., Becker A. H. (2004). The Zuker collection: a resource for the analysis of autosomal gene function in Drosophila melanogaster. Genetics 167, 203–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. B., Kim W., Lee S., Chung J. (2007). Loss of LRRK2/PARK8 induces degeneration of dopaminergic neurons in Drosophila. Biochem. Biophys. Res. Commun. 358, 534–539. [DOI] [PubMed] [Google Scholar]
- Lee S., Liu H. P., Lin W. Y., Guo H., Lu B. (2010). LRRK2 kinase regulates synaptic morphology through distinct substrates at the presynaptic and postsynaptic compartments of the Drosophila neuromuscular junction. J. Neurosci. 30, 16959–16969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B. D., Dawson V. L., Dawson T. M. (2012). Leucine-rich repeat kinase 2 (LRRK2) as a potential therapeutic target in Parkinson’s disease. Trends Pharmacol. Sci. 33, 365–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X., Parisiadou L., Gu X. L., Wang L., Shim H., Sun L., Xie C., Long C. X., Yang W. J., Ding J., et al. (2009). Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron 64, 807–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi D., Soldati T., Riederer M. A., Goda Y., Zerial M., Pfeffer S. R. (1993). Rab9 functions in transport between late endosomes and the trans Golgi network. EMBO J. 12, 677–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod D., Dowman J., Hammond R., Leete T., Inoue K., Abeliovich A. (2006). The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 52, 587–593. [DOI] [PubMed] [Google Scholar]
- MacLeod D. A., Rhinn H., Kuwahara T., Zolin A., Di Paolo G., McCabe B. D., Marder K. S., Honig L. S., Clark L. N., Small S. A., et al. (2013). RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron 77, 425–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin I., Kim J. W., Lee B. D., Kang H. C., Xu J. C., Jia H., Stankowski J., Kim M. S., Zhong J., Kumar M., et al. (2014). Ribosomal protein s15 phosphorylation mediates LRRK2 neurodegeneration in Parkinson’s disease. Cell 157, 472–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta S., Van Kolen K., da Cunha R., van den Bogaart G., Mandemakers W., Miskiewicz K., De Bock P. J., Morais V. A., Vilain S., Haddad D., et al. (2012). LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron 75, 1008–1021. [DOI] [PubMed] [Google Scholar]
- Nishida Y., Arakawa S., Fujitani K., Yamaguchi H., Mizuta T., Kanaseki T., Komatsu M., Otsu K., Tsujimoto Y., Shimizu S. (2009). Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 461, 654–658. [DOI] [PubMed] [Google Scholar]
- Orenstein S. J., Kuo S. H., Tasset I., Arias E., Koga H., Fernandez-Carasa I., Cortes E., Honig L. S., Dauer W., Consiglio A., et al. (2013). Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 16, 394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paisán-Ruíz C., Jain S., Evans E. W., Gilks W. P., Simón J., van der Brug M., López de Munain A., Aparicio S., Gil A. M., Khan N., et al. (2004). Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44, 595–600. [DOI] [PubMed] [Google Scholar]
- Parisiadou L., Xie C., Cho H. J., Lin X., Gu X. L., Long C. X., Lobbestael E., Baekelandt V., Taymans J. M., Sun L., et al. (2009). Phosphorylation of ezrin/radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. J. Neurosci. 29, 13971–13980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper R. C., Luzio J. P. (2007). Ubiquitin-dependent sorting of integral membrane proteins for degradation in lysosomes. Curr. Opin. Cell Biol. 19, 459–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plowey E. D., Cherra S. J., 3rd, Liu Y. J., Chu C. T. (2008). Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J. Neurochem. 105, 1048–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulipparacharuvil S., Akbar M. A., Ray S., Sevrioukov E. A., Haberman A. S., Rohrer J., Krämer H. (2005). Drosophila Vps16A is required for trafficking to lysosomes and biogenesis of pigment granules. J. Cell Sci. 118, 3663–3673. [DOI] [PubMed] [Google Scholar]
- Raiborg C., Stenmark H. (2009). The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 458, 445–452. [DOI] [PubMed] [Google Scholar]
- Ramirez A., Heimbach A., Gründemann J., Stiller B., Hampshire D., Cid L. P., Goebel I., Mubaidin A. F., Wriekat A. L., Roeper J., et al. (2006). Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 38, 1184–1191. [DOI] [PubMed] [Google Scholar]
- Ramonet D., Daher J. P., Lin B. M., Stafa K., Kim J., Banerjee R., Westerlund M., Pletnikova O., Glauser L., Yang L., et al. (2011). Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS ONE 6, e18568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riederer M. A., Soldati T., Shapiro A. D., Lin J., Pfeffer S. R. (1994). Lysosome biogenesis requires Rab9 function and receptor recycling from endosomes to the trans-Golgi network. J. Cell Biol. 125, 573–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi-Nakashima A., Meir J. Y., Jin Y., Matsumoto K., Hisamoto N. (2007). LRK-1, a C. elegans PARK8-related kinase, regulates axonal-dendritic polarity of SV proteins. Curr. Biol. 17, 592–598. [DOI] [PubMed] [Google Scholar]
- Satake W., Nakabayashi Y., Mizuta I., Hirota Y., Ito C., Kubo M., Kawaguchi T., Tsunoda T., Watanabe M., Takeda A., et al. (2009). Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 41, 1303–1307. [DOI] [PubMed] [Google Scholar]
- Schapansky J., Nardozzi J. D., Felizia F., LaVoie M. J. (2014). Membrane recruitment of endogenous LRRK2 precedes its potent regulation of autophagy. Hum. Mol. Genet. 23, 4201–4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman M. N. (2004). Cargo-selective endosomal sorting for retrieval to the Golgi requires retromer. J. Cell Biol. 165, 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu F., Katagiri T., Suzuki M., Watanabe T. K., Okuno S., Kuga Y., Nagata M., Fujiwara T., Nakamura Y., Takahashi E. (1997). Cloning and chromosome assignment to 1q32 of a human cDNA (RAB7L1) encoding a small GTP-binding protein, a member of the RAS superfamily. Cytogenet. Cell Genet. 77, 261–263. [DOI] [PubMed] [Google Scholar]
- Shin N., Jeong H., Kwon J., Heo H. Y., Kwon J. J., Yun H. J., Kim C. H., Han B. S., Tong Y., Shen J., et al. (2008). LRRK2 regulates synaptic vesicle endocytosis. Exp. Cell Res. 314, 2055–2065. [DOI] [PubMed] [Google Scholar]
- Simón-Sánchez J., Schulte C., Bras J. M., Sharma M., Gibbs J. R., Berg D., Paisan-Ruiz C., Lichtner P., Scholz S. W., Hernandez D. G., et al. (2009). Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 41, 1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith W. W., Pei Z., Jiang H., Moore D. J., Liang Y., West A. B., Dawson V. L., Dawson T. M., Ross C. A. (2005). Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc. Natl. Acad. Sci. USA 102, 18676–18681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradling A. C. (1993). Developmental genetics of oogenesis. In The Development of Drosophila Melanogaster, Vol. I (ed. Bate M., Arias A. M.), pp. 1–70 Plainview, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Taylor J. P., Mata I. F., Farrer M. J. (2006). LRRK2: a common pathway for parkinsonism, pathogenesis and prevention? Trends Mol. Med. 12, 76–82. [DOI] [PubMed] [Google Scholar]
- Till B. J., Reynolds S. H., Greene E. A., Codomo C. A., Enns L. C., Johnson J. E., Burtner C., Odden A. R., Young K., Taylor N. E., et al. (2003). Large-scale discovery of induced point mutations with high-throughput TILLING. Genome Res. 13, 524–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y., Yamaguchi H., Giaime E., Boyle S., Kopan R., Kelleher R. J., III, Shen J. (2010). Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc. Natl. Acad. Sci. USA 107, 9879–9884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y., Giaime E., Yamaguchi H., Ichimura T., Liu Y., Si H., Cai H., Bonventre J. V., Shen J. (2012). Loss of leucine-rich repeat kinase 2 causes age-dependent bi-phasic alterations of the autophagy pathway. Mol. Neurodegener. 7, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilariño-Güell C., Wider C., Ross O. A., Dachsel J. C., Kachergus J. M., Lincoln S. J., Soto-Ortolaza A. I., Cobb S. A., Wilhoite G. J., Bacon J. A., et al. (2011). VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 89, 162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Tang B., Zhao G., Pan Q., Xia K., Bodmer R., Zhang Z. (2008). Dispensable role of Drosophila ortholog of LRRK2 kinase activity in survival of dopaminergic neurons. Mol. Neurodegener. 3, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Schulze K. L., Hiesinger P. R., Suyama K., Wang S., Fish M., Acar M., Hoskins R. A., Bellen H. J., Scott M. P. (2007). Thirty-one flavors of Drosophila rab proteins. Genetics 176, 1307–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A., Biskup S., Leitner P., Lichtner P., Farrer M., Lincoln S., Kachergus J., Hulihan M., Uitti R. J., Calne D. B., et al. (2004). Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607. [DOI] [PubMed] [Google Scholar]
- Zimprich A., Benet-Pagès A., Struhal W., Graf E., Eck S. H., Offman M. N., Haubenberger D., Spielberger S., Schulte E. C., Lichtner P., et al. (2011). A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet. 89, 168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.