

Abstract
Mutations in the TCIRG1 gene, coding for a subunit of the osteoclast proton pump, are responsible for more than 50% of cases of human malignant autosomal recessive osteopetrosis (ARO), a rare inherited bone disease with increased bone density owing to a failure in bone resorption. A wide variety of mutations has been described, including missense, nonsense, small deletions/insertions, splice-site mutations, and large genomic deletions, all leading to a similar severe presentation. So far, to the best of our knowledge, no report of a mild phenotype owing to recessive TCIRG1 mutations is present neither in our series of more than 100 TCIRG1-dependent ARO patients nor in the literature. Here we describe an 8-year-old patient referred to us with a clinical diagnosis of ARO, based on radiological findings; of note, no neurological or hematological defects were present in this girl. Surprisingly, we identified a novel nucleotide change in intron 15 of the TCIRG1 gene at the homozygous state, leading to the production of multiple aberrant transcripts, but also, more importantly, of a limited amount of the normal transcript. Our results show that a low level of normal TCIRG1 protein can dampen the clinical presentation of TCIRG1-dependent ARO. On this basis, a small amount of protein might be sufficient to rescue, at least partially, the severe ARO phenotype, and this is particularly important when gene therapy approaches are considered. In addition, we would also recommend that the TCIRG1 gene be included in the molecular diagnosis of mild forms of human ARO. © 2014 Italian National Research Council. Journal of Bone and Mineral Research published by Wiley Periodicals, Inc. on behalf of the American Society for Bone and Mineral Research.
Keywords: AUTOSOMAL RECESSIVE OSTEOPETROSIS, TCIRG1, HYPOMORPHIC MUTATION, SPLICING DEFECT
Introduction
Autosomal recessive osteopetrosis (ARO) is a rare inherited bone disorder characterized by increased bone density owing to failure in bone resorption by the osteoclasts. The disease is genetically heterogeneous; at least seven genes are involved in its pathogenesis.1 The majority of patients bear defects in the TCIRG1 gene, encoding the osteoclast-specific a3 subunit of the vacuolar proton pump V-ATPase, which is essential for the acidification of the resorption lacuna and for vesicle trafficking and fusion in the osteoclast. A wide variety of mutations, including missense, nonsense, small deletions/insertions, splice-site mutations, and large genomic deletions, has been described by our group and others, in reports dealing with large series or single patients.2–13 All the TCIRG1-deficient ARO patients described thus far have invariably shown a homogeneous presentation, in which the bone defect is accompanied by an important hematological impairment and secondary neurological deficit (mainly blindness). Hematopoietic stem cell transplantation (HSCT) is able to rescue the disease and, owing to the severity of the phenotype, is highly recommended as therapeutic approach to be performed as soon as possible, according to the European Group for Bone Marrow Transplantation–European Society for Immunodeficiencies (EBMT-ESID) guidelines (www.ebmt.org/Contents/About-EBMT/Who-We-Are/ScientificCouncil/Documents/EBMT_ESID%20GUIDELINES%20FOR%20INBORN%20ERRORS%20FINAL%202011.pdf). Of note, no report of a mild phenotype owing to recessive TCIRG1 mutations is present in the literature to the best of our knowledge, whereas intermediate forms of osteopetrosis have been associated with biallelic or monoallelic mutations in other genes, such as CLCN714,15 and PLEKHM1.16
Here we describe the extraordinary identification of a homozygous, novel mutation in the TCIRG1 gene in a patient with a benign clinical picture, likely owing to the production of a limited amount of the normal transcript. From a translational point of view, this finding suggests that, when gene therapy approaches are considered, a small amount of protein might be sufficient to rescue the classical, severe ARO phenotype.
Case Report
An 8-year-old patient was referred to us with a clinical diagnosis of ARO. She was the only daughter of distantly related parents (fourth degree consanguinity) born at term after an uneventful pregnancy, without a familial history for the disease. The anterior fontanel closed at 5 months. At 3 years of age she experienced a traumatic bone contusion without fracture. Radiological investigation performed at that time showed a generalized increase in bone density and led to the diagnosis (see Fig. 1 for the most recent X-rays and also Supporting Fig. 1), also supported by the bone biopsy taken 1 year later. When she was 4 years old, she had an accidental tibia fracture that healed properly. Dentition occurred normally, the only dental defects at present (8 years) being caries. She displays neither growth retardation, nor hematological or sensorial defects; most recent laboratory investigations show normal blood counts and normal levels of calcium, phosphorus, alkaline phosphatase, and parathyroid hormone. She complains of bone pain but is not receiving any specific therapy and leads a normal life for her age: she attends school, plays with other children, and rides a bike, the only precaution being no sport activity. Overall, the clinical picture can be considered benign, despite the early presentation.
Fig 1.

X-rays of the patient at 5 years (upper left panel) and 7 years of age, showing a diffuse increase in bone density and bone deformities.
Materials and Methods
Samples
Clinical data and specimens, including blood and DNA samples, were collected from the patient and her parents after informed consent. The latter includes also the permission to publish her photo in this article. This research complies with the standards established by the Independent Ethical Committee of the Humanitas Clinical and Research Centre.
Molecular studies
The molecular analysis of genes known to be responsible for the different types of the disease (TCIRG1, CLCN7, PLEKHM1, RANKL, RANK, and SNX10) was performed by amplification and direct sequencing of exons and intron-exon boundaries as described.5,14,16–20 The mutation nomenclature conforms to www.hgvs.org/mutnomen.
The effect on transcript processing of the mutation found in TCIRG1 was verified at the cDNA level with the forward primer 9912F 5′-GCCTGGCTGCCAACCACTTG-3′ in exon 14 and the reverse primer 5R 5′-CATCGGAGCTCCAGCCATT-3′ in exon 17, using the following thermocycling conditions: initial denaturation step at 94°C for 3 minutes, followed by 34 cycles of denaturation at 94°C for 30 seconds, annealing at 62°C for 30 seconds, and amplification at 72°C for 30 seconds. The PCR product was cloned in the TOPO TA Cloning plasmid vector (Life Technologies, Carlsbad, CA, USA), according to the manufacturers' instructions. Individual clones were sequenced using the T7 forward primer.
In silico studies
The effect of the mutation identified was predicted using the software Alternative Splice Site Predictor ASSP (http://wangcomputing.com/assp/) and Human Splicing Finder, Version 2.4.1 (www.umd.be/HSF/).
Results
Based on the mild clinical presentation of the patient, we initially performed the molecular analysis of genes that, according to the literature, can lead to a benign phenotype or to a spectrum of severity (CLCN7, PLEKHM1, RANKL, RANK, and SNX10); however, we could not find any nucleotide change. Surprisingly, sequencing of the TCIRG1 gene led to the identification of a novel nucleotide change in intron 15 (c.1941 + 5G > A) at the homozygous state in the patient and at the heterozygous state in both of her parents (Fig. 2A). This nucleotide change was predicted to importantly affect the strength of the constitutive donor site of exon 15 using two bioinformatics tools. Indeed, in many diseases mutations at position +5 within the 5′ splice site consensus sequence are pathogenetic.21 In order to verify the in silico prediction, we cloned the PCR amplicon spanning exons 14 to 17 of TCIRG1 produced from the patient's cDNA, picked 57 independent clones, and analyzed them by direct Sanger sequencing. In this way we showed that indeed the mutated allele generated multiple aberrant transcripts through abnormal splicing: eight of them retained intron 15, 30 clones used an alternative GT donor sequence within exon 15 of which 13 skipped exon 16, and one clone used an alternative donor site within exon 14 losing part of this exon (Fig. 2B; Supporting Fig. 2). However, and more importantly, we found that a non-negligible number of clones (18/57 clones) displayed the normal sequence; overall, the amount of the normal transcript could be grossly estimated to constitute about 32% of the total transcript produced.
Fig 2.
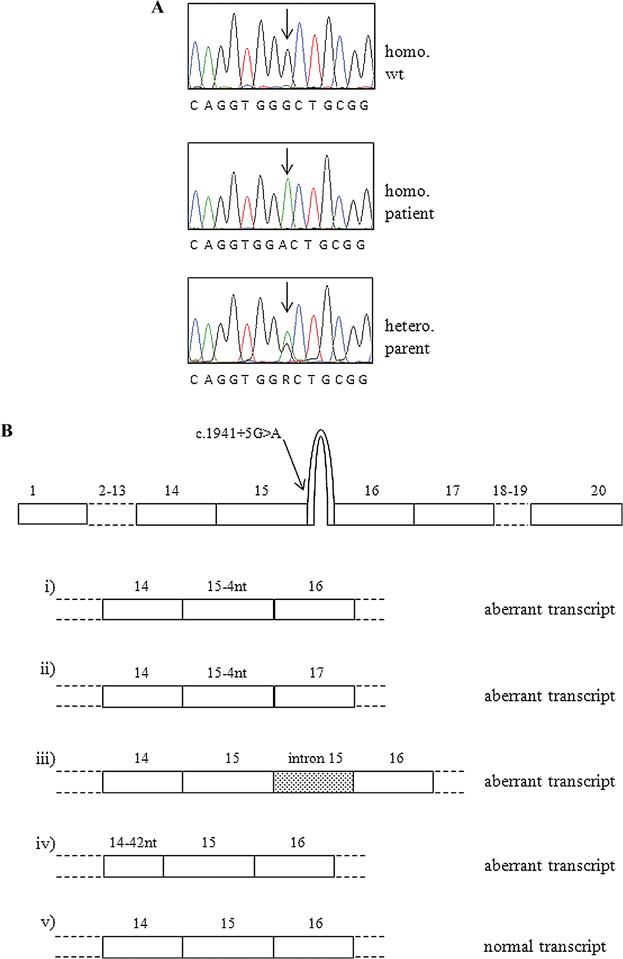
(A) Sanger sequencing chromatograms showing the nucleotide change found in the patient at the homozygous state and in her parents (only one is shown) at the heterozygous state. (B) Schematic representation of the transcripts deriving from the mutated allele; besides different products of aberrant splicing, the wild-type transcript can also be generated.
Discussion
Clinically benign osteopetrosis is usually inherited as an autosomal dominant trait (ADO) associated with mutations in either the CLCN7 or the LRP5 gene.22,23 A novel de novo heterozygous missense mutation in the TCIRG1 gene has been recently described in a Japanese patient initially reported as affected by ADO II24; however, information regarding parental consanguinity and ethnicity of the family and demonstration of the putative dominant effect of that amino acid change were not provided by the authors. On the other hand, although the data in this patient do not appear to be conclusive, they might be in agreement with a previous finding in mice.25
In the context of recessive forms of osteopetrosis, our data represent the first report of a mild phenotype due to recessive TCIRG1 mutations in the literature, to the best of our knowledge. We demonstrated that the molecular mechanism at the basis of the moderate severity of the disease in our patient consists of an incomplete splicing defect: the limited amount of correctly spliced mRNA coming from the mutated alleles would allow for the production of the small amount of protein that is sufficient for dampening the clinical outcome. A similar finding has been reported in the pathogenesis of other diseases, such as congenital factor XIII deficiency, ZAP70 deficiency, and abetalipoproteinemia.26–28 However, in TCIRG1-dependent ARO this a very rare event because, in our series of more than 100 patients bearing mutations in this gene, no such phenotype was present in more than 30 different splicing defects, including eight owing to mutations outside the canonical GT/AG consensus sequence.2,5,8,13 Our finding is even more unexpected, based on the involvement of the nucleotide position affected in spliceosomal assembly.
Our results widen the clinical spectrum that may arise from recessive mutations in TCIRG1, demonstrating that, even though extremely rare, mild forms of TCIRG1-dependent ARO exist, so this gene should also be included in the molecular workup of intermediate cases. In this regard, the possibility to exploit whole exome sequencing in the genetic diagnosis of atypical cases should be considered in order to speed up the analysis.29–31 In addition, our finding supports the hypothesis that, when gene therapy approaches are considered, a small amount of protein might be sufficient to rescue the severe ARO phenotype to a considerable extent, thus avoiding the need for obtaining high expression efficiencies. If this holds true, a satisfactory efficacy could be obtained without vector modifications aimed at achieving more robust gene expression.32 Recently, encouraging evidence of viral-mediated correction of ARO in preclinical studies has been obtained,33,34 and we hope our finding may foster implementation of this promising therapeutic approach. However, a moment of caution should be spent considering the young age of the patient and the possibility that the disease manifestations worsen with age; thus, a longer follow-up will likely allow drawing definitive conclusions.
Disclosures
All authors state that they have no conflicts of interest.
Acknowledgments
This work was supported by the Telethon Foundation (grant GGP12178 to CS); PRIN Project (200999KRFW-002 to PV; 20102M7T8X_003 to AV); Giovani Ricercatori from Ministero della Salute (grant GR-2008-1134625 to CS); Ricerca Finalizzata from Ministero della salute (RF-2009-1499,542 to AV); the European Community's Seventh Framework Program (FP7/2007-2013, SYBIL Project to AV); and PNR-CNR Aging Program 2012-2014. We are grateful to the patient's parents for their cooperation and in particular for giving the consent to publish the pictures of their daughter.
Authors' roles: Molecular studies: AP, LS, and MEC. Patient evaluation: AGML, DPVG. Drafting manuscript: CS, PV, and AV. Revising manuscript content: AP, AGML, DPVG, MEC, LS, PV, AV, and CS.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Frontal and lateral view of the patient at 8 years of age.
Alignment of the transcripts produced from the mutant TCIRG1 to the genomic sequence. Exons are depicted in black, introns in blue.
References
- 1.Sobacchi C, Schulz A, Coxon FP, Villa A, Helfrich MH. Osteopetrosis: genetics, treatment and new insights into osteoclast function. Nat Rev Endocrinol. 2013;9:522–36. doi: 10.1038/nrendo.2013.137. [DOI] [PubMed] [Google Scholar]
- 2.Frattini A, Orchard PJ, Sobacchi C, et al. Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nat Genet. 2000;25:343–6. doi: 10.1038/77131. [DOI] [PubMed] [Google Scholar]
- 3.Kornak U, Schulz A, Friedrich W, et al. Mutations in the a3 subunit of the vacuolar H(+)-ATPase cause infantile malignant osteopetrosis. Hum Mol Genet. 2000;9:2059–63. doi: 10.1093/hmg/9.13.2059. [DOI] [PubMed] [Google Scholar]
- 4.Michigami T, Kageyama T, Satomura K, et al. Novel mutations in the a3 subunit of vacuolar H(+)-adenosine triphosphatase in a Japanese patient with infantile malignant osteopetrosis. Bone. 2002;30:436–9. doi: 10.1016/s8756-3282(01)00684-6. [DOI] [PubMed] [Google Scholar]
- 5.Sobacchi C, Frattini A, Orchard P, et al. The mutational spectrum of human malignant autosomal recessive osteopetrosis. Hum Mol Genet. 2001;10:1767–73. doi: 10.1093/hmg/10.17.1767. [DOI] [PubMed] [Google Scholar]
- 6.Scimeca JC, Quincey D, Parrinello H, et al. Novel mutations in the TCIRG1 gene encoding the a3 subunit of the vacuolar proton pump in patients affected by infantile malignant osteopetrosis. Hum Mutat. 2003;21:151–7. doi: 10.1002/humu.10165. [DOI] [PubMed] [Google Scholar]
- 7.Taranta A, Migliaccio S, Recchia I, et al. Genotype-phenotype relationship in human ATP6i-dependent autosomal recessive osteopetrosis. Am J Pathol. 2003;162:57–68. doi: 10.1016/S0002-9440(10)63798-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Susani L, Pangrazio A, Sobacchi C, et al. TCIRG1-dependent recessive osteopetrosis: mutation analysis, functional identification of the splicing defects, and in vitro rescue by U1 snRNA. Hum Mutat. 2004;24:225–35. doi: 10.1002/humu.20076. [DOI] [PubMed] [Google Scholar]
- 9.Mazzolari E, Forino C, Razza A, Porta F, Villa A, Notarangelo LD. A single-center experience in 20 patients with infantile malignant osteopetrosis. Am J Hematol. 2009;84:473–9. doi: 10.1002/ajh.21447. [DOI] [PubMed] [Google Scholar]
- 10.Pangrazio A, Caldana ME, Sobacchi C, et al. Characterization of a novel Alu-Alu recombination-mediated genomic deletion in the TCIRG1 gene in five osteopetrotic patients. J Bone Miner Res. 2009;24:162–7. doi: 10.1359/jbmr.080818. [DOI] [PubMed] [Google Scholar]
- 11.Phadke SR, Fischer B, Gupta N, Ranganath P, Kabra M, Kornak U. Novel mutations in Indian patients with autosomal recessive infantile malignant osteopetrosis. Indian J Med Res. 2010;131:508–14. [PubMed] [Google Scholar]
- 12.Yuan P, Yue Z, Sun L, et al. Novel mutation of TCIRG1 and clinical pictures of two infantile malignant osteopetrosis patients. J Bone Miner Metab. 2011;29:251–6. doi: 10.1007/s00774-010-0228-6. [DOI] [PubMed] [Google Scholar]
- 13.Pangrazio A, Caldana ME, Lo Iacono N, et al. Autosomal recessive osteopetrosis: report of 41 novel mutations in the TCIRG1 gene and diagnostic implications. Osteoporos Int. 2012;23:2713–8. doi: 10.1007/s00198-011-1878-5. [DOI] [PubMed] [Google Scholar]
- 14.Frattini A, Pangrazio A, Susani L, et al. Chloride channel ClCN7 mutations are responsible for severe recessive, dominant, and intermediate osteopetrosis. J Bone Miner Res. 2003;18:1740–7. doi: 10.1359/jbmr.2003.18.10.1740. [DOI] [PubMed] [Google Scholar]
- 15.Pangrazio A, Pusch M, Caldana E, et al. Molecular and clinical heterogeneity in CLCN7-dependent osteopetrosis: report of 20 novel mutations. Hum Mutat. 2010;31:E1071–80. doi: 10.1002/humu.21167. [DOI] [PubMed] [Google Scholar]
- 16.Van Wesenbeeck L, Odgren PR, Coxon FP, et al. Involvement of PLEKHM1 in osteoclastic vesicular transport and osteopetrosis in incisors absent rats and humans. J Clin Invest. 2007;117:919–30. doi: 10.1172/JCI30328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kornak U, Kasper D, Bösl MR, et al. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell. 2001;104:205–15. doi: 10.1016/s0092-8674(01)00206-9. [DOI] [PubMed] [Google Scholar]
- 18.Sobacchi C, Frattini A, Guerrini MM, et al. Osteoclast-poor human osteopetrosis due to mutations in the gene encoding RANKL. Nat Genet. 2007;9:960–2. doi: 10.1038/ng2076. [DOI] [PubMed] [Google Scholar]
- 19.Guerrini MM, Sobacchi C, Cassani B, et al. Human osteoclast-poor osteopetrosis with hypogammaglobulinemia due to TNFRSF11A (RANK) mutations. Am J Hum Genet. 2008;83:64–76. doi: 10.1016/j.ajhg.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pangrazio A, Fasth A, Sbardellati A, et al. SNX10 mutations define a subgroup of human autosomal recessive osteopetrosis with variable clinical severity. J Bone Miner Res. 2013;28:1041–9. doi: 10.1002/jbmr.1849. [DOI] [PubMed] [Google Scholar]
- 21.De Conti L, Skoko N, Buratti E, Baralle M. Complexities of 5'splice site definition: implications in clinical analyses. RNA Biol. 2012;9:911–23. doi: 10.4161/rna.20386. [DOI] [PubMed] [Google Scholar]
- 22.Cleiren E, Bénichou O, Van Hul E, et al. Albers-Schönberg disease (autosomal dominant osteopetrosis, type II) results from mutations in the ClCN7 chloride channel gene. Hum Mol Genet. 2001;10:2861–7. doi: 10.1093/hmg/10.25.2861. [DOI] [PubMed] [Google Scholar]
- 23.Van Wesenbeeck L, Cleiren E, Gram J, et al. Six novel missense mutations in the LDL receptor-related protein 5 (LRP5) gene in different conditions with an increased bone density. Am J Hum Genet. 2003;72:763–71. doi: 10.1086/368277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wada K, Harada D, Michigami T, et al. A case of autosomal dominant osteopetrosis type II with a novel TCIRG1 gene mutation. J Pediatr Endocrinol Metab. 2013;26:575–7. doi: 10.1515/jpem-2013-0007. [DOI] [PubMed] [Google Scholar]
- 25.Ochotny N, Flenniken AM, Owen C, et al. The V-ATPase a3 subunit mutation R740S is dominant negative and results in osteopetrosis in mice. J Bone Miner Res. 2011;26:1484–93. doi: 10.1002/jbmr.355. [DOI] [PubMed] [Google Scholar]
- 26.Mikkola H, Muszbek L, Laiho E, et al. Molecular mechanism of a mild phenotype in coagulation factor XIII (FXIII) deficiency: a splicing mutation permitting partial correct splicing of FXIII A-subunit mRNA. Blood. 1997;89:1279–87. [PubMed] [Google Scholar]
- 27.Picard C, Dogniaux S, Chemin K, et al. Hypomorphic mutation of ZAP70 in human results in a late onset immunodeficiency and no autoimmunity. Eur J Immunol. 2009;39:1966–76. doi: 10.1002/eji.200939385. [DOI] [PubMed] [Google Scholar]
- 28.Di Filippo M, Créhalet H, Samson-Bouma ME, et al. Molecular and functional analysis of two new MTTP gene mutations in an atypical case of abetalipoproteinemia. J Lipid Res. 2012;53:548–55. doi: 10.1194/jlr.M020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rehm HL. Disease-targeted sequencing: a cornerstone in the clinic. Nat Rev Genet. 2013;14:295–300. doi: 10.1038/nrg3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sule G, Campeau PM, Zhang VW, et al. Next-generation sequencing for disorders of low and high bone mineral density. Osteoporos Int. 2013;24:2253–9. doi: 10.1007/s00198-013-2290-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pangrazio A, Puddu A, Oppo M, et al. Exome sequencing identifies CTSK mutations in patients originally diagnosed as intermediate osteopetrosis. Bone. 2014;59:122–6. doi: 10.1016/j.bone.2013.11.014. Feb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naldini L. Ex vivo gene transfer and correction for cell-based therapies. Nat Rev Genet. 2011;12:301–15. doi: 10.1038/nrg2985. [DOI] [PubMed] [Google Scholar]
- 33.Johansson MK, de Vries TJ, Schoenmaker T, et al. Hematopoietic stem cell-targeted neonatal gene therapy reverses lethally progressive osteopetrosis in oc/oc mice. Blood. 2007;109:5178–85. doi: 10.1182/blood-2006-12-061382. [DOI] [PubMed] [Google Scholar]
- 34.Moscatelli I, Thudium CS, Flores C, et al. Lentiviral gene transfer of TCIRG1 into peripheral blood CD34(+) cells restores osteoclast function in infantile malignant osteopetrosis. Bone. 2013;57:1–9. doi: 10.1016/j.bone.2013.07.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Frontal and lateral view of the patient at 8 years of age.
Alignment of the transcripts produced from the mutant TCIRG1 to the genomic sequence. Exons are depicted in black, introns in blue.