

Abstract
The capacity of embryonic stem (ES) cells to differentiate into cell lineages comprising the three germ layers makes them powerful tools for studying mammalian early embryonic development in vitro. The human body consists of approximately 210 different somatic cell types, the majority of which have limited proliferative capacity. However, both stem cells and cancer cells bypass this replicative barrier and undergo symmetric division indefinitely when cultured under defined conditions. Several signal transduction pathways play important roles in regulating stem cell development, and aberrant expression of components of these pathways is linked to cancer. Among signaling systems, the critical role of leukemia inhibitory factor (LIF) coupled to the Jak/STAT3 (signal transduction and activation of transcription-3) pathway in maintaining stem cell self-renewal has been extensively reviewed. This pathway additionally plays multiple roles in tumorigenesis. Likewise, the phosphatidylinositide 3-kinase (PI3K)/protein kinase B (PKB/Akt) pathway has been determined to play an important role in both stem cell maintenance and tumor development. This pathway is often induced in cancer with frequent mutational activation of the catalytic subunit of PI3K or loss of a primary PI3K antagonist, phosphatase and tensin homolog deleted on chromosome ten (PTEN). This review focusses on roles of the PI3K signal transduction pathway components, with emphasis on functions in stem cell maintenance and cancer. Since the PI3K pathway impinges on and collaborates with other signaling pathways in regulating stem cell development and/or cancer, aspects of the canonical Wnt, Ras/mitogen-activated protein kinase (MAPK), and TGF-β signaling pathways are also discussed. J. Cell. Physiol. 229: 1312–1322, 2014.
Phosphatidylinositol 3′ Kinase
Mammalian cells harbor relatively high amounts of phosphatidylinositol (Ptdlns) but only low amounts of its phosphorylated Ptdlns derivatives (PPI) within their plasma membranes. Phosphoinositide kinases generate PPI by adding phosphate groups to inositolglycerophospholipids. The individual PI3K subfamilies selectively phosphorylate different phosphoinositides, with the best studied being class PI3K-1A, the members of which are activated by insulin and polypeptide mitogen-coupled receptors to phosphorylate phosphatidyloinositol-4,5-bisphosphate (PIP2) at the D3 position of the inositol ring to generate phosphatidylionositol-3,4-5-trisphosphate (PIP3). Ligand-activated receptor tyrosine kinases (RTKs) regulate class 1 PI3K through either direct binding of their autophosphorylated phosphotyrosines to SH2 domains with the regulatory subunits of PI3K or via intermediary phosphorylation of tyrosine residues of scaffolding proteins such as insulin receptor substrate 1 (IRS1), which then bind and activate PI3K (Manning and Cantley, 2007). The PI3K product, PIP3 has high affinity for a subclass of pleckstrin homology (PH) domains and once generated induces recruitment of proteins harboring these domains to the inner leaflet of the plasma membrane resulting in the initiation of downstream signaling cascades (Fig. 1). The PH domain was first identified as a 100–120 amino acid sequence that occurs twice in the platelet protein pleckstrin, and binds with high affinity and high specificity to phosphoinositide (Haslam et al., 1993; Mayer et al., 1993). Interestingly, only 15 or approximately 10% of all known PH domains bind with high affinity to the head group of phosphoinositides, whereas the others bind phosphoinositides and inositol phosphates weakly and without specificity (Lemmon and Ferguson, 2000).
Fig 1.
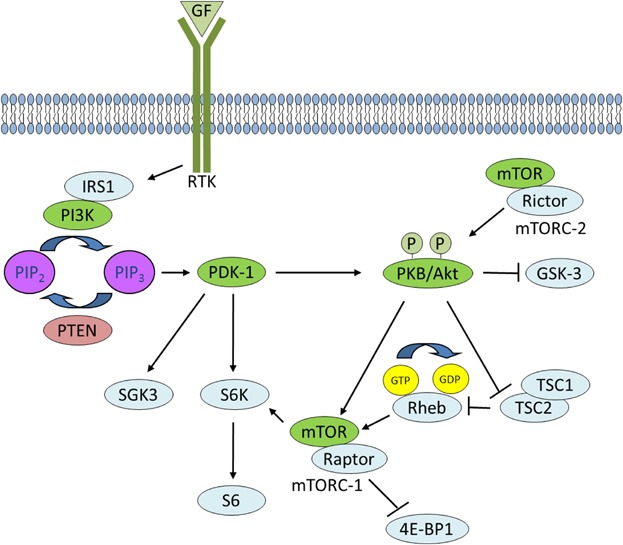
PI3K signaling. Activated RTKs activate class I PI3K through direct binding or through tyrosine phosphorylation of scaffolding adaptors, such as IRS1, which then bind and activate PI3K. PI3K phosphorylates PIP2 to generate PIP3 in a reaction that can be reversed by PTEN. PDK-1 and PKB bind to PIP3 at the plasma membrane, and signal to multiple downstream targets such as SGK3 and S6K and GSK-3, respectively. RTK signaling also activates mTORC-2 which phosphorylates PKB. PKB activates mTORC-1 through phosphorylation of TSC2 within the TCS1–TCS2 complex, which blocks the ability of TSC2 to act as a GTP-ase-activating protein for Rheb. Accumulated Rheb-GTP activates mTORC-1 which phosphorylates downstream targets such as 4E-BP1.
Phosphoinositide-Dependent Protein Kinase-1
The serine/threonine kinase 3′-phosphoinositide-dependent protein kinase 1 (PDK1) binds to PIP3 at the plasma membrane via a C-terminal PH domain. PDK1 is a single copy gene (Manning et al., 2002) and a member of the AGC family of protein kinases first reported by Cohen et al. (1997) as a critical mediator of PKB/Akt activation loop (T-loop) phosphorylation and activation. Termed a “master kinase” (Mora et al., 2004), PDK1 activates a number of downstream kinases including: PKB/Akt, serum- and glucocorticoid-inducible kinases (SGK1–3), p70 ribosomal protein S6 kinase (S6K), p90 ribosomal protein S6 kinase (RSK), p21-activated kinase-1 (PAK-1), PKC-related kinases-1 and 2 (PRK1/2), and diacylglycerol (DAG)-dependent PKCs, resulting in increased glucose uptake, protein synthesis, and inhibition of pro-apoptotic processes (Kikani et al., 2005). In addition to an N-terminal kinase domain (residues 70–359) and a C-terminal PH domain (residues 459–550) that targets PDK1 to the plasma membrane, PDK1 also contains a small phosphate binding groove in its catalytic domain called the PDK1-interacting fragment (PIF)-pocket, which is not necessary for PKB activation, but is required to activate a number of its other substrates, such as S6K and SGK (Bayascas, 2008). PDK1 knockout mice die at embryonic day 9.5 due to lack of somites, forebrain, and neural crest-derived tissues, whereas PDK1 hypomorphs are viable but 40–50% smaller due to decreased cell volume (Lawlor et al., 2002). Recently, it was reported that a basal population of PDK1 homodimers exists in cells and that this population is increased in response to PI3K signaling Formation of homodimers is strictly dependent on the PH domain of PDK1. Since monomeric PDK1 is the active form, enhanced homodimerization of PDK1 translocating to the plasma membrane may represent a negative feedback loop after stimulation to inactivate PDK1-mediated PI3K signaling, since PDK1 homodimers force an autoinhibitory conformation (Masters et al., 2010).
Protein Kinase B/Akt
PKB signaling has become increasingly complex and important in development and disease since it was first identified as a weak oncogene over two decades ago (Bellacosa et al., 1991). PKB phosphorylates many downstream substrates that play important roles in the regulation of survival, glucose uptake, metabolism, growth, proliferation, and angiogenesis. Like PDK1, PKB is also a member of the AGC family of protein kinases. There are three mammalian PKB isoforms (PKBα/Akt1, PKBβ/Akt2, and PKBγ/Akt3), which share extensive amino acid sequence identity and contain three functional domains: an N-terminal PH domain, a kinase domain and a C-terminal hydrophobic motif (HM) (reviewed in Scheid and Woodgett, 2003). PKB has two activating phosphorylation sites, Thr308 and Ser473 (PKBα residues). PDK1 is responsible for PIP3-dependent phosphorylation of PKB; its binding to PIP3 alone does not activate PKB, but it does recruit PKB to the plasma membrane and induces a conformational change permitting its phosphorylation by PDK1 at Thr308. Additional phosphorylation at Ser473 in the HM is required for full activation and this is primarily carried out by target of rapamycin complex 2 (TORC2) (Sarbassov et al., 2005).
Mammalian Target of Rapamycin (mTOR)
mTOR is a component of two independent complexes termed complex-1 (mTORC-1) and complex-2 (mTORC-2). mTOR activity is induced by intracellular nutrients and extracellular growth factors. The major effect of mTOR in complex 1 is to regulate the initiation steps of mRNA translation in protein synthesis (Gingras et al., 2001). mTOR phosphorylates p70S6K and creates a docking site for PDK1, which then phosphorylates Thr229 of p70S6K resulting in its activation. p70S6K phosphorylates a regulatory component of the 40S ribosome, S6, promoting translational efficiency (Tominaga et al., 2005). PKB activates mTORC-1 by phosphorylating and inactivating tuberous sclerosis protein 2 (TSC2) within the TSC1-TSC2 complex. As a result, Rheb-GTP accumulates since TSC2's activity as a GTPase-activating protein for Rheb is blocked by PKB phosphorylation. Rheb-GTP conformationally activates mTORC-1, which phosphorylates its downstream effectors 4E-BP1 and S6 kinases (Manning and Cantley, 2007).
Serum- and Glucocorticoid-Dependent Protein Kinases
SGKs comprise a further serine/threonine protein kinase family with three members that act downstream of PDK1 in the PI3K signaling pathway. The SGKs are structurally related to the PKB family except they lack a PH domain at the amino-terminus. SGK3 contains a PX domain which preferentially binds to PI(3)P and targets the proteins to endosomes (Tessier and Woodgett, 2006a). SGK3 can be phosphorylated at two regulatory sites: Thr320 (activation loop) and Ser486 (hydrophobic motif). Interestingly, in contrast to PKB, phosphorylation of SGK3 by PDK1 is inhibited at the plasma membrane. Instead, targeting SGK3 to endosomes via the PX domain is required for activation of the protein. However, once the hydrophobic motif of SGK3 is phosphorylated, the PX domain is no longer required for activation. Thus, a constitutively active form of SGK3 can be generated by replacing the hydrophobic motif of SGK3 with the phosphomimetic hydrophobic motif of PRK2 (Tessier and Woodgett, 2006b). SGKs share some substrates with PKB/Akt but since their primary sequence preferences are distinct, tend to phosphorylate distinct residues.
Glycogen Synthase Kinase-3
GSK-3 was among the first direct targets of PKB to be identified (Cross et al., 1995) and comprises two isoforms encoded by separate genes, GSK-3α and GSK-3β and is active under resting conditions. GSK-3 is a promiscuous kinase with many substrates, and a tendency to phosphorylate and inactivate its downstream substrates (reviewed in Woodgett, 2005). Upon PI3K activation, PKB inhibits GSK-3 by phosphorylating Ser21 in GSK-3α and Ser9 in GSK-3β, resulting in dephosphorylation and activation of certain substrates of GSK-3 (Doble and Woodgett, 2003), including glycogen synthase and eIF2B, that regulate the rates of glycogen metabolism and protein synthesis, respectively (Patel et al., 2004).
PTEN
The actions of PI3K are reversed by PTEN, a phosphatidylinositol 3′ phosphatase that selectively catalyzes dephosphorylation of the 3′ phosphate of the inositol ring of PIP3, resulting in the bisphosphate product PIP2 (Stambolic et al., 1998). PTEN was first identified in 1997 as a tumor suppressor mutated in a large number of cancers (Li et al., 1997). In that study, mutations of PTEN were detected in 17% of primary glioblastoma, 31% of glioblastoma cell lines and xenografts, all prostate cancer cell lines tested, and 6% of breast cancer cell lines and xenografts. PTEN comprises a tensin-like domain and a catalytic domain, and possesses dual protein and phospholipid phosphatase activity. Tumor suppressor activity is dependent on lipid phosphatase activity, and requires both the phosphatase (catalytic) domain and the C2 (lipid membrane-binding) domain. Targeted disruption of PTEN exons 3–5 in mice leads to PTEN null embryos that die by E9.5 and display abnormal patterning and extensive overgrowth of the cephalic and caudal regions (Stambolic et al., 1998; Suzuki et al., 1998). The early embryonic lethality precluded the functional analysis of PTEN in adult tissues and organs. However, several groups generated viable tissue-specific PTEN knockout mice using the Cre-loxP system (Knobbe et al., 2008). Conditional PTEN-deficient mice develop teratomas, cholangiocellular carcinomas, leiomyosarcomas, prostate, pancreas, breast, and thyroid cancers (Suzuki et al., 2008). Non-cancerous phenotypes include increased cell proliferation, resistance to apoptosis, stem cell renewal, centromeric instability, DNA double-strand breaks, increased angiogenesis, and autoiummune disease (Suzuki et al., 2008). In addition to its role at the plasma membrane, dephosphorylating PIP3, PTEN plays a critical role in DNA damage responses within the nucleus (Ming and He, 2012).
Role of PI3K Signaling in Cancer
PTEN is one of the most commonly lost tumor suppressors in human cancers. Frequent genetic inactivation of PTEN occurs in glioblastoma, melanoma, endometrial, bladder, kidney, colon, and prostate cancer; and reduced expression is found in many other tumor types such as leukemia, thyroid, lung, liver, and breast cancer (Hollander et al., 2011). There is a 50% mortality rate in PTEN heterozygous mice in their first year and these animals develop mammary, thyroid, endometrial, and prostate cancers, as well as T-cell lymphomas—a spectrum that closely resembles the neoplasias in humans with PTEN mutations (Suzuki et al., 2008). Inactivation of PTEN results in unrestrained signaling of the PI3K/PKB signaling cascade, which plays a critical role in cancer by giving tumor cells survival and growth advantages. Indeed, it was the discovery of PTEN as a tumor suppressor that directly linked PI3K to human cancer and since then, mutations causing activation of the PI3K signaling pathway have been found to be amongst the most frequent in human cancers (reviewed in Liu et al., 2009).
Oncogenic mutations in the catalytic subunit of PI3K (PI3KCA) have been found in various types of solid tumors (Samuels and Velculescu, 2004), frequently occurring in the helical domain (E545K and E542K) in exon 9 and the kinase domain (H1047R) in exon 20 (Wong et al., 2010). These somatic missense mutations yield increased PIP3 levels, activate PKB signaling, and promote cellular transformation in vitro and in vivo (Bader et al., 2005; Isakoff et al., 2005; Samuels et al., 2005). Generally, mutations in PDK1 are rarely detected in human cancer (Hunter et al., 2006), but 20% of breast cancers show amplification or overexpression of PDK1 (Brugge et al., 2007). The PKB/Akt isoforms play critical roles in tumorigenesis including tumor initiation, progression, and metastasis (Stambolic and Woodgett, 2006; Hemmings and Restuccia, 2012). Amplification of PKBα and PKBβ has been identified in a variety of cancers including breast, colon, ovarian, lung, gastric, pancreas, and head and neck (Liu et al., 2009). In 2007, Carpten et al. (2007) reported a rare transforming mutation in the PH domain of PKBα (E17K) in melanoma, breast, colorectal, and ovarian cancers, which increased PKB phosphorylation via growth factor-independent constitutive association with the plasma membrane (Yuan and Cantley, 2008). Shortly thereafter, the equivalent mutation was found in PKBγ in human melanoma tumors and melanoma cell lines (Davies et al., 2008). Due to the widespread nature of PI3K pathway activation in cancer a number of PI3K, PKB, and mTOR inhibitors are undergoing clinical trials and at least three mTOR antagonists, Rapamune, Temsirolimus, and Everolimus, have been approved (Liu et al., 2009), although another (Ridaforolimus) recently failed.
Role of PI3K Signaling in ES Cell Maintenance
Evidence that PI3K signaling may be important for maintenance of mouse ES cell self-renewal emerged using LY294002 (Paling et al., 2004), an inhibitor of PI3K, and forced expression of a dominant-negative mutant (Welham et al., 2011). Both loss-of-function approaches demonstrated reduced capacity of mouse ES cells to maintain an undifferentiated state. Overexpression of the pluripotency-associated transcription factor Nanog was shown to be sufficient to maintain ES cell self-renewal without LIF (Chambers et al., 2003), and gene expression profiling of mouse ES cells treated with LY294002 revealed suppression of Nanog and other known regulators of ES cell pluripotency (Storm et al., 2014). Interestingly, during retinoic acid (RA)-induced differentiation of F9 embryonic carcinoma cells, LY294002 decreased Nanog expression in the early time points, but had no effect in later stages despite inhibiting PKB phosphorylation throughout, suggesting that Nanog expression is differentially regulated by the PI3K/PKB pathway depending on differentiation state (Kim et al., 2010).
Prior to work by the Welham group, the PI3K signaling pathway had been implicated in ES cell development by the observation that PTEN null mouse ES cells displayed increased cell cycle progression and cell survival due to accelerated G1/S transition accompanied by down-regulation of p27KIP1, a major inhibitor of G1 cyclin-dependent kinases, and PKB activation which led to increased Bad phosphorylation, respectively (Sun et al., 1999). A role for PI3K in controlling ES cell proliferation was further supported by Takahashi et al. (2005), who showed that overexpression of an active, membrane-bound form of PI3K p110α could rescue the proliferative defects of mouse ES cells lacking ERas (embryonic stem cell-expressed Ras), which is localized to the plasma membrane via a CAAX motif, and promotes the formation and proliferation of teratomas by mouse ES cells. Moreover, treatment with LY294002 markedly increased the G0/G1 phase in ES cells demonstrating the role of PI3K signaling in cell-cycle control of ES cells (Jirmanova et al., 2002).
Though PDK1−/− ES cells grow normally, IGF1 cannot activate PKBα, S6K, and SGK1 in the absence of PDK1 (Williams et al., 2002; Collins et al., 2003). PTEN−/− cells exhibit accelerated growth, but the fact that PTEN and PKBα double knockout ES cells grow much more slowly than wildtype ES cells suggests that PKB functions as a downstream effector of PI3K in ES cell survival and cell proliferation (Stiles et al., 2002; Takahashi et al., 2005). Watanabe et al. (2006) demonstrated that activation of PKB signaling is sufficient to maintain pluripotency in mouse and monkey ES cells. In this study, expression of a myristoylated and activated form of PKB in ES cells maintained the stem cell markers: Nanog, Oct-3/4, PGC7/Stella, and Rex-1 8 days after LIF withdrawal. Typically, expression of these markers is lost 24–36 h following removal of LIF. Reciprocal roles for PKB and Oct-3/4 in positively regulating each other in embryonal carcinoma cells (ECCs), the malignant equivalent of ES cells, has also emerged (Lin et al., 2012). PKB was shown to phosphorylate and positively regulate the protein stability, nuclear localization, and transcriptional activity of Oct-3/4. On the other hand, unphosphorylated Oct-3/4 bound to the PKBα promoter and acted to inhibit its expression. Recently, we have built on these studies by generating ES cell lines that stably express activated alleles of components of the PI3K pathway (Ling et al., 2013). ES cells expressing active forms of PDK1 or PKB/AKT exhibited LIF-independent maintenance of Oct-4 expression and pluripotency. When these cells were treated with PKB inhibitors, both Oct-4 expression and pluripotency were lost. However, treatment with rapamycin did not affect Oct-4 expression or pluripotency, suggesting that these effects are independent of mTOR signaling. Furthermore, when grown in the presence of a mutant form of GSK-3 that is unresponsive to PKB, again Oct-4 expression and pluripotency were unaffected, suggesting that these effects are also independent of GSK-3 signaling. Interestingly, Turner et al. (2014) reported that ES cells exhibit a neuroectodermal fate as they exit pluripotency and the potential for an endomesodermal fate rises with time as the expression of Nanog decreases, Oct-4 is lost, and β-catenin increases. Consistent with this finding, we observed a preference for neural tissue in teratomas generated from activated PDK1 or PKB ES cells (unpublished results) and a complete absence of neural tissue in teratomas generated from GSK-3 null ES cells (Doble et al., 2007).
Sidebar 1. Essential Transcription Factors in Pluripotency.
To date, three intrinsic transcriptional regulators have been found to be essential for ES cell identity: Oct-4, Sox-2, and Nanog. These nuclear factors regulate ES cell pluripotency through a proposed self-sustaining gene-regulatory network (Niwa, 2007). Oct-4 is a mammalian POU domain homeobox transcription factor associated with the establishment of the inner cell mass (ICM). Oct-4 expression is maintained in the epiblast of pre- and post-implantation embryos, and is later restricted to migratory primordial germ cells where it can be found until at least formation of the genital ridges (Chambers and Smith, 2004; Hattori et al., 2004). Precise Oct-4 levels are required for pluripotency, since too much expression leads to differentiation into primitive endoderm and mesoderm (Niwa et al., 2000), and too little (through loss of function) results in differentiation into trophoectoderm (Nichols et al., 1998). Oct-4 can also bind to Sox-2, an HMG-box transcription factor, in the nucleus to regulate pluripotency and lineage specification. Sox-2 is expressed in oocytes, ICM, epiblast, germ cells, multipotent cells of extraembryonic ectoderm, neural lineage cells, brachial arches and gut endoderm (Boyer et al., 2006). Masui et al. (2007) observed that expression of Oct-4 rescued loss of pluripotency in ES cells depleted of Sox-2, supporting a role for Sox-2 in promoting ES cell potentiality through its ability to maintain levels of Oct-4. Nanog is a homeodomain transcription factor that plays a crucial role in maintaining the pluripotent epiblast and preventing differentiation to primitive endoderm (Pan and Thomson, 2007). It can serve as a marker of pluripotency since Nanog mRNA is not found in differentiated cells. Chambers et al. (2003) showed that Nanog acts in parallel with STAT-3 in driving ES cell self-renewal, and constitutive expression of Nanog is sufficient for clonal expansion of ES cells, bypassing STAT-3 in maintaining Oct-4 levels, enabling renewal of ES cells. However, recent studies have found that Nanog expression fluctuates and is dynamic within populations of ES cells (Chambers et al., 2007), and in fact, ES cell populations themselves are heterogeneous (Canham et al., 2010). Interestingly, Chambers et al. (2007) found that although the cells are prone to differentiate, mouse ES cells can self-renew indefinitely in the total absence of Nanog. Thus, of the three master regulators comprising the core pluripotent transcriptional network, Oct-4 acts as the most robust marker of pluripotency.
PI3K Signaling Components Crosstalk With Other Pathways in ES Cell Maintenance: LIF/Jak/STAT Signaling Pathway
The presence of pluripotent stem cells in vivo is fleeting, and these cells only exist in the mouse embryo until the early post-implantation stage. In vitro, daughter cells of ES cells can be made to self-renew and remain pluripotent by symmetric division under appropriate tissue culture conditions. Mouse ES cells are routinely grown in vitro by co-culturing with a layer of fibroblasts that secrete LIF, and/or with the addition of LIF supplementation to the culture medium. LIF is a member of the IL-6 family of cytokines and binding of LIF to its receptor, LIFR, recruits the transmembrane co-receptor, gp130 and leads to the phosphorylation and nuclear translocation of STAT-3, thereby activating the canonical Jak/STAT pathway (Chambers and Smith, 2004) (Fig. 2). In the absence of LIF, stem cells in culture appear to differentiate haphazardly into an assortment of cell types representing the three germ layers. Although LIF signals to activate at least three signaling pathways—including Jak/STAT, PI3K/PKB, and Erk/MAPK pathways (Akira et al., 1994; Boeuf et al., 1997; Niwa et al., 1998, 2009; Matsuda et al., 1999; Watanabe et al., 2006; Welham et al., 2007; Griffiths et al., 2011). STAT-3 is the critical mediator of LIF-mediated mouse ES cell maintenance as STAT-3 activity can sustain ES cell self-renewal even in the absence of LIF (Burdon et al., 1999; Matsuda et al., 1999). Interestingly, in pluripotent human ES cells, STAT-1, -3, and -5 are not phosphorylated, and LIF does not support human ES cell pluripotency when grown in the absence of a feeder layer (Noggle et al., 2005). Identifying the various factors that sustain self-renewal and preserve multi-lineage differentiation of both human and mouse stem cells is an active area of stem cell research. Several signaling pathways have been shown to play important roles in stem cell and cancer stem cell development including: PI3K (Welham et al., 2007), Wnt (Sato et al., 2004), fibroblast growth factor (FGF) (Dvorak et al., 2006), bone morphogenic protein (BMP) (Ying et al., 2003), transforming growth factor (TGF)-β (James et al., 2005) Notch (Carlson and Conboy, 2007), and Hedgehog (Peacock et al., 2007). PI3K pathway components cross-talk with each of these other pathways and several of these interactions are described in more detail below as they relate to ES cell maintenance and cancer.
Fig 2.
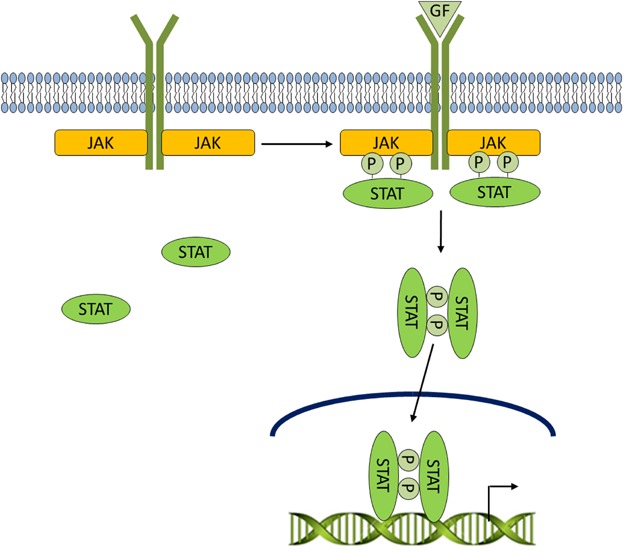
LIF/Jak/STAT signaling. Cells can communicate with each other through the secretion of cytokines, small (8–30 kDa) soluble proteins. Upon binding of cytokines and growth factors to their cognate receptors, receptor-associated Jaks phosphorylate tyrosine residues of STATs. Phosphorylated STATs form homodimers, shuttle to the nucleus, and participate in transcriptional regulation of a variety of genes.
Canonical Wnt Signaling
The Wnt pathway is highly conserved throughout evolution in multicellular organisms and is mutationally activated in 90% of colon cancers through loss of adenomatous polyposis coli (APC) or mutation of negatively acting phosphorylation sites in β-catenin (Saunders and Wallace, 2007). Likewise, activating mutations in β-catenin are also found in 48% of small intestinal adenocarcinomas and 64% of gastric polyps (Giles et al., 2003). The Wnt signaling pathway is classified as canonical, which signals via β-catenin, or non-canonical, which operates in a β-catenin-independent manner (Davis and Zur Nieden, 2008). Canonical signaling by the Wnt family of secreted glycolipoproteins plays a central role in embryonic development and adult diseases such as cancer (Huang and He, 2008). Wnts 1, 3a, and 8 bind to two receptors: Frizzled-1 (Fz-1), a serpentine receptor, and low-density lipoprotein-related protein (LRP) 5 or 6. The binding of Wnt promotes phosphorylation of residues in the cytoplasmic portion of the LRP6 co-receptor by casein kinase-1 (CK-1) and GSK-3, allowing it to act as a high affinity docking site for Axin, a scaffolding protein which is normally associated with APC, CK-1, and GSK-3 in what is termed the β-catenin destruction complex. Under resting conditions, this complex acts as an efficient processing machine that phosphorylates free β-catenin, targeting it for ubiquitinylation and subsequent degradation by the 26S proteasome. As a result of Wnt-induced reorganization of Axin, this destruction complex is destabilized, and newly synthesized β-catenin escapes degradation: hence its cytosolic concentration increases. The accumulating β-catenin enters the cell nucleus, binds members of the T cell factor/lymphoid enhancer-binding factor (TCF/LEF) family of transcription factors and triggers expression of a portfolio of growth promoting genes but also including axin-2 which acts to switch off the pathway (reviewed in Huang and He, 2008; MacDonald et al., 2009; Wu et al., 2009) (Fig. 3).
Fig 3.
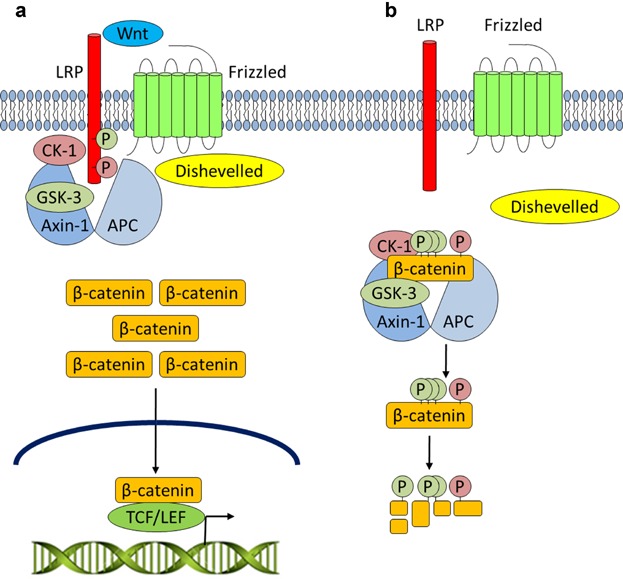
Canonical Wnt signaling. a: Upon Wnt stimulation, Dishevelled is engaged, the destruction complex is disrupted, and CK-1 and GSK-3 activities are diverted to LRP at the cell membrane. Unphosphorylated β-catenin may accumulate and enter the nucleus to regulate gene expression upon binding to TCF/LEF DNA-binding proteins. b: In the absence of Wnt signaling, the destruction complex comprised of APC, Axin-1, CK-1, and GSK-3 promotes the phosphorylation and subsequent ubiquitin-mediated degradation of β-catenin.
GSK-3 has many substrates and is involved in several physiological processes. Sato et al. (2004) identified a role for GSK-3 in stem cell maintenance upon finding that the addition of 6-bromo-indirubin-3′-oxime (BIO), a potent and selective GSK-3 inhibitor, to mouse and human ES cells enhanced self-renewal and pluripotency. Subsequently, Bone et al. (2009, 2011) reported robust enhancement of mouse ES cell self-renewal with the treatment of highly selective bisindolylmaleimides that specifically inhibit GSK-3 in the presence of LIF and serum. After screening for various small molecule inhibitors and factors that could promote pluripotency, Austin Smith's group described a “ground state” of ES cells by showing that two small molecule inhibitors—PD0325901, a MEK inhibitor which blocked differentiation-inducing signaling and CHIR99021, a GSK-3 inhibitor that enhanced cell growth capacity and viability—could also sustain expression of Oct-4 and Nanog and ES cell self-renewal (Ying et al., 2008). Further evidence of the important role that GSK-3 plays in stem cell development derives from ES cells in which all four alleles of GSK-3α and GSK-3β have been genetically inactivated (Doble et al., 2007). These double knockout ES cells fail to differentiate and exhibit hyperactivated Wnt/β-catenin signaling. Knockdown of β-catenin rescued the differentiation defect of GSK-3-null cells, but surprisingly, the effects of β-catenin on pluripotency do not appear to be dependent on TCF-mediated signaling (Kelly et al., 2011). Rather, stabilization of β-catenin induces the formation of β-catenin/Oct-4 complexes, which enhance Oct-4 activity and reinforce pluripotency (Kelly et al., 2011). There is evidence that PI3K-mediated signaling promotes ES cell self-renewal by regulating the activity and localization of GSK-3 (Bechard and Dalton, 2009). This study suggested that upon LIF withdrawal, PI3K/PKB signaling declines and active GSK-3 accumulates in the nucleus, where it targets c-Myc through phosphorylation at Thr58, earmarking it for degradation. However, when PI3K signaling is active, nuclear GSK-3 is rapidly exported back into the cytoplasm in a PKB-dependent manner, and thus c-Myc is maintained at the necessary levels to sustain self-renewal (Bechard and Dalton, 2009).
In addition to being a well-established downstream component of the PI3K signaling pathway, GSK-3 negatively regulates the canonical Wnt/β-catenin signaling pathway and the Notch and Hedgehog pathways (Foltz et al., 2002; Jia et al., 2002; Price and Kalderon, 2002; Espinosa et al., 2003; Wu and Pan, 2010; McNeill and Woodgett, 2010). Whether GSK-3-mediated crosstalk actually exists between these pathways has been a controversial topic. For example, the emergence of several recent studies suggesting that PKB-mediated inhibition of GSK-3 promotes β-catenin accumulation has sparked the debate once again (He et al., 2004; Ishibe et al., 2006; Gu et al., 2007; Kobielak et al., 2007; Maes et al., 2010; Voskas et al., 2010). Physiological levels of GSK-3 do not limit the capacity of the destruction complex to mediate β-catenin degradation; only a small fraction (<10%) of the total GSK-3 in a cell is associated with Axin and actively engaged in canonical Wnt signaling (Lee et al., 2003; Benchabane et al., 2008). Other pools of GSK-3 are additionally sequestered in several other pathways. For example, upon insulin stimulation and consequent PI3K activation, PKB phosphorylates GSK-3 and negatively regulates its kinase activity (Cross et al., 1995). A similar mechanism of negative regulation of GSK-3 has been demonstrated in other growth factor pathways, and though regulation of β-catenin has been demonstrated in many of these pathways (He et al., 2004; Ishibe et al., 2006; Gu et al., 2007; Kobielak et al., 2007; Maes et al., 2010), the direct convergence of Wnt and growth factor pathways on β-catenin regulation remains a subject of debate (Voskas et al., 2010). Consistent with several other studies (Ding et al., 2000; McManus et al., 2005; Ng et al., 2009), we did not observe an effect of activated PI3K on β-catenin signaling in ES cells (Ling et al., 2013). Neither active PDK-1 nor active PKB led to enhanced β-catenin levels in ES cells. This was the case whether these ES cells were grown in the presence of a wildtype or mutant form of GSK-3 that is unresponsive to PKB.
Several human cancers tend to involve separate mutations of both pathways (Wu et al., 2007). If PI3K activation alone was sufficient to activate Wnt signaling, then one would question why additional mutations that lead to β-catenin stabilization would be required. Several studies have also shown that growth factor stimulation which leads to GSK-3 inhibition through PI3K signaling does not result in stabilization of β-catenin in the cell (Ding et al., 2000; McManus et al., 2005; Ng et al., 2009). Furthermore, GSK-3 molecules with mutations in the PKB phosphorylation sites, and thus insensitive to inhibition by PI3K signaling, are still inhibited by Wnt signaling (McManus et al., 2005; Doble et al., 2007; Ng et al., 2009). Despite the fact that PI3K and Wnt signaling pathways share GSK-3 as a core regulatory protein, it would appear that the insulation of these pathways is sufficient to prevent cross-talk (McNeill and Woodgett, 2010). As demonstrated by Ng et al. (2009), Axin may shield the associated GSK-3 within the destruction complex from protein kinases such as PKB that would otherwise phosphorylate and inactivate GSK-3. Although GSK-3 that is not directly associated with the complex may still be phosphorylated and inhibited, sufficient levels remain bound to the Axin complex to phosphorylate and suppress the accumulation of β-catenin (Voskas et al., 2010).
Ras/MAPK and TGF-β Signaling Pathways
In the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) signaling pathway (Fig. 4), activating mutations of K-Ras are found in 45% of colon and 90% of pancreatic cancers, and Raf mutations have been observed in approximately 66% of melanomas (Sebolt-Leopold and Herrera, 2004). The Ras/MAPK pathway is activated by growth factor receptor tyrosine kinases. Upon growth factor stimulation, the adaptor protein, growth factor receptor binding protein 2 (Grb2), binds to specific phospho-tyrosine residues on activated receptors via its SH2 domain. Grb2 also binds to the guanine nucleotide exchange factor (GEF) Son of sevenless (SOS) through its two SH3 domains. This recruits the Grb2/SOS complex to the plasma membrane where it docks to the phosphorylated receptor and activates SOS, which then catalyzes the removal of guanosine diphosphate (GDP) from the Ras subfamily of small GTPases. Ras then rapidly binds guanosine triphosphate (GTP) and adopts an active conformation. Activated Ras recruits the serine/threonine kinase c-Raf to the membrane leading to Raf kinase activation and resulting in phosphorylation and activation of mitogen-activated protein/extracellular signal-regulated kinase (MEK) on two serine residues within its activation segment (T-Loop). Activated MEK in turn phosphorylates ERK on a tyrosine and threonine residue within its T-Loop, such that it subsequently targets its nuclear and non-nuclear substrates (described in Gomperts et al., 2009; Cakir and Grossman, 2009).
Fig 4.
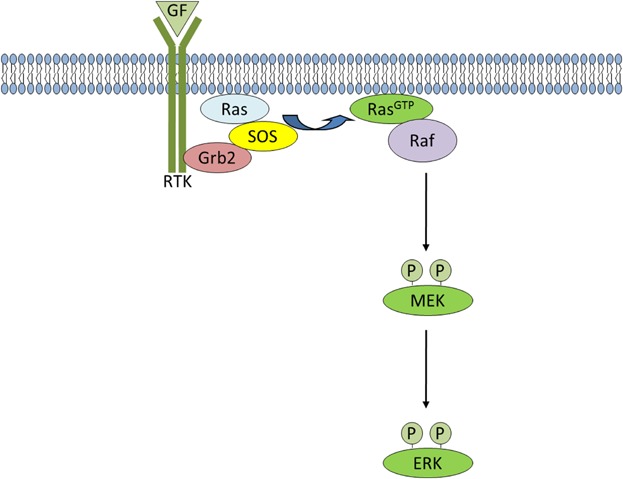
Ras/MAPK signaling. Upon growth factor stimulation, the adaptor protein, Grb2, binds to specific phospho-tyrosine residues on activated receptors via its SH2 domain. Grb2 also binds to SOS and activates it at the plasma membrane, which then catalyzes the removal of GDP. Ras then rapidly binds GTP and adopts an active conformation. Activated Ras recruits Raf to the membrane leading to Raf kinase activation and resulting in phosphorylation and activation of MEK. Activated MEK, in turn phosphorylates ERK, so that it can subsequently activate its nuclear and non-nuclear substrates.
The TGF-β superfamily of ligands include: bone morphogenetic proteins (BMPs), growth and differentiation factors (GDFs), anti-müllerian hormone (AMH), Activin, Nodal, and TGF-βs. TGF-β signaling normally suppresses cell proliferation, and is inactivated through loss of the effector protein, SMAD4, in over half of pancreatic carcinomas (Bardeesy et al., 2006). The pathway is activated when a TGF-β superfamily ligand binds to a serine/threonine TGF-β type II receptor dimer, which recruits and phosphorylates a type I receptor dimer forming a hetero-tetrameric complex with the ligand which then, according to the receptor specificity leads to SMAD phosphorylation, nuclear translocation and activation of gene expression (Attisano and Wrana, 1996) (Fig. 5).
Fig 5.
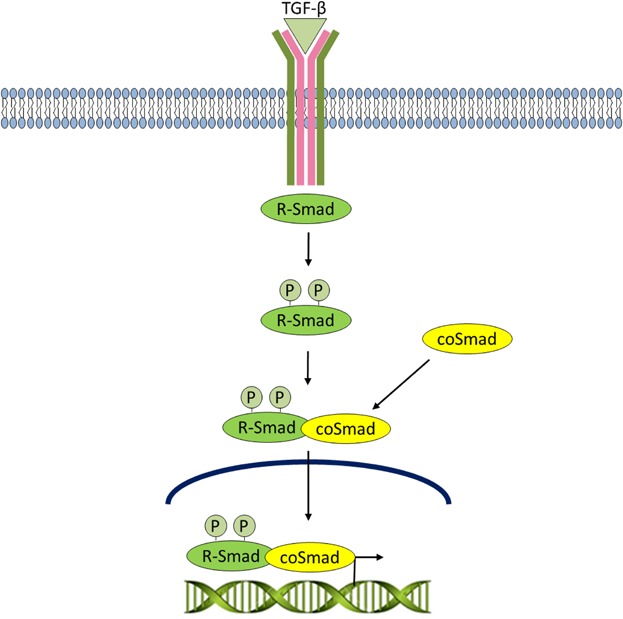
TGF-β signaling. The pathway is activated when aTGF-β superfamily ligand binds to a TGF-β type II receptor dimer, which recruits and phosphorylates a type I receptor dimer forming a hetero-tetrameric complex with the ligand. The type I receptor goes on to phosphorylate R-Smads. Phosphorylated R-SMADs have a high affinity for the effector protein, coSMAD, and consequently forms a complex with coSMAD. R-SMAD/coSMAD complexes accumulate and enter the nucleus where they activate transcription.
Unlike human ES cells which require FGF and TGF-β/Activin signaling to sustain ES cell self-renewal (James et al., 2005; Vallier et al., 2005), mouse ES cells instead depend on LIF and BMP4 (Ying et al., 2003; Qi et al., 2004). FGF/MAPK signaling induces ES cell differentiation in mice (Kunath et al., 2007) whereas BMP4 inhibits this MAPK-mediated differentiation (Qi et al., 2004). Interestingly, PI3K and MAPK signaling have each been implicated downstream of at least two of the three major pathways that control ES cell fate—LIF, FGF, and TGF-β/BMP (Ying et al., 2008; Niwa et al., 2009); however, they appear to counteract each other's downstream activities in mouse ES cells. Downstream of LIF, for example, PI3K positively regulates T-box 3 (Tbx3), a pluripotency factor (Ivanova et al., 2006), whereas MAPK negatively regulates it (Niwa et al., 2009). Although PI3K and MAPK are co-regulated in multiple pathways, the prevalence of one activity over another appears to depend on the relative concentrations of upstream factors that stimulate LIF, FGF, and TGF-β/BMP pathways in mouse ES cells. Such an effect was recently demonstrated in a study by Singh et al. (2012) where an intricate balance of PI3K, MAPK, and Wnt signaling was shown to converge on SMAD2/3 to promote either the self-renewal or differentiation of human ES cells. Clearly, nature has fine-tuned the means by which early cell fate is influenced and the normal requirement for multiple inputs of precise timing and magnitude likely acts as a combination lock to ensure robust transition from the “ground state” to the complexity of a developing organism.
Summary
Although embryonic stem cells possess pluripotentiality only transiently in vivo, this remarkable state can be artificially captured in vitro through growth factors and/or small molecule inhibitors. It is unsurprising that multiple signals can orchestrate this precarious cell state as the early stages of development require enormous plasticity to ensure correct proportions and consequences within a tiny number of cells which are largely discriminated by space and time. What is perhaps more surprising is that there are clearly multiple, unsubtle mechanisms by which pluripotency can be maintained. This reveals a vulnerability that is clearly exploited in tumor etiology where de-differentiation or inappropriate expansion of progenitor cells that retain replicative potential is driven by mutational induction of similar signaling systems that are so potent in early development. Of course, cells have multiple safeguards and lineage-specification and differentiation is usually locked in once it has occurred. However, the commonality shared by many transforming events is that they target genes that have key roles in early embryogenesis. It is beguiling that nature has maintained such variety of backdoor mechanisms that allow reversal of differentiation. Clearly, this capacity has a strong beneficial drive as the risk is huge. Knowledge of the mechanisms by which cells can be maintained and expanded prior to differentiation into desired cell types offers much hope for regenerative and molecular corrective medicine. This has only accelerated with the advent of induced pluripotential stem cells and the myriad of paths by which these can be derived. There is also reason to expect that greater knowledge of the molecular mechanisms that control the transcriptional circuitry underpinning stem cellness will lead to new therapeutic approaches for a variety of cancers that rely upon the usurping developmental controls.
Acknowledgments
Supported by grants to J.R.W. from the Terry Fox Foundation and Canadian Institutes of Health Research (MOP 74711 and 12858).
Literature Cited
- Akira S, Nishio Y, Inoue M, Wang XJ, Wei S, Matsusaka T, Yoshida K, Sudo T, Naruto M, Kishimoto T. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell. 1994;77:63–71. doi: 10.1016/0092-8674(94)90235-6. [DOI] [PubMed] [Google Scholar]
- Attisano L, Wrana JL. Signal transduction by members of the transforming growth factor-beta superfamily. Cytokine Growth Factor Rev. 1996;7:327–339. doi: 10.1016/s1359-6101(96)00042-1. [DOI] [PubMed] [Google Scholar]
- Bader AG, Kang S, Zhao L, Vogt PK. Oncogenic PI3K deregulates transcription and translation. Nat Rev Cancer. 2005;5:921–929. doi: 10.1038/nrc1753. [DOI] [PubMed] [Google Scholar]
- Bardeesy N, Cheng KH, Gerger JH, Chu GC, Pahler J, Olson P, Hezel AF, Horner J, Lauwers GY, Hanahan D, DePinho RA. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006;20:3130–3146. doi: 10.1101/gad.1478706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayascas JR. Dissecting the role of the 3-phosphoinositide-dependent protein kinase-1 (PDK1) signaling pathways. Cell Cycle. 2008;19:2978–2982. doi: 10.4161/cc.7.19.6810. [DOI] [PubMed] [Google Scholar]
- Bechard M, Dalton S. Subcellular localization of glycogen synthase kinase 3β controls embryonic stem cell self-renewal. Mol Cell Biol. 2009;29:2092–2104. doi: 10.1128/MCB.01405-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellacosa A, Testa JR, Stall SP, Tsichlis PN. A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-likeregion. Science. 1991;254:274–277. doi: 10.1126/science.254.5029.274. [DOI] [PubMed] [Google Scholar]
- Benchabane H, Hughes EG, Takacs CM, Baird JR, Ahmed Y. Adenomatous polyposis coli is present near the minimal level required for accurate graded responses to the Wingless morphogen. Development. 2008;135:963–971. doi: 10.1242/dev.013805. [DOI] [PubMed] [Google Scholar]
- Boeuf H, Hauss C, Graeve FD, Baran N, Kedinger C. Leukemia inhibitory factor-dependent transcriptional activation in embryonic stem cells. J Cell Biol. 1997;138:1207–1217. doi: 10.1083/jcb.138.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bone HK, Daminao T, Bartlett S, Perry A, Letchford J, Sanchez, Ripoll Y, Nelson AS, Welham MJ. Involvement of glycogen synthase kinase-3 in regulation of murine embryonic stem cell self-renewal revealed by a series of bisindolymaleimides. Chem Biol. 2009;16:15–27. doi: 10.1016/j.chembiol.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Bone HK, Nelson AS, Goldring CE, Tosh D, Welham MJ. A novel chemically directed route for the generation of definitive endoderm from human embryonic stem cells based on inhibition of GSK-3. J Cell Sci. 2011;124:1992–2000. doi: 10.1242/jcs.081679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer LA, Mathur D, Jaenisch R. Molecular control of pluripotency. Curr Opin Genet Dev. 2006;16:455–462. doi: 10.1016/j.gde.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Brugge J, Hung M, Mills MC. A new mutational AKTivation in the PI3K pathway. Cancer Cell. 2007;12:104–107. doi: 10.1016/j.ccr.2007.07.014. [DOI] [PubMed] [Google Scholar]
- Burdon T, Stracey C, Chambers I, Nichols J, Smith A. Suppression of SHP-2 and ERK signalling promotes self-renewal of mouse embryonic stem cells. Dev Biol. 1999;210:30–43. doi: 10.1006/dbio.1999.9265. [DOI] [PubMed] [Google Scholar]
- Cakir M, Grossman AB. Targeting MAPK (Ras/ERK) and PI3K/Akt pathways in pituitary tumorigenesis. Expert Opin Ther Targets. 2009;13:1121–1134. doi: 10.1517/14728220903170675. [DOI] [PubMed] [Google Scholar]
- Canham MA, Sharov AA, Ko MS, Brickman JM. Functional heterogeneity of embryonic stem cells revealed through translational amplification of an early endodermal transcript. PLoS Biol. 2010;8:e1000379. doi: 10.1371/journal.pbio.1000379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson ME, Conboy IM. Regulating the Notch pathway in embryonic, adult and old stem cells. Curr Opin Pharmacol. 2007;7:303–309. doi: 10.1016/j.coph.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage S, Uhlik M, Lin A, Du J, Qian YW, Zeckner DJ, Tucker-Kellogg G, Touchman J, Patel K, Mousses S, Bittner M, Schevitz R, Lai MH, Blanchard KL, Thomas JE. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- Chambers I, Colby D, Robertson M, Nichols J, Lee S, Tweedie S, Smith A. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. 2003;113:643–655. doi: 10.1016/s0092-8674(03)00392-1. [DOI] [PubMed] [Google Scholar]
- Chambers I, Smith A. Self-renewal of teratocarcinoma and embryonic stem cells. Oncogene. 2004;23:7150–7160. doi: 10.1038/sj.onc.1207930. [DOI] [PubMed] [Google Scholar]
- Chambers I, Silva J, Colby D, Nichols J, Nijmeijer B, Robertson M, Vrana J, Jones K, Grotewold L, Smith A. Nanog safeguards pluripotency and mediates germline development. Nature. 2007;450:1230–1234. doi: 10.1038/nature06403. [DOI] [PubMed] [Google Scholar]
- Cohen P, Alessi DR, Cross DA. PDK1, one of the missing links in insulin signal transduction? FEBS Lett. 1997;410:3–10. doi: 10.1016/s0014-5793(97)00490-0. [DOI] [PubMed] [Google Scholar]
- Collins FJ, Deak M, Aurthur JS, Armit LJ, Alessi DR. In vivo role of PIF-binding docking site of PDK1 defined by knock-in mutation. EMBO J. 2003;22:4202–4211. doi: 10.1093/emboj/cdg407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Davies MA, Stemke-Hale K, Tellez C, Calderone TL, Deng W, Prieto VG, Lazar AJ, Gershenwald JE, Mills GB. A novel AKT3 mutation in melanoma tumors and cell lines. Br J Cancer. 2008;99:1265–1268. doi: 10.1038/sj.bjc.6604637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis LA, Zur Nieden NI. Mesodermal fate decisions of a stem cell: The Wnt switch. Cell Mol Life Sci. 2008;65:2658–2674. doi: 10.1007/s00018-008-8042-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding VW, Chen RH, McCormick F. Differential regulation of glycogen synthase kinase 3β by insulin and Wnt signaling. J Biol Chem. 2000;275:32475–32481. doi: 10.1074/jbc.M005342200. [DOI] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR. GSK-3: Tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble BW, Patel S, Wood GA, Kockeritz LK, Woodgett JR. Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev Cell. 2007;12:857–871. doi: 10.1016/j.devcel.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorak P, Dvorakova D, Hampl A. Fibroblast growth factor signaling in embryonic and cancer stem cells. FEBS Lett. 2006;580:2869–2874. doi: 10.1016/j.febslet.2006.01.095. [DOI] [PubMed] [Google Scholar]
- Espinosa L, Ingles-Esteve J, Aguilera C, Bigas A. Phosphorylation by glycogen synthase kinase-3 beta down-regulates Notch activity, a link for Notch and Wnt pathways. J Biol Chem. 2003;278:32227–32235. doi: 10.1074/jbc.M304001200. [DOI] [PubMed] [Google Scholar]
- Foltz DR, Santiago MC, Berechid BE, Nye JS. Glycogen synthase kinase-3beta modulates notch signaling and stability. Curr Biol. 2002;12:1006–1011. doi: 10.1016/s0960-9822(02)00888-6. [DOI] [PubMed] [Google Scholar]
- Giles RH, van Es JH, Clevers H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta. 2003;1653:1–24. doi: 10.1016/s0304-419x(03)00005-2. [DOI] [PubMed] [Google Scholar]
- Gingras AC, Raught B, Sonenberg N. Regulation of translationinitiation by FRAP/mTOR. Genes Dev. 2001;15:807–826. doi: 10.1101/gad.887201. [DOI] [PubMed] [Google Scholar]
- Gomperts BD, Kramer IM, Tatham PER. Signal transduction. 2nd edition. San Diego: Elsevier, Inc; 2009. pp. 327–337. [Google Scholar]
- Griffiths DS, Li J, Dawson MA, Trotter MW, Cheng YH, Smith AM, Mansfield W, Liu P, Kouzarides T, Nichols J, Bannister AJ, Green AR, Göttgens B. LIF-independent JAK signalling to chromatin in embryonic stem cells uncovered from an adult stem cell disease. Nat Cell Biol. 2011;13:13–21. doi: 10.1038/ncb2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu D, Yu B, Zhao C, Ye W, Lv Q, Hua Z, Ma J, Zhang Y. The effect of pleiotrophin signaling on adipogenesis. FEBS Lett. 2007;581:382–388. doi: 10.1016/j.febslet.2006.12.043. [DOI] [PubMed] [Google Scholar]
- Haslam RJ, Koide HB, Hemmings BA. Pleckstrin domain homology. Nature. 1993;363:309–310. doi: 10.1038/363309b0. [DOI] [PubMed] [Google Scholar]
- Hattori N, Nishino K, Ko YG, Hattori N, Ohgane J, Tanaka S, Shiota K. Epigenetic control of mouse Oct-4 gene expression in embryonic stem cells and trophoblast stem cells. J Biol Chem. 2004;279:17063–17069. doi: 10.1074/jbc.M309002200. [DOI] [PubMed] [Google Scholar]
- He XC, Zhang J, Tong WG, Tawfik O, Ross J, Scoville DH, Tian Q, Zeng X, He X, Wiedemann LM, Mishina Y, Li L. BMP signaling inhibits intestinal stem cell self-renewal through suppression of Wnt-beta-catenin signaling. Nat Genet. 2004;36:1117–1121. doi: 10.1038/ng1430. [DOI] [PubMed] [Google Scholar]
- Hemmings BA, Restuccia DF. PI3K-PKB/Akt pathway. Cold Spring Harb Persp Biol. 2012;4:a011189. doi: 10.1101/cshperspect.a011189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer. 2011;11:289–301. doi: 10.1038/nrc3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, He X. 2008. Wnt/beta-catenin signaling: New (and old) players and new insights. Curr Opin Cell Biol. 2008;20:119–125. doi: 10.1016/j.ceb.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter C, Smith R, Cahill DP, Stephens P, Stevens C, Teague J, Greenman C, Edkins S, Bignell G, Davies H, O'Meara S, Parker A, Avis T, Barthorpe S, Brackenbury L, Buck G, Butler A, Clements J, Cole J, Dicks E, Forbes S, Gorton M, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Jenkinson A, Jones D, Kosmidou V, Laman R, Lugg R, Menzies A, Perry J, Petty R, Raine K, Richardson D, Shepherd R, Small A, Solomon H, Tofts C, Varian J, West S, Widaa S, Yates A, Easton DF, Riggins G, Roy JE, Levine KK, Mueller W, Batchelor TT, Louis DN, Stratton MR, Futreal PA, Wooster R. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after akylator chemotherapy. Cancer Res. 2006;66:3987–3991. doi: 10.1158/0008-5472.CAN-06-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isakoff SJ, Engelman JA, Irie HY, Luo J, Brachmann SM, Pearline RV, Cantley LC, Brugge JS. Breast cancer-associated PI3KCA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005;65:10992–11000. doi: 10.1158/0008-5472.CAN-05-2612. [DOI] [PubMed] [Google Scholar]
- Ishibe S, Haydu JE, Togawa A, Marlier A, Cantley LG. Cell confluence regulates hepatocyte growth factor-stimulated cell morphogenesis in a beta-catenin-dependent manner. Mol Cell Biol. 2006;26:9232–9243. doi: 10.1128/MCB.01312-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova N, Dobrin R, Lu R, Kotenko I, Levorse J, DeCoste C, Schafer X, Lun Y, Lemischka IR. Dissecting self-renewal in stem cells with RNA interference. Nature. 2006;442:533–538. doi: 10.1038/nature04915. [DOI] [PubMed] [Google Scholar]
- James D, Levine AJ, Besser D, Hammati-Brivanlou A. TGFbeta/activin/nodal signaling is necessary for the maintenance of pluripotency in human embryonic stem cells. Development. 2005;132:1273–1282. doi: 10.1242/dev.01706. [DOI] [PubMed] [Google Scholar]
- Jia J, Amanai K, Wang G, Tang J, Wang B, Jiang J. Shaggy/GSK3 antagonizes Hedgehog signalling by regulating Cubitus interruptus. Nature. 2002;416:548–552. doi: 10.1038/nature733. [DOI] [PubMed] [Google Scholar]
- Jirmanova L, Afanassieff M, Gobert-Gosse S, Markossian S, Savatier P. Differential contributions of ERK and PI3-kinase to the regulation of cyclin D1 expression and to the control of the G1/S transition in mouse embryonic stem cells. Oncogene. 2002;21:5515–5528. doi: 10.1038/sj.onc.1205728. [DOI] [PubMed] [Google Scholar]
- Kelly KF, Ng DY, Jayakumaran G, Wood GA, Koide H, Doble BW. β-catenin enhances Oct-4 activity and reinforces pluripotency through a TCF-independent mechanism. Cell Stem Cell. 2011;8:214–227. doi: 10.1016/j.stem.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikani CK, Dong LQ, Liu F. “New”-clear functions of PDK1: Beyond a master kinase in the cytosol? J Cell Biochem. 2005;96:1157–1162. doi: 10.1002/jcb.20651. [DOI] [PubMed] [Google Scholar]
- Kim JS, Kim BS, Kim J, Park CS, Chung, IY The phosphoinositide-3-kinase/Akt pathway mediates the transient increase in Nanog expression during differentiation of F9 cells. Arch Pharm Res. 2010;33:1117–1125. doi: 10.1007/s12272-010-0719-y. [DOI] [PubMed] [Google Scholar]
- Knobbe CB, Lapin V, Suzuki A, Mak TW. The roles of PTEN in development, physiology, and tumorigenesis in mouse. Oncogene. 2008;27:5398–5415. doi: 10.1038/onc.2008.238. [DOI] [PubMed] [Google Scholar]
- Kobielak K, Stokes N, de la Cruz J, Polak L, Fuchs E. Loss of a quiescent niche but not follicle stem cells in the absence of bone morphogenetic protein signaling. Proc Natl Acad Sci. 2007;104:10063–10068. doi: 10.1073/pnas.0703004104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunath T, Saba-El-Leil MK, Almousailleakh M, Wray J, Meloche S, Smith A. FGF stimulation of the Erk1/2 signalling cascade triggers transition of pluripotent embryonic stem cells from self-renewal to lineage commitment. Development. 2007;134:2895–2902. doi: 10.1242/dev.02880. [DOI] [PubMed] [Google Scholar]
- Lawlor MA, Mora A, Ashby PR, Williams MR, Murray-Tait V, Malone L, Prescott AR, Lucocq JM, Alessi DR. Essential role of PDK1 in regulating cell size and development in mice. EMBO J. 2002;21:3728–3738. doi: 10.1093/emboj/cdf387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Salic A, Kruger R, Heinrich R, Kirschner MW. The roles of APC and Axin derived from experimental and theoretical analysis of the Wnt pathway. PLoS Biol. 2003;1:E10. doi: 10.1371/journal.pbio.0000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon MA, Ferguson KM. Signal-dependent membrane targeting by pleckstrin homology (PH) domains. Biochem J. 2000;350:1–18. [PMC free article] [PubMed] [Google Scholar]
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- Lin Y, Yang Y, Li W, Chen Q, Li J, Pan X, Zhou L, Liu C, Chen C, He J, Cao H, Yao H, Zheng L, Xu X, Xia Z, Ren J, Xiao L, Li L, Shen B, Zhou H, Wang YJ. Reciprocal regulation of Akt and Oct4 promotes the self-renewal and survival of embryonal carcinoma cells. Mol Cell. 2012;48:627–640. doi: 10.1016/j.molcel.2012.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling LS, Voskas D, Woodgett JR. Activation of PDK-1 maintains mouse embryonic stem cell self-renewal in a PKB-dependent manner. Oncogene. 2013;32:5397–5408. doi: 10.1038/onc.2013.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–643. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald BT, Tamai K, He X. Wnt/β-catenin signaling: Components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes C, Goossens S, Bartunkova S, Drogat B, Coenegrachts L, Stockmans L, Moermans K, Nyabi O, Haigh K, Naessens M, Haenebalcke L, Tuckermann JP, Tjwa M, Carmeliet P, Mandic V, David JP, Behrens A, Nagy A, Carmeliet G, Haigh JJ. Increased skeletal VEGF enhances β-catenin activity and results in excessively ossified bones. EMBO J. 2010;29:424–441. doi: 10.1038/emboj.2009.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10:151–162. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: Navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters TA, Calleja V, Armoogum DA, Marsh RJ, Applebee CJ, Laguerre M, Bain AJ, Larijani B. Regulation of-phosphoinositide-dependent protein kinase 1 activity by homodimerization in live cells. Sci Signal. 2010;3:ra78. doi: 10.1126/scisignal.2000738. [DOI] [PubMed] [Google Scholar]
- Masui S, Nakatake Y, Toyooka Y, Shimosato D, Yagi R, Takahashi K, Okochi H, Okuda A, Matoba R, Sharov AA, Ko MS, Niwa H. Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nat Cell Biol. 2007;9:625–635. doi: 10.1038/ncb1589. [DOI] [PubMed] [Google Scholar]
- Matsuda T, Nakamura T, Nakao K, Arai T, Katsuki M, Heike T, Yokota T. STAT3 activation is sufficient to maintain an undifferentiated state of mouse embryonic stem cells. EMBO J. 1999;18:4261–4269. doi: 10.1093/emboj/18.15.4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer BJ, Ren R, Clark KL, Baltimore D. A putative modular domain present in diverse signaling molecules. Cell. 1993;73:629–630. doi: 10.1016/0092-8674(93)90244-k. [DOI] [PubMed] [Google Scholar]
- McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, Alessi DR. Role that phosphorylation of GSK3 plays in insulin and Wnt signaling defined by knockin analysis. EMBO J. 2005;24:1571–1583. doi: 10.1038/sj.emboj.7600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill H, Woodgett JR. When pathways collide: Collaboration and connivance among signaling proteins in development. Nat Rev Mol Cell Biol. 2010;11:404–413. doi: 10.1038/nrm2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming M, He YY. PTEN in DNA damage repair. Cancer Lett. 2012;319:125–129. doi: 10.1016/j.canlet.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora A, Komander D, van Aalten DM, Alessi DR. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol. 2004;15:161–170. doi: 10.1016/j.semcdb.2003.12.022. [DOI] [PubMed] [Google Scholar]
- Ng SS, Mahmoudi T, Danenberg E, Bejaoui I, de Lau W, Korswagen HC, Schutt M, Clevers H. Phosphatidylinositol 3-kinase signaling does not activate the Wnt cascade. J Biol Chem. 2009;284:35308–35313. doi: 10.1074/jbc.M109.078261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols J, Zevnik B, Anastassiadis K, Niwa H, Klewe-Nebenius D, Chambers I, Schöler H, Smith A. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell. 1998;95:379–391. doi: 10.1016/s0092-8674(00)81769-9. [DOI] [PubMed] [Google Scholar]
- Niwa H, Burdon T, Chambers I, Smith A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 1998;12:2048–2060. doi: 10.1101/gad.12.13.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H, Miyazaki J, Smith AG. Quantitative expression of Oct3/4 defines differentiation, dedifferentiationor self-renewal of ES cells. Nat Genet. 2000;24:372–376. doi: 10.1038/74199. [DOI] [PubMed] [Google Scholar]
- Niwa H. How is pluripotency determined and maintained? Development. 2007;134:635–646. doi: 10.1242/dev.02787. [DOI] [PubMed] [Google Scholar]
- Niwa H, Ogawa K, Shimosato D, Adachi K. A parallel circuit of LIF signaling pathways maintains pluripotency of mouse ES cells. Nature. 2009;460:118–122. doi: 10.1038/nature08113. [DOI] [PubMed] [Google Scholar]
- Noggle SA, James D, Brivanlou AH. A molecular basis for human embryonic stem cell pluripotency. Stem Cell Rev. 2005;1:111–118. doi: 10.1385/SCR:1:2:111. [DOI] [PubMed] [Google Scholar]
- Paling NR, Bone HK, Welham MJ. Regulation of embryonic stem cell self-renewal by phosphoinositide 3-kinase-dependent signaling. J Biol Chem. 2004;279:48063–48070. doi: 10.1074/jbc.M406467200. [DOI] [PubMed] [Google Scholar]
- Pan G, Thomson JA. Nanog and transcriptional networks in embryonic stem cell pluripotency. Cell Res. 2007;17:42–49. doi: 10.1038/sj.cr.7310125. [DOI] [PubMed] [Google Scholar]
- Patel S, Doble B, Woodgett JR. Glycogen synthase-3 in insulin and Wnt signaling: A double-edged sword. Biochem Soc Trans. 2004;32:803–808. doi: 10.1042/BST0320803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peacock CD, Wang Q, Gesell GS, Corcoran-Schwartz IM, Jones E, Kim J, Devereux WL, Rhodes JT, Huff CA, Beachy PA, Watkins DN, Matsui W. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci. 2007;104:4048–4053. doi: 10.1073/pnas.0611682104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MA, Kalderon D. Proteolysis of the Hedgehog signaling effector Cubitus interruptus requires phosphorylation by glycogen synthase kinase 3 and casein kinase 1. Cell. 2002;108:823–835. doi: 10.1016/s0092-8674(02)00664-5. [DOI] [PubMed] [Google Scholar]
- Qi X, Li TG, Hao J, Hu J, Wang J, Simmons H, Miura S, Mishina Y, Zhao GQ. BMP4 supports self-renewal of embryonic stem cells by inhibiting mitogen-activated protein kinase pathways. Proc Natl Acad Sci. 2004;101:6027–6032. doi: 10.1073/pnas.0401367101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels Y, Velculescu VE. Oncogenic mutations of PI3KCA in human cancers. Cell Cycle. 2004;3:1221–1224. doi: 10.4161/cc.3.10.1164. [DOI] [PubMed] [Google Scholar]
- Samuels Y, Diaz LA, Jr, Schmidt-Kittler O, Cummins JM, Delong L, Cheong I, Rago C, Huso DL, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Mutant PI3KCA promotes cell growth and invasion of human cancer cells. Cancer Cell. 2005;7:561–573. doi: 10.1016/j.ccr.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Sato N, Meijer L, Skaltsounis L, Greengard P, Brivanlou AH. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat Med. 2004;10:55–63. doi: 10.1038/nm979. [DOI] [PubMed] [Google Scholar]
- Saunders FR, Wallace HM. Polyamine metabolism and cancer prevention. Biochem Soc Trans. 2007;35:364–368. doi: 10.1042/BST0350364. [DOI] [PubMed] [Google Scholar]
- Scheid MP, Woodgett JR. Unravelling the activation mechanisms of protein kinase B/Akt. FEBS Lett. 2003;546:108–112. doi: 10.1016/s0014-5793(03)00562-3. [DOI] [PubMed] [Google Scholar]
- Sebolt-Leopold JS, Herrera R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat Rev Cancer. 2004;4:937–947. doi: 10.1038/nrc1503. [DOI] [PubMed] [Google Scholar]
- Singh AM, Reynolds D, Cliff T, Ohtsuka S, Mattheyses AL, Sun Y, Menendez L, Kulik M, Dalton S. Signaling network crosstalk in human pluripotent cells: A Smad2/3-regulated switch that controls the balance between self-renewal and differentiation. Cell Stem Cell. 2012;10:312–326. doi: 10.1016/j.stem.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- Stambolic V, Woodgett JR. Functional distinctions of protein kinase B/Akt isoforms defined by their influence on cell migration. Trends Cell Biol. 2006;16:461–466. doi: 10.1016/j.tcb.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Stiles B, Gilman V, Khanzenzon N, Lesche R, Li A, Qiao R, Liu X, Wu H. Essential role of AKT-1/protein kinase B alpha in PTEN-controlled tumorigenesis. Mol Cell Biol. 2002;22:3842–3851. doi: 10.1128/MCB.22.11.3842-3851.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm MP, Kumpfmueller B, Thompson B, Kolde R, Vilo J, Hummel O, Schulz H, Welham MJ. Characterization of the phosphoinositide 3-kinase-dependent transcriptome in murine embryonic stem cells: Identification of novel regulators of pluripotency. Stem Cells. 2014;27:764–775. doi: 10.1002/stem.3. [DOI] [PubMed] [Google Scholar]
- Sun H, Lesche R, Li DM, Liliental J, Zhang H, Gao J, Gavrilova N, Mueller B, Liu X, Wu H. PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc Natl Acad Sci. 1999;96:6199–6204. doi: 10.1073/pnas.96.11.6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, de la Pompa JL, Stambolic V, Elia AJ, Sasaki T, del Barco Barrantes, Ho A, Wakeham A, Itie A, Khoo W, Fukumoto M, Mak TW. High cancer susceptibility and embryonic lethality associated with mutation of thePTEN tumor suppressor gene in mice. Curr Biol. 1998;8:1169–1178. doi: 10.1016/s0960-9822(07)00488-5. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Nakano T, Mak TW, Sasaki T. Portrait of PTEN: Messages from mutant mice. Cancer Sci. 2008;99:209–213. doi: 10.1111/j.1349-7006.2007.00670.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Nakagawa M, Young SG, Yamanaka S. Differential membrane localization of ERas and Rheb, two Ras-related proteins involved in the phosphatidylinositol 3-kinase/mTOR pathway. J Biol Chem. 2005;280:32768–32774. doi: 10.1074/jbc.M506280200. [DOI] [PubMed] [Google Scholar]
- Tessier M, Woodgett, JR Serum and glucocorticoid-regulated protein kinases: Variations on a theme. J Cell Biochem. 2006a;98:1391–1407. doi: 10.1002/jcb.20894. [DOI] [PubMed] [Google Scholar]
- Tessier M, Woodgett JR. Role of the Phox homology domain and phosphorylation in activation of serum and glucocorticoid-regulated kinase-3. J Biol Chem. 2006b;281:23978–23989. doi: 10.1074/jbc.M604333200. [DOI] [PubMed] [Google Scholar]
- Tominaga Y, Tamguney T, Kolesnichenko M, Bilanges B, Sokoe D. Translational deregulation in PDK1−/− embryonic stem cells. Mol Cell Biol. 2005;25:8465–8475. doi: 10.1128/MCB.25.19.8465-8475.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DA, Trott J, Hayward P, Rué P, Martinez-Arias A. An interplay between extracellular signalling and the dynamics of the exit from pluripotency drives cell fate decisions in mouse ES cells. BioRxiv. 2014 doi: 10.1242/bio.20148409. DOI: 10.1101/000653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallier L, Alexander M, Pedersen RA. Activin/Nodal and FGF pathways cooperate to maintain pluripotency of human embryonic stem cells. J Cell Sci. 2005;118:4495–4509. doi: 10.1242/jcs.02553. [DOI] [PubMed] [Google Scholar]
- Voskas D, Ling LS, Woodgett JR. Does GSK-3 provide a shortcut for PI3K activation of Wnt signaling? F1000. Biol Rep. 2010;2:82. doi: 10.3410/B2-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Umehara H, Murayama K, Okabe M, Kimura T, Nakano T. Activation of Akt signaling is sufficient to maintain pluripotency in mouse and primate embryonic stem cells. Oncogene. 2006;25:2697–2707. doi: 10.1038/sj.onc.1209307. [DOI] [PubMed] [Google Scholar]
- Welham MJ, Kingham E, Bone HK. Phosphoinositide 3-kinases and regulation of embryonic stem cell fate. Biochem Soc Trans. 2007;35:225–228. doi: 10.1042/BST0350225. [DOI] [PubMed] [Google Scholar]
- Welham MJ, Kingham E, Sanchez-Ripoll Y, Kumpfmueller B, Storm M, Bone H. Controlling embryonic stem cell proliferation and pluripotency: The role of PI3K- and GSK-3-dependent signaling. Biochem Soc Trans. 2011;39:674–678. doi: 10.1042/BST0390674. [DOI] [PubMed] [Google Scholar]
- Williams MR, Authur JS, Balendra A, van der Kaay J, Poli V, Cohen P, Alessi DR. The role of 3-phosphoinositide-dependent protein kinase 1 in activating AGC kinases defined in embryonic stem cells. Curr Biol. 2002;10:439–448. doi: 10.1016/s0960-9822(00)00441-3. [DOI] [PubMed] [Google Scholar]
- Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev. 2010;20:87–90. doi: 10.1016/j.gde.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodgett JR. Recent advances in the protein kinase B signaling pathway. Curr Opin Cell Biol. 2005;17:150–157. doi: 10.1016/j.ceb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Wu R, Hendrix-Lucas N, Kuick R, Zhai Y, Schwartz DR, Akyol A, Hanash S, Misek DE, Katabuchi H, Williams BO, Fearon ER, Cho KR. Mouse model of human ovarian endometrioid adenocarcinoma based on somatic defects in the Wnt/beta-catenin and PI3K/Pten signaling pathways. Cancer Cell. 2007;11:321–333. doi: 10.1016/j.ccr.2007.02.016. [DOI] [PubMed] [Google Scholar]
- Wu G, Huang H, Garcia Abreu J, He X. Inhibition of GSK3 phosphorylation of beta-catenin via phosphorylated PPPSPXS motifs of Wnt coreceptor LRP6. PLoS ONE. 2009;4:e4926. doi: 10.1371/journal.pone.0004926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Pan W. GSK3: A multifaceted kinase in Wnt signaling. Trends Biochem Sci. 2010;35:161–168. doi: 10.1016/j.tibs.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying QL, Nichols J, Chambers I, Smith A. BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell. 2003;115:281–292. doi: 10.1016/s0092-8674(03)00847-x. [DOI] [PubMed] [Google Scholar]
- Ying QL, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, Cohen P, Smith A. The ground state of embryonic stem cell self-renewal. Nature. 2008;453:519–523. doi: 10.1038/nature06968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan TL, Cantley LC. PI3K pathway alterations in cancer: Variations on a theme. Oncogene. 2008;27:5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]