

Abstract
Striated muscle contraction is regulated by an interaction network connecting the effects of troponin, Ca2+, and myosin-heads to the azimuthal positioning of tropomyosin along thin filaments. Many missense mutations, located at the actin-tropomyosin interface, however, reset the regulatory switching mechanism either by weakening or strengthening residue-specific interactions, leading to hyper- or hypo-contractile pathologies. Here, we compute energy landscapes for the actin-tropomyosin interface and quantify contributions of single amino acid residues to actin-tropomyosin binding. The method is a useful tool to assess effects of actin and tropomyosin mutations, potentially relating initial stages of myopathy to alterations in thin filament stability and regulation. Landscapes for mutant filaments linked to hyper-contractility provide a simple picture that describes a decrease in actin-tropomyosin interaction energy. Destabilizing the blocked (relaxed)-state parallels previously noted enhanced Ca2+-sensitivity conferred by these mutants. Energy landscapes also identify post-translational modifications that can rescue regulatory imbalances. For example, cardiomyopathy-associated E62Q tropomyosin mutation weakens actin-tropomyosin interaction, but phosphorylation of neighboring S61 rescues the binding-deficit, results confirmed experimentally by in vitro motility assays. Unlike results on hyper-contractility-related mutants, landscapes for tropomyosin mutants tied to hypo-contractility do not present a straightforward picture. These mutations may affect other components of the regulatory network, e.g., troponin-tropomyosin signaling.
Keywords: Actin, myosin, tropomyosin, cardiomyopathy, muscle regulation
Introduction
Muscle contractility depends critically on the role played by the coiled-coil regulatory protein tropomyosin to cooperatively activate and relax the myofibrillar thin filament. In turn, crossbridge cycling by the myosin motor on actin and thus muscle performance is also controlled cooperatively [1–3]. Tropomyosin, the lynchpin in this on-off switching process, relies on Ca2+ binding to troponin and myosin binding to actin to define its azimuthal position on successive actin subunits along the thin filament. In this way, tropomyosin sterically blocks or alternatively exposes myosin-binding sites, and thereby controls myosin motor activity and contractility [1–8]. End-to-end linked tropomyosin molecules form a continuous cable that hovers about 10 Å over the helical array of actin-subunits comprising the thin filament [8–12]. Since the cable is semi-rigid, local azimuthal movement at one site evoked by troponin or myosin is propagated along actin filaments, thus amplifying cooperative switching signals [13].
The axial position of tropomyosin along the actin filament is defined by residue-specific electrostatic interactions between amino acids within the “α-zones” of tropomyosin pseudo-repeating domains (seven each in cardiac and skeletal muscle) and complementary targets on successive actin subunits [3,10–12]. The large radial separation between actin and tropomyosin ensures that these interactions, while residue-specific, are relatively weak and thus compatible with tropomyosin’s azimuthal movements on actin at low energy cost. Indeed, tropomyosin only binds to F-actin effectively by linking head-to-tail along F-actin to form a continuous cable with high collective binding strength [9,12]. Hence, locally tropomyosin affinity is low, while globally it is high.
The actin-tropomyosin assembly has been examined in atomic detail by Li et al. [10] who built a structural model of the troponin-free filament by selecting the position of tropomyosin on the F-actin surface that optimized electrostatic interactions. The near equivalence between the coordinates of this structure and the density profile of actin and tropomyosin in EM reconstructions validated the model [10]. Orzechowski et al. [11] then extended this work by mapping energy landscapes to determine interaction energy terms for tropomyosin located at different positions on F-actin that covered the entire surface of actin explored by tropomyosin. The lowest energy position of tropomyosin on actin was found to localize in a relatively shallow energy basin and, as in Li et al. [10], very close to tropomyosin’s low-Ca2+ blocking-site on troponin-regulated thin filaments. This arrangement was corroborated by Spudich and colleagues using other models of the filament [14]. It follows that the role of Ca2+-free troponin during muscle regulation is to decrease the azimuthal dynamics of tropomyosin and to fix tropomyosin at its energetic minimum on actin, blocking access of myosin to actin. In contrast, Ca2+-saturated troponin and then myosin-binding itself function to distort the energy landscape and hence bring about tropomyosin movement to the “open” M-state position and thin filament activation [15, 16]. Thus, mutations in thin filament proteins, particularly ones that strengthen or others that weaken interfacial electrostatic contacts between actin and tropomyosin, will deform the filament energy landscape and thus bias tropomyosin toward or away from the filament blocked-state. Indeed, actin mutants D292V and K326N have been shown to strongly alter the actin-tropomyosin landscape profile and interfere with thin filament regulation [17].
It is well known that point mutations found on practically all myofibrillar proteins profoundly affect cardiac and skeletal muscle contractility and lead to cardiomyopathies and skeletal muscle disease syndromes of varying severity. Over 30 of these mutations localize to residues present on the actin – tropomyosin interface and, as in the case of the actin mutants already mentioned, many may modify the actin-tropomyosin energy landscape, influence tropomyosin positioning on actin, and perturb cooperative interactions between actin, tropomyosin, troponin and myosin [17–23]. In order to investigate these possibilities further, the net energetic contributions made by each wild-type actin and tropomyosin residue to the interaction was first measured. Residues contributing most to actin-tropomyosin interaction were highlighted (Fig. 1) and then compared to the residues in missense mutants associated with myopathies [reviewed in references 18–23]. Energy landscapes were then generated for each mutant tropomyosin on F-actin.
Figure 1.

Striated muscle α-tropomyosin sequence annotation. Figure adapted from Brown et al. [3] to emphasize tropomyosin heptad repeats (a,b,c,d,e,f,g) with the beginning residue of each repeat numbered. The so-called α-zone residues of tropomyosin [3] that locate close to actin sub-domain 1 [10] are shaded. Acidic and basic residues are indicated by black type, while others are in light blue. Residues noted in the present study that form strong electrostatic interactions with oppositely charge ones on actin are circled in black (cf. [10]), and ones associated with hyper- or hypo-contractile characteristics colored green or red respectively [cf. 17,18].
The information acquired in this study may prove to be useful as a diagnostic and/or predictive tool to assess effects of actin and tropomyosin mutations by relating the critical initial stages of disease development to alterations in thin filament stability and regulation. We find that the landscapes for mutant filaments associated with hyper-contractility, for example those linked to hypertrophic cardiomyopathy (HCM), skeletal muscle arthrogryposis and congenital fiber-type disproportion (CFTD), provide a simple picture. In these cases, most of the mutations examined are associated with a decrease in actin-tropomyosin interaction energy that will tend to destabilize the blocked (relaxed)-state of the thin filament. Our measurements parallel previously noted enhanced Ca2+-sensitivity conferred by these mutants [17–23]. We show, in addition, that energy landscape computation in combination with known actin-tropomyosin sequence and structural information can be used prospectively to identify potential effects of post-translational modifications to rescue regulatory imbalances. For instance, our in silico methodology shows that HCM-associated E62Q tropomyosin mutation weakens actin-tropomyosin interaction, but that phosphorylation of neighboring S61 rescues the binding deficit, a result that we then validate experimentally. In marked contrast to our work on regulatory imbalances that are expected to contribute to hyper-contractility, we find that landscapes for tropomyosin mutants tied to less well-characterized cases of hypo-contractility, associated for example with dilated cardiomyopathy (DCM), do not present a straightforward picture. It is not clear whether or not these mutations alter actin-tropomyosin interactions or affect other components of the regulatory network, such as troponin-tropomyosin signaling and/or actin-myosin association. Thus, mutation-induced distortions in energy landscape contours appear to directly prefigure HCM and skeletal muscle hyper-contractility, but not the DCM phenotype. We discuss this apparent inconsistency.
Materials and Methods
Computation of electrostatic interactions between actin and tropomyosin
Coulombic interactions were computed between actin subunits and individual tropomyosin residues to generate electrostatic energy landscapes as described by Orzechowski et al. [11]. This method was previously developed to determine landscapes for the entire tropomyosin molecule with F-actin during its transition between the blocking and open regulatory positions of tropomyosin on thin filaments.
Initial structure formulation
The recently proposed all-atom model of the tropomyosin cable on F-actin [11,12] was used as a starting model. The model consists of two tandem head-to-tail tropomyosin molecules bound to a 34 subunit long F-actin filament and poised at a 39 to 40 Å radius. Tropomyosin pseudo-rotation and side chain orientations were taken from Li et al. [10]. The molecule wraps around the F-actin helix matching the F-actin superhelical twist [10, 24] and is linked end-to-end by the recently described head-to-tail conformation [12, 25]. The overall actin-tropomyosin structure is consistent with EM, NMR and crystallographic information and was used as a starting reference point in grid searches.
Grid search protocol
A grid search approach was used to determine Coulombic interactions of individual residues over a comprehensive series of positions of tropomyosin on F-actin. Tropomyosin was treated as a rigid body and shifted from its lowest energy position as in Orzechowski et al. [11] across the F-actin interface in 2° increments and in the longitudinal direction along the axis of the F-actin in 2 Å steps. The total displacement of tropomyosin on actin covered a 42 Å by 30° grid over all points between the blocked and open positions [11].
Energy minimization
Actin-tropomyosin structures were energy minimized at every grid point position as previously [11]. After the minimization, non-bonded interactions were evaluated using the distance-dependent dielectric constant documented in CHARMM version c35b2 [26].
Actin-tropomyosin mutant models
Point mutations in either cardiac or skeletal muscle tropomyosin that have been characterized by charge reversal or neutralization were chosen for study [17,18]. All-atom actin-tropomyosin models were constructed by replacing wild-type residues with mutated ones; thus constructing 20 new models each with one or another single amino acid substitution. The following tropomyosin mutations were studied: E40K, E41K, E54K, D58H, E62Q, K70T, D84N, R91G, E117K, E138K, R160H, R167H, K168E, D175N, L185R, E180G, E192K, D230N, R244G, K248E. Limiting our analysis to point mutations located between residues 40 and 248 was conducive to relatively rapid energy minimization since a relatively small system could be built for a single tropomyosin molecule truncated at its ends by 20 residues and an F-actin model consisting of 20 actin monomers. In this way, Coulombic interaction energy landscapes were determined, and energy differences between the wild-type and mutants calculated for any region of the grid. Models of tropomyosin containing phosphorylated serine 61 were generated using the CHARMM SP32 patch.
Protein preparation for motility assays
DNA construction and protein purification
Human striated α-tropomyosin cDNA, containing the code for the N-terminal extension Met-Ala-Ser in the vector pGOV4 (Gene Oracle, Mountain View, CA), was excised using Nco I and BamH I and inserted into pET-3d for further manipulation and expression. Primers (shown below with mutated codons in bold) were designed using NEBaseChanger (New England BioLabs, Ipswich, MA) and PCR-based mutagenesis reaction performed using Q5® Site-Directed Mutagenesis Kit (New England BioLabs, Ipswich, MA). E62Q mutant tropomyosin was generated using the primer pair: 5′ GGCACTGAAAGATGCCCAAGAG3′ (FORWARD PRIMER) and 5′ TGGCTATACTTGTCCAGTTCATCTTC3′ (REVERSE PRIMER). Similarly the S61D and S61D/E62Q mutant tropomyosin constructs were generated with the following primer pairs respectively 5′ TGAGGCACTGAAAGATGCCCAAG3′ (FORWARD PRIMER) and 5′ TCATACTTCTCCAGTTCATCTTCTG3′ (REVERSE PRIMER); 5′ TCAGGCACTGAAAGATGCCCAAG 3′ (FORWARD PRIMER) and 5′ TCATACTTCTCCAGTTCATCTTCTG3′ (REVERSE PRIMER).
The desired codon mutations were confirmed via sequencing and the mutant tropomyosin cDNA’s were transformed into Rosetta cells for expression. Purification of bacterially expressed tropomyosin was performed as described in Coulton et al. [27]. where the final purification column was substituted with a HiTrap Q HP column (GE Healthcare, Piscataway, NJ) and a salt gradient from 100 mM KCl to 900 mM KCl used.
Tissue purified porcine skeletal muscle myosin was prepared according to Margossian and Lowey [28]. Tissue porcine cardiac troponin complex was prepared as in Potter [29].
Reconstituted thin filaments
Thin filaments were reconstituted by incubating TRITC labeled actin (1 μM) with 0.6 μM tropomyosin (wild-type or mutant) overnight at 40°C. Troponin was then added to the actin-tropomyosin complex and the mixture incubated for at least one hour. Reconstituted thin filaments were stored at 4°C and used within five days of preparation.
Motility assays
Solutions
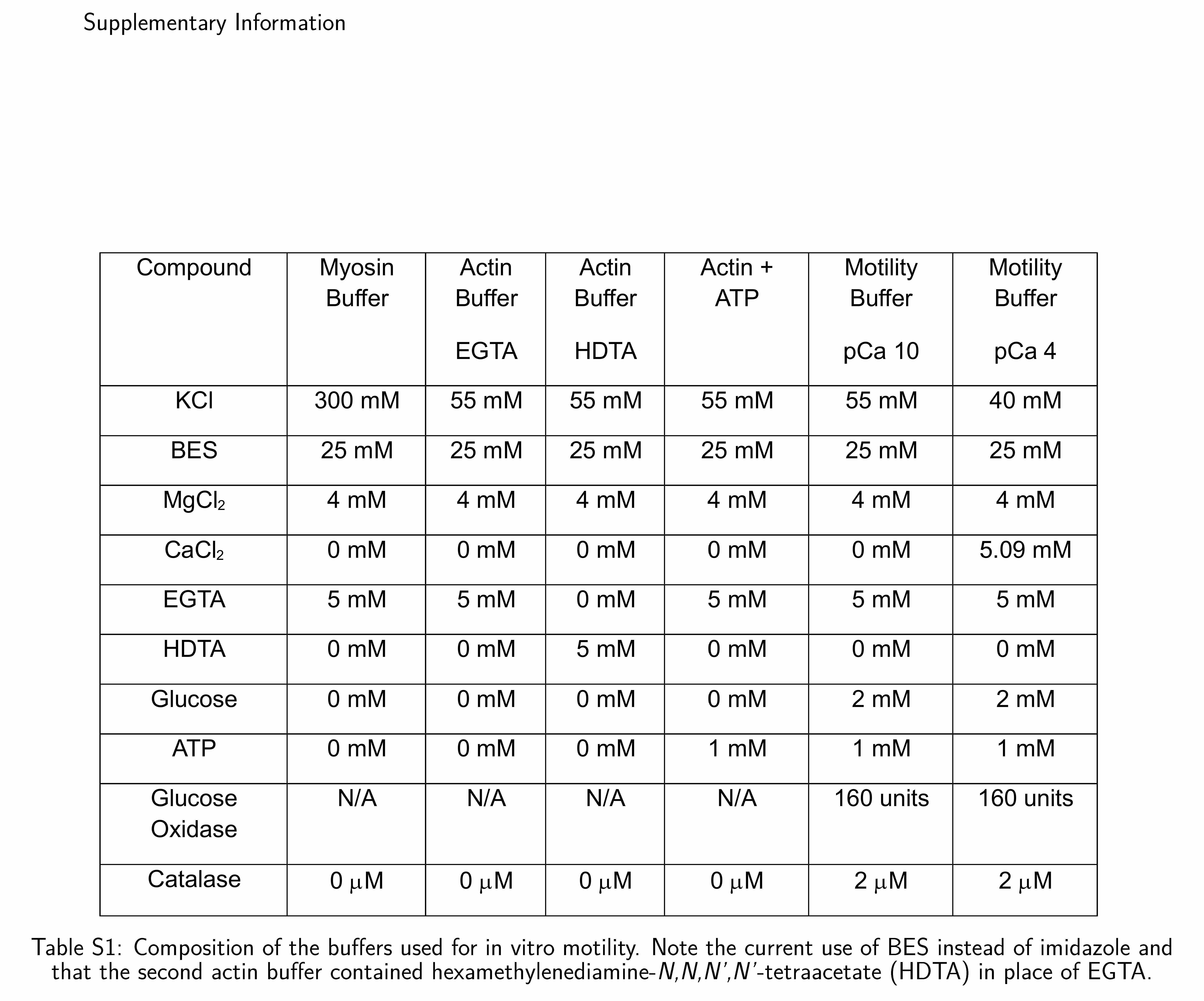
All solutions used for in vitro motility assays were prepared as described previously [30,31] with some modifications (see Table S1 in the Supplementary Information). Imidazole was replaced by BES buffer, and the actin solution contained HDTA. HDTA, a weak calcium binding agent, was used instead of EGTA to ensure that Ca2+ concentration was constant following addition of motility buffer to the flow cell for measuring motility.
Calcium regulated motility
The velocity of reconstituted thin filaments propelled by porcine skeletal muscle myosin was performed as described previously [30] with some minor changes. Reconstituted thin filaments were first diluted to 10 nM in actin buffer containing HDTA (Table S1). The filaments were incubated with myosin for 3 minutes followed by a 3 minute incubation with extra 300 nM troponin and tropomyosin (to prevent dissociation of bound tropomyosin at low filament concentrations assayed in the flow cell). In vitro motility was initiated by the addition of motility buffer containing 1 mM ATP at Ca2+ concentrations ranging from pCa 10 to 4. In vitro motility was followed as previously [30,31]. Significance of data points in pCa/velocity plots was determined by F-test analysis using the program Prism 6 (GraphPad 7825, La Jolla, CA 92037 USA), taking p-values less than 0.05 as significant.
Data Acquisition and Analysis
Movies of filament motility were acquired at five frames per second on an intensified CCD camera (IC300; PTI, Birmingham, NJ, USA) using software (XCAP) and a PCIe card (PIXCI SV7) purchased from EPIX, Inc. (381 Lexington Drive Buffalo Grove, IL 60089). They were analyzed with an ImageJ plug-in, wrMTRCK (written by and available from J. S. Pedersen (jsp@phage.dk)). The plug-in to ImageJ is a centroid-tracking program that automatically locates, tracks and reports the velocity of objects in the movie. Moving filaments in each movie were then averaged to give a single value. 25 to 30 such movies were made for the non-phosphomimetic tropomyosins and 15 to 20 movies for phosphomimetic tropomyosins, where 75 to 90 filaments were studied in each to obtain reliable values at higher pCa values.
Results and Discussion
Dissecting the Actin-Tropomyosin Energy Landscape
Energy contributions of individual tropomyosin residues
The energetics of tropomyosin movement over the surface of F-actin was previously explored by determining Coulombic interaction energy terms for full-length tropomyosin and F-actin. To accomplish this, tropomyosin was repositioned computationally between blocked- and open-state locations on actin to sample potential interactions. Hence, these energy landscapes measured the composite electrostatic contributions from all seven tropomyosin pseudo-repeats, each interacting with a different actin subunit along F-actin (Fig. 2a, cf. reference [11]). However, since amino acid sequences of the seven pseudo-repeats are not identical, the contribution of each component may vary. Therefore in the previous study [11], the landscape was dissected by focusing on energetic input from each pseudo-repeat to individual actin subunits. In the current study, we have now carried this analysis further, by determining interaction terms of every tropomyosin amino acid residue over all possible targets on actin. 284 landscapes were generated, i.e. one for each tropomyosin residue. A match between the position of the energy minima in the new residue-specific landscapes and the minimum in the previously determined composite map thus identifies tropomyosin residues that contribute to the overall attractive interactions (Figs. 1, 2b, Fig. S1 in the Supplementary Material) (and the sum of these interactions produces to the composite map in Figure 2a). Conversely, residue-specific landscapes lacking such correlation label amino acids that make little or no contribution to actin-tropomyosin interaction.
Figure 2.

Energy landscapes showing contributions of individual tropomyosin and actin residues to the collective electrostatic interaction of the two proteins on thin filaments, with the energy levels (in kcal/mol) shown by color scales. For each point measured, tropomyosin was repositioned longitudinally and azimuthally over actin (see Materials and Methods) and the respective Coulombic energy determined. The [0, 0] position corresponds to the minimum energy position occupied by full-length tropomyosin on F-actin as described by Li et al. [10] and the point indicated by the x marks a minor adjustment in the location of this position based on a refinement by Orzechowski et al. [11]. The energy landscape for the entire tropomyosin molecule moved over actin is shown in (a) (i.e. accounting for the combined energies of all interacting residues), and then this landscape was decomposed to show contributions derived from single tropomyosin (b) and single actin (c) residues, here showing landscapes only for those tropomyosin and actin residues that individually contribute to strong (> 10 kcal/mol) Coulombic interactions (residues numbered), whereas, in Figures S1 and S2, landscapes for all residues, regardless of interaction strength are shown. Note that the color scales differ in (a), (b) and (c).
It is not surprising that residues with charged side chains (Arg, Lys, Asp, Glu) which interact strongly with actin are localized mostly to tropomyosin α-zones, that is to repeating regions of tropomyosin in contact with actin subdomain 1 [3, 10]. The tropomyosin residues identified by this procedure (Fig. 1), in fact, match up well to those suggested previously by EM reconstruction to make close contacts with oppositely charged residues on actin (Fig. 2b, cf. Li et al. [11]). Hence, mutations causing charge reversal or neutralization of these or immediately adjacent residues should change the local binding affinity of tropomyosin for actin at positions close to or at the blocked state.
Energy contributions of individual actin residues
Contributions made by individual actin residues to the composite landscape were also assessed by the above method, here measuring interaction terms of every actin amino acid over all possible targets on the surface of tropomyosin. A cluster of positively charged actin residues (Arg147, Lys326 and Lys328) project from the base of subdomain 1 on actin [10, 16] and these make large individual contributions to actin-tropomyosin interactions detected in the energy landscape (Figs. 2c, S2). The contributions made by Lys326 and Lys328 are particularly favorable. These two residues are well-separated from other charged residues on actin that might obscure Coulombic interactions required for tropomyosin binding. Neighboring Arg147 sits within 8 Å of oppositely charged residues Glu167 and Asp292 on actin which may diminish the overall attractive interaction between Arg147 and tropomyosin. Residues Asp25 and Lys28, located at the top of actin subdomain 1, also contribute to the interaction but in a complex way, since they not only interact with residues on tropomyosin, but with each other and with surrounding charged residues on actin. Again, contacts made by actin residues with tropomyosin outlined here mirror those suggested by EM reconstruction [10], reemphasizing that mutations in tropomyosin interfering with these links will be deleterious.
Mapping Mutations to the Actin-Tropomyosin Energy Landscape
As mentioned, analysis of the contributions of single amino acid residues to the overall energy landscape highlights residues that contribute most to the composite binding energetics required for thin filament assembly (Figs. 1, 2, S1). It is striking that many point mutations on tropomyosin leading to disease outcomes map to these or closely neighboring residues, and may act by altering electrostatic interactions that determine actin-tropomyosin regulatory state balance (Fig. 1). Indeed, mutations leading to hyper-contractility show widespread distribution within the tropomyosin α-zones required for actin-binding (13 of 20), whereas those associated with hypo-contractility have no obvious pattern (Fig. 1). 24 of 30 charged residues on tropomyosin that interact strongly with actin locate to b and f heptad positions (amino acids facing actin), but only 2 of 20 mutations associated with hypercontractility and 3 of 15 linked to hypocontractility appear in these positions. Thus, mutations that directly affect strongly interacting sites are relatively rarely observed, suggesting that mutations in residues near to the strongly interacting sites have a secondary yet significant electrostatic effect or otherwise perturb regulatory function.
In order to quantify the impact of tropomyosin mutations on actin-tropomyosin interaction, additional energy landscapes were computed for the mutants. The position of the energy minima in control and mutant landscapes were compared and magnitude of the change in interaction energy measured.
Mutations in Tropomyosin Leading to Hyper-contractility
Energy landscapes were first computed for twelve tropomyosin mutants associated with hyper-contractile phenotypes. In each case, a wild-type residue was replaced by a mutant amino acid, and then a new energy landscape was generated by moving the mutant tropomyosin molecule axially and azimuthally over the surface of actin, and the Coulombic interactions were measured at each point, as previously done for control tropomyosin. The location of energy minima in most of the mutant actin-tropomyosin energy landscapes examined is identical to that of the control tropomyosin’s position; in some, the minimum is slightly shifted, but by no greater than by 4° azimuthally and 2 Å axially (data not shown). Thus, none of the single point mutations significantly affected the energetically optimal position of tropomyosin on F-actin. This is not surprising, since the interaction energy contributions from the other 20 or so residues interacting normally on six of the remaining seven pseudo-repeating elements in tropomyosin are unaffected by any one single amino acid substitution in the seventh repeat. While the minima all locate to virtually the same spot in the landscapes (i.e. close to or at the blocked state regulatory position for tropomyosin), the minima invariably are less negative than that displayed by the control (i.e. the interaction becomes energetically less favored after mutation). This effect is most evident by inspection of difference maps made by subtracting landscape maps for the various mutants from the wild-type landscapes, revealing that all the mutants examined destabilize interactions, as indicated by a positive value in the difference plot, close to or at tropomyosin’s blocked state on actin (i.e. at or near the energy minimum site), without generating new local minima of any obvious significance. However, the magnitude of the energy differences between mutants and control values varies greatly and thus the degree to which mutations influence the stability of tropomyosin in the blocked state at the minima must also vary (Figs. 3, 4, S3). Mutants E62Q and K248E cause the largest change in interaction energy (>40 kcal/mol) and each is caused by a charge reversal. It is noteworthy that these residues and oppositely charged ones on actin normally interact with each other electrostatically (the former to actin R147, K326 and K328 and the latter to D25); the corresponding mutations would therefore be expected to diminish these interactions, as would the E58H mutation with R147. Mutations in amino acids 21, 22, 63, 70, 91, 168, 180, and 185 lie immediately next to sites on tropomyosin that make primary electrostatic contacts with actin. Thus all of the above mutants are likely to perturb normal actin-tropomyosin interactions in the blocked state leading to hyper-contractility (Figs. 3, 4, S3). In fact, with the exception of mutation K70T, all of the mutations tested in this group showed less favorable interactions with actin as indicated by an increase in the electrostatic interaction energy when compared to control values, fully consistent with a contribution of these mutants to the development of a hyper-contractile phenotype, while K70T causes a slight decrease (and may affect coiled-coil stability or interactions with TnT and myosin). As a proof-of-concept trial, we also tested a synthetic mutant E138K (Figs. 3, 4, S3). Residue 138 lies next to E139, which is known to bind strongly to oppositely charged residues on actin. As expected, the charge reversal decreases the attractive interaction of tropomyosin for actin, just as for example the well-studied E180G mutation reduces the attraction of E181 for actin.
Figure 3.

Difference maps generated comparing mutant and wild-type landscapes. Landscapes (left column) measuring wild-type interactions between four individual tropomyosin residues and actin, (a) E62, (b) E138, (c) E180, (d) K248 (as Fig. 2). Corresponding landscapes of mutated forms of these residues that result in major imbalances of the electrostatic interaction energy, viz. E62Q, E138K, E180G and K248E (center column). Difference maps generated by subtracting the wild-type landscapes from mutant ones (right column). The positive values in the difference plots surrounding the [0,0] region reveals that these mutants will destabilize actin-tropomyosin interactions in the blocked-state of the thin filament. Note that each of the residues evaluated normally either forms a strong interaction with actin or is immediately adjacent to such a residue, and that mutations at residues 62, 180 and 248 are associated with hyper-contractile phenotypes. Individuals with mutations in residue 138 have not been described.
Figure 4.

Histograms revealing changes in Coulombic binding interactions between tropomyosin and actin due to tropomyosin mutations associated with hyper-contractility. Differences in electrostatic binding interaction surrounding the energy minimum site (averaged over an approximately 16 Å2 region as indicated by white squares in Figure 2) between wild-type and mutant tropomyosin landscapes quantified by subtracting one from the other. The positive values noted indicate a decrease in strength of binding by the mutants.
On the other hand, residues 160, 175, and 192 are also associated with hyper-contractile phenotypes but locate further from consensus binding sites. They therefore would be expected to have a small direct effect on binding, consistent with landscape comparisons, and here the hyper-contractile phenotype evoked may relate to possible effects on troponin and myosin interactions with tropomyosin or to tropomyosin stiffness changes. Mutations in uncharged residues (95, 172, 215, 281) associated with HCM were not evaluated, since they are likely to affect the stability of the hydrophobic tropomyosin core and not the charged actin-tropomyosin interface.
Interchain salt bridges between residues (at heptad positions e and g (Fig. 1)) normally stabilize coiled-coil tropomyosin (3). Hence, mutations associated with charge changes at these sites (e.g. at residues 70, 91, 168, 175, 180) may increase local coiled-coil flexibility, and thus modify tropomyosin position on actin filaments [32, 33]. This could amplify effects noted above that weaken actin-tropomyosin interaction and thereby exacerbate aberrant responses to troponin and myosin.
Rescuing the Effect of Mutations in Tropomyosin Leading to Hyper-contractility
The E62Q tropomyosin mutation causes a comparatively large increase in the interaction energy value between actin and tropomyosin (Figs. 3, 4, 5c), and therefore would be expected to produce a corresponding large weakening of actin – tropomyosin binding in the blocked-state configuration. In contrast, Ser61, the residue lying next to residue 62, is a potential phosphorylation target on tropomyosin [34] and phosphorylation would be expected to increase actin – tropomyosin interaction energy by augmenting the local negative charge, as indicated by energy landscape measurements (Fig. 5b). Moreover, the corresponding landscape of the E62Q coupled to S61-phosphorylation shows that the presence of extra negative charge reverses the effect of the E62Q mutation to yield a landscape close to that of the control (Fig. 5d).
Figure 5.
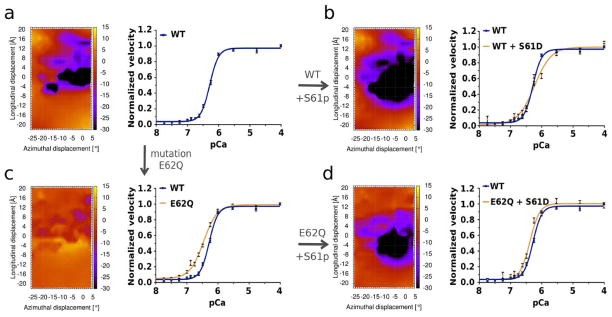
Rescuing the effect of E62Q tropomyosin mutation by phosphorylation. (a) energy landscape for wild-type tropomyosin E62 on actin (as in Figs. 2a, 3a) and corresponding plot of the Ca2+-dependence of wild-type troponin-tropomyosin-based thin filament motility. (b) energy landscape for phosphorylated-S61 tropomyosin and corresponding plot of Ca2+-dependent thin filament motility (here thin filaments composed of F-actin, phosphomimetic S61D tropomyosin and wild-type troponin). Compared to control maps, energy landscapes show an increase in binding strength between the phosphorylated S61 and actin, and the motility assays show a corresponding rightward shift in Ca2+-sensitivity (tan curve) relative to that of the control (blue curve). (c) energy landscape of E62Q tropomyosin on actin (as in Fig. 3a) and corresponding plot of Ca2+-dependent thin filament motility (thin filaments composed of F-actin, E62Q tropomyosin and wild-type troponin). Note the leftward shift in the E62Q related Ca2+-sensitivity (tan curve) relative to that of the control (blue curve). (d) energy landscape for the E62Q mutant tropomyosin containing phosphorylated S61 and corresponding plot of Ca2+-dependent thin filament motility (thin filaments composed of F-actin, double mutant E62Q/S61D tropomyosin and wild-type troponin). Here, energy landscapes and in vitro motility show an almost complete restoration of control relationships (tan and blue curves for double mutant vs. wild-type preparations). Plots of in vitro motility in (a-d) are normalized. Raw data showed that filament velocity for each preparation was maximum at pCa > 6.5, with respective values (in μm/sec) for wild-type, and E62Q, S61D, E62Q/S61D mutants of 2.8 ± 0.04, 2.9 ± 0.04, 2.6* ± 0.03, and 2.5* ± 0.05, and pCa50 values of 6.3 ± 0.01, 6.5* ± 0.02, 6.0* ± 0.01, and 6.4* ± 0.03 (asterisks indicate significant differences from wild-type at p values < 0.05). Hill coefficients (n) were 3.1 ± 0.2, 2.0* ± 0.1, 1.7* ± 0.2, and 2.8 ± 0.2 for the four preparations.
To validate these results experimentally, the myosin-propelled sliding velocity of tropomyosin and troponin-decorated thin filaments was measured as a function of added calcium for thin filaments containing the E62Q, phosphomimetic S61D and E62Q-S61D mutant tropomyosins. In vitro motility assays show that the velocity of thin filaments reconstituted with these expressed mutants increases sigmoidally as a function of Ca2+ - concentration (Fig. 5); however, actin associated with the single point mutant tropomyosin, E62Q, requires lower Ca2+ to fully activate troponin-tropomyosin regulated filaments when compared to activation by wild-type tropomyosin (Fig. 5c)(as expected from earlier steady-state acto-S1 ATPase work [19], and from the destabilization of the actin-tropomyosin interaction noted above). In contrast, the double mutant E62Q-S61D restores Ca2+-sensitivity toward normal while slightly reducing sliding velocity (Fig. 5d). The shift in Ca2+-sensitivity by E62Q and subsequent reversal by S61D parallel the blocked-state phosphorylation-dependent stabilization of tropomyosin indicated by our corresponding landscape calculations above. Likewise, the contrasting decrease in Ca2+-sensitivity produced by the single mutant S61D likely resulted from blocked site stabilization (Fig. 5b). The slight alterations in velocity observed (noted in the legend to Fig. 5) may represent alterations in tropomyosin-myosin interactions [35], which are not part of our energy landscape calculations. Notably, the shifts observed in pCa50 for the mutant tropomyosins could be predicted from the in silico calculation of energy landscapes. Thus, we have demonstrated that our methods can be implemented to predict the outcomes of mutations and to propose post-translational modifications that potentially can rescue regulatory imbalances.
Mutations Leading to Hypo-contractility
The mutations discussed above which are linked to hyper-contractility share a common mechanism involving a local disruption of contacts between actin and tropomyosin and a corresponding weakening of the blocking configuration interactions of tropomyosin on actin, leading presumably to diminished relaxation and enhanced activation by Ca2+ and myosin. Hence, the effect represents a potential “loss” of tropomyosin’s inhibitory function. In contrast, an enhanced tropomyosin inhibition, stabilizing tropomyosin in the off-state, as appears to be the effect of the engineered S61D tropomyosin construct tested above, would represent a “gain” of tropomyosin function. Such a gain of inhibitory function, enhancing the actin-tropomyosin interaction, in turn, would produce loss of contractile function by diminishing the tropomyosin-response to Ca2+-binding to troponin and therefore myosin-binding to actin. DCM in cardiac muscle and diminished tone in skeletal muscle would result. (N.B. The inherently confusing terms “gain” and “loss of function” are mentioned to be consistent with current usage.)
Tropomyosin mutants E54K, R167H, D230N and R244G, linked to the development of DCM and skeletal muscle weakness, represent sites on tropomyosin that interact electrostatically with oppositely charged residues on actin. However, energy landscape measurements show that they do not increase the strength of the interaction as would be expected if the DCM resulted strictly from an imbalance in actin-tropomyosin electrostatic interactions. Contrary to expectation, these mutations in fact diminish such interactions (Fig. 6), as we have observed for mutations causing hyper-contractility. Thus any diminished contractility seen experimentally would not appear to be caused by a simple stabilization of tropomyosin on actin in the blocked-state. Additional tropomyosin mutations, associated with DCM and hypo-contractility in skeletal muscles are found on residues 8, 15, 92, 201, 239, 277 and elsewhere, but these locate far from residues on tropomyosin that interface with actin and most are part of the tropomyosin core (Fig. 1). In contrast, it is possible that side-chains of DCM mutants E40K and E41K may collectively form stabilizing interactions with oppositely charged partners on actin (Fig. 6), yet the separation between these side-chains from potential targets on actin are 9 Å or greater, whereas attractive linkages between actin and tropomyosin side-chains are usually 2 to 7 Å apart. The same considerations also apply to the D84K mutant tested.
Figure 6.

Histograms revealing changes in Coulombic binding interactions between tropomyosin and actin due to tropomyosin mutations associated with hypo-contractility. Differences in electrostatic binding interactions surrounding the energy minimum site (again averaged over an approximately 16 Å2 region) between wild-type and mutant tropomyosin landscapes quantified by subtracting one from the other. Note that both negative and positive differences are observed.
Tropomyosin-Based Hyper-contractile vs. Hypo-contractile Phenotypes
The tropomyosin molecule appears to have evolved to optimize its interactions with actin. Thus a mutation in tropomyosin producing a “gain” of regulatory function by enhancing actin-tropomyosin interaction might be rare or non-existent. Indeed, mutations might more readily corrupt the structure/function relationship of a protein like tropomyosin that has been 500 to 600 million years in the making (as mutations do in HCM) than upgrade it (i.e. to enhance blocked state actin-tropomyosin binding, promoting relaxation and thereby causing DCM). This view seems to be borne out by our own analysis of the tropomyosin mutants that have been linked to the development of DCM and to skeletal muscle hypo-contractility, since these do not seem to obviously enhance actin-tropomyosin interaction, yet still lead to hypo-contractility. Thus, there is no obvious direct relationship between the hypo-contractile phenotype engendered by many tropomyosin mutants and actin-tropomyosin interfacial interactions. This lack of correlation suggests that hypo-contractility has a separate origin and is not directly related to electrostatic interactions between tropomyosin and actin. Most likely, the “loss of tropomyosin function” results from a failure of tropomyosin to bind or respond to troponin or myosin properly. Our analysis suggests strongly that these alternatives should be examined experimentally.
Conclusions
Energy landscapes measuring the electrostatic interactions between tropomyosin and F-actin have been shown to be a useful means of determining residues that contribute to the binding stability of the thin filament, particularly ones that determine the blocked-state tropomyosin position on F-actin. This type of analysis has confirmed the specificity of amino acid side-chain – side-chain associations proposed by Li et al. [10], which are a determinant of actin-tropomyosin interactions. In addition, the expanded methodology presented here has revealed the effect of amino acids surrounding primary electrostatic interaction sites that previously was not obvious.
The current study shows that a weakening in the Coulombic interactions between actin and tropomyosin is a good prospective indicator for the development of hyper-contractile myopathic phenotypes. However, strict correlations between the magnitude of altered interaction energy, the molecular pathway(s) leading to myopathy and the severity of phenotypic outcomes are not obvious. The impact of a particular mutation no doubt depends on the collective effects of multiple interrelated interactions that govern the transitions between regulatory states and the compensations that counter the changed physiology.
Our energy landscape measurements show that tropomyosin is positioned on actin over a shallow energy basin. The arrangement sets up a delicate regulatory balance between myofibrillar activity and relaxation. Hence, minor alterations brought about by mutations can tip this balance slightly in one or another direction, just as experimentally imposed perturbations to manipulate actin-tropomyosin chemistry or conditions applied in vitro that strain actin or the tropomyosin cable can alter the regulatory balance [36, 37]. In any case, the effects of some of the mutations studied here may appear to be modest and can be tolerated in many human subjects for years. Mutations that dramatically affect strong actin-tropomyosin interactions presumably are not well tolerated.
Mutations that decrease the affinity of tropomyosin for actin generally are associated with hyper-contractile phenotypes, and typically these mutations result in a loss of tropomyosin inhibitory function. In a case like actin-tropomyosin, inspection of structure combined with evaluation of energy landscapes can then be used to predict the outcomes of post-translation modification, as done here for the E62Q mutation. Thus, the analytical power of our computational approach supported by in vitro functional characterization can be drawn on to develop strategies to potentially reverse critical early stage cardiomyopathies and corresponding skeletal muscle diseases. Connections between many of the mutations in tropomyosin and hyper-contractile phenotypes are evident in our work; in contrast, the relationship between hypo-contractility and the energetics of actin-tropomyosin interaction are not obvious and corresponding phenotypic connections are obscure. In the latter cases, tropomyosin mutation may affect tropomyosin-troponin or tropomyosin-myosin interactions that impede contractile responses, and/or interfere with muscle energy consumption [38, 39]. These responses may result in a cascade of systemic compensations, which cannot be evaluated simply by single molecule studies examined in the current study. Experiments therefore should be designed to test these under-investigated possibilities.
Supplementary Material
{kind=link}
Highlights.
Interaction energies between mutant tropomyosins and actin were calculated.
Missense mutations reset the switching mechanism controlling muscle contraction.
Mutations distort energy landscapes in ways that explain many phenotypic traits.
Outcomes of mutations can be predicted in silico and validated experimentally.
Post-translational interventions can rescue regulatory imbalances.
Acknowledgments
These studies were supported by NIH grants R37HL036153 (to W.L.) and R01HL077280 (to J.R.M.). The Massachusetts Green High Performance Computing Center provided computational resources.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tobacman LS. Thin filament-mediated regulation of cardiac contraction. Annu Rev Physiol. 1996;58:447–481. doi: 10.1146/annurev.ph.58.030196.002311. [DOI] [PubMed] [Google Scholar]
- 2.Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiol Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 3.Brown JH, Cohen C. Regulation of muscle contraction by tropomyosin and troponin: how structure illuminates function. Adv Protein Chem. 2005;71:121–159. doi: 10.1016/S0065-3233(04)71004-9. [DOI] [PubMed] [Google Scholar]
- 4.Haselgrove JC. X-ray evidence for a conformational change in actin-containing filaments of vertebrate striated muscle. Cold Spring Harbor Symp Quant Biol. 1972;37:341–352. [Google Scholar]
- 5.Huxley HE. Structural changes in actin and myosin-containing filaments during contraction. Cold Spring Harbor Symp Quant Biol. 1972;37:361–376. [Google Scholar]
- 6.Parry DAD, Squire JM. Structural role of tropomyosin in muscle regulation: analysis of the X-ray diffraction patterns from relaxed and contracting muscles. J Mol Biol. 1973;75:33–55. doi: 10.1016/0022-2836(73)90527-5. [DOI] [PubMed] [Google Scholar]
- 7.Lehman W, Craig R, Vibert P. Ca2+-induced tropomyosin movement in Limulus thin filaments revealed by three-dimensional reconstruction. Nature. 1994;368:65–67. doi: 10.1038/368065a0. [DOI] [PubMed] [Google Scholar]
- 8.Poole KJ, Lorenz M, Evans G, Rosenbaum G, Pirani A, Tobacman LS, Lehman W, Holmes KC. A comparison of muscle thin filament models obtained from electron microscopy reconstructions and low-angle X-ray fibre diagrams from non-overlap muscle. J Struct Biol. 2006;155:273–284. doi: 10.1016/j.jsb.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 9.Hitchcock-DeGregori SE. Tropomyosin: Function follows form. Adv Exp Med Biol. 2008;644:60–72. doi: 10.1007/978-0-387-85766-4_5. [DOI] [PubMed] [Google Scholar]
- 10.Li XE, Tobacman LS, Mun JY, Craig R, Fischer S, Lehman W. Tropomyosin position on F-actin revealed by EM reconstruction and computational chemistry. Biophys J. 2011;100:1005–1013. doi: 10.1016/j.bpj.2010.12.3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orzechowski M, Moore JR, Fischer S, Lehman W. Tropomyosin movement on F-actin during muscle activation explained by energy landscapes. Arch Biochem Biophys. 2014;545:63–68. doi: 10.1016/j.abb.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Orzechowski M, Li XE, Fischer S, Lehman W. An atomic model of the tropomyosin cable on F-actin. Biophys J. 2014;107:694–699. doi: 10.1016/j.bpj.2014.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li XE, Holmes KC, Lehman W, Jung H, Fischer S. The shape and flexibility of tropomyosin coiled coils: implications for actin filament assembly and regulation. J Mol Biol. 2010;395:327–339. doi: 10.1016/j.jmb.2009.10.060. [DOI] [PubMed] [Google Scholar]
- 14.Sunitha SM, Mercer JA, Spudich JA, Sowdhamini R. Integrative structural modelling of the cardiac thin filament: energetics at the interface and conservation patterns reveal a spotlight on period 2 of tropomyosin. Bioinform Biol Insights. 2012;6:203–223. doi: 10.4137/BBI.S9798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lehman W, Galińska-Rakoczy A, Hatch V, Tobacman LS, Craig R. Structural basis for the activation of muscle contraction by troponin and tropomyosin. J Mol Biol. 2009;388:673–681. doi: 10.1016/j.jmb.2009.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lehman W, Orzechowski M, Li XE, Fischer S, Raunser S. Gestalt-binding of tropomyosin on actin during thin filament activation. J Muscle Res Cell Motility. 2013;34:155–163. doi: 10.1007/s10974-013-9342-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marston S, Memo M, Messer MA, Papadaki M, Nowak K, McNamara E, Ong R, El-Mezgueldi M, Li X, Lehman W. Mutations in repeating structural motifs of tropomyosin cause gain of function in skeletal muscle myopathy patients. Hum Mol Genet. 2013;22:4978–4987. doi: 10.1093/hmg/ddt345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Redwood C, Robinson CP. Alpha-tropomyosin mutations in inherited cardiomyopathies. J Muscle Res Cell Motility. 2013;34:285–294. doi: 10.1007/s10974-013-9358-5. [DOI] [PubMed] [Google Scholar]
- 19.Chang AN, Harada K, Ackerman MJ, Potter JD. Functional consequences of hypertrophic and dilated cardiomyopathy-causing mutations in alpha-tropomyosin. J Biol Chem. 2005;280:34343–34349. doi: 10.1074/jbc.M505014200. [DOI] [PubMed] [Google Scholar]
- 20.Clarke NF, Waddell LB, Sie LTL, van Bon BWN, Mclean C, Kornberg A, Lammens M, North KN. Mutations in TPM2 and congenital fibre type disproportion. Neuromuscul Disord. 2012;22:955–958. doi: 10.1016/j.nmd.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 21.Ochala J, Gokhin DS, Pénisson-Besnier I, Quijano-Roy S, Monnier N, Lunardi J, Romero NB, Fowler VM. Congenital myopathy-causing tropomyosin mutations induce thin filament dysfunction via distinct physiological mechanisms. Hum Mol Genet. 2012;21:4473–4485. doi: 10.1093/hmg/dds289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bai F, Wang L, Kawai M. A study of tropomyosin’s role in cardiac function and disease using thin-filament reconstituted myocardium. J Muscle Res Cell Motil. 2013;34:295–310. doi: 10.1007/s10974-013-9343-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Memo M, Marston S. Skeletal muscle myopathy mutations at the actin tropomyosin interface that cause gain- or loss-of-function. J Muscle Res Cell Motil. 2013;34:165–169. doi: 10.1007/s10974-013-9344-y. [DOI] [PubMed] [Google Scholar]
- 24.Oda T, Iwasa M, Aihara T, Maéda Y, Narita A. The nature of the globular- to fibrous-actin transition. Nature. 2009;457:441–445. doi: 10.1038/nature07685. [DOI] [PubMed] [Google Scholar]
- 25.Li XE, Orzechowski M, Lehman W, Fischer S. Structure and flexibility of the tropomyosin overlap junction. Biochem Biophys Res Commun. 2014;446:304–308. doi: 10.1016/j.bbrc.2014.02.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brooks BR, Brooks CL, MacKerell AD, Nilsson L, Petrella RJ, Roux B, et al. CHARMM: The biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coulton AT, Koka K, Lehrer SS, Geeves MA. Role of the head-to-tail overlap region in smooth and skeletal muscle beta-tropomyosin. Biochemistry. 2008;47:388–397. doi: 10.1021/bi701144g. [DOI] [PubMed] [Google Scholar]
- 28.Margossian S, Lowey S. Preparation of myosin and its subfragments from rabbit skeletal muscle. Methods Enzymol. 1982;85(Part B):55–71. doi: 10.1016/0076-6879(82)85009-x. [DOI] [PubMed] [Google Scholar]
- 29.Potter JD. Preparation of troponin and its subunits. Methods Enzymol. 1982;85(Part B):241–263. doi: 10.1016/0076-6879(82)85024-6. [DOI] [PubMed] [Google Scholar]
- 30.Greenberg MJ, Mealy TR, Watt JD, Jones M, Szczesna-Cordary D, Moore DJR. The molecular effects of skeletal muscle myosin regulatory light chain phosphorylation. Am J Physiol Regul Integr Comp Physiol. 2009;297:R265–274. doi: 10.1152/ajpregu.00171.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greenberg MJ, Kazmierczak K, Szczesna-Cordary D, Moore DJR. Cardiomyopathy-linked myosin regulatory light chain mutations disrupt myosin strain-dependent biochemistry. Proc Natl Acad Sci U S A. 2010;107:17403–17408. doi: 10.1073/pnas.1009619107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li XE, Suphamungmee W, Janco M, Geeves MA, Marston SB, Fischer S, Lehman W. The flexibility of two tropomyosin mutants, D175N and E180G, that cause hypertrophic cardiomyopathy. Biochem Biophys Res Commun. 2012;424:493–496. doi: 10.1016/j.bbrc.2012.06.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loong CK, Zhou HX, Chase PB. Familial hypertrophic cardiomyopathy related E180G mutation increases flexibility of human cardiac α-tropomyosin. FEBS Lett. 2012;586:3503–3507. doi: 10.1016/j.febslet.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Naga Prasad SV, Jayatilleke A, Madamanchi A, Rockman HA. Protein kinase activity of phosphoinositide 3-kinase regulates beta-adrenergic receptor endocytosis. Nature Cell Biol. 2005;7:785–796. doi: 10.1038/ncb1278. [DOI] [PubMed] [Google Scholar]
- 35.Behrmann E, Müller M, Penczek PA, Mannherz HG, Manstein DJ, Raunser S. Structure of the rigor actin-tropomyosin-myosin complex. Cell. 2012;150:327–338. doi: 10.1016/j.cell.2012.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lehman W, Hatch V, Korman V, Rosol M, Thomas L, Maytum R, Geeves MA, Van Eyk JE, Tobacman LS, Craig R. Tropomyosin and actin isoforms modulate the localization of tropomyosin strands on actin filaments. J Mol Biol. 2000;302:593–606. doi: 10.1006/jmbi.2000.4080. [DOI] [PubMed] [Google Scholar]
- 37.Sousa DR, Stagg SM, Stroupe ME. Cryo-EM structures of the actin:tropomyosin filament reveal the mechanism for the transition from C- to M-state. J Mol Biol. 2013;425:4544–4555. doi: 10.1016/j.jmb.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moore JR, Leinwand L, Warshaw DM. Understanding cardiomyopathy phenotypes based on the functional impact of mutations in the myosin motor. Circ Res. 2012;111:375–385. doi: 10.1161/CIRCRESAHA.110.223842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spudich JA. Hypertrophic and dilated cardiomyopathy: four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys J. 2014;106:1236–1249. doi: 10.1016/j.bpj.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.