

Abstract
Evidence linking prostatitis and prostate cancer development is contradictory. To study this link, the POET3 mouse, an inducible model of prostatitis, was crossed with a Pten-loss model of prostate cancer (Pten+/−) containing the ROSA26 luciferase allele to monitor prostate size. Prostatitis was induced, and prostate bioluminescence was tracked over 12 months, with lesion development, inflammation, and cytokine expression analyzed at 4, 8, and 12 months and compared with mice without induction of prostatitis. Acute prostatitis led to more proliferative epithelium and enhanced bioluminescence. However, 4 months after initiation of prostatitis, mice with induced inflammation had lower grade pre-neoplastic lesions. A trend existed toward greater development of carcinoma 12 months after induction of inflammation, including one of two mice with carcinoma developing perineural invasion. Two of 18 mice at the later time points developed lesions with similarities to proliferative inflammatory atrophy, including one mouse with associated carcinoma. Pten+/− mice developed spontaneous inflammation, and prostatitis was similar among groups of mice at 8 and 12 months. Analyzed as one cohort, lesion number and grade were positively correlated with prostatitis. Specifically, amounts of CD11b+Gr1+ cells were correlated with lesion development. These results support the hypothesis that myeloid-based inflammation is associated with lesion development in the murine prostate, and previous bouts of CD8-driven prostatitis may promote invasion in the Pten+/− model of cancer.
The importance of the immune system and chronic inflammation in the pathogenesis of cancer development has been recognized by recent inclusion of inflammation as an enabling characteristic of cancer formation.1 Inflammation has been shown to promote tumor formation in multiple tissues, with some of the most notable examples in the gastrointestinal tract, including reflux esophagitis and esophageal cancer, Helicobacter-associated gastritis and gastric cancer, and inflammatory bowel disease and colorectal cancer.2–4
Both prostate cancer and prostatitis are common diseases in American men; prostate cancer is the most common malignancy in American men, and the prevalence of prostatitis is as high as 9% by age 79 years according to one Minnesota-based study.5 Given the high prevalence of both diseases and the association between chronic inflammation and cancer, a causal relation between the two diseases seems plausible. However, current epidemiologic and clinical evidence about the relation between inflammation and carcinogenesis in the prostate is contradictory.
Within the past year, a meta-analysis including 20 relatively recent case-control and cohort studies examining the link between prostatitis and prostate cancer found a positive association between the presence of inflammation and prostate cancer development.6 In addition in 2013, a study of prostatitis and benign prostatic hyperplasia found a relevant association between prostatitis and prostate cancer and between benign prostatic hyperplasia and cancer development in Asian men, a population with lower overall incidence of prostate cancer.7 Concurrently, two biopsy-based studies were published that identified and scored both acute and chronic inflammation in prostatic biopsies that were negative for carcinoma on initial screening. Both studies, one performed in Finland, the other in the United States, found that the presence of acute or chronic inflammation on the initial biopsy was negatively associated with development of prostate cancer in follow-up biopsies.8,9 Interestingly, in both studies the presence of acute prostatitis on the initial screening resulted in a lower cumulative risk for development of prostate cancer in subsequent screenings.
Many epidemiologic studies about anti-inflammatory treatment and prostate cancer risk have also been conducted, with similarly conflicting results. In several studies, taking aspirin is associated with decreased prostate cancer risk, whereas other nonsteroidal anti-inflammatories either increase prostate cancer risk or have no effect.10,11 Other studies show that aspirin treatment has no protective effect on prostate cancer development, and other nonsteroidal anti-inflammatories have only a modest effect.12 History of sexually transmitted disease has also been variably associated with prostate cancer development. In results from a recent prospective study of men's health in California, the presence of prostatitis was positively associated with prostate cancer development, but previous infection with a sexually transmitted disease increased the risk of prostate cancer development only in certain ethnic groups, a finding at odds with previous studies of sexually transmitted diseases and prostate cancer risk.13 Taken together, the published epidemiologic and clinical data linking prostatitis and cancer development are mixed, with strong recent evidence to suggest that the presence of histologically confirmed prostatitis is protective for prostate cancer development.
Other approaches have also been used to examine this potential link, including in vitro studies using prostate cancer cell lines and treatment with inflammatory cytokines and in vivo studies using animal models. As one example of the in vitro evidence linking prostatitis and cancer development, studies examining the effects of IL-6 on prostate cancer cell line growth confer a growth advantage and enhanced vascular endothelial growth factor production to chronically treated LNCaP cells.14,15 Animal models of prostatitis have been developed, with a few used to investigate inflammation's effect on carcinogenesis in vivo. With the use of uropathogenic Escherichia coli infection of the prostate as a model of prostate inflammation, previously infected murine prostates showed enhanced hyperplasia and dysplasia with higher degrees of oxidative damage to epithelial cell DNA, suggesting inflammation can lead to the early steps of cellular transformation.16 In another mouse model, inhibitor of NF-κB (IκB) kinase 2 was constitutively activated in the prostate of mice with prostate-specific deletion of the tumor suppressor Pten. IκB kinase 2 activation mimics chronic inflammation through downstream activation of NF-κB, a master transcription factor for several proinflammatory pathways. Here, mice with both activation of IκB kinase 2 and loss of one allele of Pten had larger epithelial lesions and increased fibrous stroma in the prostate compared with mice with loss of Pten only but no development of invasive lesions.17 Thus, pieces of in vitro and in vivo evidence suggest a positive association between inflammation and prostate carcinogenesis.
We are interested in investigating the effects of prostatitis on prostate cancer development in an animal model relevant to both diseases. Prostatitis is currently classified as acute bacterial, chronic bacterial, asymptomatic inflammatory, or chronic prostatitis/chronic pelvic pain syndrome (CP/CPPS), which can be either inflammatory or noninflammatory.18 Approximately 90% of all cases of prostatitis are diagnosed as CP/CPPS, which has evidence for an autoimmune component as part of the syndrome's complex pathogenesis.19 Because many cases of prostatitis may have an autoimmune component, the POET-3 mouse model of prostatitis was developed and previously characterized by our laboratory.20 This model makes use of the availability of tools already in place by using ovalbumin as a prostate-specific model antigen. Prostatitis is induced in a controlled manner via adoptive transfer of preactivated CD8+ OT-I T cells, which bear a transgenic T-cell receptor recognizing a peptide in the ovalbumin protein. Acute prostatitis is followed by a chronic phase, with detection of enhanced concentrations of leukocytes up to 80 days after induction of inflammation.20 This model was crossed with the C57/Luc/Pten+/− mouse model of prostate carcinogenesis, which uses the Cre-loxP system driven by the prostate-specific Probasin promoter to inactivate one allele of Pten in the prostate.21 The prostate-specific expression of luciferase allows for in vivo imaging of the prostate over time.
Using this model, two episodes of prostate-specific inflammation were induced to study the effects of inflammation in a model that develops subtle epithelial lesions over several months. The studies addressed the hypothesis that induced prostatitis would enhance the development of precancerous epithelial lesions. In the acute phase, prostate epithelium was highly proliferative and showed enhanced bioluminescence. However, by 4 months after inflammation, mice with previous bouts of inflammation showed similar numbers of lesions and slightly decreased average grade of lesions compared with those mice in which inflammation was not induced, although spontaneous inflammation was observed in noninduced mice. A trend toward invasion was observed at later time points in mice with induced inflammation, but average lesion grade, size of lesions, and number of lesions were unchanged between the induced and noninduced mice. Taking into consideration that Pten+/− mice develop spontaneous prostatitis over time, the mice were also analyzed as a single cohort, and a positive association between myeloid cell infiltration and lesion development was observed.
Materials and Methods
Mice and Genotyping
Animal experiments described in this study were approved by the Purdue University Animal Care and Use Committee. The C57/Luc/Pten−/− mouse model resulted from interbreeding PB-Cre4+ mice [B6.Cg-Tg(Pbsn-cre)4Prb], Ptenfl/fl mice (C;129S4-Ptentm1Hwu/J), and ROSA26-LSL-Luc mice [FVB.129S6 (B6)-Gt (ROSA)26Sortm1(Luc)Kael/J], and backcrossing the resulting mice with albino C57BL/6 mice (C57BL/6J-Tyrc-2J/J), as previously described.21 These mice were interbred with POET-3 mice, a previously described model of prostatitis, to obtain P3+Pten−/− mice containing the POET-3 transgene; PB Cre, floxed alleles of Pten; and prostate luciferase expression.20 Genotypes needed for this study were generated via the speed expansion service at The Jackson Laboratory (Bar Harbor, ME), where sperm from C57/Luc/Pten−/− and C57/Luc/Pten+/− mice also hemizygous for the POET-3 transgene were used to fertilize ova from albino C57BL/6 mice (C57BL/6J-Tyrc-2J/J). The resulting mice were either hemizygous (P3+) or negative (P3−) for the POET-3 transgene, had two intact Pten alleles (Pten+/+) or one allele of Pten floxed (Pten+/−), were hemizygous for PB-Cre (PB-Cre+), and were hemizygous for ROSA26-LSL-Luc (Luc+). Mice were genotyped for the presence of all genetic mutations, as previously described.21
In Vivo Bioluminescence Imaging
Mice for in vivo imaging were anesthetized with a combination of ketamine and xylazine and were dosed intraperitoneally with 0.15 mg/g of body weight D-Luciferin firefly potassium salt (Gold Biotechnology, St. Louis, MO) dissolved in Dulbecco's phosphate-buffered saline. Mice were placed in dorsal recumbency, and bioluminescent signal was imaged with the IVIS Lumina II (Caliper Life Sciences, Hopkinton, MA). Bioluminescence was collected by using a 2-minute exposure at 5-minute intervals for up to 30 minutes. Bioluminescence was measured in photons flux/second within a region of interest, defined as 8% maximum signal by using the Living Image software version 4.0 (Caliper Life Sciences). The single, peak bioluminescent signal obtained over the 30-minute imaging session for each mouse was used in data analysis.
Induction of Prostatitis
Splenocytes were isolated from male Rag1−/− OT-I mice [a cross between B6.129S7-Rag1tm1Mom/J mice obtained from The Jackson Laboratory and Tg(TcraTcrb) 1100Mjb mice obtained as a gift from Dr. William Heath (Walter and Eliza Hall Institute, Melbourne, Australia)] and cultured at 5 × 105/mL with 1 μg/mL SIINFEKL (ova peptide 357-264; American Peptide Company Inc., Sunnyvale, CA) for 48 hours in RPMI 1640 medium with 10% fetal bovine serum, 5% sodium pyruvate, 5% penicillin-streptomycin, and 5% 1 mol/L HEPES. Live cells were purified by Fico/Lite (Atlanta Biologicals, Norcross, GA), and 5 × 106 cells were injected intravenously.
Flow Cytometry
Single-cell suspensions of prostates were obtained by mincing the tissues in a solution of RPMI 1640 medium with 2 μg/mL collagenase D (Roche Diagnostics, Indianapolis, IN) and digesting the tissue at 37°C for approximately 1 hour. Single-cell suspensions of splenocytes were also collected from each mouse. Cells were filtered via 35-μm mesh cell strainer caps, washed with medium, and treated with TruStain fcX anti-CD16/32 antibody (catalog 101319; BioLegend, San Diego, CA), then labeled with the following directly conjugated anti-mouse antibodies according to the manufacturer’s instructions: BioLegend peridinin chlorophyll anti-CD45 (catalog 103129), fluorescein isothiocyanate (FITC) anti-CD45 (catalog 103107), phosphatidylethanolamine (PE)/cyanine 7 anti-CD45R/B220 (catalog 103221), FITC anti-CD4 (catalog 100405), PE anti-CD11b (catalog 101207), PE/cyanine 7 anti-CD11b (catalog 101215), allophycocyanin anti–Gr-1 (catalog 108411), FITC anti-F4/80 (catalog 123107), PE anti-FcεRIα (catalog 134307), and allophycocyanin anti-CD4 (catalog 17-0042-81;eBioscience, San Diego, CA). Aliquots of sample were separated and labeled with the appropriate isotype control antibodies (BioLegend). Labeling for the intracellular antigen FOXP3 was performed with the PE-conjugated anti-FOXP3 Flow Kit (BioLegend). Analysis was performed with the FACSCanto II (BD Biosciences, San Jose, CA), and data were further analyzed with FlowJo software version 9 (TreeStar Inc., Ashland, OR).
RNA Isolation, cDNA Synthesis, and RT-PCR
Sections of each lobe of the prostate were flash frozen in liquid nitrogen and stored at −80°C before homogenization. For RNA extraction, tissues were submerged in 2 mL of TRK lysis buffer (Omega Bio-Tek Inc., Norcross, GA) and homogenized with the Tissue Tearor (BioSpec Products, Inc., Bartlesville, OK). Lysates were centrifuged, and the supernatant was collected. RNA was isolated from the supernatant by using the Total RNA isolation kit I (Omega Bio-Tek Inc.), and cDNA synthesis was performed with qScript cDNA SuperMix (Quanta BioSciences, Inc., Gaithersburg, MD). RT-PCR was performed with LightCycler 96 Real-Time PCR System (Roche Diagnostics) and PrimeTime qPCR probes (Integrated DNA Technologies, Inc., Coralville, IA) for the following genes: Il1b (assay, Mm.PT.56a.41616450), Il2 (assay, Mm.PT.56a.11478202), Il4 (assay, Mm.PT.56a.32703659), Il5 (assay, Mm.PT.56a.41498972), Il6 (assay, Mm.PT.56a.10005566), Il10 (assay, Mm.PT.56a.13531087), Il13 (assay, Mm.PT.56a.31366752), and Il6ra (assay, Mm.PT.58.31166746). TaqMan qPCR probes were used to detect the following genes: Ifng (assay, Mm00801778_m1), Tnf (assay, Mm00443258_m1), and Tgfb1 (assay, MM00441724_m1). As an endogenous control the 18s rRNA gene was detected via TaqMan Ribosomal RNA Control reagents (catalog 4308329; Applied Biosystems, Foster City, CA) and used for normalization. Relative amounts of mRNA were calculated as 2−(Ct gene of interest−Ct 18s rRNA), where Ct is the threshold cycle value.
Histology and Digital Slide Analysis
Prostate tissue was fixed with 10% neutral-buffered formalin for 24 to 48 hours, followed by routine tissue processing and paraffin embedding. A representative 5-μm section of each of the four lobes was obtained and stained with hematoxylin and eosin (H&E). Slides were digitized with an Aperio Digital Slide Scanner (Leica, Wetzler, Germany), and slides were analyzed by two board-certified veterinary pathologists (G.N.B. and P.W.S.) and annotated with ImageScope software version 11.2.0.780 (Leica). Individual mouse prostatic intraepithelial neoplasia (mPIN) and carcinoma lesions were outlined and measured with the ImageScope software. Immunohistochemical analysis was performed by using either the nuclear version 9 or the membrane version 9 algorithms, depending on the localization of the labeling. Average lesion grade was obtained by calculating as follows:
| (1) |
A grading scheme modified from previously published schemes was developed and used to separate mPIN lesions into three separate grades (PIN I, II, III); criteria for carcinoma were adapted from the Bar Harbor meeting of the Mouse Models of Human Cancer Consortium Prostate Pathology Committee.22–24 A three-tier grading scheme for histologic presence of prostatic inflammation was modified from an existing grading scheme; each lobe was scored individually, and a sum of the scores was used as the total prostate inflammation.25
Immunohistochemistry
For anti–Ki-67 and anti–Bcl-2, tissue sections were deparaffinized, rehydrated, submerged in decloaker solution (Biocare Medical, Concord, CA), and heated to 96°C for 20 minutes by using a laboratory microwave (Ted Pella, Inc., Redding, CA). Sections were then treated with 3% hydrogen peroxide, protein block solution (Dako, Carpinteria, CA), primary antibody (anti–Ki-67 sp6 clone, anti–Bcl-2 E17 clone; Abcam Inc., Cambridge, MA), peroxidase-linked polymeric anti-rabbit antibody (Dako), and finally 3,3′-diaminbenzidine substrate; washes with tris-buffered saline that contained 0.5% Tween 20 were conducted between each step. Slides were counterstained with hematoxylin. For anti–p-AKT, tissue sections were deparaffinized, rehydrated, submerged in antigen unmasking solution (Vector Laboratories, Burlingame, CA), heated to 121°C in a benchtop autoclave (2100-Retriever; Prestige Medical, Northridge, CA), and allowed to cool to room temperature. Sections were pretreated with 3% H2O2, then incubated with primary antibody (anti–p-AKT serine 473; Cell Signaling Technology Inc., Danvers, MA) and anti-rabbit secondary antibody (BA-1000; Vector Laboratories). Sections were incubated with Vectastain Elite ABC reagent (Vector Laboratories) and peroxidase substrate solution (Vector Laboratories), followed by hematoxylin counterstain. Appropriate target-specific positive and negative control tissues were used.
Cell Sorting
A previously published protocol for isolation of prostate epithelia was followed.26 Briefly, minced prostate tissue was digested with 1 μg/mL collagenase-I in RPMI 1640 medium containing 10% fetal bovine serum for 2 hours, after trypsinization at 37°C to isolate cells. The suspensions were passed through 20-gauge needles three to five times and 40-μm cell strainers. To enrich prostate epithelial cells, isolated cells were incubated with fluorescence-conjugated specific antibodies as follows: Sca1-allophycocyanin (catalog 108112; BioLegend), CD49f-PE (catalog 12-0495-83; eBioscience), CD45-FITC (catalog 103108; BioLegend), and CD31-FITC (catalog 553372; BD Biosciences).27 CD45−CD31−Sca1+CD49f+ (prostate progenitors), CD45−CD31−Sca1−CD49f− (luminal cells), and CD45−CD31−Sca1−CD49f+ (basal cells) were collected via live cell sorting with the use of a FACSAria (BD Biosciences).
Western Blot Analysis
Cell lysates were prepared by addition of RIPA lysis buffer (Rockland Immunochemicals, Inc., Gilbertsville, PA) containing 1× protease inhibitor cocktail (catalog P8340; Sigma-Aldrich, St. Louis, MO), 1× PhosphataseArrest I (G-Biosciences, St. Louis, MO), 1 mmol/L phenylmethylsulfonyl fluoride, and 0.1% Triton X-100. Protein concentrations were determined with Bradford reagent (catalog B6916; Sigma-Aldrich) according to the manufacturer's instructions by using dilutions of 2 mg/mL bovine serum albumin (Thermo Scientific, Waltham, MA) to produce a standard curve for each experiment. Approximately 30 to 40 μg of protein per sample was loaded into wells for electrophoresis. Western blot analyses were performed by probing polyvinylidene difluoride membranes with the following antibodies: anti–β-actin clone 8H10D10, anti–phospho-IκB (14D4) rabbit monoclonal antibody 2859, and anti–phospho-IκB (44D4) rabbit monoclonal antibody 4812 (Cell Signaling Technology Inc.).
Statistical Analysis
Statistical analyses were performed with Prism version 5.04 (GraphPad Inc., La Jolla, CA). Student's t-test and analysis of variance were used in cases of data with expected normal distribution, with Mann-Whitney test used in cases of non-normal distribution. Correlations between sets of continuous data were analyzed by fitting simple linear regression. Statistical significance was considered with P < 0.05.
Results
Bioluminescence and Cell Proliferation Increase during the Acute Phase of Prostatitis
To study prostatitis and its potential effects on prostate cancer formation, a previously described model of inducible CD8+ T-cell–driven prostatitis, the POET-3 mouse,20,28 was crossed with a prostate-specific Pten-deficient model of prostate cancer formation that included the ROSA26 luciferase allele to allow in vivo monitoring of prostate size21 (Figure 1A). Both mouse models were previously backcrossed onto the C57Bl/6 background to ensure genetic homogeneity. Once the POET-3/C57/Luc/Pten−/− mice were available, adequate numbers of mice for experimentation were generated with The Jackson Laboratory's speed expansion service. Speed expansion generated four genotypes for study: POET-3+/C57/Luc/Pten+/− (designated P3+Pten+/−), POET-3−/C57/Luc/Pten+/− (designated P3−Pten+/−), POET-3+/C57/Luc/Pten+/+ (designated P3+Pten+/+), and POET-3−/C57/Luc/Pten+/+ (designated P3−Pten+/+). Thus, mice were generated with the ability to form epithelial lesions (Pten+/−) and with and without the transgene that allows inducible inflammation (POET-3). For comparison, mice with intact Pten+/+ with and without the POET-3 transgene were also generated.
Figure 1.

Bioluminescence increased with induction of prostatitis. A: A schematic shows the two major mouse models that were crossed for this study: the C57/Luc/Pten−/− model of prostate carcinogenesis that contains Cre recombinase driven by the prostate-specific promoter probasin, luciferase expression in the prostate; and prostate-specific inactivation of Pten was crossed with the POET-3 model, allowing for controlled induction of autoimmune prostatitis. B: Experimental timeline shows ages of treatments and timing of tissue harvests. C: Bioluminescence of P3+Pten+/+ mice increased transiently after induction of prostatitis. D: Bioluminescence increased in the weeks after induction of prostatitis in P3+Pten+/− mice. Data are expressed as means ± SEM (C and D). n = 12 to 14 early time points (C); n = 4 later time points (C); n = 15 to 22 early time points (D); n = 4 to 5 later time points (D). ∗∗P < 0.01, ∗∗∗P < 0.001, and ∗∗∗∗P < 0.0001, Student's t-test.
At 12 weeks of age, all genotypes of mice had similar amounts of bioluminescence, which increased gradually over time (Figure 1, C and D). At weeks 13 and 18 all mice received activated OT-I cells via adoptive transfer (Figure 1B). Prostatitis was induced only in POET-3+ mice as previously described.20 Groups of 10 mice (5 P3+Pten+/−, 5 P3−Pten+/−) or 8 mice (4 P3+Pten+/+ and 4 P3−Pten+/+) were euthanized and evaluated at 4, 8, and 12 months after induction of prostatitis (Figure 1B). Bioluminescent imaging was performed every 1 to 2 weeks during the study. P3+Pten+/+ mice with induced prostatitis had significantly increased (P < 0.001) bioluminescence in the 2 weeks after each instance of adoptive transfer of OT-I T cells; however, bioluminescence normalized soon after each episode and remained similar in each group for the remainder of the study (Figure 1C). In contrast, P3+Pten+/− mice had significantly increased bioluminescence (P < 0.001) immediately after the first adoptive transfer of OT-I T cells at 13 weeks, which persisted until the second adoptive transfer of T cells at 18 weeks (Figure 1D). Interestingly, the second episode of prostatitis resulted in a greater increase in bioluminescence compared with the first, suggesting enhanced epithelial cell proliferation after repeated episodes of inflammation (Figure 1D). Prostatitis was verified via histopathology, which showed diffuse interstitial infiltration of lymphocytes, with fewer numbers of macrophages and neutrophils throughout prostates of P3+Pten+/− mice after adoptive transfer of OT-I T cells, with no detectable inflammation in P3+Pten+/− mice before adoptive transfer and P3−Pten+/− mice after adoptive transfer (Figure 2A). Immunohistochemical labeling for Ki-67 showed a significant increase in epithelial cell proliferation in all lobes of the prostates of P3+Pten+/− mice (P < 0.0001) compared with both P3+Pten+/− mice before adoptive transfer of OT-I T cells and P3−Pten+/− also challenged with OT-I T cells (Figure 2B). Approximately 50% of prostate epithelial cells labeled positively with Ki-67 in P3+Pten+/− mice after induction of inflammation, suggesting CD8+ T-cell–driven prostatitis was a strong signal for epithelial cell proliferation. Immunohistochemical labeling of phosphorylated Akt (p-Akt) was used as an indicator of lesion development in these mice, as previously described, and areas of p-Akt+ epithelium were increased (P < 0.01) in the lateral lobes of mice during the acute phase of prostatitis (Figure 2C).21 Some differences were observed in cell proliferation, with decreased proliferation in the ventral lobe, and Akt labeling, with increased labeling in the lateral lobe. Differences were likely not due to the amount of inflammation, because all lobes were shown to have similar inflammatory infiltrate in this model.20 Differences could be a result of an as yet undescribed difference in epithelial cell biology between different prostate lobes in the mouse. Taken together, the above results indicated that severe prostatitis in P3+Pten+/− mice lead to epithelial cell hyperplasia in the acute phase of inflammation and initial early expansion of multifocal areas of epithelium.
Figure 2.

Prostatitis increased the percentage of dividing epithelial cells and number of cells with enhanced Akt signaling. A: P3+Pten+/− before adoptive transfer of OT-I T cells at 12 weeks of age and P3−Pten+/− mice after adoptive transfer of OT-I T cells at 13 weeks of age showed no prostatitis by H&E staining, low amounts of cell proliferation by anti–Ki-67 IHC, and few cells with enhanced phosphorylation of Akt by anti–p-Akt IHC. P3+Pten+/− mice 6 days after adoptive transfer of OT-I T cells at 13 weeks of age showed severe prostatitis by H&E staining, high amounts of cell proliferation by anti–Ki-67 IHC, and larger areas of cells with enhanced phosphorylation of Akt by anti–p-Akt IHC. Anterior lobes shown in H&E, Ki-67 panels; lateral lobes shown in p-Akt panels. B: Quantification of anti–Ki-67 IHC in the three groups of mice represented in A. C: Quantification of anti–p-Akt IHC in the three groups of mice represented in A. Data are expressed as means ± SEM (B and C). n = 5 mice each group (B); n = 5 mice in Pre and P3− (C); n = 4 mice in P3+ (C). ∗∗P < 0.01, ∗∗∗∗P < 0.0001, one-way analysis of variance or Student's t-test. IHC, immunohistochemistry.
Experimentally Induced Prostatitis Does Not Translate into Increased Incidence, Size, or Grade of Epithelial Precursor Lesions
To test our hypothesis that induced inflammation promotes enhanced carcinogenesis in an environment permissive to cell proliferation, five P3+Pten+/− and five P3−Pten+/− mice were sacrificed at 4, 8, and 12 months after induction of prostatitis. At each time point, prostate tissue was collected and examined histologically. A grading scheme modified from previously published schemes was developed and used to separate mPIN lesions into the following three separate grades: PIN I, II, and III, with PIN III being the most severe of the precursor lesions observed.22,23 Biological differences in the grading scheme were validated via immunohistochemical labeling for Ki-67, in which the proliferative index was increased with higher grades (see PIA-Like Lesions Are Present in Areas of Inflammation and Fibrosis). Lesions were counted, graded, and measured in each lobe. As expected from previously published data that used the C57/Luc/Pten+/−, lesion development was slow, with few instances of mPIN at the 4-month time point (30 weeks of age) and only rare instances of carcinoma developing in the 12-month group (60 weeks of age) (Figure 3A).21 At 4 months after induction of prostatitis, the incidence of epithelial lesions did not differ between P3+Pten+/− and P3−Pten+/− mice (Figure 3B), and the average grade of mPIN lesions was lower in P3+Pten+/− mice (P = 0.02), suggesting that prostatitis may have influenced or slowed lesion development in these mice (Figure 3C). By 8 and 12 months after inflammation, incidence of epithelial lesions and average grade of lesions were statistically similar in both groups of mice (Figure 3, B and C). Similarly, the size of PIN I and PIN II lesions did not differ between the two groups of mice at 4, 8, or 12 months after inflammation (Figure 3D and data not shown). No differences were detected between the sizes of PIN III lesions, although there were small total numbers of PIN III lesions and large variation between PIN III sizes (data not shown).
Figure 3.

Experimentally induced prostatitis did not result in increased numbers or severity of epithelial lesions. A: Examples of epithelial lesions in P3+Pten+/− and P3−Pten+/− mice 30 weeks of age (4 months after induction of prostatitis), 46 weeks of age (8 months after induction), and 60 weeks of age (12 months after induction). The photomicrographs taken from H&E-stained sections represent lesions considered PIN I in the lateral lobe (in both genotypes at 30 weeks, indicated with arrows; adjacent PIN II lesion indicated with asterisk), PIN III in the dorsal lobe (in both genotypes at 46 weeks), and PIN III with adjacent foci of invasion in the lateral lobe (both genotypes at 60 weeks). Foci of carcinoma are indicated with arrows in 60 weeks column. Inset at 60 weeks shows p-Akt IHC, showing epithelial cells invading surrounding stroma. B: Total lesion number did not differ between P3+Pten+/− and P3−Pten+/− mice at 4, 8, and 12 months after induction of inflammation. Lesions from all four prostate lobes in each mouse were tallied and combined. C: Average lesion grade for all prostate lobes was obtained for each mouse. Mice with induction of prostatitis have decreased average lesion grade at 4 months after induction compared with mice without induction of inflammation. Both groups had similar lesion grades at 8 and 12 months after induction. D: Size of PIN I lesions in the dorsal lobe of the prostate did not differ between P3+Pten+/− and P3−Pten+/− mice at 4, 8, and 12 months. Size of other lesion types in the other prostate lobes showed similar results. E: Percentage of mice with each of the mPIN lesion grades and carcinoma was similar at 12 months after induction of prostatitis (60 weeks), with more P3+Pten+/− showing carcinoma. Data are expressed as means ± SEM (B and C); error bars represent SEM (D). n = 4 to 5 mice each group (B–D); n = 5 mice each group (E). ∗P < 0.05. Mann-Whitney test. IHC, immunohistochemistry.
In contrast to mice with loss of one allele of Pten, only rare instances of epithelial lesions were observed the P3+Pten+/+ and P3−Pten+/+ mice (Supplemental Figure S1A). However, an interesting trend toward the presence of hyperplastic lesions was observed in the P3+Pten+/+ mice, with one of three P3+ mice developing at least one instance of hyperplasia at 4 months after inflammation and no mice developing lesions in the P3− group at 4 months (data not shown). Hyperplastic lesions were observed in a single P3+Pten+/+ mouse at 8 months after inflammation, but lesions were not observed in the P3−Pten+/+ mouse group (data not shown). By 12 months after induction of inflammation, three mice in the P3−Pten+/+ group had instances of hyperplasia, with no hyperplasia observed in P3+Pten+/+ mice; however, at least one instance of PIN I was seen in the previously inflamed mice (Supplemental Figure S1A).
Experimentally Induced Prostatitis May Promote Development of Invasive Lesions
Although experimental induction of prostatitis did not result in an increased incidence or higher grade of epithelial lesions as hypothesized, several noteworthy histologic observations were made in P3+Pten+/− prostates. First, a trend toward increased carcinoma development was present in P3+Pten+/− mice compared with P3−Pten+/− mice. Carcinoma was observed only in the mice evaluated at 12 months after prostatitis induction, with two of five P3+Pten+/− mice and one of five P3−Pten+/− mice developing carcinoma (Figure 3E). In one P3+Pten+/− mouse with carcinoma, the mouse was removed from the study 2 months earlier than scheduled because of acute onset of lethargy and inappetence. Grossly the prostate was enlarged, and the urinary bladder was swollen, with multiple areas of serosal hemorrhage (Figure 4A). Microscopically there were multiple mPIN lesions throughout the prostate and periprostatic suppurative inflammation. Within the wall of the urinary bladder, longitudinal sections of peripheral nerve were circumscribed by a pleomorphic population of round-to-polygonal epithelial cells with anisocytosis, anisokaryosis, and multiple mitotic figures, interpreted as prostatic carcinoma with perineural invasion (PNI) and extraprostatic extension (Figure 4B).
Figure 4.
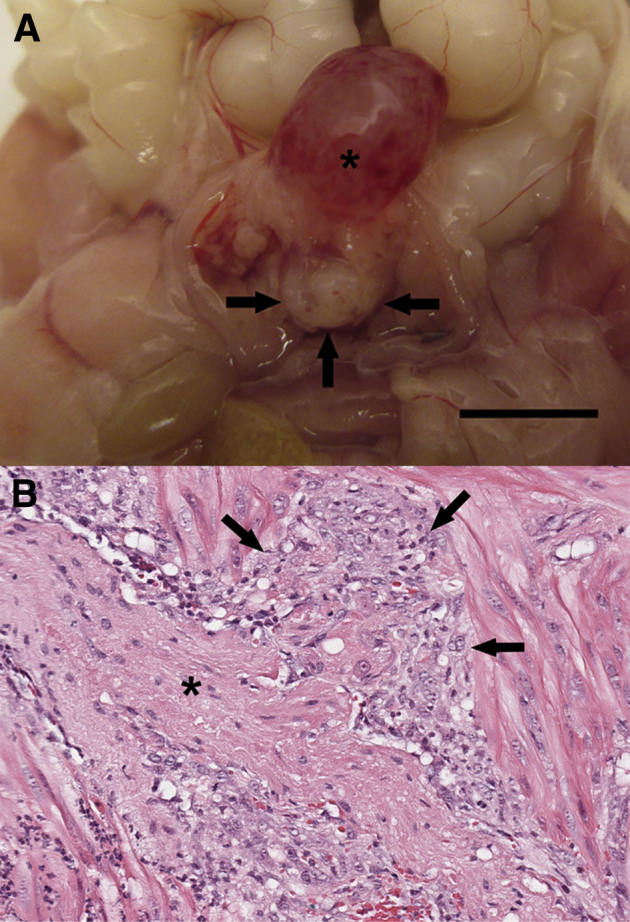
PNI of prostate carcinoma occurred in a single P3+Pten+/− mouse approximately 10 months after induction of inflammation. A: Gross image demonstrates hemorrhagic cystitis (asterisk) and prostatomegaly with hemorrhage (arrows) in a P3+Pten+/− mouse. B: Photomicrograph of a H&E-stained section of prostate and adjacent urinary bladder wall shows a peripheral nerve (asterisk) surrounded by a monomorphic population of carcinomatous epithelial cells (arrows) exhibiting nuclear pleomorphism, anisokaryosis, anisocytosis, and increased mitotic rate. Scale bar = 1 cm.
PIA-Like Lesions Are Present in Areas of Inflammation and Fibrosis
In addition, 1 of 8 Pten+/− mice at 8 months and 1 of 10 Pten+/− mice at 12 months contained regions of the lateral lobe of the prostate in which multiple prostate glands were lined by flattened, atrophic epithelial cells (Figure 5A). Glands lined by atrophic epithelium were surrounded by extensive interstitial fibrosis and inflammation. Some affected glands were ectatic. Interestingly, some glands contained mPIN-like lesions or, in one instance, a microinvasive carcinoma lesion, despite their quiescent morphologic appearance (Figure 5A). Given the morphologic similarity of these lesions to the previously described proliferative inflammatory atrophy (PIA) lesions observed in human prostates, further characterization of the nature of these lesions was initiated.29,30 PIA lesions in men have been shown to have increased proliferative capacity and labeling of antiapoptotic proteins, such as Bcl-2.29 Immunohistochemical labeling for Ki-67, Bcl-2, and p-Akt was performed on sections of prostate containing the PIA-like lesions (data not shown and Figure 5A). Although Bcl-2 was not expressed in these lesions, the proliferative index of affected epithelium was found to be approximately seven times higher than normal epithelium, was higher than all grades of mPIN, and was equally as high as instances of carcinoma (Figure 5B). Individual glands in other lobes of prostates from P3+Pten+/− and P3−Pten+/− mice in the 8- and 12-month time points had focal areas of atrophied epithelium but were differentiated from the aforementioned lesions by decreased proliferative indices and lack of surrounding interstitial fibrosis and inflammation.
Figure 5.

PIA-like lesions developed in two Pten+/− mice, one at 8 months and one at 12 months after induction of inflammation. A: Photomicrographs of H&E-stained sections of lateral prostate from a P3+Pten+/− mouse (left column). Epithelium lining dilated glands shows features of PIA, with flattened, low cuboidal cells that contain scant cytoplasm. In the top panels, epithelial cells show the early stages of microinvasion, dissecting through the basement membrane and underlying layers of collagen. H&E, top right; anti–p-Akt, top left. Stretches of affected epithelium have a high proliferation rate, with increased cells positive for anti–Ki-67 (bottom right panel). B: PIA-like lesions show higher proliferative rates than normal epithelium and various amounts of precancerous epithelium lesions from mice 12 months after induction of inflammation. Multiple lesions were sampled for each category in each mouse. Data are expressed as means ± SEM (B). n = 10 mice in WNL and PIN groups (B); n = 1 mouse in carcinoma group (B); n = 2 mice in PIA group (B). WNL, normal epithelium.
Experimentally Induced Prostatitis Leads to Increased Numbers of Lymphoid Cells in Early Time Points; Spontaneous Prostatitis Tends to Include Increased Numbers of Myeloid Cells
To better understand the type and severity of prostatitis occurring at these different time points, prostate homogenates were analyzed via flow cytometry with the use of phenotypic markers of leukocyte subsets. Four months after induction of prostatitis, P3+Pten+/− mice had a trend toward increased percentage of CD45+ cells in the prostate (P = 0.06), with the percentage of CD45+ cells becoming more even between the two groups at 8 and 12 months after inflammation, with the exception of a single outlier in the P3− 12-month group (Figure 6A). The percentage of lymphoid subsets infiltrating prostates at 4 months after induction of inflammation was increased for CD8+ T cells in mice that were previously inflamed (P = 0.055); when correcting for the percentage of CD45 infiltration in each mouse, trends toward increased numbers of CD8+, CD4+, and B220+ cells in P3+ mice were observed, with significantly increased numbers of CD4+ cells (P = 0.03) in previously inflamed mice (Supplemental Figure S2A and Figure 6B). The percentage of Gr1+CD11b− cells was significantly increased in P3−Pten+/− mouse prostates compared with P3+ mice (P = 0.028), with trends toward increased CD11b+Gr1− and CD11b+Gr1+ cells in P3− mice as well at the 4-month time point (Supplemental Figure S2B). Taken together, these results suggest that mice with repeated episodes of inflammation have an infiltrate biased toward lymphoid cells, whereas the P3− mice with spontaneous prostatitis had relatively greater percentages of myeloid cell infiltration. At both 8 and 12 months after induction of prostatitis, no statistically significant differences between percentages or numbers of infiltrating leukocytes existed between the P3+ and P3− groups, suggesting that inflammation had normalized between both groups (Supplemental Figure S2, C and D, and Figure 6, B and C). However, total numbers of myeloid cells, specifically Gr1+CD11b+ cells, increased over time in both groups (Figure 6C and Supplemental Figure S2D).
Figure 6.

Lesion development was positively correlated with myeloid cell infiltration. A: Percentage of total CD45+ events was measured in prostate homogenates of P3+Pten+/− and P3−Pten+/− mice at various time points via flow cytometry. B: CD45+ events were further classified according to various subsets of lymphoid cells. Data are displayed as number of cells bearing the indicated antigens per amount of CD45+ cells. At 4 months after induction of prostatitis (30 weeks), P3+Pten+/− mice showed a trend toward greater numbers of lymphoid cells, with significantly more CD4+ cells. C: Myeloid subsets were also investigated; data are displayed as number of cells bearing the indicated antigens per amount of CD45+ cells. P3+Pten+/− and P3−Pten+/− mice showed increases in all myeloid subsets over time, with significantly more Gr1+ and Gr1+CD11b+ cells at 12 months after induction of prostatitis. B and C: The first two bars in each subset represent the 30-week timepoint, while the last two bars in each subset represent the 60-week timepoint. D: Histologically determined inflammation score was positively correlated with lesion development in a combined cohort of P3+Pten+/− and P3−Pten+/− mice at all time points. P = 0.0035, R2 = 0.2932, linear regression analysis. E: Lesion development was positively correlated with the percentage of total leukocytes (CD45+ cells) bearing the myeloid markers CD11b and Gr-1. P = 0.0002, R2 = 0.4635, linear regression analysis. F: Lesion development was negatively correlated with the percentage of total leukocytes (CD45+ cells) bearing CD4, CD8, or B220. P = 0.0168, R2 = 0.2158, linear regression analysis. Data are expressed as means ± SEM (A–C). n = 4 to 5 mice each group (A–C). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗∗P < 0.0001, Student's t-test.
Lesion Development across Both P3+Pten+/− and P3−Pten+/− Mice Is Positively Correlated with Infiltration of Myeloid Cells
Although the development of PNI may be associated with previous episodes of experimentally induced prostatitis, the development of lesions, including PIN III, PIA, and carcinoma, occurred in both P3+Pten+/− and P3−Pten+/− mice. As previously reported, we observed the development of prostatitis and interstitial fibrosis in close proximity to many of these lesions.21 We documented a steady increase in the percentage of total CD45+ cells recovered from whole prostate homogenates in both groups of mice, indicating that spontaneous prostatitis occurs in Pten+/− mice even without previous episodes of induced prostatitis (Figure 6A). Given the lack of association of lesion incidence or grade with induction of inflammation, we hypothesized that the severity of lesions was linked to the presence of inflammation generally, not just the CD8-driven inflammation induced in mice with the POET-3 transgene. Thus, mice were analyzed post hoc as a single cohort without regard to the presence or absence of POET-3, via simple linear regression. In this study, both lesion incidence and lesion grade were positively correlated with inflammation, whether measured by flow cytometry and the presence of CD45 or by histologic grade as inflammation score (P = 0.0035, R2 = 0.2932; data not shown) (Figure 6D). As mentioned in the previous subsection, changes in the cellular composition of prostatitis occurred over time, with P3+Pten+/− mice gradually moving from a more lymphocyte-based prostatitis to inflammation that includes some lymphocytes and various myeloid subsets. Literature linking inflammation and cancer supports the idea that myeloid-based inflammation, which includes macrophages and neutrophils, may be the more detrimental type of inflammation in reference to promotion of carcinogenesis.31,32 When a model was fit by using myeloid cell infiltration (any cell with the presence of CD11b, Gr1, or both), lesion incidence was positively correlated (P = 0.0015, R2 = 0.3604) with the percentage of myeloid cell infiltrate in the prostate (Figure 6E). The best fit (P = 0.0002, R2 = 0.4635) was obtained if only those myeloid cells bearing both CD11b and Gr1 were used (data not shown). Conversely, when considering lymphoid infiltrate (any cell bearing CD8, CD4, or B220), lesion incidence was negatively correlated (P = 0.0168, R2 = 0.2158) (Figure 6F). Considering previous data indicating lower lesion grade and higher numbers of CD4+ lymphocytes in the early 4-month postinflammation group, these results suggested high-grade epithelial lesions in the prostate may be associated with the presence of myeloid cell infiltrate.
Cytokine expression was also considered in the context of inflamed versus less-inflamed mice, as measured via histopathologic grading; this analysis was done with a combined cohort including mice with induced and noninduced inflammation. Although no statistically significant differences were observed between P3+Pten+/− and P3−Pten+/− mice for cytokine expression at the 4-, 8-, and 12-month time points, mice with histologically more severe inflammation (those mice with an inflammation score of 4 or higher) had a trend toward increased IL-6 expression compared with those mice with less inflammation and fewer lesions (Supplemental Figure S3A). Cytokines that characterized the type 1 or acute inflammatory response had similar expression between inflamed and less-inflamed mice, and amounts of cytokines that typically associated with type 2 inflammation were typically lower in inflamed mice compared with less-inflamed mice at the 12-month time point (Supplemental Figure S3B). The IL-6 receptor subunit α did not follow the trend of IL-6, with similar mRNA expression between inflamed and less-inflamed mice at 12 months of age (data not shown). Thus, as previously reported in other studies of cytokines and prostate carcinogenesis, IL-6 may play a role in the development of prostate cancer.
Acute and Chronic Inflammation in the Prostates of P3+Pten+/− Mice Leads to Decreased IκB
Given that p-Akt has been linked to activation of the transcription factor NF-κB in a PTEN-null prostate cancer-derived epithelial cell line and that nuclear localization of NF-κB has been linked to biochemical recurrence of prostate cancer and tumor grade in human prostate cancer, investigation into whether NF-κB was activated in the PTEN loss mouse model of prostate cancer may be important.33–35 Little has been done to link NF-κB activation to cancer development in mouse models of prostate cancer, although constitutive NF-κB activity has been shown in the transgenic adenocarcinoma of the mouse prostate model.36 To address this question, acute prostate inflammation was induced via OT-I injection in two young (2 to 3 months old) P3+Pten+/− mice. Nonleukocytic, nonstromal cells were collected via fluorescent-activated cell sorting in these inflamed mice, and in two young (2 to 3 months old) P3+Pten+/− mice given phosphate-buffered saline (uninflamed) and two older (12 months old) P3+Pten+/− mice that had not been dosed with OT-I cells. Given previous results in Figures 2 and 6, acute inflammation is present in young P3+Pten+/− mice dosed with OT-I cells, chronic inflammation is present in older P3+Pten+/− mice, and no inflammation was present in young phosphate-buffered saline-dosed P3+Pten+/− mice. Western blot analysis showed decreased IκB in both inflamed young and chronically inflamed older P3+Pten+/− mice compared with young noninflamed mice, suggesting IκB degradation and resulting NF-κB nuclear localization (Figure 7A). Decreased IκB corresponded with increased phosphorylated IκB in inflamed young mice but not in chronically inflamed older mice (Figure 7A). Furthermore, IL6, a downstream target gene of activated NF-κB, showed a many-fold increase in mRNA expression in sorted nonleukocytic, nonstromal prostate cells from young inflamed mice compared with young naive mice (Figure 7B). Taken together, these results suggested that the transcription factor NF-κB is activated in prostate epithelium after acute, and perhaps chronic, inflammation in P3+Pten+/− mice.
Figure 7.

Inflammation in this model lead to decreased IκB protein expression and IL-6 mRNA expression in prostate epithelial cells. A: Western blot analyses showed decreased IκB protein in young mice with OT-I–induced inflammation and in chronically inflamed older mice. Phosphorylated IκB concentrations were increased in young mice with OT-I–induced inflammation. B: IL-6 mRNA expression was increased in prostate epithelial cells from young mice with OT-I–induced inflammation. Data represented a pooled sample of sorted epithelial cells from two mice in each group.
Discussion
This study was initiated to address the current discrepancies about the relation between prostatitis and prostate cancer development in the biomedical literature and to fill a need for animal models to address this question. The POET-3 mouse specifically models T-cell–driven prostate autoimmunity, whereas the C57/Luc/Pten+/− mouse models a slow progression to prostate cancer, using the genetic alteration most commonly found in human prostate cancer.37 Advantages of this particular model include prostate-specific inflammation and Pten gene deletion, acute episodes of T-cell–driven inflammation followed by CP to mimic autoimmunity, progression of epithelial lesion development that is similar to human prostate cancer, ability to monitor prostate size in vivo, and a homogenous genetic background (C57BL/6).20,21,38
Studies that use rodent models of prostatitis and prostate cancer development have suggested that inflammation promotes development or progression of epithelial lesions in the prostate. Coupled with additional epidemiologic evidence, these studies informed our hypothesis that prostatitis will promote the progression of epithelial lesions in this mouse model.16,17 The results of our study were mixed and suggest a more complex relation between inflammation and cancer development in the prostate. In the simplest setting, the P3+Pten+/+ mouse, early time points after induction of inflammation suggest that induced prostatitis may promote the development of hyperplastic lesions, because one of three inflamed P3+Pten+/+ mice at 4 months and one of four mice at 8 months after induction of prostatitis had hyperplastic epithelial lesions (Supplemental Figure S1A). Mice in the uninflamed control group did not develop histologically recognizable epithelial lesions until 12 months after treatment, suggesting that prostatitis might induce hyperplasia more quickly in normal prostate epithelium. These results are consistent with a role for inflammation in benign prostatic hyperplasia, as suggested by previous investigators.39–42
Interpretation of inflammation in the P3−Pten+/− and P3+Pten+/− mice was more complicated, because results differed between early and late time points. In P3+Pten+/− mice, the week immediately after induction of prostatitis had highly proliferative prostate epithelium, via both bioluminescence and Ki-67 immunolabeling (Figures 1 and 2). Although bioluminescence in inflamed mice remained increased above the unchallenged mice for several weeks, the mice examined at 4 months after induction of prostatitis did not have increased incidence, size, or grade of mPIN lesions. (Figure 3) In fact, when comparing the average grade of epithelial lesions across all prostate lobes between inflamed and uninflamed mice, previously inflamed mice had a lower average lesion grade. The explanation of this observation likely lies in the type of inflammation that was induced, and likely persisted, in the prostates of previously inflamed mice. Flow cytometric analysis showed trends toward higher numbers of lymphoid subsets (CD8+, CD4+, B220+), with statistically significantly more CD4+ T cells in 4-month prostates of P3+ mice (Figure 6). Previous characterization of the POET-3 model shows significantly enhanced expression of T helper cells type 1 (Th1) and acute inflammatory cytokines such as IL-2, IL-12, and tumor necrosis factor-α.20 These early episodes of inflammation likely influenced recruitment of Th1 CD4+ cells, as supported by increased numbers of CD4+ cells at 4 months after inflammation, and may have biased the prostate microenvironment toward a less cancer-promoting type 1 inflammation. Inflammation that developed spontaneously over this same time frame in P3−Pten+/− mice was biased toward myeloid-based inflammation, which suggests a more tumor-promoting type 2 inflammatory microenvironment. Indeed, when mice from all time points are analyzed without regard for POET-3 status, lymphoid-based prostatitis was significantly negatively correlated with lesion development, whereas myeloid-based inflammation was significantly positively correlated (Figure 6). These results together suggest that episodes of Th1-based prostatitis in the P3+Pten+/− mouse may be protective in reference to early mPIN development and may lend support to recently published evidence showing prostatitis as protective for development of prostate cancer.8,9 Much of the literature before these referenced studies relied on physician-diagnosed cases of prostatitis, of which 90% were likely cases of CP/CPPS.19 Evidence suggests that approximately one-third of CP/CPPS cases are characterized by biopsy-determined prostate inflammation, and recent studies suggest that the presence of histologically defined prostatitis is similar between men with and without symptoms of CP/CPPS.25,43 Many of the patients in previous studies of prostatitis and cancer development were likely diagnosed on the basis of pelvic pain and may have lacked histologic prostatitis, which makes the recent biopsy-based studies in the United States and Finland much stronger when considering true leukocytic infiltration and its relation to cancer development.
Considering the later time points in our study, 8 and 12 months after induction of prostatitis, the incidence, size, and average grade of mPIN lesions was similar across both groups of mice. However, a trend toward invasion was noted in P3+Pten+/− mice, with more mice developing carcinoma. One of the weaknesses of this study was the lack of statistical power for rare events, such as carcinoma development, leading to speculation about whether previous bouts of prostatitis might have promoted invasion at later points. Because the C57/Luc/Pten+/− phenotype includes the development of spontaneous inflammation over time, we had the opportunity to also analyze lesion development in light of development of prostatitis in general, whether it was noninduced (spontaneous) or induced inflammation. In these analyses, lesion incidence and lesion grade were clearly correlated with inflammation, with the most relevant correlation coming when infiltration of myeloid cells bearing both CD11b and Gr1 was considered. As previously mentioned, cancer development and progression are associated with specific leukocyte subsets and specific cytokine environments. Macrophages, neutrophils, and myeloid-derived suppressor cells (MDSCs) all produce substances that are considered damaging to host cell membranes and DNA, such as reactive oxygen and nitrogen species. These components might be helpful to the host if directed at tumor cells themselves, but when considering the early development of malignancy and the progression from lower grade mPIN lesions to higher grade precursor lesions and eventually invasive lesions, the products often associated with myeloid cells might be considered detrimental and tumor promoting. Specifically, leukocytes bearing the CD11b+Gr1+ phenotype might be considered either activated neutrophils or MDSCs, both of which are known to produce damaging free radicals that promote cancer progression.44 In addition, MDSCs may interfere with antitumor immunity via suppression of cytotoxic T cells directed toward tumor antigens.45 Previous work by our group has shown that cells sorted from prostates of Pten−/− mice have the capacity to suppress T-cell proliferation.21 We also evaluated Pten+/− mice for MDSC-mediated suppression at 7 months of age and observed strong inhibition of T-cell proliferation (data not shown). On the basis of these observations, a portion of the CD11b+Gr1+ infiltrating cells in this report are considered MDSCs that may have contributed to lesion progression.21
In addition to the previous findings, two interesting and unique lesions were noted in mice from this study: PNI and PIA. Although PNI is a common occurrence in prostate cancer and has been linked to poorer prognosis in other cancers, the role of PNI in predicting prostate cancer outcome is not clear.46–48 PNI has been documented in a chemically induced rat model of prostate cancer, two mouse models using the SV40-Tag transgene, a single mouse with overexpression of human 15-lipoxygenase-1, and mice with double knockout of Pten and TP53, but it has not been documented in mice with loss of only Pten, to our knowledge.49–53 Lesions resembling PIA have not been described in mouse models of prostate cancer. A consensus report relating to interpretation of lesions in transgenic mouse models have described epithelial atrophy in mice after castration but it has not related these lesions to previous episodes of inflammation or characterized them according to their proliferative capacity.22 Interestingly, these lesions developed in prostates of Pten+/− mice that had severe prostatitis, either from previous induction with OT-I T cells or spontaneous development. At least one report in rats and one report in mice both dosed with 2-amino-1-methy-6-phenylimidazo (4,5-b) pyridine show development of prostatitis, followed by atrophic epithelial lesions, suggesting that this particular lesion may develop as a result of inflammation and partial destruction of the epithelial lining.54,55 Atrophic lesions in the treated rats and mice developed after prostatitis and had some degree of cellular atypia in the rats; however, proliferative indices of these lesions were not specifically assessed.54,55 The morphology of these lesions in Pten+/− mice certainly seems to suggest previous injury and cell loss, as in the rat model, with flattening of epithelial cells to cover the basement membrane. With only two mice having these lesions, determination of their cause is speculative. Phosphorylation of Akt in the Pten+/− microenvironment during the acute phase of prostatitis (Figure 2C) or during the development of spontaneous inflammation later on likely leads to phosphorylation of Bad and subsequently allows Bcl-2 to inhibit epithelial cell apoptosis during severe inflammation in our model, as occurs in animal models of prostate cancer and prostate cancer cell lines.56,57 Although the pathogenesis of PIA in men is unknown, overexpression of Bcl-2 has been shown in human PIA lesions, suggesting that previous Akt signaling and/or loss of Pten, accumulation of Bcl-2, and development of PIA may be linked.
Perhaps a more interesting observation related to these lesions is that in one instance, epithelial cells arising from a PIA-like gland dissect through the basement membrane and underlying layers of collagen to exhibit early microinvasion. PIA lesions and their spatial and temporal association with high-grade PIN and prostate adenocarcinoma in men have been well documented; however, only a single publication shows a direct morphologic link between atrophic glands and carcinoma in human prostate tissue, and the lesions are not currently considered true precursors of adenocarcinoma.29,30,58,59 The cancer-like properties of the epithelium, including their increased proliferative capacity and resistance to apoptosis, at least in men, make them suspect as cancer precursors. Although the lesions described here differ from the human lesions in their expression of an antiapoptotic protein, their highly proliferative nature and morphologic similarities suggest that they may be analogous entities. Documented early microinvasion of the basement membrane suggests that atrophic epithelial lesions with highly proliferative capacity may indeed represent a precursor lesion to prostate carcinoma.
A final part of this study considered expression of cytokines in whole prostate homogenates. Although no statistically significant differences were observed between P3−Pten+/− and P3+Pten+/− mice, a trend toward increasing IL-6 expression in mice with a histologic inflammation score of 4 or greater was observed when mice of all genotypes and time points were examined. As previously mentioned, IL-6 has been one of the cytokines most indicated in prostate carcinogenesis, with evidence coming from in vitro studies of IL-6 and prostate cancer cell lines, increases in IL-6 in sera of patients at risk of prostate cancer development, and genetic polymorphisms in the IL-6 and IL-6 receptor genes conferring decreased risk of prostate cancer development.14,60,61 Although the trends we observed were not statistically significant, they support previous literature suggesting a role for IL-6 in prostate carcinogenesis. Interestingly, IL-6 has been shown to activate the phosphatidylinositol 3-kinase/Akt pathway and trigger expression of cyclin A1, with positive effects on survival in prostate cancer cell lines.62 Thus, chronic inflammation may promote Akt pathway activity in the prostate. Given that loss of Pten, and subsequent increased Akt activity, without additional inflammatory stimuli (ie, no OT-I injection) leads to prostate inflammation in this model, the fact that IL-6 can lead to further Akt stimulation could potentially establish a cycle of constant inflammation and Akt stimulation.
To add information about the pathogenesis of inflammation and cancer development in this particular model, additional experiments were undertaken to understand whether the transcription factor NF-κB was activated in prostate epithelial cells in these mice. Protein analysis showed decreased IκB in acutely inflamed young P3+Pten+/− mice and chronically inflamed older P3+Pten+/− mice, suggesting degradation of this protein and subsequent release of NF-κB to allow nuclear localization in prostate epithelial cells. Thus, inflammation in this model seems to promote NF-κB activation, which in turn would allow a positive feed-forward process to further promoting inflammation and epithelial cell survival and proliferation.63 Furthermore, IL6, a NF-κB target gene implicated in the progression of prostate cancer, was also found to be up-regulated in prostate epithelial cells during episodes of acute inflammation. This finding further supports that NF-κB is activated in our model after inflammation, but it also lends some insight into the source of IL-6 in the prostate microenvironment, because IL-6 was found to be up-regulated in prostate homogenates with greater inflammation (Figure 6).
Conclusions
Our study suggests that the relation between inflammation and prostate cancer is complex and depends largely on the type of inflammatory microenvironment present in the prostate and the state of the epithelium (normal or loss of Pten). Inflammation dominated by T cells and type 1 cytokines likely leads to slower progression of epithelial precursor lesions, even if epithelial proliferation is high after episodes of inflammation. In contrast, inflammation that includes more myeloid cells, especially CD11b+Gr1+ cells, is associated with increased incidence of mPIN and higher grade epithelial lesions. In addition, previous bouts of T-cell–driven prostatitis or development of spontaneous inflammation may lead to focal damage of epithelium that later develops into atrophic epithelium with a high rate of proliferation and the potential for invasion. Finally, IL-6, a cytokine produced both in acute inflammation and by type 2-skewed macrophages and CD4+ T cells, is likely important for the development of epithelial lesions in the P3+Pten+/− prostate. Further studies using this model would be beneficial to advance our understanding of the cellular and molecular components of the inflammatory response that are associated with the development and progression of prostate cancer.
Acknowledgments
We thank Renee E. Vickman, Hsing-Hui Wang, Sandra Torregrosa-Allen, and Tripti Bera for assistance with tissue harvests, mouse treatments, and mouse genotyping; Aaron Taylor for expertise in conducting bioluminescence studies; Tracy Wiegand and Carol Bain for tissue processing, slide preparation, and slide digitization; and the members of the Ratliff laboratory for helpful advice and thoughtful conversation throughout the duration of the study.
Footnotes
Supported by NIH grants R01 DK84454 (T.L.R.), R21 CA154126 (M.D.H. and T.L.R.), and 5T32OD011122 (salary support for G.N.B.) and Purdue University Center for Cancer Research grant P30 CA023168 (T.L.R.).
Disclosures: None declared.
Supplemental Data
Examples of rare epithelial lesions in P3+Pten+/+ and P3−Pten+/+ mice. Hyperplastic foci characterized by multiple cribiform structures lined by a single layer of epithelial cells with benign cytologic features and few mitotic figures are present in P3+Pten+/+ mice at 30 and 46 weeks (4 and 8 months after induction of inflammation) but are not observed in P3−Pten+/+ mice until 60 weeks (12 months after induction of inflammation). A single lesion suggestive of PIN I, with pleomorphic cells containing enlarged nuclei, was observed in one P3+Pten+/+ mouse at 60 weeks.
Spontaneous inflammation at 4 months after induction of prostatitis is skewed toward myeloid cells. A: Percentage of leukocytes (total CD45+ cells) in prostates from mice 4 months after induction of prostatitis show increased CD8+ cells. B: Percentage of leukocytes (total CD45+ cells) in prostates from mice 4 months after induction of prostatitis show trends toward increased myeloid cells, with significantly more Gr1+CD11b− cells. C: Total lymphoid infiltrate into prostates 8 months after induction of inflammation show no differences between P3+Pten+/− and P3−Pten+/− mice but an overall decrease in T cells and an increase in B cells from the 4-month time point. D: Total myeloid infiltrate into prostates 8 months after induction of inflammation show no significant differences between P3+Pten+/− and P3−Pten+/− mice, but an overall increase in CD11b+Gr1+ cells from the 4-month time point. Data are expressed as means ± SEM (A–D). n = 4 to 5 mice each group (A and B); n = 4 mice each group (C and D). ∗P < 0.05, Student's t-test.
Expression of Il-6 mRNA is higher in prostates with greater inflammatory infiltrate, as measured by histologic analysis, at each of the examined time points (A) and compared with other cytokines at 12 months after induction of inflammation (B). Data are expressed as means ± SEM (A and B). n = 8 in 4 months 0 to 3 group (A); n = 1 in 4 months 4+ group (A); n = 4 in 8 months 0 to 3 group (A); n = 4 in 8 months 4+ group (A); n = 2 in 12 months 0 to 3 group (A); n = 7 in 12 months 4+ group (A). IFN-γ, interferon-γ; TGF-β, transforming growth factor-β; TNF-α, tumor necrosis factor-α.
References
- 1.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Abdel-Latif M.M., Duggan S., Reynolds J.V., Kelleher D. Inflammation and esophageal carcinogenesis. Curr Opin Pharmacol. 2009;9:396–404. doi: 10.1016/j.coph.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 3.Chiba T., Marusawa H., Ushijima T. Inflammation-associated cancer development in digestive organs: mechanisms and roles for genetic and epigenetic modulation. Gastroenterology. 2012;143:550–563. doi: 10.1053/j.gastro.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 4.Terzic J., Grivennikov S., Karin E., Karin M. Inflammation and colon cancer. Gastroenterology. 2010;138:2101–2114.e5. doi: 10.1053/j.gastro.2010.01.058. [DOI] [PubMed] [Google Scholar]
- 5.Roberts R.O., Lieber M.M., Rhodes T., Girman C.J., Bostwick D.G., Jacobsen S.J. Prevalence of a physician-assigned diagnosis of prostatitis: the Olmsted County Study of Urinary Symptoms and Health Status Among Men. Urology. 1998;51:578–584. doi: 10.1016/s0090-4295(98)00034-x. [DOI] [PubMed] [Google Scholar]
- 6.Jiang J., Li J., Yunxia Z., Zhu H., Liu J., Pumill C. The role of prostatitis in prostate cancer: meta-analysis. PLoS One. 2013;8:e85179. doi: 10.1371/journal.pone.0085179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hung S.C., Lai S.W., Tsai P.Y., Chen P.C., Wu H.C., Lin W.H., Sung F.C. Synergistic interaction of benign prostatic hyperplasia and prostatitis on prostate cancer risk. Br J Cancer. 2013;108:1778–1783. doi: 10.1038/bjc.2013.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yli-Hemminki T.H., Laurila M., Auvinen A., Maattanen L., Huhtala H., Tammela T.L., Kujala P.M. Histological inflammation and risk of subsequent prostate cancer among men with initially elevated serum prostate-specific antigen (PSA) concentration in the Finnish prostate cancer screening trial. BJU Int. 2013;112:735–741. doi: 10.1111/bju.12153. [DOI] [PubMed] [Google Scholar]
- 9.Moreira D.M., Nickel J.C., Gerber L., Muller R.L., Andriole G.L., Castro-Santamaria R., Freedland S.J. Baseline prostate inflammation is associated with a reduced risk of prostate cancer in men undergoing repeat prostate biopsy: results from the REDUCE study. Cancer. 2014;120:190–196. doi: 10.1002/cncr.28349. [DOI] [PubMed] [Google Scholar]
- 10.Shebl F.M., Sakoda L.C., Black A., Koshiol J., Andriole G.L., Grubb R., Church T.R., Chia D., Zhou C., Chu L.W., Huang W.Y., Peters U., Kirsh V.A., Chatterjee N., Leitzmann M.F., Hayes R.B., Hsing A.W. Aspirin but not ibuprofen use is associated with reduced risk of prostate cancer: a PLCO study. Br J Cancer. 2012;107:207–214. doi: 10.1038/bjc.2012.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Veitonmaki T., Tammela T.L., Auvinen A., Murtola T.J. Use of aspirin, but not other non-steroidal anti-inflammatory drugs is associated with decreased prostate cancer risk at the population level. Eur J Cancer. 2013;49:938–945. doi: 10.1016/j.ejca.2012.09.030. [DOI] [PubMed] [Google Scholar]
- 12.Mahmud S.M., Franco E.L., Turner D., Platt R.W., Beck P., Skarsgard D., Tonita J., Sharpe C., Aprikian A.G. Use of non-steroidal anti-inflammatory drugs and prostate cancer risk: a population-based nested case-control study. PLoS One. 2011;6:e16412. doi: 10.1371/journal.pone.0016412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng I., Witte J.S., Jacobsen S.J., Haque R., Quinn V.P., Quesenberry C.P., Caan B.J., Van Den Eeden S.K. Prostatitis, sexually transmitted diseases, and prostate cancer: the California Men's Health Study. PLoS One. 2010;5:e8736. doi: 10.1371/journal.pone.0008736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steiner H., Godoy-Tundidor S., Rogatsch H., Berger A.P., Fuchs D., Comuzzi B., Bartsch G., Hobisch A., Culig Z. Accelerated in vivo growth of prostate tumors that up-regulate interleukin-6 is associated with reduced retinoblastoma protein expression and activation of the mitogen-activated protein kinase pathway. Am J Pathol. 2003;162:655–663. doi: 10.1016/S0002-9440(10)63859-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steiner H., Berger A.P., Godoy-Tundidor S., Bjartell A., Lilja H., Bartsch G., Hobisch A., Culig Z. An autocrine loop for vascular endothelial growth factor is established in prostate cancer cells generated after prolonged treatment with interleukin 6. Eur J Cancer. 2004;40:1066–1072. doi: 10.1016/j.ejca.2003.11.033. [DOI] [PubMed] [Google Scholar]
- 16.Elkahwaji J.E., Zhong W., Hopkins W.J., Bushman W. Chronic bacterial infection and inflammation incite reactive hyperplasia in a mouse model of chronic prostatitis. Prostate. 2007;67:14–21. doi: 10.1002/pros.20445. [DOI] [PubMed] [Google Scholar]
- 17.Birbach A., Eisenbarth D., Kozakowski N., Ladenhauf E., Schmidt-Supprian M., Schmid J.A. Persistent inflammation leads to proliferative neoplasia and loss of smooth muscle cells in a prostate tumor model. Neoplasia. 2011;13:692–703. doi: 10.1593/neo.11524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krieger J.N., Nyberg L., Jr., Nickel J.C. NIH consensus definition and classification of prostatitis. JAMA. 1999;282:236–237. doi: 10.1001/jama.282.3.236. [DOI] [PubMed] [Google Scholar]
- 19.Pontari M.A. Etiology of chronic prostatitis/chronic pelvic pain syndrome: psychoimmunoneurendocrine dysfunction (PINE syndrome) or just a really bad infection? World J Urol. 2013;31:725–732. doi: 10.1007/s00345-013-1061-z. [DOI] [PubMed] [Google Scholar]
- 20.Haverkamp J.M., Charbonneau B., Crist S.A., Meyerholz D.K., Cohen M.B., Snyder P.W., Svensson R.U., Henry M.D., Wang H.H., Ratliff T.L. An inducible model of abacterial prostatitis induces antigen specific inflammatory and proliferative changes in the murine prostate. Prostate. 2011;71:1139–1150. doi: 10.1002/pros.21327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Svensson R.U., Haverkamp J.M., Thedens D.R., Cohen M.B., Ratliff T.L., Henry M.D. Slow disease progression in a C57BL/6 pten-deficient mouse model of prostate cancer. Am J Pathol. 2011;179:502–512. doi: 10.1016/j.ajpath.2011.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shappell S.B., Thomas G.V., Roberts R.L., Herbert R., Ittmann M.M., Rubin M.A., Humphrey P.A., Sundberg J.P., Rozengurt N., Barrios R., Ward J.M., Cardiff R.D. Prostate pathology of genetically engineered mice: definitions and classification. The consensus report from the Bar Harbor meeting of the Mouse Models of Human Cancer Consortium Prostate Pathology Committee. Cancer Res. 2004;64:2270–2305. doi: 10.1158/0008-5472.can-03-0946. [DOI] [PubMed] [Google Scholar]
- 23.Berman-Booty L.D., Sargeant A.M., Rosol T.J., Rengel R.C., Clinton S.K., Chen C.S., Kulp S.K. A review of the existing grading schemes and a proposal for a modified grading scheme for prostatic lesions in TRAMP mice. Toxicol Pathol. 2012;40:5–17. doi: 10.1177/0192623311425062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park J.H., Walls J.E., Galvez J.J., Kim M., Abate-Shen C., Shen M.M., Cardiff R.D. Prostatic intraepithelial neoplasia in genetically engineered mice. Am J Pathol. 2002;161:727–735. doi: 10.1016/S0002-9440(10)64228-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nickel J.C., Roehrborn C.G., O'Leary M.P., Bostwick D.G., Somerville M.C., Rittmaster R.S. Examination of the relationship between symptoms of prostatitis and histological inflammation: baseline data from the REDUCE chemoprevention trial. J Urol. 2007;178:896–900. doi: 10.1016/j.juro.2007.05.041. discussion 900–901. [DOI] [PubMed] [Google Scholar]
- 26.Lukacs R.U., Goldstein A.S., Lawson D.A., Cheng D., Witte O.N. Isolation, cultivation and characterization of adult murine prostate stem cells. Nat Protoc. 2010;5:702–713. doi: 10.1038/nprot.2010.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lawson D.A., Xin L., Lukacs R.U., Cheng D., Witte O.N. Isolation and functional characterization of murine prostate stem cells. Proc Natl Acad Sci U S A. 2007;104:181–186. doi: 10.1073/pnas.0609684104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lees J.R., Charbonneau B., Hayball J.D., Diener K., Brown M., Matusik R., Cohen M.B., Ratliff T.L. T-cell recognition of a prostate specific antigen is not sufficient to induce prostate tissue destruction. Prostate. 2006;66:578–590. doi: 10.1002/pros.20307. [DOI] [PubMed] [Google Scholar]
- 29.De Marzo A.M., Marchi V.L., Epstein J.I., Nelson W.G. Proliferative inflammatory atrophy of the prostate: implications for prostatic carcinogenesis. Am J Pathol. 1999;155:1985–1992. doi: 10.1016/S0002-9440(10)65517-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Putzi M.J., De Marzo A.M. Morphologic transitions between proliferative inflammatory atrophy and high-grade prostatic intraepithelial neoplasia. Urology. 2000;56:828–832. doi: 10.1016/s0090-4295(00)00776-7. [DOI] [PubMed] [Google Scholar]
- 31.DeNardo D.G., Andreu P., Coussens L.M. Interactions between lymphocytes and myeloid cells regulate pro- versus anti-tumor immunity. Cancer Metastasis Rev. 2010;29:309–316. doi: 10.1007/s10555-010-9223-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grivennikov S.I., Greten F.R., Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dan H.C., Cooper M.J., Cogswell P.C., Duncan J.A., Ting J.P., Baldwin A.S. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008;22:1490–1500. doi: 10.1101/gad.1662308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fradet V., Lessard L., Begin L.R., Karakiewicz P., Masson A.M., Saad F. Nuclear factor-kappaB nuclear localization is predictive of biochemical recurrence in patients with positive margin prostate cancer. Clin Cancer Res. 2004;10:8460–8464. doi: 10.1158/1078-0432.CCR-04-0764. [DOI] [PubMed] [Google Scholar]
- 35.Shukla S., MacLennan G.T., Fu P., Patel J., Marengo S.R., Resnick M.I., Gupta S. Nuclear factor-kappaB/p65 (Rel A) is constitutively activated in human prostate adenocarcinoma and correlates with disease progression. Neoplasia. 2004;6:390–400. doi: 10.1593/neo.04112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shukla S., Maclennan G.T., Marengo S.R., Resnick M.I., Gupta S. Constitutive activation of P I3 K-Akt and NF-kappaB during prostate cancer progression in autochthonous transgenic mouse model. Prostate. 2005;64:224–239. doi: 10.1002/pros.20217. [DOI] [PubMed] [Google Scholar]
- 37.Phin S., Moore M.W., Cotter P.D. Genomic rearrangements of PTEN in prostate cancer. Front Oncol. 2013;3:240. doi: 10.3389/fonc.2013.00240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ittmann M., Huang J., Radaelli E., Martin P., Signoretti S., Sullivan R., Simons B.W., Ward J.M., Robinson B.D., Chu G.C., Loda M., Thomas G., Borowsky A., Cardiff R.D. Animal models of human prostate cancer: the consensus report of the New York meeting of the Mouse Models of Human Cancers Consortium Prostate Pathology Committee. Cancer Res. 2013;73:2718–2736. doi: 10.1158/0008-5472.CAN-12-4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kramer G., Steiner G.E., Handisurya A., Stix U., Haitel A., Knerer B., Gessl A., Lee C., Marberger M. Increased expression of lymphocyte-derived cytokines in benign hyperplastic prostate tissue, identification of the producing cell types, and effect of differentially expressed cytokines on stromal cell proliferation. Prostate. 2002;52:43–58. doi: 10.1002/pros.10084. [DOI] [PubMed] [Google Scholar]
- 40.Steiner G.E., Stix U., Handisurya A., Willheim M., Haitel A., Reithmayr F., Paikl D., Ecker R.C., Hrachowitz K., Kramer G., Lee C., Marberger M. Cytokine expression pattern in benign prostatic hyperplasia infiltrating T cells and impact of lymphocytic infiltration on cytokine mRNA profile in prostatic tissue. Lab Invest. 2003;83:1131–1146. doi: 10.1097/01.lab.0000081388.40145.65. [DOI] [PubMed] [Google Scholar]
- 41.Robert G., Descazeaud A., Nicolaiew N., Terry S., Sirab N., Vacherot F., Maille P., Allory Y., de la Taille A. Inflammation in benign prostatic hyperplasia: a 282 patients' immunohistochemical analysis. Prostate. 2009;69:1774–1780. doi: 10.1002/pros.21027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gandaglia G., Briganti A., Gontero P., Mondaini N., Novara G., Salonia A., Sciarra A., Montorsi F. The role of chronic prostatic inflammation in the pathogenesis and progression of benign prostatic hyperplasia (BPH) BJU Int. 2013;112:432–441. doi: 10.1111/bju.12118. [DOI] [PubMed] [Google Scholar]
- 43.True L.D., Berger R.E., Rothman I., Ross S.O., Krieger J.N. Prostate histopathology and the chronic prostatitis/chronic pelvic pain syndrome: a prospective biopsy study. J Urol. 1999;162:2014–2018. doi: 10.1016/S0022-5347(05)68090-1. [DOI] [PubMed] [Google Scholar]
- 44.Gabrilovich D.I., Ostrand-Rosenberg S., Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gabrilovich D.I., Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liebig C., Ayala G., Wilks J.A., Berger D.H., Albo D. Perineural invasion in cancer: a review of the literature. Cancer. 2009;115:3379–3391. doi: 10.1002/cncr.24396. [DOI] [PubMed] [Google Scholar]
- 47.Katz B., Srougi M., Dall'Oglio M., Nesrallah A.J., Sant'anna A.C., Pontes J., Jr., Antunes A.A., Reis S.T., Viana N., Sanudo A., Camara-Lopes L.H., Leite K.R. Perineural invasion detection in prostate biopsy is related to recurrence-free survival in patients submitted to radical prostatectomy. Urol Oncol. 2013;31:175–179. doi: 10.1016/j.urolonc.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 48.Elharram M., Margel D., Finelli A., Trachtenberg J., Evans A., van der Kwast T.H., Sweet J.M., Fleshner N. Perineural invasion on prostate biopsy does not predict adverse pathological outcome. Can J Urol. 2012;19:6567–6572. [PubMed] [Google Scholar]
- 49.Bosland M.C., Prinsen M.K., Dirksen T.J., Spit B.J. Characterization of adenocarcinomas of the dorsolateral prostate induced in Wistar rats by N-methyl-N-nitrosourea, 7,12-dimethylbenz(a)anthracene, and 3,2′-dimethyl-4-aminobiphenyl, following sequential treatment with cyproterone acetate and testosterone propionate. Cancer Res. 1990;50:700–709. [PubMed] [Google Scholar]
- 50.Garabedian E.M., Humphrey P.A., Gordon J.I. A transgenic mouse model of metastatic prostate cancer originating from neuroendocrine cells. Proc Natl Acad Sci U S A. 1998;95:15382–15387. doi: 10.1073/pnas.95.26.15382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matzuk M.M., Brown C.W., Kumar T.R. Humana Press; Totowa, NJ: 2001. Transgenics in endocrinology. [Google Scholar]
- 52.Kelavkar U.P., Parwani A.V., Shappell S.B., Martin W.D. Conditional expression of human 15-lipoxygenase-1 in mouse prostate induces prostatic intraepithelial neoplasia: the FLiMP mouse model. Neoplasia. 2006;8:510–522. doi: 10.1593/neo.06202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martin P., Liu Y.N., Pierce R., Abou-Kheir W., Casey O., Seng V., Camacho D., Simpson R.M., Kelly K. Prostate epithelial Pten/TP53 loss leads to transformation of multipotential progenitors and epithelial to mesenchymal transition. Am J Pathol. 2011;179:422–435. doi: 10.1016/j.ajpath.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Borowsky A.D., Dingley K.H., Ubick E., Turteltaub K.W., Cardiff R.D., Devere-White R. Inflammation and atrophy precede prostatic neoplasia in a PhIP-induced rat model. Neoplasia. 2006;8:708–715. doi: 10.1593/neo.06373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li G., Wang H., Liu A.B., Cheung C., Reuhl K.R., Bosland M.C., Yang C.S. Dietary carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine-induced prostate carcinogenesis in CYP1A-humanized mice. Cancer Prev Res (Phila) 2012;5:963–972. doi: 10.1158/1940-6207.CAPR-12-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang H., Cheville J.C., Pan Y., Roche P.C., Schmidt L.J., Tindall D.J. PTEN induces chemosensitivity in PTEN-mutated prostate cancer cells by suppression of Bcl-2 expression. J Biol Chem. 2001;276:38830–38836. doi: 10.1074/jbc.M103632200. [DOI] [PubMed] [Google Scholar]
- 57.Majumder P.K., Febbo P.G., Bikoff R., Berger R., Xue Q., McMahon L.M., Manola J., Brugarolas J., McDonnell T.J., Golub T.R., Loda M., Lane H.A., Sellers W.R. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med. 2004;10:594–601. doi: 10.1038/nm1052. [DOI] [PubMed] [Google Scholar]
- 58.Wang W., Bergh A., Damber J.E. Morphological transition of proliferative inflammatory atrophy to high-grade intraepithelial neoplasia and cancer in human prostate. Prostate. 2009;69:1378–1386. doi: 10.1002/pros.20992. [DOI] [PubMed] [Google Scholar]
- 59.Woenckhaus J., Fenic I. Proliferative inflammatory atrophy: a background lesion of prostate cancer? Andrologia. 2008;40:134–137. doi: 10.1111/j.1439-0272.2007.00831.x. [DOI] [PubMed] [Google Scholar]
- 60.Terracciano D., Bruzzese D., Ferro M., Autorino R., di Lorenzo G., Buonerba C., Mariano A., Macchia V., Altieri V., di Carlo A. Soluble interleukin-6 receptor to interleukin-6 (sIL6R/IL-6) ratio in serum as a predictor of high Gleason sum at radical prostatectomy. Oncol Lett. 2011;2:861–864. doi: 10.3892/ol.2011.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kwon E.M., Salinas C.A., Kolb S., Fu R., Feng Z., Stanford J.L., Ostrander E.A. Genetic polymorphisms in inflammation pathway genes and prostate cancer risk. Cancer Epidemiol Biomarkers Prev. 2011;20:923–933. doi: 10.1158/1055-9965.EPI-10-0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wegiel B., Bjartell A., Culig Z., Persson J.L. Interleukin-6 activates PI3K/Akt pathway and regulates cyclin A1 to promote prostate cancer cell survival. Int J Cancer. 2008;122:1521–1529. doi: 10.1002/ijc.23261. [DOI] [PubMed] [Google Scholar]
- 63.Nguyen D.P., Li J., Yadav S.S., Tewari A.K. Recent insights into NF-kappaB signalling pathways and the link between inflammation and prostate cancer. BJU Int. 2014;114:168–176. doi: 10.1111/bju.12488. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Examples of rare epithelial lesions in P3+Pten+/+ and P3−Pten+/+ mice. Hyperplastic foci characterized by multiple cribiform structures lined by a single layer of epithelial cells with benign cytologic features and few mitotic figures are present in P3+Pten+/+ mice at 30 and 46 weeks (4 and 8 months after induction of inflammation) but are not observed in P3−Pten+/+ mice until 60 weeks (12 months after induction of inflammation). A single lesion suggestive of PIN I, with pleomorphic cells containing enlarged nuclei, was observed in one P3+Pten+/+ mouse at 60 weeks.
Spontaneous inflammation at 4 months after induction of prostatitis is skewed toward myeloid cells. A: Percentage of leukocytes (total CD45+ cells) in prostates from mice 4 months after induction of prostatitis show increased CD8+ cells. B: Percentage of leukocytes (total CD45+ cells) in prostates from mice 4 months after induction of prostatitis show trends toward increased myeloid cells, with significantly more Gr1+CD11b− cells. C: Total lymphoid infiltrate into prostates 8 months after induction of inflammation show no differences between P3+Pten+/− and P3−Pten+/− mice but an overall decrease in T cells and an increase in B cells from the 4-month time point. D: Total myeloid infiltrate into prostates 8 months after induction of inflammation show no significant differences between P3+Pten+/− and P3−Pten+/− mice, but an overall increase in CD11b+Gr1+ cells from the 4-month time point. Data are expressed as means ± SEM (A–D). n = 4 to 5 mice each group (A and B); n = 4 mice each group (C and D). ∗P < 0.05, Student's t-test.
Expression of Il-6 mRNA is higher in prostates with greater inflammatory infiltrate, as measured by histologic analysis, at each of the examined time points (A) and compared with other cytokines at 12 months after induction of inflammation (B). Data are expressed as means ± SEM (A and B). n = 8 in 4 months 0 to 3 group (A); n = 1 in 4 months 4+ group (A); n = 4 in 8 months 0 to 3 group (A); n = 4 in 8 months 4+ group (A); n = 2 in 12 months 0 to 3 group (A); n = 7 in 12 months 4+ group (A). IFN-γ, interferon-γ; TGF-β, transforming growth factor-β; TNF-α, tumor necrosis factor-α.