

Abstract
The mitochondrial presequence protease (PreP) is a member of the pitrilysin class of metalloproteases. It degrades the mitochondrial targeting presequences of mitochondria-localized proteins as well as unstructured peptides such as amyloid-β-peptide. The specific activity of PreP is reduced in Alzheimer patients and animal models of Alzheimer disease. The loss of activity can be mimicked in vitro by exposure to oxidizing conditions, and indirect evidence suggested that inactivation was due to methionine oxidation. We performed peptide mapping analyses to elucidate the mechanism of inactivation. None of the 24 methionine residues in recombinant human PreP was oxidized. We present evidence that inactivation was due to oxidation of cysteine residues and consequent oligomerization through intermolecular disulfide bonds. The most susceptible cysteine residues to oxidation are Cys34, Cys112, and Cys119. Most, but not all, of the activity loss is restored by the reducing agent, dithiothreitol. These findings elucidate a redox mechanism for regulation of PreP and also provide a rational basis for therapeutic intervention in conditions characterized by excessive oxidation of PreP.
Keywords: Presequence protease, protein oxidation, peptide degradation, cysteine oxidation, methionine sulfoxide
Human presequence protease (hPreP), previously known as human metalloprotease 1, is a protease localized in the mitochondrial matrix that degrades peptides up to 70 amino acids [1]. It belongs to a class of metalloproteases, the pitrilysins, that also includes insulin degrading enzyme [2]. Both hPreP and insulin degrading enzyme contain an inverted zinc motif at the active site and can degrade peptides. Insulin degrading enzyme, as its name implies, can degrade insulin; in contrast, hPreP does not degrade insulin. hPreP degrades the mitochondrial targeting peptide, or presequence, after it is cleaved from the precursor protein. hPreP also degrades unstructured peptides including amyloid-β [3, 4]. The crystal structure of a substrate bound Arabidopsis thaliana PreP has been solved with the substrate trapped in a chamber formed by two domains of the protease. In the absence of substrate, electrostatic repulsive forces from acidic patches in the domains are thought to prevent closing of the chamber. A peptide enriched in basic residues, such as the presequence of a mitochondrial targeted protein, neutralizes the repulsive charges, allowing the chamber to close on the substrate. Proteolytic attack on the substrate then ensues [5].
hPreP is important in the maintenance of mitochondrial function. Mop112 is the yeast homologue of hPreP and degrades peptides generated in the mitochondrial matrix [6]. Disrupting Mop112 impairs respiratory growth on non-fermentable substrates but not on glucose. Arabidopsis thaliana has two isoforms of PreP, PreP1 and PreP2. Single knockouts do not have significant phenotypes but deletion of both causes chlorosis and mitochondrial dysfunction early in development and a 40% reduction in biomass throughout development [7, 8]. This phenotype is thought to be due to accumulation in mitochondria of amphipathic presequence peptides that disrupt mitochondrial membrane integrity and uncouple respiration [9, 10].
Reduced hPreP activity has been reported in mitochondria isolated from the brain of patients with Alzheimer disease and in Alzheimer disease mouse models [11]. The amount of hPreP in the brain was not decreased, suggesting that the decreased activity was due to conformational changes or to post-translational modifications of the enzyme. Oxidative stress occurs in neurodegenerative disorders [12, 13], creating a cellular environment that may promote oxidative inactivation of hPreP. As outlined above, a loss of hPreP activity may lead to accumulation of presequences and other peptides in mitochondria, thereby disrupting normal mitochondrial function. Exposure of purified hPreP to 500 μM hydrogen peroxide led to loss of hPreP activity [14], providing a convenient in vitro model of in vivo oxidative inactivation. In earlier studies of the oxidatively modified hPreP, we found that dithiothreitol (DTT) treatment led to a modest recovery of activity and that incubation with both DTT and methionine sulfoxide reductase A supported substantially greater recovery of activity than did DTT alone (Fig. 5A in [14]). The effect of methionine sulfoxide reductase A suggested that oxidation of methionine contributed to oxidative inactivation of hPreP. The investigations reported here were aimed at identifying the residues in hPreP that are oxidized upon exposure to hydrogen peroxide.
Experimental Procedures
Reagents and recombinant proteins
Hydrogen peroxide was Sigma H-1009 (St. Louis, MO, USA). Substrate V was #ES005 from R&D Systems (Minneapolis, MN, USA). DTT was Pierce product 20291 (Thermo Scientific, Rockford, IL, USA). HEPES as the free acid was product 101926 from MP Biomedicals (Santa Ana, CA, USA). Mallinckrodt supplied Na2HPO4 (#7914-04) and KOH (#6984 St. Louis, MO, USA) while KH2PO4 was Aldrich #342416 (St. Louis, MO, USA). MgCl2 was Fisher Scientific product M33-500 (Pittsburgh, PA, USA). Amicon supplied the10 kDa cutoff centrifuge tubes (UFC501816, EMD Millipore, Billerica, MA, USA).
Production and purification of hPreP proteins and of mouse methionine sulfoxide reductase A proteins were performed as described previously [14, 15].
Inactivation of hPreP by hydrogen peroxide
hPreP was diluted to 2.4 μM (0.27 μg/μl) with 50 mM HEPES-KOH buffer, pH 8.2, and incubated with 500 μM hydrogen peroxide for 0, 30, 60, 90, 120, or 240 min at room temperature. At each time point, 100 μl was sampled and 8 μl were immediately assayed for activity assay. The remaining 92 μl was made 5 mM in DTT to stop the oxidation process. After standing 10 min, these samples were dried in a vacuum centrifuge for subsequent peptide mapping as described below. hPreP activity was measured with substrate V as described [14]. Briefly, 0.9 μmol hPreP (1 μg; ~ 4 μl) was added to 96 μl of pre-warmed reaction buffer containing 50mM HEPES and 10 mM MgCl2, pH 8.2 in a 96-well plate kept at 37 °C. Each sample was assayed in duplicate. The reaction was started by addition of 1 μg of substrate V (0.2 μl) and the fluorescence measured at 1 s intervals for 40 s. The plate reader was a SpectraMaxGerminiEM (Molecular Devices, Sunnyvale, CA, USA) set to an excitation wavelength of 320 nm and an emission wavelength of 405 nm.
Alkylated hPreP was prepared as follows. To reduce any pre-existing disulfide bonds, 0.9 μM hPreP (1 μg/μl) in 50 mM HEPES-KOH, pH 8.2 was incubated with 5 mM DTT for 30 min at 37 °C with agitation at 300 rpm. The sample was then divided in half. One half was incubated with 20 mM iodoacetamide and the other half with buffer for 30 min at 37 °C with agitation at 300 rpm. To remove the excess iodoacetamide, samples were filtered through a 10 kDa cutoff ultrafilter and washed 6 times with the HEPES buffer. Incubation with hydrogen peroxide was then carried out as described above.
hPreP activity rescue assay by methionine sulfoxide reductase A
1.9 μM hPreP was oxidized by 500 μM hydrogen peroxide for 4 h as described above. To remove hydrogen peroxide without perturbing the thiol-disulfide status of the protein, the solution was immediately filtered through a 10k Da cutoff ultrafilter and washed 4 times with 450 μl 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.3. The hPreP concentration was determined with the Bradford assay [16] after which the hPreP was diluted to 2.4 μM (0.27 μg/μl with the phosphate buffer. hPreP was then incubated with 1 μM methionine sulfoxide reductase A or BSA and 10 mM DTT for 3 h at 37 °C. Activity was then assayed as described above. The experiment was repeated on different days and the mean and standard error of the mean (SEM) were calculated from the separate days’ results.
Reducing and non-reducing SDS-PAGE gel electrophoresis
The concentrated loading solution was 10% glycerol, 2% SDS, 0.05% bromophenol, and 50 mM Tris, pH 6.8. For non-reducing conditions, 0.9 μmol hPreP (1 μg; 4 μl) was mixed with 1 μl of loading solution and loaded. For reducing conditions, 9 volumes of loading solution were mixed with 1 volume of β-mecaptoethanol. 0.9 μmol hPreP (1 μg; 4 μl) was mixed with 1 μl of the β-mecaptoethanol containing loading solution and heated at 95°C for 10 min before loading. The samples were electrophoresed on 10–20% Tris-Glycine SDS-PAGE gels (Novagen). Proteins were visualized by staining with Coomassie Blue G250. The amount of hPreP in a band was quantitated from the fluorescence of Coomassie Blue [17], scanned with an Odyssey Infrared Imager (Li-Cor Biosciences, Lincoln, NE, USA) and processed by Odyssey Image Studio, version 3.1. Background was corrected for by subtracting the mean pixels located within 2 pixels on the left and right of the band.
Peptide mapping for methionine oxidation status
Dried samples were redisolved in 100 μl 6 M guanidine, 1 mM DTPA, 100 mM Tris, pH 8.5 buffer, and any disulfides present were reduced by incubation with 5 mM DTT for 30 min at 37°C with agitation at 300 rpm. They were then alkylated by 20 mM iodoacetamide for 30 min in the dark at 37°C with agitation at 300 rpm. Excess iodoacetamide was scavenged by addition of another 10 mM DTT followed by 5 min incubation at room temperature. Proteins were precipitated with 10% trichloroacetic acid, then washed twice with 1 ml 1:1 mixture of ethanol:ethyl acetate. The trichloroacetic acid also precipitates guanidine, but it redisolves in the ethanol:ethyl acetate. The samples were dried in the vacuum centrifuge, then dissolved in 20 μl 100 mM Tris, 1 mM DTPA, pH 8.5 and digested with 1 μg of trypsin overnight at 37° C. Solutions were then made 0.05% in trifluoroacetic acid, the concentration of trifluoroacetic acid in the HPLC solutions. (Our current protocol is to acidify to pH 2–3 with 10% trifluoroacetic acid.) Peptide mapping by HPLC-mass spectrometry was performed as described [18], with an acetonitrile gradient from 0 to 50%, developed at 1%/min. Peptides were identified both by their mass and by de novo sequencing using Agilent Masshunter software version 5 (Agilent Technologies, Santa Clara, CA, USA) and PEAKS versions 6 and 7 (Bioinformatics Solutions, Waterloo, ON, Canada).
Peptide mapping for cysteine oxidation status by double alkylation
In this procedure, hPreP was first alkylated with iodoacetamide to label cysteines with a free thiol. Then disulfide bonds were reduced and the newly reduced thiols were labeled with acrylamide. Those cysteines which had a free thiol have a mass increase of 57 Da while those that were in disulfide linkage had an increase of 71 Da, allowing calculation of the oxidized fraction of each cysteine containing peptide. ~30 μg hPreP (control or 4 h oxidized) were dried in the vacuum centrifuge and then redisolved in 100 of μl 6 M guanidine, 1 mM DTPA, 100 mM Tris, pH 8.5. The solution was made 20 mM in iodoacetamide and incubated for 30 min in the dark at 37° C with agitation at 300 rpm. Protein was precipitated with 10% trichloroacetic acid, then washed twice with 1 ml 1:1 mixture of ethanol:ethyl acetate. After drying in the vacuum centrifuge, samples were again redisolved in 100 μl 6 M guanidine, 1 mM DTPA, 100 mM Tris, pH 8.5 with 10 mM DTT and incubated for 30 min at 37° C with agitation at 300 rpm. Trichloroacetic acid precipitation and ethanol:ethyl acetate wash was repeated to remove DTT. After drying, samples were redisolved in 100 of μl 6 M guanidine, 1 mM DTPA, 100 mM Tris, pH 8.5 with of 20 mM acrylamide and incubated for 30 min in the dark at 37 °C with agitation at 300 rpm. Protein was again precipitated with 10% trichloroacetic acid, then washed twice with 1 ml 1:1 mixture of ethanol:ethyl acetate. After drying, samples were subjected to trypsin digestion and HPLC-mass spectrometric mapping as described above.
Statistical analysis
Student’s t-test with the Bonferroni correction for multiple comparisons was utilized to identify significant differences. With the Bonferroni correction, significance is accepted when the p value is less than 0.05/n, where n is the number of comparisons. The analyses were performed with PRISM software versions 5 and 6 (GraphPad Software, LaJolla, CA, USA).
Results
Determination of methionine oxidation in hPreP
Incubation of hPreP with 500 μM hydrogen peroxide caused inactivation that followed first order kinetics (Fig. 1). After 240 min incubation, activity was decreased by 60%. Aliquots were taken at 0, 30, 60, 90, 120, and 240 min for HPLC-mass spectrometric mapping to determine which, if any, of the 24 methionines in hPreP underwent an oxidation that correlated with inactivation. We were able to determine the status of all 24 methionines at each time point and found that none were significantly oxidized during the incubation. The results for the 240 min analyses are shown in Supplementary Fig. 1.
Fig. 1.
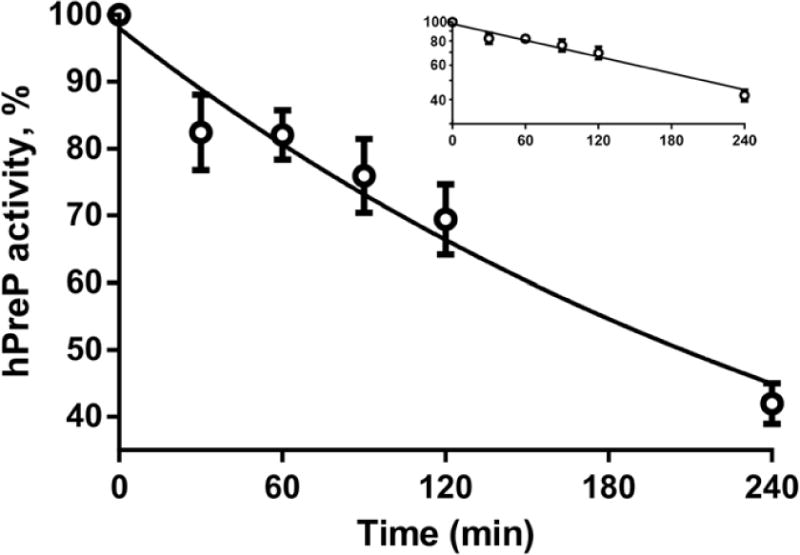
Kinetics of inactivation of hPreP by 500 μM hydrogen peroxide at pH 8.2. The plotted points are the mean and SEM for 3 experiments performed on separate days. The insert plots the same data as a semilogarithmic plot and demonstrates that inactivation is first order. The semilogarithmic plot was fit by linear regression and drawn as a solid line in both plots. Log(hPreP activity) = 1.99 – 0.00141(min), R2=0.86. The time to half-inactivation is 3.4 h.
Methionine sulfoxide reductase A can reduce the S epimer of methionine sulfoxide to methionine. Although thioredoxin is the physiological reductant that drives the reductase cycle, DTT is routinely used for convenience. In our previous study, incubation with methionine sulfoxide reductase A and DTT partially reversed the inactivation of hPreP while DTT alone had little effect [14]. Since the peptide mapping results rule out oxidation of methionines in hPreP, we reinvestigated the effect of methionine sulfoxide reductase A. We reproducibly observed reactivation of hPreP by DTT alone (Fig. 2A). However, addition of wild-type reductase had no effect. The same results were observed when the wild-type reductase was replaced by a catalytically inactive variant (C72A) or by a variant in which all 4 cysteine residues were mutated to serine (Fig. 2). These observations were readily replicated in experiments performed on 4 separate days. In addition, we prepared another batch of hPreP and observed the same effect of DTT and lack of additional effect of methionine sulfoxide reductase A in experiments performed on 3 separate days (Fig. 2B). We conclude that methionine oxidation is not responsible for inactivation of hPreP by hydrogen peroxide.
Fig. 2.
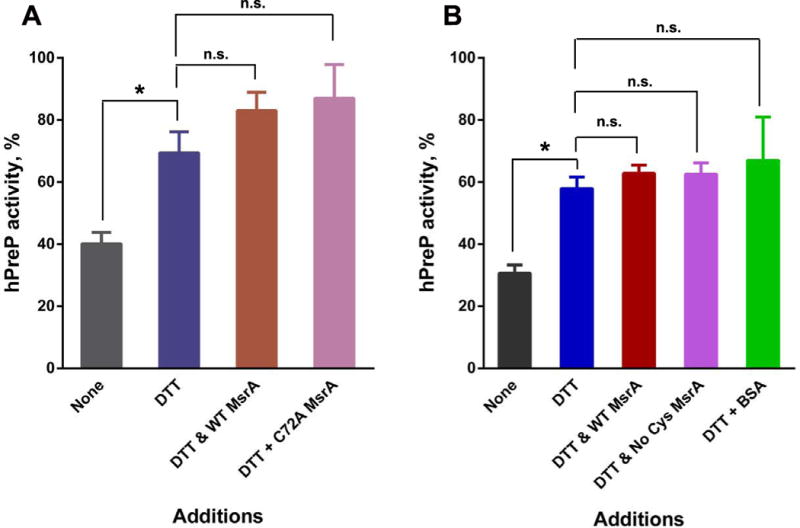
Recovery of hPreP activity by reduction. Four h oxidized hPreP, 2.4 μM, was incubated for 3 h at 37 °C with the indicated additions and then assayed for activity. The activity of unoxidized hPreP was set to 100%. When added, DTT was 10 mM and methionine sulfoxide reductase A was 1 μM. Panels A and B are results from two separate preparations of hPreP. The results are plotted as the mean and SEM with n=4 in panel A and n=3 in panel B. Comparisons were assessed by Student’s t test with the Bonferroni correction for multiple comparisons. Pairs whose p values are less than 0.05 are marked with an asterisk, and those whose p values were ≥ 0.05 are marked n.s. (not significant).
Further characterization of the inactivation reaction
Varying the pH between 6.0 and 9.0 has little effect on the rate of methionine oxidation while cysteine oxidation is distinctly increased at higher pH because it is the thiolate form of cysteine that is susceptible to oxidation by hydrogen peroxide [19]. We examined the relationship between pH and inactivation of hPreP and found that it increased with increasing pH (Fig. 3B). However, the increase in rate over the pH range of 6–9 is only about 2 fold, suggesting that the pKa of the oxidized cysteine residue(s) is high (over 9.5), which is not unusual for a folded protein [20]. The pH dependence and the effect of DTT on reactivation are consistent with oxidation of cysteine as the underlying mechanism of inactivation. Further, SDS gel electrophoresis under non-reducing conditions showed a loss of intensity of the hPreP monomer coupled with appearance of higher molecular weight forms, including some that remained in the stacking gel (Fig. 3A). The high molecular weight form was reverted to the monomer by DTT treatment, consistent with oligomerization caused by disulfide formation. Notably, the fractional loss of activity matched the fraction of protein that was oligomerized (Fig. 3B). To directly test whether cysteine oxidation was actually causing inactivation, we alkylated accessible thiols in hPreP by treatment with iodoacetamide. This form of hPreP was resistant to inactivation (Fig. 4).
Fig. 3.
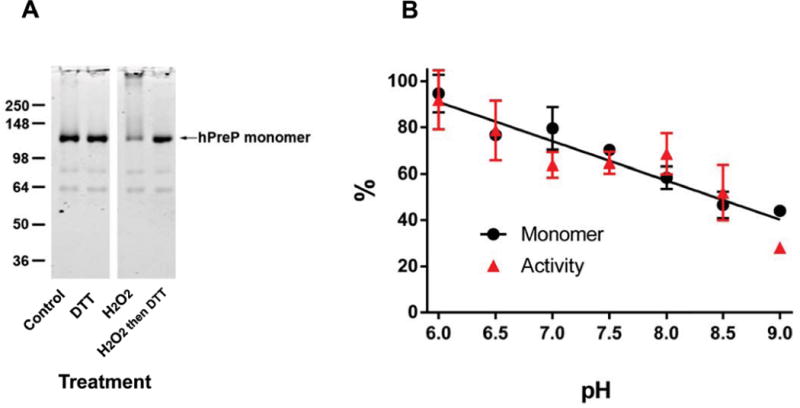
Oxidation of hPreP induces oligomerization whose extent matches the loss of activity. (A) Non-reducing SDS-PAGE gel of hPreP subjected to varying treatments. 1 μg hPreP was loaded in each lane. Oligomerization was induced by hydrogen peroxide and reversed by subsequent incubation with DTT. (B) Oligomerization and inactivation are quantitatively linked. hPreP was oxidized in 50 mM HEPES at varying pH. Regression lines fit separately to oligomerization and inactivation did not differ from each other (p=0.62) and thus a single regression line is shown.
Fig. 4.
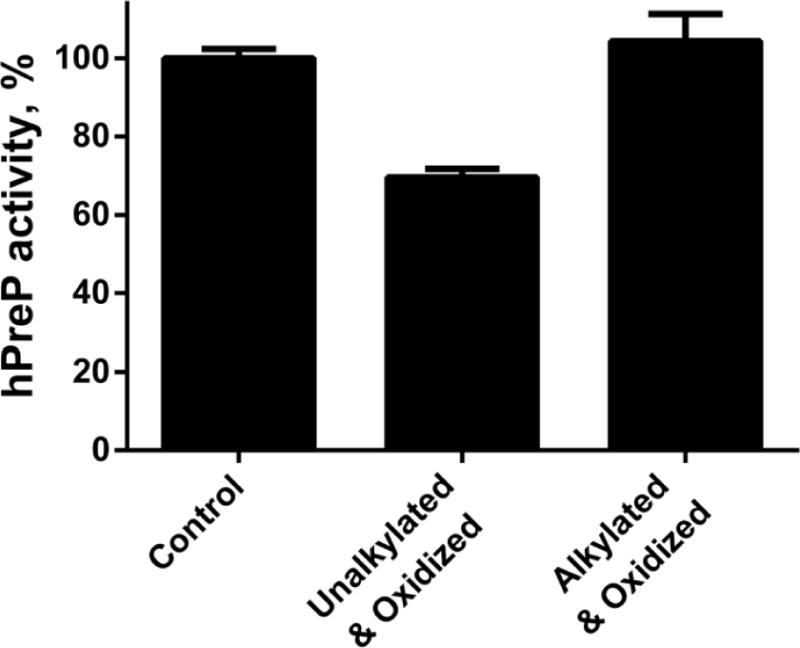
Alkylation of accessible cysteines prevents hydrogen peroxide-mediated inactivation of hPreP. Control and alkylated hPreP were incubated with 500 μM hydrogen peroxide at pH 7.4 for 4 h and then assayed for hPreP activity. Each bar plots the mean and SEM of 3 independent experiments.
Quantitation of disulfide bond formation of each cysteine residue
hPreP has 16 cysteine residues, and we were able to determine the status of all but one by HPLC-mass spectrometric peptide mapping (Fig. 5). Recovery of the peptide containing Cys588 was low and variable, so we could not assess its status. None of the 15 cysteines were in disulfide linkage in the unoxidized protein, while 4 h oxidation induced some disulfide formation by all cysteine residues except Cys454. However, the fraction forming disulfide bridges was relatively low except for Cys34, Cys112, and Cys119 for which disulfide formation was ~80%. Further, these disulfides were fully reduced by DTT (not shown). We conclude that Cys34, Cys112, and Cys119 are particularly prone to disulfide formation, leading to linkages between hPreP molecules that can account for the oligomerization observed during oxidation.
Fig. 5.
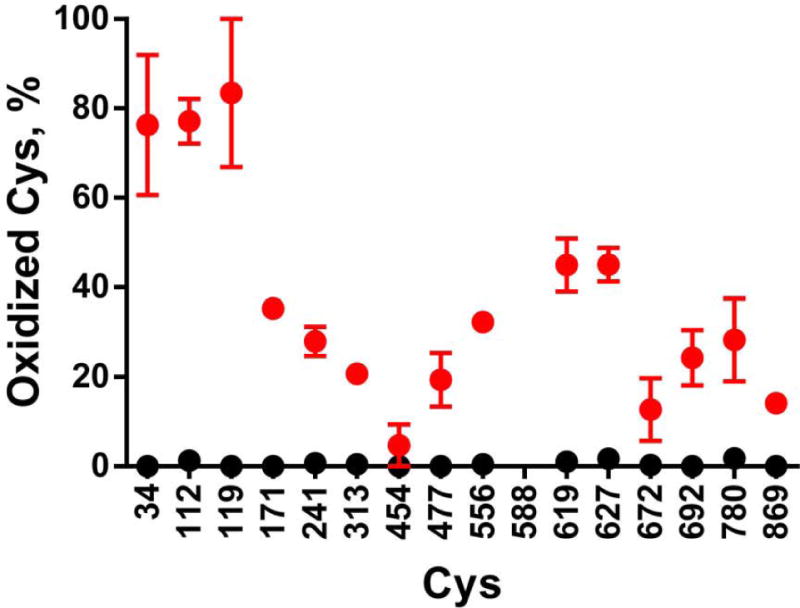
Quantitation of cysteine oxidation. hPreP was oxidized by 500 μM hydrogen peroxide for 4 h in HEPES buffer at pH 8.2. The oxidation state of each cysteine was determined by peptide mapping as described in Experimental Procedures. Black circles plot the control not exposed to hydrogen peroxide, and red circles plot the hPreP incubated with hydrogen peroxide. The points are the mean and SEM from 3 independent experiments.
Discussion
Hydrogen peroxide causes an oxidative inactivation of hPreP, although the rate of reaction is slow. Incubation with 500 μM peroxide caused a loss of ~50% activity in 4 h (Fig. 1). In our previous study, we suggested that methionine oxidation was responsible for the inactivation of hPreP, although that study provided no direct evidence for methionine oxidation [14]. The present study was undertaken to identify the specific residues that were oxidized under the conditions of the earlier study, and we therefore used 500 μM peroxide because that was the concentration employed in the earlier study.
We recognize that the use of a higher than physiological concentration of oxidant could support oxidations that effectively do not occur at lower concentrations. However, the use of experimentally convenient concentrations of oxidants has allowed elucidation of the targets of physiologically relevant oxidations. For example, when glutamine synthetase was found to undergo an in vivo oxidative inactivation, a non-physiological in vitro model system was established that consisted of 15–25 mM ascorbate, 25 μM iron, and oxygen from room air [21]. The oxidized protein was shown to be carbonylated and subsequent model studies demonstrated that carbonyl groups were derived primarily from oxidation of lysine residues to δ-aminoadipic semialdehyde [22]. Later, several laboratories established that these are indeed the major in vivo carbonyl moieties in proteins [23].
The accumulation of oxidatively modified proteins occurs during aging in many organisms, notably during the last third of lifespan [24]. As mentioned above, loss of hPreP activity has been reported in Alzheimer patients and in mouse models of Alzheimer Disease [11]. The development of Alzheimer Disease is a very slow process, but the effective study of the process requires model systems that accelerate potentially causal mechanisms. The effective study of the process requires model systems that accelerate potentially causal mechanisms. Of course, additional studies must be undertaken to determine whether the results from our current model are applicable to the in vivo situation.
Our earlier paper implicated methionine oxidation in the mechanism of inactivation of hPreP [14], while the data in our current study rule out such a mechanism. What accounts for the difference? The data implicating methionine oxidation comes from one experiment in the first publication: methionine sulfoxide reductase A partially re-activated hPreP. During the 2 years in which the present work was conducted, multiple replications of that experiment have not reproduced the earlier reported effect of methionine sulfoxide reductase A. Additional studies with mutant reductase and extensive, reproducible peptide mapping firmly establish that there is no significant oxidation of methionine residues in our system. Both laboratories worked diligently to reveal a difference in experimental procedure that could account for the dichotomy, but we could not identify such a difference. While we cannot explain the earlier results, we are confident that the extensive characterization reported in the present work rules out a role for methionine oxidation in the inactivation of hPreP.
In our earlier report we showed that mutation of the Met206 to leucine markedly increased the rate of inactivation of hPreP by hydrogen peroxide. Met206 is located within the active site of hPreP, and this methionine is conserved in PreP from plants to yeast and to mammals [14]. The increased susceptibility of Met206Leu could have been due to increased oxidation of the active site zinc-binding histidine residues as a consequence of loss of antioxidant defense provided by Met206 [25]. However, an alternative explanation is that methionine and leucine are not simply interchangeable hydrophobic residues, and recent work makes clear that the sulfur of methionine plays an important structural role in many proteins by forming a hydrophobic bond whose strength is similar to that of an electrostatic bond [26]. The analyses in this paper demonstrate that no methionine residues in hPreP are oxidized by hydrogen peroxide, favoring the structural rather than antioxidant role of Met206.
Alkylation of cysteine residues before exposure to hydrogen peroxide prevented oxidative inactivation of hPreP, supporting the conclusion that hydrogen peroxide inactivates hPreP by oxidizing cysteine residues and inducing disulfide bond formation. We found that Cys34, Cys112, and Cys119 are particularly susceptible to oxidation. Cys34, Cys112, and Cys119 are conserved among PreP sequences from mammals while only Cys 112 is conserved in the fungal and plant sequences (Fig. 6). A 2 Å resolution structure of hPreP has recently been published [27] and was deposited in the Protein Data Bank as 4L3T. We should point out that the hPreP in this structure was modified, with some lysine residues derivatized to N-dimethyl-lysine and some cysteine residues derivatized to S-(dimethylarsenic)-cysteine. These modifications will likely introduce some differences in structure compared to the unmodified protein. In addition, the crystal structure was the best fit to two co-crystallized proteins with different side chain modifications. Inspection of the structure revealed that the thiol of Cys34 is fully exposed on the surface of the protein and readily available for intermolecular disulfide formation (Fig. 6). While Cys119 was one of the cysteine residues particularly prone to disulfide formation, Cys119 is not required for oxidative inactivation of hPreP [14]. Cys112 is sited in the vicinity of the active site so that formation of a disulfide between Cys112 and any other cysteine residue is likely to interfere with substrate binding. Also, hPreP is thought to undergo a substantial conformational change at a hinge region which opens and closes the proteolytic chamber [5]. We hypothesize that disulfide formation is likely to inhibit hPreP activity by preventing proper opening and closing of the chamber.
Fig. 6.
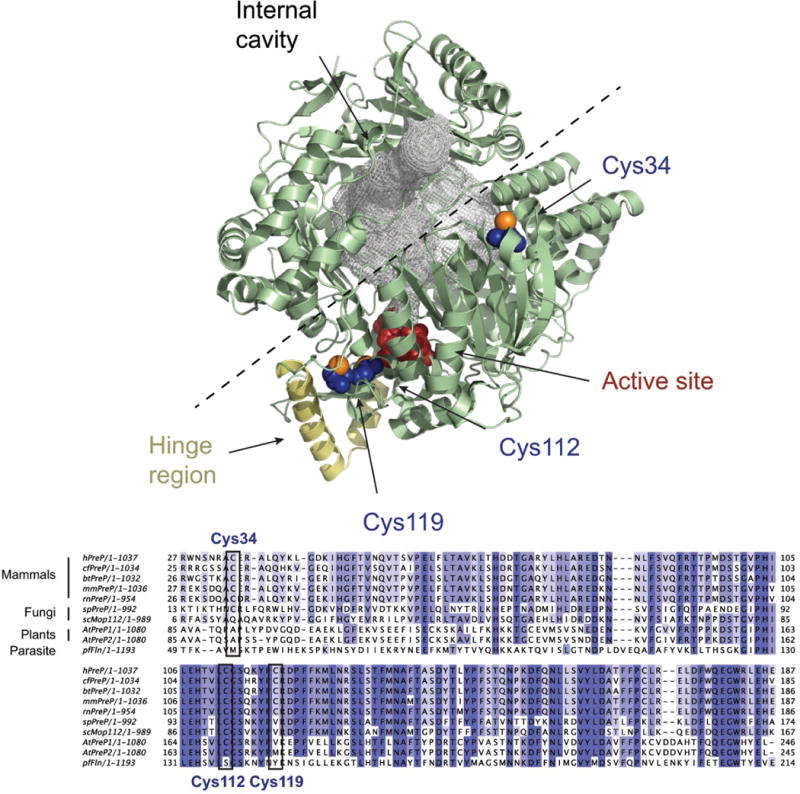
Crystal structure of hPreP. Upper panel. Overview of the hPreP structure [27] (4L3T, pale green) with the residues composing the active site shown in red and the hinge region colored in yellow. The internal cavity in hPreP is filled with a gray mesh, and the dashed line shows the proposed path of opening. The oxidation-susceptible residues Cys34, Cys112, and Cys119 are shown in blue, except for the sulfur atoms which are orange. The figure was prepared using PyMOL [30].
Lower panel. Alignment of PreP sequences from Homo sapiens (hPreP), Canis familiaris (cfPreP), Bos taurus (btPreP), Mus musculus (mmPreP), Rattus norvergicus (rnPreP), Schizosaccharomyces pombe (spPreP), S. cerevisiae (scMop112), A. thaliana (AtPreP1 and AtPreP2) and Plasmodium falciparum (pfFln). The alignment was generated with Clustal Omega [31] and is restricted to the region around Cys34 to Cys119.
Treatment of the oxidized hPreP with the reducing agent DTT reversed most of the loss of activity caused by oxidation. The residual inhibition observed in Fig. 2 may be a consequence of buried disulfide formation that is not accessible to DTT. It might also result from conformational changes induced by disulfide formation that cannot be reversed simply by reduction of the disulfide, or to oxidations that cannot be reversed by DTT. With regard to the latter possibility, we did not detect any oxidation of cysteine residues to the sulfinic or sulfonic acids, nor were any of the active site histidine residues oxidized.
Redox-mediated regulation by reversible oligomerization has been studied in detail for the 2-Cys peroxiredoxins (reviewed in [28, 29]). This important antioxidant enzyme scavenges hydrogen peroxide via a mechanism in which the active site cysteine is oxidized to the sulfenic acid. If the sulfenic acid is further oxidized to the sulfinic acid, oligomerization is induced. The oligomer loses peroxidatic activity but gains protein chaperone activity. The sulfinic acid can be reduced back to the thiol by sulferedoxin. Reduction causes dissociation of the oligomer with loss of chaperone activity and return of peroxidase activity. The finding that disulfide formation with oligomerization is the mechanistic basis of oxidative inactivation of hPreP leads us to speculate that the oligomer may, as for the peroxiredoxins, gain a novel activity. Knowledge of the mechanism of hPreP inactivation also provides a rational basis for therapeutic intervention in conditions where loss of hPreP activity is deleterious.
Supplementary Material
Highlights.
Presequence protease (PreP) is susceptible to reversible oxidative inactivation.
We determined the status of methionine and cysteine in oxidized PreP.
We detected no oxidation of methionine residues in PreP.
Inactivation is due to oligomerization mediated by intermolecular disulfide bonds.
Acknowledgments
Funding: This study was supported by the Intramural Research Program of the National Heart, Lung, and Blood Institute (JC, RLL) and by grants from the Swedish Research Council (621-2012-4713) and the Swedish Alzheimer Foundation (03-067). (to EG).
ABBREVIATIONS
- BSA
Bovine serum albumin
- DTT
Dithiothreitol
- DTPA
Diethylene triamine pentaacetic acid
- HEPES
N-(2-Hydroxyethyl)piperazine-N′-2-ethanesulfonic Acid
- hPreP
Human presequence protease
- MsrA
Methionine sulfoxide reductase A
- PreP
Presequence protease
- SEM
Standard error of the mean
- WT
Wild Type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mzhavia N, Berman YL, Qian Y, Yan L, Devi LA. Cloning, expression, and characterization of human metalloprotease 1: a novel member of the pitrilysin family of metalloendoproteases. DNA and cell biology. 1999;18:369–380. doi: 10.1089/104454999315268. [DOI] [PubMed] [Google Scholar]
- 2.Glaser E, Alikhani N. The organellar peptidasome, PreP: A journey from Arabidopsis to Alzheimer’s disease. Biochimica et Biophysica Acta (BBA) – Bioenergetics. 2010;1797:1076–1080. doi: 10.1016/j.bbabio.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 3.Falkevall A, Alikhani N, Bhushan S, Pavlov PF, Busch K, Johnson KA, Eneqvist T, Tjernberg L, Ankarcrona M, Glaser E. Degradation of the Amyloid β-Protein by the Novel Mitochondrial Peptidasome, PreP. Journal of Biological Chemistry. 2006;281:29096–29104. doi: 10.1074/jbc.M602532200. [DOI] [PubMed] [Google Scholar]
- 4.Pinho CM, Teixeira PF, Glaser E. Mitochondrial import and degradation of amyloid-beta peptide. Biochim Biophys Acta. 2014 doi: 10.1016/j.bbabio.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 5.Johnson KA, Bhushan S, Stahl A, Hallberg BM, Frohn A, Glaser E, Eneqvist T. The closed structure of presequence protease PreP forms a unique 10 000 A3 chamber for proteolysis. EMBO J. 2006;25:1977–1986. doi: 10.1038/sj.emboj.7601080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kambacheld M, Augustin S, Tatsuta T, Müller S, Langer T. Role of the Novel Metallopeptidase MoP112 and Saccharolysin for the Complete Degradation of Proteins Residing in Different Subcompartments of Mitochondria. Journal of Biological Chemistry. 2005;280:20132–20139. doi: 10.1074/jbc.M500398200. [DOI] [PubMed] [Google Scholar]
- 7.Nilsson Cederholm S, Bäckman H, Pesaresi P, Leister D, Glaser E. Deletion of an organellar peptidasome PreP affects early development in Arabidopsis thaliana. Plant Mol Biol. 2009;71:497–508. doi: 10.1007/s11103-009-9534-6. [DOI] [PubMed] [Google Scholar]
- 8.Kmiec B, Teixeira PF, Glaser E. Phenotypical consequences of expressing the dually targeted Presequence Protease, AtPreP, exclusively in mitochondria. Biochimie. 2014;100:167–170. doi: 10.1016/j.biochi.2013.12.012. [DOI] [PubMed] [Google Scholar]
- 9.Hugosson M, Andreu D, Boman HG, Glaser E. Antibacterial peptides and mitochonrial presequences affect mitochonrial coupling, respiration and protein import. European Journal of Biochemistry. 1994;223:1027–1033. doi: 10.1111/j.1432-1033.1994.tb19081.x. [DOI] [PubMed] [Google Scholar]
- 10.Nicolay K, Laterveer F, Laurens van Heerde W. Effects of amphipathic peptides, including presequences, on the functional integrity of rat liver mitochondrial membranes. J Bioenerg Biomembr. 1994;26:327–334. doi: 10.1007/BF00763104. [DOI] [PubMed] [Google Scholar]
- 11.Alikhani N, Guo L, Yan S, Du H, Pinho CM, Chen JX, Glaser E, Yan SS. Decreased Proteolytic Activity of the Mitochondrial Amyloid-β Degrading Enzyme, PreP Peptidasome, in Alzheimer’s Disease Brain Mitochondria. Journal of Alzheimer’s Disease. 2011;27:75–87. doi: 10.3233/JAD-2011-101716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giordano S, Darley-Usmar V, Zhang J. Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox Biology. 2014;2:82–90. doi: 10.1016/j.redox.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sultana R, Butterfield DA. Oxidative modification of brain proteins in Alzheimer’s disease: perspective on future studies based on results of redox proteomics studies. Journal of Alzheimer’s disease : JAD. 2013;33(Suppl 1):S243–251. doi: 10.3233/JAD-2012-129018. [DOI] [PubMed] [Google Scholar]
- 14.Filipe Teixeira P, Moreira Pinho C, Branca RM, Lehtiö J, Levine RL, Glaser E. In vitro oxidative inactivation of human presequence protease (hPreP) Free Radical Biology and Medicine. 2012;53:2188–2195. doi: 10.1016/j.freeradbiomed.2012.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim G, Cole NB, Lim JC, Zhao H, Levine RL. Dual Sites of Protein Initiation Control the Localization and Myristoylation of Methionine Sulfoxide Reductase A. Journal of Biological Chemistry. 2010;285:18085–18094. doi: 10.1074/jbc.M110.119701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 17.Luo S, Wehr NB, Levine RL. Quantitation of protein on gels and blots by infrared fluorescence of Coomassie blue and Fast Green. Anal Biochem. 2006;350:233–238. doi: 10.1016/j.ab.2005.10.048. [DOI] [PubMed] [Google Scholar]
- 18.Liu X, Lee DY, Cai S, Yu S, Shu S, Levine RL, Korn ED. Regulation of the actin-activated MgATPase activity of Acanthamoeba myosin II by phosphorylation of serine 639 in motor domain loop 2. Proc Natl Acad Sci USA. 2013;110:E23–32. doi: 10.1073/pnas.1219713110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barton JP, Packer JE, Sims RJ. Kinetics of reaction of hydrogen-peroxide with cysteine and cysteamine. J Chem Soc Perkin Trans. 1973;2:1547–1549. [Google Scholar]
- 20.Lim JC, Gruschus JM, Kim G, Berlett BS, Tjandra N, Levine RL. A Low pKa cysteine at the active site of mouse methionine sulfoxide reductase A. J Biol Chem. 2012;275:25596–25601. doi: 10.1074/jbc.M112.369116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levine RL. Oxidative modification of glutamine synthetase. II. Characterization of the ascorbate model system. J Biol Chem. 1983;258:11828–11833. [PubMed] [Google Scholar]
- 22.Amici A, Tsai L, Levine RL, Stadtman ER. Conversion of amino acid residues in proteins and amino acid homopolymers to carbonyl derivatives by metal-catalyzed reactions. J Biol Chem. 1989;264:3341–3346. [PubMed] [Google Scholar]
- 23.Requena JR, Chao CC, Levine RL, Stadtman ER. Glutamic and aminoadipic semialdehydes are the main carbonyl products of metal-catalyzed oxidation of proteins. Proc Natl Acad Sci USA. 2001;98:69–74. doi: 10.1073/pnas.011526698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levine RL, Stadtman ER. Oxidative modification of proteins during aging. Exp Gerontol. 2001;36:1495–1502. doi: 10.1016/s0531-5565(01)00135-8. [DOI] [PubMed] [Google Scholar]
- 25.Levine RL, Mosoni L, Berlett BS, Stadtman ER. Methionine residues as endogenous antioxidants in proteins. Proc Natl Acad Sci USA. 1996;93:15036–15040. doi: 10.1073/pnas.93.26.15036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valley CC, Cembran A, Perlmutter JD, Lewis AK, Labello NP, Gao J, Sachs JN. The Methionine-aromatic Motif Plays a Unique Role in Stabilizing Protein Structure. J Biol Chem. 2012;287:34979–34991. doi: 10.1074/jbc.M112.374504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.King JV, Liang WG, Scherpelz KP, Schilling AB, Meredith SC, Tang WJ. Molecular Basis of Substrate Recognition and Degradation by Human Presequence Protease. Structure. 2014;22:996–1007. doi: 10.1016/j.str.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wood ZA, Schroder E, Robin Harris J, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends in biochemical sciences. 2003;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 29.Barranco-Medina S, Lazaro JJ, Dietz KJ. The oligomeric conformation of peroxiredoxins links redox state to function. FEBS Lett. 2009;583:1809–1816. doi: 10.1016/j.febslet.2009.05.029. [DOI] [PubMed] [Google Scholar]
- 30.Schrodinger LLC. The PyMOL Molecular Graphics System, Version 1.3r1. 2010 [Google Scholar]
- 31.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular systems biology. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.