

Chronic neutrophilic leukemia (CNL) and atypical chronic myeloid leukemia (aCML) are rare entities grouped into the World Health Organisation (WHO) categories myeloproliferative and myelodysplastic/myeloproliferative neoplasms (MPN and MDS/MPN overlap), respectively. According to the WHO 2008 classification,1 both entities are characterized by leukocytosis and a hypercellular bone marrow, predominantly consisting of granulocytic cells.
Chronic neutrophilic leukemia is diagnosed by the expansion of neutrophils in the peripheral blood, the exclusion of an elevated blast count, and hepatosplenomegaly. In contrast to other MPNs, before 2013, no molecular marker was known to prove clonality or could shed light on the molecular nature of the disease. Therefore, CNL had been diagnosed by a number of exclusion criteria eliminating evidence for other neoplasms or myelodysplastic syndromes.
Atypical CML is diagnosed by a similar approach, since diagnosis according to WHO has featured an increased number of neutrophil precursors, a defined threshold of blasts and monocytes, and dysplasia in the granulocytic lineage. A distinctive feature for differential diagnosis of CNL and aCML is the proportion of immature neutrophils (≥10% in aCML and <10% in CNL). As in CNL, other neoplasms and myelodysplastic syndromes should be excluded.
In addition, also chronic myelomonocytic leukemia (CMML) shares several of these characteristics and, therefore, needs to be discriminated from the other two entities, especially by the absolute number of monocytes for clinical decision making.
In the last three years, important markers have been identified for the diagnosis and differential diagnosis in these entities. ASXL1, SRSF2, and TET2 were found to be frequently mutated in CMML.2,3 SETBP1 was identified to be frequently mutated in aCML, which was shown to co-occur frequently with mutations in ASXL1 and CBL.4,5 CSF3R mutations were found to associate with CNL and aCML.6,7
In Philadelphia negative MPNs, cytogenetic abnormalities occur, but the frequency differs and no specific abnormality has been defined in the different entities so far.8 Therefore, the aim of our study was to determine the frequencies of the new armamentarium of genes, i.e. ASXL1, CBL, CSF3R, SETBP1, SRSF2, and TET2 mutations in CNL, aCML, and CMML, to help guide the diagnosis and clinical decisions of these three, in part overlapping, entities. A total of 218 patients were diagnosed according to the WHO 2008 criteria, including 14 cases with CNL, 58 with aCML, and 146 with CMML (for more clinical details see Online Supplementary Table S1). Cytogenetics was available in 211 (97%) cases. In all cases, BCR-ABL1 was excluded by RT-PCR and/or FISH, and JAK2V617F mutation was analyzed by melting curve analyses, as were JAK2 exon 12 and MPL mutations in JAK2wild-type (wt) patients. CALR mutations were analyzed in JAK2wt CNL and aCML patients by Sanger sequencing. Presence of PDGFR-rearrangements was excluded in CNL by expression analyses of PDGFRA and PDGFRB. In all patients the mutational hot spot regions of ASXL1, CBL, CSF3R, SETBP1, and SRSF2 were analyzed by Sanger sequencing. The complete coding region of TET2 was analyzed by next generation sequencing in 217 of 218 cases. For more details see Online Supplementary Appendix.
Cytogenetic aberrations were detected in 54 of 211 cases (26%); the most frequent were trisomie 8 (n=14), deletion of the Y chromosome (n=7), del(20q) (n=3), and i(17)(q10) (n=3). However, there was no association to one of these entities.
Mutational analyses showed that ASXL1 was frequently mutated in all three diseases, resulting in mutation frequencies of 57% in CNL (8 of 14), 66% in aCML (38 of 58), and 45% in CMML (66 of 146), respectively (Figures 1 and 2). A similar frequency of ASXL1 mutations has previously been published in CMML.9 However, the frequency in aCML was higher than the 23% reported by Piazza et al.5 This finding of frequent appearance in CNL was surprising since little is known about clonality markers in addition to CSF3R in CNL. Furthermore, in CMML, MDS, and also PMF there is evidence that mutations in ASXL1 provide prognostic information, with faster leukemic transformation.9–12 CBL mutations clustered mostly in CMML patients (21%, 31 of 146), were less frequent in aCML (10%, 6 of 58), and not found in CNL (0 of 14), matching reported data.13 In contrast, CSF3R was often mutated in CNL patients (43%, 6 of 14), but rarely in aCML and CMML cases, with only 2 patients each harboring a CSF3R mutation (3%, 2 of 58; 1%, 2 of 146). However, these 4 cases with CSF3R mutation in aCML and CMML showed neutrophil counts below 80% and increased monocyte numbers above 1000/μL in CMML cases and more than 10% neutrophilic precursors in aCML. Although a higher mutation frequency was reported for CNL patients, the rare occurrence of CSF3R mutations in aCML and CMML is in accordance with other reports.6,7 Two mutation types have been identified in CSF3R, the membrane proximal mutations and truncating mutations.6,7 While the membrane proximal mutations are mostly missense mutations in exon 14, with p.Thr618Ile as most prominent representative, the truncating mutations are mostly frameshift and nonsense mutation in exon 17. Maxson et al. demonstrated in cell culture experiments that membrane proximal mutations are sensitive to JAK inhibitors while truncating mutations are more sensitive to SRC kinase inhibitors.6 This might indicate a potential therapy for these patients.14 Surveying the two mutation types of CSF3R showed that, in our cohort, 4 cases carried a membrane proximal and 2 cases a truncation mutation, while 4 cases were affected by both types. There was no relation of any type to any morphological entity (Online Supplementary Figure S1). CSF3R mutations were thus significantly associated with CNL (P<0.001). On the other hand, mutations in SETBP1 were also differentially distributed within the three entities (P<0.001) entities and correlated to aCML, where 33% (19 of 58) patients were SETBP1 mutated, while in CNL and CMML the mutation frequencies were lower with 14% (2 of 14) and 6% (9 of 146), respectively, in line with other published cohorts.6,7,14 SRSF2 mutations were detected at a high frequency within the CMML group (51%, 74 of 146), as described previously.2,3 Here we could show that a nearly as high proportion of SRSF2 mutations was observed in aCML patients (40%, 23 of 58), and also a notable number of CNL patients was SRSF2 mutated (21%, 4 of 14). Although SRSF2 was mutated in all three entities, this molecular marker was distributed differentially and associated mostly with CMML (P=0.06). This was even more prominent in combination with mutations in TET2. TET2 was most frequently mutated in CMML cases (59%, 86 of 146), followed by aCML cases (41%, 24 of 58) and also in CNL (29%, 4 of 14), fitting the already reported frequencies for CMML or aCML.15 Both mutated TET2 alone and concomitant mutations with SRSF2 associated significantly with CMML (P=0.012 and P=0.004, respectively). Remarkably, in CNL, a concomitant detection of mutated SRSF2 and TET2 was not observed in any case; however, this relationship was not statistically significant.
Figure 1.
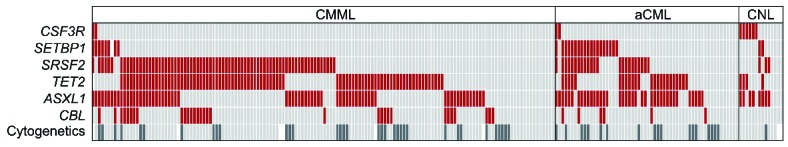
Mutational screening of CNL, aCML, and CMML. Alignment of gene mutations, cytogenetics, and entity information. Each column represents one of the 218 patients analyzed for CSF3R, SETBP1, SRSF2, TET2, ASXL1, CBL, and karyotype (shown in rows). Upper rows: red: mutated gene, light gray: non-mutated gene. Cytogenetics: dark gray: aberrant karyotype (n=54), light gray: normal karyotype (n=157). White: no data available.
Figure 2.
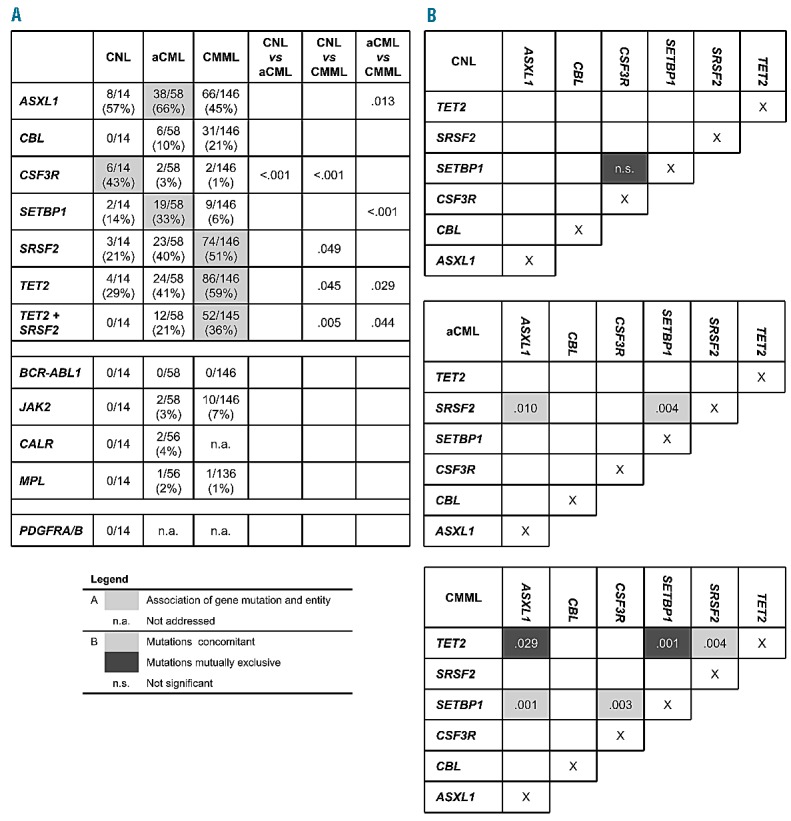
Association of gene mutations. (A) Table showing the mutation frequencies of the analyzed genes for each entity separately. Significant associations are filled in gray. (B) Scheme showing association of gene mutations within each entity. Positive association (concomitant): light gray, negative association (mutually exclusive): dark gray. P-values for significant associations are given; P<0.05 was considered significant.
Focusing on co-occurrence of gene mutations showed in CNL patients an equal distribution of mutated genes, without any significant co-occurrence of gene mutations (Figures 1 and 2). However, although not significant, SETBP1 and CSF3R mutations were mutually exclusive in CNL patients (0 of 6), while 3 of the 4 CSF3R mutated cases (75%) within the two other entities showed an additional SETBP1 mutation. In aCML patients, this was different. In this entity SETBP1 mutations were more often associated with SRSF2 mutations (P=0.004). Additionally, SRSF2 mutations also often co-occurred with mutated ASXL1 (P=0.010). Even more associations were found in CMML patients, where mutated TET2 and mutated SRSF2, as well as mutated SETBP1 and ASXL1, occurred more frequently together (P=0.004 and P=0.001, respectively). In contrast, TET2 and ASXL1 as well as TET2 and SETBP1 rarely showed co-occurring mutations in CMML (P=0.029 and P=0.001, respectively). All these correlations were also analyzed and confirmed in the total cohort (n=218) (Online Supplementary Figure S2). Looking at clinical data in these three entities regarding differences in cases with wild-type or mutated marker genes showed that CNL patients carrying a CSF3R mutation were more often male (5 of 6 vs. 2 of 8 CSF3Rwt). In aCML, SETBP1 mutated patients showed a higher hemoglobin level compared to SETBP1wt patients (12.0 vs. 9.9 g/dL; P=0.016). Comparing CMML patients with combined TET2 and SRSF2 mutation (TET2mut/SRSF2mut) with patients having either no mutation (TET2wt/SRSF2wt) or only one mutation in these two genes (TET2mut/SRSF2wt or TET2wt/SRSF2mut) showed that patients with TET2 and SRSF2 mutations had higher white blood cell counts (16.2 vs. 13.0 ×109/L; P=0.013), a less pronounced monocytosis (4750 vs. 5382/μL; P=0.008), and were more often male (P=0.043) (Online Supplementary Table S3).
In conclusion, the mutational landscape of ASXL1, CBL, CSF3R, SETBP1, SRSF2, and TET2 shows some common features, but also indicates characteristic and individual molecular patterns in CNL, aCML, and CMML. Taking the new mutations into account, mutational analyses of JAK2, CALR, and MPL should be considered when there is an increase in polymorphonuclear leukocytes, to strengthen the diagnosis of MPN. Alterations in CSF3R could suggest a diagnosis of CNL, while in SETBP1 mutations could suggest a diagnosis of aCML. On the other hand, an ongoing monocytosis would direct the mutational analyses to SRSF2 and CBL that would rather indicate CMML in mutated cases. Independently, the investigation of ASXL1 and TET2 as frequently mutated genes in all entities could help to prove clonality and to distinguish malignant diseases from reactive changes. Furthermore, mutation in ASXL1 is a negative prognostic marker in AML, MDS, and PMF and should, therefore, be tested for its prognostic impact in MPN.
Acknowledgments
We thank all clinicians for sending samples to our laboratory for diagnostic purposes and for providing clinical information and follow-up data. In addition, we would like to thank all the co-workers at the MLL Munich Leukemia Laboratory, especially the technical assistance of Rabea Konietschke and Carina Schrauder, who contributed to sequencing analyses in this study.
Footnotes
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed Lyon: International Agency for Research on Cancer (IARC), 2008. [Google Scholar]
- 2.Meggendorfer M, Roller A, Haferlach T, Eder C, Dicker F, Grossmann V, et al. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML). Blood. 2012;120(15):3080–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478(7367):64–9. [DOI] [PubMed] [Google Scholar]
- 4.Meggendorfer M, Bacher U, Alpermann T, Haferlach C, Kern W, Gambacorti-Passerini C, et al. SETBP1 mutations occur in 9% of MDS/MPN and in 4% of MPN cases and are strongly associated with atypical CML, monosomy 7, isochromosome i(17)(q10), ASXL1 and CBL mutations. Leukemia. 2013;27(9):1852–60. [DOI] [PubMed] [Google Scholar]
- 5.Piazza R, Valletta S, Winkelmann N, Redaelli S, Spinelli R, Pirola A, et al. Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat Genet. 2013;45(1):18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maxson JE, Gotlib J, Pollyea DA, Fleischman AG, Agarwal A, Eide CA, et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med. 2013;368(19):1781–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pardanani A, Lasho TL, Laborde RR, Elliott MA, Hanson CA, Knudson RA, et al. CSF3R T618I is a highly prevalent and specific mutation in chronic neutrophilic leukemia. Leukemia. 2013;27(9):1870–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bacher U, Haferlach T, Kern W, Hiddemann W, Schnittger S, Schoch C. Conventional cytogenetics of myeloproliferative diseases other than CML contribute valid information. Ann Hematol. 2005;84(4):250–7. [DOI] [PubMed] [Google Scholar]
- 9.Gelsi-Boyer V, Brecqueville M, Devillier R, Murati A, Mozziconacci MJ, Birnbaum D. Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. J Hematol Oncol. 2012;512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Itzykson R, Kosmider O, Renneville A, Gelsi-Boyer V, Meggendorfer M, Morabito M, et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol. 2013;31(19):2428–36. [DOI] [PubMed] [Google Scholar]
- 11.Thol F, Friesen I, Damm F, Yun H, Weissinger EM, Krauter J, et al. Prognostic significance of ASXL1 mutations in patients with myelodysplastic syndromes. J Clin Oncol. 2011;29(18):2499–506. [DOI] [PubMed] [Google Scholar]
- 12.Vannucchi AM, Lasho TL, Guglielmelli P, Biamonte F, Pardanani A, Pereira A, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013;27(9):1861–9. [DOI] [PubMed] [Google Scholar]
- 13.Reiter A, Invernizzi R, Cross NC, Cazzola M. Molecular basis of myelodysplastic/myeloproliferative neoplasms. Haematologica. 2009;94(12):1634–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gotlib J, Maxson JE, George TI, Tyner JW. The new genetics of chronic neutrophilic leukemia and atypical CML: implications for diagnosis and treatment. Blood. 2013;122(10):1707–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kohlmann A, Grossmann V, Klein HU, Schindela S, Weiss T, Kazak B, et al. Next-Generation Sequencing Technology Reveals a Characteristic Pattern of Molecular Mutations in 72.8% of Chronic Myelomonocytic Leukemia by Detecting Frequent Alterations in TET2, CBL, RAS, and RUNX1. J Clin Oncol. 2010;28(24):3858–65. [DOI] [PubMed] [Google Scholar]